
Thermally accessible triplet state of π-nucleophiles does exist. Evidence from first principles study of ethylene interaction with copper species†
Sergey V. Bondarchuk *a and
Boris F. Minaevab
*a and
Boris F. Minaevab
aDepartment of Organic Chemistry, Bogdan Khmelnitsky Cherkasy National University, blvd. Shevchenko 81, 18031 Cherkasy, Ukraine. E-mail: bondchem@cdu.edu.ua; Fax: +380-472-37-21-42; Tel: +380-472-37-65-76
bDepartment of Theoretical Chemistry and Biochemistry, Royal Institute of Technology, AlbaNova, S-106 91 Stockholm, Sweden
First published on 8th January 2015
Abstract
Three different models of ethylene interaction with copper species, namely, the Cu(100) surface, odd-numbered copper clusters C2H4/Cun (where n = 3, 7, 11, 15, 17, 19, 21, 25 and 27) and atomic copper C2H4/Cu were studied theoretically. It was found that the ethylene molecule possesses three different types of bonding depending on the presence of the unpaired spin on the reacting copper atom. These bonding structures demonstrate different types of band gap (bulk) or SOMO–LUMO gaps (cluster/atom), where SOMO stands for the singly occupied and LUMO means the lowest unoccupied molecular orbitals of the copper species. The obtained results are in good agreement with the previous experimental and computational results on the structural, spectral and energetic properties of the studied species. The bulk copper and sub-nanosized clusters (n > 7) build up the mono-π-bonded ground state complexes with ethylene where the latter species possesses the C2v symmetry. The single-atom complex C2H4/Cu forms the CS-symmetrical ground state ![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 2A′ and the excited 2B2 and 4B state complexes of the C2v and C2 symmetry, respectively. The 2A′ state complex is mono-σ-bonded and involves the singlet ethylene moiety. The more tightly bound excited 2B2 complex has the di-σ-bonded structure and corresponds to the triplet ethylene. The adiabatic energy difference between the 2B2 and 2A′ states is equal to 10.8 kcal mol−1 and can be ascribed to the singlet–triplet splitting of the ethylene moiety interacting with copper. The QTAIM analysis supports the coordination type of the Cu–C bonds in all the studied complexes. Formation of the C2H4/Cu(100), C2H4/Cun and C2H4/Cu species is in accord with the well-known Dewar–Chatt–Duncanson model, in such a way that the opposing σ-donation step yields the ground state complex (2A′), while the subsequent more expensive supporting π*-back donation step provides the excited 2B2 state complex. In the present paper we have developed a computational procedure to optimize the latter complex.
2A′ and the excited 2B2 and 4B state complexes of the C2v and C2 symmetry, respectively. The 2A′ state complex is mono-σ-bonded and involves the singlet ethylene moiety. The more tightly bound excited 2B2 complex has the di-σ-bonded structure and corresponds to the triplet ethylene. The adiabatic energy difference between the 2B2 and 2A′ states is equal to 10.8 kcal mol−1 and can be ascribed to the singlet–triplet splitting of the ethylene moiety interacting with copper. The QTAIM analysis supports the coordination type of the Cu–C bonds in all the studied complexes. Formation of the C2H4/Cu(100), C2H4/Cun and C2H4/Cu species is in accord with the well-known Dewar–Chatt–Duncanson model, in such a way that the opposing σ-donation step yields the ground state complex (2A′), while the subsequent more expensive supporting π*-back donation step provides the excited 2B2 state complex. In the present paper we have developed a computational procedure to optimize the latter complex.
1. Introduction
All chemical reactions are usually divided into two most common types – thermal and photochemical processes. The photochemical reactions are considered as those proceeding via the excited electronic state reactants, while the thermal ones are assumed to involve only the ground electronic state. Even vibrationally “hot” ground states of typical π-nucleophiles (alkenes, alkynes and alkadienes) lie much lower than the first vibrational level of the first excited (triplet) state.1 Indeed, according to the Boltzmann distribution, a noticeable occupation of the first excited (triplet) state at 298 K occurs only when the singlet–triplet energy splitting is lower than 1 eV;1 therefore, the latter value may be specified as the energy cutoff level. This means that the excited states, which appear below 1 eV could be involved in principle into the thermal “dark” reactions at ambient temperature.2Our recent B3LYP/6-31+G(d,p) calculations for a number of π-nucleophiles suggest that the singlet–triplet (S–T) energy splitting (ΔT–S) values are about 2 eV (for alkadienes), 3 eV (for alkenes) and about 4 eV (for alkynes).3a,b In contrast, the ΔT–S values for the aryl cations rarely exceed 1 eV and in a number of cases have negative values as it follows from our recent B3LYP/6-311++G(2d,2p) calculations of 25 various mono-, di-, three substituted phenyl cations, as well as their ortho-, meta-, and para-substituted derivatives.3c Thus, chemical reactions involving these reactive intermediates are difficult to ascribe to a specific type, at least, on the basis of the above criterion unsatisfied. The thermal and photochemical classification is only useful for those compounds, which have energetically well separated electronic S–T states. This does not touch the particular experimental method of the reaction initiation but only the most useful way in terms of the above general classification.
