
Growth pathways of silver nanoplates in kinetically controlled synthesis: bimodal versus unimodal growth†
Abstract
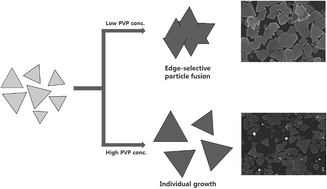
Silver nanoplates were synthesized by reducing silver nitrate (AgNO3) with poly(vinyl pyrrolidone) (PVP) in N,N-dimethylformamide (DMF). In the synthesis, the hydroxyl end groups of the PVP served as a mild reductant in the kinetically controlled synthesis, and DMF played a critical role as a reaction medium (solvent) for the formation and growth of the nanoplates. In the present study, the growth of the nanoplates proceeded along different pathways, as evidenced by variations in the shape evolution, depending on the PVP to AgNO3 weight ratio. When the concentration of PVP is below a certain value, a large number of small nanoplates with different sizes were initially formed and then fused together along their lateral planes, leading to the formation of a secondary large nanoplate (bimodal particle growth). When the PVP concentration became higher and the surface capping was enhanced, the small nanoplates continued to grow individually without fusing (unimodal particle growth). This study not only advanced our understanding of the role played by PVP in the kinetically controlled synthetic reaction but also allows us to produce Ag nanoplates with a high aspect ratio in a single step.
 Please wait while we load your content...
Please wait while we load your content...

