
Metal–organic framework-derived nickel phosphides as efficient electrocatalysts toward sustainable hydrogen generation from water splitting†
Abstract
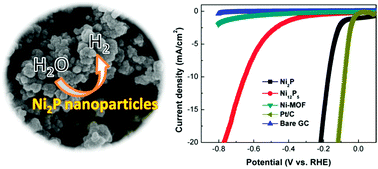
Developing robust earth-abundant electrocatalysts for the hydrogen evolution reaction (HER) is an ongoing scientific challenge. The cheap and active metal phosphides have emerged as new candidates for electrochemical HER. Herein, we report on the scalable synthesis of nickel phosphides (Ni2P and Ni12P5) via directly phosphatizing a Ni-based metal–organic framework (MOF) for the first time. The MOF-derived Ni2P nanoparticles exhibited high-performance for electrochemical HER, as manifested by a low overpotential and a large cathodic current density.
- This article is part of the themed collection: Nanoscience and nanotechnology in electrochemistry
 Please wait while we load your content...
Please wait while we load your content...