Earlier, we have proposed that the radical/electrophilic addition to a multiple C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or C
C or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond can involve the triplet state of the corresponding π-nucleophile.3a This was a mere qualitative proposal based on the possibility of the triplet state formation due to the exchange interaction with a paramagnetic species4 – the aryl radical, cation or the catalyst (Cu2+, Fe2+ ions, etc.). These catalysts are used in the Meerwein addition to the double bond and other similar processes.5 Later on, we have shown that the triplet states of π-nucleophiles are not directly involved into the reaction at a particular kinetic stage but contribute in some non-explicit way.3b The results revealed a multireference nature of the transition state (TS) of the nucleophile–electrophile addition reaction with a significant contribution of the so-called “double–triplet” singlet state electronic configuration. This configuration is arising from the combination of two triplet reagents (electrophile and nucleophile) which have the total zero spin in the collision complex and participates in configuration interaction (CI) mixing with other singlet states including the ground closed-shell wavefunction of the whole reacting system. In the TS region the CI mixing of such zero-spin states produces an avoiding crossing and essential deformation of the TS open-shell wavefunction.6 This formal approach has proven itself to be a quite convenient way for the barrier height calculations.3b
C bond can involve the triplet state of the corresponding π-nucleophile.3a This was a mere qualitative proposal based on the possibility of the triplet state formation due to the exchange interaction with a paramagnetic species4 – the aryl radical, cation or the catalyst (Cu2+, Fe2+ ions, etc.). These catalysts are used in the Meerwein addition to the double bond and other similar processes.5 Later on, we have shown that the triplet states of π-nucleophiles are not directly involved into the reaction at a particular kinetic stage but contribute in some non-explicit way.3b The results revealed a multireference nature of the transition state (TS) of the nucleophile–electrophile addition reaction with a significant contribution of the so-called “double–triplet” singlet state electronic configuration. This configuration is arising from the combination of two triplet reagents (electrophile and nucleophile) which have the total zero spin in the collision complex and participates in configuration interaction (CI) mixing with other singlet states including the ground closed-shell wavefunction of the whole reacting system. In the TS region the CI mixing of such zero-spin states produces an avoiding crossing and essential deformation of the TS open-shell wavefunction.6 This formal approach has proven itself to be a quite convenient way for the barrier height calculations.3b
Despite the fact that a huge body of recent literature on the ethylene interaction with the transition metals and other surfaces is presented,7,8 we were surprised to find no data so far regarding the ethylene triplet state formation on interaction with surfaces, except one for ethylene8a and one for acetylene.8b Meanwhile, it is a rather obvious conclusion arising upon account of the exchange mixing between the singlet and triplet states induced by an external spin. This general mechanism was first proposed by Hojitink4a and further developed by Chiu4b for the paramagnetic species (doublets, triplets, etc.). In the case of the transition metal atom or ion as an external paramagnetic species, the above mechanism can be represented in terms of the well-known “opposing σ-donation and supporting π*-backdonation” or the Dewar–Chatt–Duncanson (DCD) model.7g,k,x,9 This can be illustrated schematically by Fig. 1.
 | ||
Fig. 1 The “opposing σ-donation” (a) and “supporting π*-backdonation” (b) of the electron as initiation of the S0 ![[long arrow, wavy then straight]](https://www.rsc.org/images/entities/char_e0f6.gif) T1 transition in the ethylene molecule; D, S and T denote the doublet, singlet and triplet spin state of the copper and ethylene moieties of the whole reacting system, respectively. T1 transition in the ethylene molecule; D, S and T denote the doublet, singlet and triplet spin state of the copper and ethylene moieties of the whole reacting system, respectively. | ||
The aforementioned exchange mixing should cause decrease of the singlet–triplet energy splitting (ΔT–S) of the ethylene molecule. According to the recent high-level computations using the CCSD and Monte Carlo methods, the ΔT–S value for ethylene was found to be 65.8 and 66.0 kcal mol−1, respectively;10 this is in a good agreement with our recent B3LYP/6-31+G(d,p) results (60.1 kcal mol−1)3b being even closer to the experimental value (58 ± 3 kcal mol−1).10b At the same time, upon interaction with copper species, the ΔT–S value should be expected to fall down below 1 eV allowing the triplet state of ethylene to be accessible under the thermal activation. This means that the transition metal can act as a catalyst, which opens an additional reaction channel (the triplet one) leading to lowering the total activation energy.3a,b
To check this supposition one should calculate the C2H4/Cu complexes possessing the spin population which corresponds to combinations of the 1 and 3 spin multiplicity inside the ethylene moiety in Fig. 1 being the total doublet state. The first of them is the ground state complex, while the second (Fig. 1, combination 3) should be considered as an excited state when an electron passes from the π(b3u) molecular orbital (MO) to the π*(b2g)-MO of ethylene. To form a stable complex where the ethylene moiety possess the singlet state, the copper atom must spin polarize the π(b3u)-MO and to pair with one of the spins appeared at the nearest carbon atom. Thus, a polarized non-zero spin population is expected on the carbon atom, which is not bound with copper (Fig. 1). In the triplet complex the both spin combinations between the copper and carbon atoms are spin-polarized. Thus, the two covalent types of bonding are expected to be formed in this case and the residual spin population should appear mainly on the carbon atoms (Fig. 1).
It is worthwhile noting that the calculations of such complexes are faced with the known problem of the isoelectronic systems. For instance, the following systems: C2H4/Cu0, C2H4˙−/Cu+, C2H42−/Cu2+ are indistinguishable in terms of single-reference DFT. Therefore, one should keep this point in mind and monitor the final atomic charges on the copper and ethylene moieties. In this paper we have applied three models corresponding to ethylene interaction with the bulk copper, sub-nanosized clusters Cun (n = 3, 7, 11, 15, 17, 19, 21, 25 and 27) and, finally, with atomic copper. These are depicted as C2H4/Cu(100), C2H4/Cun and C2H4/Cu, respectively. Such approach, which includes three interaction models, was used to verify a simple atomistic model as reliable one for description of realistic interaction of the ethylene molecule with fine copper particles (catalyst).
2. Computational details
2.1 Periodic DFT calculations
The modeling of ethylene interaction with the bulk copper was performed using Cambridge Serial Total Energy Package (CASTEP) module11 implemented in Materials Studio 5.5 program suite.12 Exchange and correlation interactions were described using the generalized gradient approximation (GGA) approach with the functional parameterized by Perdew–Burke–Ernzerhof (PBE).13 Ultrasoft pseudopotential was used to describe the electron–core interactions and its representation was performed in a reciprocal space. For geometry optimization, the electronic wave functions were expanded in a plane wave basis set with an energy cutoff of 300 eV. Then a more extended basis set (with 400 eV energy cutoff) was used for a single-point energy evaluation. Such accuracy is enough for an adequate description of similar adsorbate–adsorbent system.7g,14 A Monkhorst–Pack k-point sampling scheme with a 5 × 5 × 1 k-point grid for supercell was specified during the geometry optimization, while the SCF tolerance was set to 1 × 10−6 eV per atom.To model the surface, a copper face-centered cubic crystal of the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group (a = 3.615 Å) was built. Thereafter, the Cu(100) surface was cleaved and a four-layer slice was extracted and combined into a 2 × 2 periodic slab. This surface is completely enough to sufficiently accommodate the ethylene molecule. A 15 angstrom vacuum slab was then added to avoid unphysical interactions between the neighboring layers. Thus, the final supercell was obtained with the following dimensions: a = b = 5.112 Å and c = 22.377 Å, respectively. Nuclear positions of the copper atoms in two deeper layers were constrained, while the upper layers of the surface was allowed to relax. The ethylene molecule was aligned in atop orientation.
m space group (a = 3.615 Å) was built. Thereafter, the Cu(100) surface was cleaved and a four-layer slice was extracted and combined into a 2 × 2 periodic slab. This surface is completely enough to sufficiently accommodate the ethylene molecule. A 15 angstrom vacuum slab was then added to avoid unphysical interactions between the neighboring layers. Thus, the final supercell was obtained with the following dimensions: a = b = 5.112 Å and c = 22.377 Å, respectively. Nuclear positions of the copper atoms in two deeper layers were constrained, while the upper layers of the surface was allowed to relax. The ethylene molecule was aligned in atop orientation.
To take into account the long-range electron correlations, the Tkatchenko–Scheffler scheme was applied during the single-point energy calculation.15 This presents an accurate nonempirical method to obtain molecular C6 coefficients from the ground-state electron density and reference values for the free atoms operating with polarizability and volume.15 Thus, the C6 coefficients are calculated as the following:
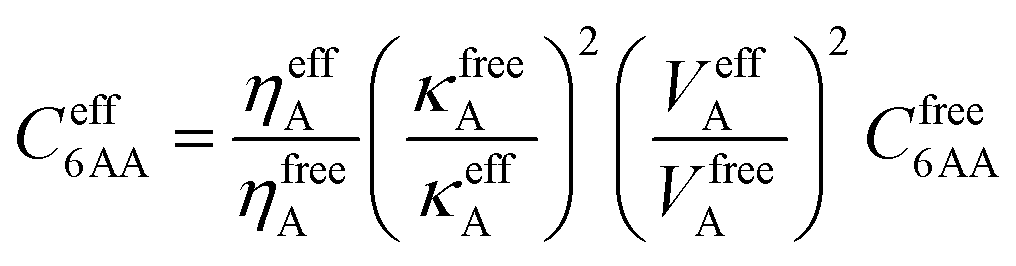 | (1) |
Finally, the adsorption energy (Eads) is defined as:
| Eads = EC2H4/Cu(100) − (ECu(100) + EC2H4), | (2) |
2.2 Molecular DFT calculations
This part of the study includes calculation of the C2H4/Cun and C2H4/Cu complexes. All the geometry optimizations and spectra calculations have been performed using Gaussian09 program package.16 We have selected Minnesota exchange–correlation energy functional, namely, the M06.17 This is a versatile functional for non-covalent interactions, transition metal thermochemistry and for organometallic compounds. The calculations of the C2H4/Cun complexes were carried out using the Pople's split-valence triple-ζ basis set (6-311G) with addition of both polarization (2d,2p) and diffuse (++) functions for the carbon and hydrogen atoms.18 The copper atoms were treated with a pseudopotential, namely, the LANL2DZ basis set.19 In the case of the C2H4/Cu complex the ethylene moiety was treated using the less extended basis set, namely, the 6-311+G(d,p). The reason for such basis set reduction will be described in the following section. To justify the obtained geometries as the minima (number of imaginary frequencies NIMAG = 0) or the first order saddle points (NIMAG = 1), the vibrational frequency analysis was subsequently performed. The latter structures (TS) were calculated using quadratic synchronous transit (QST3) method.20 To verify the obtained TS as a structure which is connecting the reagents and products, the intrinsic reaction coordinate (IRC) calculations were subsequently performed.21To describe the studied interactions as a realistic feature of condensed phase, the polarizable continuum model (PCM) simulations have been included.22 Ethanol was chosen as a model medium because it is used as a solvent in the copper-catalyzed reaction between olefins and aryl diazonium tosylates; the latter reaction is of current synthetic interest for our research group. To define cavities the universal force field (UFF) radii were used.22 The overlap index and a minimum radius of the spheres were specified as 0.8 and 0.5 Å, respectively. Electronic spectra were calculated in terms of Time-Dependent Density Functional Theory (TD DFT).23 The DFT calculations of the closed-shell species were performed using the spin-restricted Kohn–Sham formalism, while the open-shell species were calculated in terms of the spin-unrestricted Kohn–Sham approach. The nature of chemical bonding was analyzed using the Quantum Theory of Atoms in Molecules (QTAIM).24 These calculations were performed using the AIMQB program within the AIMStudio suite by means of the Proaim basin integration method.25 Additionally, some part of the topological parameters was obtained from the post-SCF analyses, which was conducted using a recently developed Multiwfn program package.26
For description of chemical bonding in the studied structures we have analyzed such QTAIM parameters as electron density (ρ) and its Laplacian (∇2ρ). These quantities are important since they are verifiable by the precision X-ray diffraction analysis. When expressed in terms of the Abramov gradient decomposition, the aforementioned values yield three important parameters, namely, potential (v), Lagrangian kinetic (g) and Hamiltonian kinetic (he) energy densities.27
Recently Lu and Chen have proposed a useful molecular property called the Laplacian bond order (LA,B).28 For atoms A and B this can be simply written as:
 | (3) |
3. Results and discussion
3.1 Adsorption of ethylene on the copper (100) surface
Ethylene is a perfect model compound for the present study. On one hand, this molecule possesses one of the highest singlet–triplet energy gaps among alkenes.3b This guaranties that if the triplet 3(ππ*) state of ethylene is achievable under the certain conditions, the rest alkenes should be achievable too. On the other hand, the ethylene molecule is the smallest one among all its derivatives that simplifies the calculations. The latter fact became the reason why adsorption of ethylene on various copper surfaces has been extensively studied both experimentally and theoretically. In particular, we have found references which are related to ethylene interaction with the Cu(100),7g,29 Cu(110),7g,q,8b Cu(111)7g,v and Cu(210)7h surfaces. All these studies suggest that the adsorbed ethylene molecule undergoes an elongation of the CC bond and the partial sp2-to-sp3 rehybridization. The extent of rehybridization depends on the adsorption site.7g There are six possible adsorption sites on Cu(100) surface: atop-bridge, atop-hollow, bridge, cross-bridge, diag-hollow and cross-hollow (see Fig. S1 in the ESI†).
Experimentally, only two adsorption sites were found to be operational, namely, cross-bridge and diag-hollow.29b,c The adsorption energy Eads, the CC bond length (lCC) and adsorption height (hads) values were found to be 0.35 eV, 1.44 Å and 1.3/1.5 Å, respectively.29b,c Recently, Hanke et al.7g have examined several density functionals, which include van der Waals interactions and dispersion correction term. It was found that all the applied functionals predict the top adsorption site of ethylene. Moreover, the PBE-vdwDF functional provides the best fit of the energy with experiment, but dramatically underestimates the lCC and overestimates the hads values.7g In contrast, the semi-local functional (PBE) behaves opposite. It provides the best structural results, but poor energies. The latter, however, can be significantly improved by a simple addition of a dispersion correction (Tkatchenko–Scheffler or Grimme).7g In the present work we chose the atop adsorption site because such ethylene orientation corresponds to those of the odd-numbered clusters and single atom.
Our structural results are presented in Fig. 2. As one can see in Fig. 2a and b, the ethylene adsorption leads to a significant perturbation of the copper surface. The copper atom, which coordinates with ethylene, lies out of the surface plane by 10.6°. Meanwhile, the ethylene molecule also undergoes drastic deformations. The CC bond elongates up to 1.374 Å and the hydrogen atoms move away from the copper surface by 8.3°. This results in a partial rehybridization of the carbon atoms and the ethylene moiety structure becomes the C2v-symmetrical instead of the D2h symmetry point group (Fig. 2a and b). The question then arises, “Why do these structural changes happen and what is the nature of the bonding?” To answer this question one should consider three key factors, namely, (a) band gap (or the SOMO–LUMO gap in clusters and single atom), (b) frontier molecular orbital nature and (c) the presence of unpaired spin. Obviously, the bulk copper, clusters and atom possess different combinations of the aforementioned factors (a, b and c). Thus, we will further discuss our results in terms of the latter three factors.
 | ||
| Fig. 2 Two projections of the optimized supercell together with several structural parameters (a and b); the supercell orientation on the Cu(100) surface covered by ethylene (c); plot of partial and total density of states (d). | ||
Let us first analyze the QTAIM parameters at the bond critical point (BCP) between the ethylene moiety and copper surface (Table 1). There is only one BCP between these fragments (see Fig. S2 in the ESI†). Thus, it becomes clear that there is only one bond formed during adsorption rather than two bonds. According to the Cremer–Kraka bonding criteria this bond corresponds to the coordination type (∇2ρ > 0 and he(r) < 0).30 This conclusion is in accord with generally accepted DCD complex formation model.7g,k,x,9 Formation of the C2H4/Cu(100) complex proceeds via only opposing σ-donation from the b3u-MO of ethylene to the 4s-MO of copper (Fig. 1). The π*-backdonation requires much more energy because of the relatively high level of the b2g-MO of ethylene. But we should stress that the latter process depends on the band gap (or the SOMO–LUMO gap) due to the shift of the 3d level of copper regarding to the b2g-MO of ethylene. Taking into account the latter statement, one can conclude that the triplet ethylene formation should require much less energy when proceeds on the bulk metal surfaces, which are characterized by the absence of the band gap.
C) as well as between ethylene and surface (Cu–Et)
| Bond order | ρ(r) | ∇2ρ(r) | v(r) | g(r) | he(r) | |
|---|---|---|---|---|---|---|
| CC |
Cu–Et | |||||
| 1.3437 | 0.0716 | 0.0593 | 0.2698 | −0.0695 | 0.0685 | −0.0010 |
Indeed, due to the negative value of the band gap and overlapping of valence and conduction bands, the d-electrons always occupy the latter to some extent (Fig. 2d). Moreover, a simple thermal activation can promote an effective occupation of the conduction band; this will result in formation of the triplet ethylene due to vibronic coupling between vibrationally “hot” ground state and the first excited state corresponding to the triplet ethylene molecule.2 The calculated electronic band structure of clean Cu(100) surface and one covered by ethylene are presented in Fig. S3 in the ESI.† The triplet state formation by the exchange mixing mechanism is possible only when the unpaired spin is present. Obviously, the bulk copper has not an unpaired spin and this term is not applicable for macroscopic objects in the explicit manner. On the other hand, the electron gas in conduction band can play role of the unpaired spin. We speculate that the π-electron system of ethylene perturbs and polarizes electron gas; thus, a part of explicit spin appears on the interacting copper atom allowing the exchange mixing mechanism (Fig. 2).4,8 It is worthwhile noting that our present structural and energetic results are in a good agreement with the recent results of Hanke et al.7g Thus, the calculated Eads is equal to 0.47 eV (versus 0.48 eV), but the predicted hads value is lower by 0.09 Å than the former results7g and equals to 2.234 Å.
Though the bulk copper is the most realistic model for the present study, the prediction of the excited triplet state of ethylene is a rather challenging task here because the calculations proceed for the ground electronic state of the total system. Therefore, we present further the study of ethylene interaction with the systems which have discrete energy levels – clusters and atoms.
3.2 Interaction of ethylene with odd-numbered copper clusters C2H4/Cun (n = 3, 7, 11, 15, 17, 19, 21, 25 and 27)
Interaction of ethylene with various metal clusters has been widely described in the literature.7j,k,p,q,s,x,8,9a Quite recently, Lyalin and Taketsugu have calculated adsorption of ethylene on neutral, anionic, and cationic gold clusters consisting of up to 10 atoms.7k They have found that ethylene adsorbs by two different configurations, corresponding to the π- and di-σ-bonded species. The authors pointed out that the di-σ-bonded ethylene is activated more strongly in comparison with the π-bonded structure.7k We declare here the di-σ-bonded ethylene corresponds to the triplet excited state. To monitor the electronic state of the ethylene moiety one should take into account the spin polarization effects during the complex formation, which were omitted in the former studies.In the present work we have studied the odd-numbered clusters because they have a paramagnetic character. This allows exchange mixing of the singlet and triplet spin states inside the ethylene moiety, but, at the same time, such treatment is challenging from the methodological point of view. On one hand, the single-determinant approaches, like DFT/UM06 is unable to distinguish the spin combinations 1 and 3 illustrated in Fig. 1. On the other hand, treatment of diamagnetic systems, like even-numbered copper clusters, encounters another spin contamination problem, which is presented in Scheme 1.
 | ||
| Scheme 1 Different spin combinations in the diamagnetic complex C2H4/Cu2. | ||
Due to the periodic group similarity, the Cu and Au atoms should exhibit the same binding performance. It is shown recently that in the case of the C2H4/Au complex only one σ-bond is formed.7k In the following section we will show that the C2H4/Cu complex in the ground 2A′ state has the similar nature. But we have developed a procedure for obtaining the di-σ-bonded state of the C2H4/Cu complex corresponding to the excited state with the triplet ethylene moiety. The simple ground (singlet) state calculations of the C2H4/Cu2 and C2H4/Au2 complexes (1A state) yield the pure sp3-hybridization of the carbon atoms. The optimized geometry is, therefore, similar to the staggered ethane.7k Meanwhile, the triplet 3B2 state of the C2H4/Cu2 complex remains the cyclic geometry with complete delocalization of the two unpaired spins onto the copper (0.518 on each) and carbon (0.521 on each) atoms. Thus, since the spin states of the ethylene moiety are rather hard to interpret in the diamagnetic complexes with even-numbered clusters, we have studied only paramagnetic species.
The optimized structures of the C2H4/Cun complexes together with the selected structural parameters are illustrated in Fig. 3. The optimized structures of the pure clusters are presented in Fig. S4 in the ESI.† As one can see in Fig. 3 all the obtained complexes are mono-π-bonded. The QTAIM analysis yields only one BCP similar to the C2H4/Cu(100) system (Fig. 2). The bond type has the same nature (coordination bond), which follows from the calculated ∇2ρ and he(r) values (Table 2). The distortions of the ethylene moiety in the C2H4/Cun complexes, which indicates the sp2-to-sp3 rehybridization are lower than in the bulk model. This qualitatively follows from the values of the C–(CH2) dihedral angle (ϕ). Stuve and Madix have proposed a quantitative characteristic of the sp2-to-sp3 rehybridization, namely, the πσ parameter.31a This is based on the earlier proposal of Powell et al.31b about account of the ν(CC) and δ(CH2) vibrational frequencies, which were called as the band I and II, respectively:
 | (4) |
 | ||
| Fig. 3 The structure of the C2H4/Cun complexes optimized by the UM06 method in ethanol; blue numbers correspond to the CC bond distances, red numbers indicate the Cu–C interatomic distances; ϕ is the C–(CH2) dihedral angle. | ||
C) and δ(CH2) vibrational frequencies, namely, band I and II (cm−1), the πσ parameter, Laplacian bond orders, spin density at the copper atom interacting with ethylene (ρSEt), the QTAIM parameters at the BCP between the cluster and the ethylene moiety
| n | ν(CC) |
δ(CH2) | πσ | Bond order | ρSEt | ρ(r) | ∇2ρ(r) | v(r) | g(r) | he(r) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1–C2 | Cu–C1 | Cu–C2 | ||||||||||
| 3 | 1531 | 1257 | 0.328 | 1.4021 | 0.1036 | 0.1055 | 0.459 | 0.0709 | 0.3334 | −0.0938 | 0.0886 | −0.0053 |
| 7 | 1576 | 1303 | 0.159 | 1.5025 | 0.0704 | 0.0709 | 0.324 | 0.0616 | 0.2866 | −0.0753 | 0.0735 | −0.0018 |
| 11 | 1552 | 1284 | 0.238 | 1.4445 | 0.0654 | 0.0756 | 0.212 | 0.0613 | 0.2724 | −0.0727 | 0.0704 | −0.0023 |
| 15 | 1569 | 1297 | 0.183 | 1.4981 | 0.0609 | 0.0526 | 0.246 | 0.0558 | 0.2433 | −0.0624 | 0.0616 | −0.0008 |
| 17 | 1582 | 1307 | 0.140 | 1.5119 | 0.0588 | 0.0541 | 0.077 | 0.0560 | 0.2518 | −0.0638 | 0.0634 | −0.0004 |
| 19 | 1592 | 1312 | 0.113 | 1.5150 | 0.0627 | 0.0627 | 0.049 | 0.0658 | 0.2829 | −0.0821 | 0.0764 | −0.2257 |
| 21 | 1610 | 1322 | 0.063 | 1.5634 | 0.0650 | 0.0648 | 0.011 | 0.0600 | 0.2788 | −0.0723 | 0.0710 | −0.0013 |
This parameter is calibrated in such a way that it equals to zero for isolated ethylene and equals to unit for dibromoethane.31a The calculated πσ parameters for the C2H4/Cun complexes are listed in Table 2. As one can see in Table 2, the values of πσ parameter decrease when moving from smaller to larger clusters. Note that the lCC bond length varies rather randomly (Fig. 3), while the Laplacian bond order (LCC) behaves linearly and slightly increases with rising n (Table 2). The correlations of ν(CC), δ(CH2), LCC and πσ parameter with the number of copper atoms are illustrated in Fig. S5† and the calculated values of the SOMO and LUMO energies of the studied copper clusters are listed in Table S1 in the ESI.† Though the QTAIM analysis predicts only one π-bond between the ethylene species and clusters (Fig. 3), we have estimated the LCC values for the Cu–C atomic pair; the latter decreases with rising of the cluster size.
The binding nature in the C2H4/Cun complexes depends on the presence of unpaired spin on the reacting copper atom. When the number of copper atoms rises, the local spin density decreases (Table 2). Therefore, the mono-π-bonded character of the complexes with the ground singlet state of the ethylene moiety prevails. The local spin density at the reacting copper atom also depends on the number of copper atoms in the cluster. The correlation plot is illustrated in Fig. S5 in the ESI.† The large clusters behave as purely diamagnetic species, therefore we could not obtain the di-σ-bonded complexes when n > 7. Meanwhile, Lyalin and Taketsugu have described the similar complexes with Au3, Au5, Au7 and Au9.7k This is due to the planar shape of the gold clusters used. We have built our clusters trying to form a spherical shape as much as possible (Fig. 3). For large clusters (n > 7) we have found that one-third of the unpaired spin is usually located inside the cluster, while the rest is spread onto the outer atoms. It is important that the CC bond length in the complexes with Au3, Au5, Au7 and Au9 remains almost unchanged: 1.45, 1.47, 1.47 and 1.46 Å, respectively.7k This result is in a complete agreement with our calculation of the triplet ethylene in the 2B2 state of the C2H4/Cu complex (1.466 Å). This item will be described in more details in the following section. The calculated binding energies of the C2H4/Cun complexes are presented in Table 3; the coordination numbers and free valence values of the carbon atoms in the complexes are listed in Table S2 in the ESI.†
| n | 3 | 7 | 11 | 15 | 17 | 19 | 21 | 25 | 27 |
| Ebind | −25.1 | −7.1 | −16.0 | −15.4 | −25.1 | −18.4 | −14.7 | −17.6 | −17.3 |
Summing up, the copper clusters are characterized by different combination of the aforementioned key factors affecting the binding mode of ethylene. On one hand, the clusters have positive SOMO–LUMO gap, which narrows when n goes to infinity (see Fig. S5 in the ESI†). On the other hand, a lack of the unpaired spin occurs simultaneously with the cluster growth. Thus, the ground state optimization of the ethylene complexes with small paramagnetic clusters (n < 11) provides both the mono-π-bonded and di-σ-bonded complexes corresponding to the singlet and triplet ethylene moieties, respectively. Treatment of the bigger clusters leads only to the mono-π-bonded complexes. Finally, the di-σ-bonded models can be obtained by the exited state optimization when the proper excited state corresponding to the π*-backdonation step, is defined by means of TD DFT or CASSCF methods. We avoid such study because this is a very expensive computational procedure accompanied with the known difficult problem of orbital relaxation. Instead, we concentrate our attention on the simplest system.
3.3 The single-atom model C2H4/Cu
The metal–organic coordination complexes of ethylene and acetylene are known for a wide variety of metals. Thus, such complexes with Cu,9f,32 Co9g,32c,33a Pd,9f,33b Au,7k,32f Ag,9g,32f Cr,9d,32c Fe,32c,33c Ni,32c,33d Ti,9c,32c V,32c,33e Pt,9f Mn,32c Na,33f Sc,33g Rh,33h Nb,33e Ta,33e Zn9c and Hf9c atoms were studied both experimentally and theoretically. The syntheses of the (C2H4)nCu and (C2H4)mCu2 complexes (where n = 1–3 and m = 4 or 6) together with their IR and UV-vis spectral investigation were first reported by Ozin et al. in far 1977.32e At the same time, the first attempts to study these complexes were made also theoretically.32e,f Unfortunately, a lack of reliable theoretical methods in 1970s became the reason why the authors could not describe correctly the structure and spectra of the synthesized species.32e,f That time, the di-σ-bonded (C2v) structure of the C2H4/Cu complex was proposed on the ground of the single-point energy calculation and the electronic spectrum tentative assignment. Just recently, Lyalin and Taketsugu have fully optimized geometry and calculated IR spectrum of the C2H4/Au complex; they have found that the species has the CS symmetry point group and represents the mono-σ-bound compound.7kSince the gold and copper atoms have the periodic group similarity the same point group is expected for the analogous copper complex. Indeed, our present ground state calculation of the C2H4/Cu species has provided the CS-symmetrical mono-σ-bound structure. The optimized geometry and selected structural parameters are given in Fig. 4a. As we have mentioned in the previous section the binding mode strongly depends on the presence of the unpaired spin on the interacting copper atom. When a whole spin is present (Cu atom) the mono-σ-bonded complex is formed. If spin population on the copper atom is equal to zero (Cu+ ion) the purely mono-π-bonded complex occurs. Detachment of the second electron (Cu2+ ion) returns the previous binding mode – the CS-symmetrical mono-σ-bound complex. The optimized geometries of the C2H4/Cu+ and C2H4/Cu2+ complexes together with selected structural and QTAIM parameters are presented in Fig. S6 in the ESI.†
 | ||
| Fig. 4 Structures of the 2A′, 2B2 and 4B complexes of ethylene with the copper atom optimized by the DFT(UM06) method in ethanol (a), 3D maps of spin density plotted with isovalue of 0.015 a.u. (b) and frontier molecular orbital levels (c). | ||
Ozin et al.32e have found experimentally that the three most intense bands in the IR spectrum of the C2H4/Cu complex correspond to the ν(CC) (1475 cm−1), δ(CH2) (1156/1138 cm−1) and ρW(CH2) (840 cm−1). Meanwhile, the UV-vis spectrum consists of two bands at 420 and 381 nm.32e We have calculated these spectra for the 2A′ and 2B2 states of the C2H4/Cu complex. The corresponding plots are illustrated in Fig. S7 and S8 in the ESI.† It becomes clear that the experimental IR and UV-vis spectra of the synthesized C2H4/Cu complex correspond to the ground 2A′ state. Note that we have checked various DFT functionals in order to find the most reliable one. The results are listed in Table S3 in the ESI.† No doubt that the M06 functional occurs to be the best one among the functionals studied. We also should stress that the calculated UV-vis spectrum of the 2A′ state complex contains the longwave absorption band at 658 nm (Fig. S8 in the ESI†). The experimental spectrum of the synthesized C2H4/Cu complex also includes the broad band near 500 nm.32e The authors, however, ascribed this band to the (C2H4)2Cu or (C2H4)3Cu complexes, which probably appeared upon the matrix annealing.32e Taking into account the good agreement of the IR spectral data, we suggest that this band characterizes the C2H4/Cu complex, but further more detailed study of the structure and spectral properties of the (C2H4)2Cu or (C2H4)3Cu complexes is also important.
As we have mentioned above, the 2A′ state of the C2H4/Cu complex corresponds to the singlet state inside the ethylene moiety. In the present paper, however, we have developed a procedure to obtain the excited state C2H4/Cu complex, which corresponds to the triplet state ethylene. To calculate the latter we have applied the SOMO and LUMO mixing during the geometry optimization. First, we have calculated the quartet 4B state of the C2H4/Cu complex (Fig. 4a). In such complex the ethylene moiety corresponds to the triplet state, which can be seen from the color highlighting of the ethylene and copper fragments (Fig. 4a). The obtained geometry was then used as the starting structure for the doublet state calculation accounting the SOMO–LUMO mixing. Such orientation of the interacting moieties provides the proper MOs which fit the best overlap between fragments. Thus, the SCF procedure leads to a minimum corresponding to the 2B2 state of the C2H4/Cu complex with the triplet ethylene fragment inside. The subsequent frequency analysis has proven the global minimum of this complex. To monitor the resulting spin population we have plotted 3D maps of the spin density (Fig. 4b). As one can see in Fig. 4b the spin density maps are in a complete agreement with the qualitative scheme illustrated in Fig. 1.
The peculiarities of the SOMO–LUMO mixing procedure make a restriction on the basis set expansion. As we have stated in the Computational Details, the copper and hydrogen atoms have been treated by the 6-311+G(d,p) basis set, while the C2H4/Cun complexes were calculated using the 6-311++G(2d,2p) basis set for the latter atoms. Actually, we have not calculated the ethylene–cluster complexes using SOMO–LUMO mixing procedure because this is impossible due to a different orbital pattern. Thus, only the ground state C2H4/Cun complexes could be obtained. Concerning the 2B2 state of the C2H4/Cu complex, the more extended basis set also provides different orbital pattern when the Cu–C distance is near 3 Å. Therefore, we have decided to use 6-311+G(d,p) basis set and we believe that the energies obtained such way are still reliable.
Of course, the use of the pseudopotential even for the complex with a single copper atom can raise some doubt about accuracy of the results in principle. Frozen core electrons and only the double-split basis for valence shell, lacking some part of diffuse and polarization functions restrict the accuracy of the method and the credit to major conclusions. Unfortunately, we cannot calculate the C2H4/Cu complex with an extended all-electron basis set on copper atom using the SOMO–LUMO mixing procedure. Any extension of the basis set leads to occurrence of artificial vacant orbitals and LUMO does not represent the 3d-AO on copper which is necessary for the proper simulation of the physically reasonable spin arrangement in the complex. The present work is done entirely in terms of DFT approach and inside this scheme we cannot extend the basis set. We have to stress that the relativistic effective core potential basis set LANL2DZ has proved to be physically reasonable in many applications including the triplet excited state description in various organometallic systems.34a–c
To clarify the mechanism of the triplet ethylene formation we have performed relaxed scans of the potential energy surfaces (PESs) profiles corresponding to the 2A′, 2B2 and 4B states of the C2H4/Cu complex (Fig. 5). As one can see in Fig. 5, the 2A′ state complex form a rather shallow minimum (−7.2 kcal mol−1), while the 2B2 state complex form a deep minimum lying by 10.8 kcal mol−1 higher in energy than the previous one. We estimate this energy difference as the singlet–triplet energy splitting of ethylene reacting with copper. Since this value is more than twice lower than 1 eV, the excited 2B2 complex can be achievable under the thermal activation accompanied with vibronic coupling.1,2 We speculate that the ethylene derivatives are expected to have even smaller singlet–triplet splitting than 10.8 kcal mol−1, when interacting with the copper atoms/surfaces, because their isolated molecules already have.3b
 | ||
| Fig. 5 The potential energy surfaces (PESs) profiles relaxed scans of the C2H4/Cu complexes in the 2A′, 2B2 and 4B states (step 0.25 Å). Blue curve corresponds to the singlet state of the ethylene moiety, while the red and black curves reflect behavior of the triplet ethylene in the complexes. | ||
Let us return to Fig. 5. The 2B2 state complex, corresponding to the triplet ethylene fragment, is remains to be bound until the C–Cu interatomic distances do not exceed ∼3 Å (red curve). In this range the exchange mixing between the singlet and triplet states of ethylene and copper atom does operate. The more weakly bound complex does not possess S–T mixing and starts to decay. This accompanies the geometry change in the ethylene moiety, which acquires the D2d symmetry instead of the C2v. Due to the same spin multiplicity, the vertical transitions 2A′ → 2B2 between the ground and excited state complexes are allowed within the C–Cu interatomic distance ranged in 2–3 Å. Beyond this range the exchange mixing becomes too small, therefore the 2B2 complex has the geometry similar to the quartet 4B complex, which has very shallow and broad minimum at 3 Å. Further increasing the C–Cu interatomic distance makes the structural and energetic differences between the 2B2 and 4B states almost negligible (Fig. 5). Finally, they converge to one energy level, which corresponds to the singlet–triplet splitting of pure ethylene.3b
To illustrate how the SOMO–LUMO mixing procedure yields the triplet ethylene moiety inside the excited 2B2 complex we have plotted the energies of frontier MOs as functions of the C–Cu interatomic distance (Fig. S9 in the ESI†). As on can see in Fig. S9,† when mixing procedure is not applied the two crossing points between the SOMO and LUMO appear. Thus, in the range of the C–Cu interatomic distance of 2–3 Å the SOMO and LUMO have different symmetry. As a result, one obtains the ground state complex of the C2v symmetry instead of the CS one. It is important to note that the SOMO(b2), which reflects the π*-backdonation step in the DCD model, has very diffuse nature (Fig. S9 in the ESI†). The overlap between the copper and ethylene proceeds through the far slices of the d orbital. This results in a weak overlap and, hence, in a rather small dissociation energy of the complex (57.1 kcal mol−1). We also should stress, that the π*-backdonation step in the DCD model is usually illustrated through the closer slices of the d orbital7g,k,x,9 being a principal mistake. Such efficient overlap is characteristic for strong covalently bound molecules, like ethylene oxide or sulfide, where the p orbital of oxygen or sulfur interact with the b2g-MO of ethylene. Therefore, the dissociation energy of such molecules is much higher than that of the 2B2 complex C2H4/Cu.
Finally, we have performed the TS search to ensure that the 2A′ and 2B2 states are not lying on the same PES. The results are illustrated in Fig. S10 in the ESI.† The obtained TS has the CS symmetry and does correspond to a local minimum (v1 = 457i cm−1). The IRC scan, however, exhibits that the obtained TS corresponds to a simple rotation of the CH2 group (Fig. S10 in the ESI†). Thus, we can conclude again that the 2A′ and 2B2 states do lie on different PESs and the vertical transition 2A′ → 2B2 (10.8 kcal mol−1) corresponds to the singlet–triplet splitting of the ethylene molecule, which interact with copper.
3.4 The QTAIM analysis of chemical bonding in the studied complexes
The results of the critical point search in the2A′, 2B2 and 4B state complexes are illustrated in Fig. 6. The calculated data of the QTAIM parameters at the corresponding critical points are listed in Tables 4–6. It is known that a good criterion of the bond types are the values of electron density ρ(r) and its Laplacian ∇2ρ(r) at the BCP.24 If ρ(r) is close to 0.1 and ∇2ρ(r) ≪ 0 than the given bond is shared (covalent or polar).24 In contrast, if ρ(r) is about 0.01 and ∇2ρ(r) > 0 than the considered bond corresponds to a closed-shell interaction (the ionic bond).24 As one can see in Tables 4–6, no any shared chemical bonds between copper and ethylene in the studied complexes. All of them correspond to closed-shell interaction (coordination bonds). According to the Espinosa formula, the potential energy density at the BCP in the weak complexes correlates with the bond energy.35 Thus, the 2B2 state complex is more than 5.3 times stronger than the 2A′ complex and more than 13.5 times stronger than the 4B one (Tables 4–6). It is interesting that the latter complex also forms the ring, but it is extremely weak, like the whole complex (2.8 kcal mol−1), see Fig. 5.
 | ||
| Fig. 6 The symmetry unique bond (magenta) and ring (green) critical points in which the local descriptors are estimated. | ||
2A′ state complexa
| CP | ρ(r) | ∇2ρ(r) | ε(r) | v(r) | g(r) | he(r) | Ī(r) | S(r) | Ω(r) | LOL(r) | ELF(r) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a ε is ellipticity of electron density; Ī is local ionization energy; S is local information entropy; Ω is sign(λ2)ρ; LOL and ELF are localized orbital locator and electron localization function, respectively. | |||||||||||
| 1 | 0.2763 | −0.9139 | 0.0098 | −0.3088 | 0.0402 | −0.2687 | 0.5107 | 0.3822 | −0.2763 | 0.8934 | 0.9860 |
| 2 | 0.3221 | −0.8992 | 0.2446 | −0.4672 | 0.1212 | −0.3460 | 0.5998 | 0.4315 | −0.3221 | 0.7820 | 0.9279 |
| 3 | 0.2733 | −0.8959 | 0.0140 | −0.3045 | 0.0403 | −0.2642 | 0.5123 | 0.3790 | −0.2733 | 0.8914 | 0.9854 |
| 4 | 0.0615 | 0.1875 | 0.0224 | −0.0605 | 0.0537 | −0.0068 | 0.45223 | 0.0111 | −0.0615 | 0.3386 | 0.2091 |
| CP | ρ(r) | ∇2ρ(r) | ε(r) | v(r) | g(r) | he(r) | Ī(r) | S(r) | Ω(r) | LOL(r) | ELF(r) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.2618 | −0.5930 | 0.1943 | −0.3094 | 0.0807 | −0.2288 | 0.5326 | 0.0366 | −0.2618 | 0.7925 | 0.9358 |
| 2 | 0.0830 | 0.3251 | 1.2758 | −0.1064 | 0.0938 | −0.0125 | 0.5140 | 0.0143 | −0.0830 | 0.3260 | 0.1911 |
| 3 | 0.2693 | −0.8573 | 0.0436 | −0.3072 | 0.0465 | −0.2608 | 0.4824 | 0.0375 | −0.2693 | 0.8741 | 0.9797 |
| 4 | 0.0803 | 0.3617 | −4.2685 | −0.1081 | 0.0992 | −0.0088 | 0.5516 | 0.0140 | 0.0803 | 0.3020 | 0.1577 |
| CP | ρ(r) | ∇2ρ(r) | ε(r) | v(r) | g(r) | he(r) | Ī(r) | S(r) | Ω(r) | LOL(r) | ELF(r) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.0116 | 0.0263 | −3.7395 | −0.0074 | 0.0070 | −0.0004 | 0.3549 | 0.0027 | 0.0116 | 0.1975 | 0.0645 |
| 2 | 0.2783 | −0.6997 | 0.0089 | −0.3372 | 0.0812 | −0.2561 | 0.5721 | 0.3844 | −0.2783 | 0.8076 | 0.9463 |
| 3 | 0.0119 | 0.0246 | 1.3192 | −0.0079 | 0.0070 | −0.0009 | 0.3447 | 0.0027 | −0.0119 | 0.2046 | 0.0712 |
| 4 | 0.2684 | −0.8540 | 0.0298 | −0.2998 | 0.0432 | −0.2567 | 0.5035 | 0.0374 | −0.2684 | 0.8814 | 0.9822 |
| 5 | 0.2702 | −0.8663 | 0.0271 | −0.3034 | 0.0434 | −0.2600 | 0.5078 | 0.0376 | −0.2702 | 0.8820 | 0.9824 |
Such low values of the bonding energy in the 2A′ and 4B complexes make them quite unstable. Simple thermal activation enables either decay of the 2A′ state complex, or its excitation to the 2B2 state complex, which is rather stable and might be responsible for further ethylene reactivity.
4. Conclusions
In the present survey, we have searched for the low-lying excited complexes of π-nucleophiles (ethylene) with transition metals (copper), which are characterized by the triplet nature of the π-nucleophile moiety. Herein, the low-lying means that the excitation energy should be lower than 1 eV.1,2 Thus, the excited 2B2 state lies higher in energy than the ground2A′ state complex by 10.8 kcal mol−1 (0.47 eV). This excited state can be achievable under the dark conditions and can play an important role in the addition reaction to the double bond (AE and AR). Indeed, the triplet π-nucleophiles posses a twisted (D2d) ethylene fragment and almost completely broken π-bond.3b,10 Thus the triplet ethylene can add the paramagnetic species with no barrier. We have offered this mechanism earlier for the aryl cation (AE) or radical (AR) addition to the double bond.3a,b,36 The aryl cations are characterized by a low singlet–triplet energy splitting and often exist in the triplet ground state.3c,37
The present developed procedure for the 2B2 state complex optimization provides only a semiquantitative description of the π-nucleophiles triplet state reactivity. Quite important information can be revealed from the modeling of π-nucleophile reaction with radical or electrophilic species on the copper surface. We have found that the 2B2 state complex C2H4/Cu exists until the C–Cu interatomic distance not exceeds of about 3 Å. Within this range ethylene conserves the triplet nature (C2v) and is bonded with copper. Thus, we speculate that a comprehensive theoretical study of the triple complexes [copper⋯π-nucleophile⋯radical/electrophile] will gain valuable results on the kinetics of the triplet π-nucleophiles reactivity and deserves a further study. It does not mean that only the triplet π-nucleophiles should be considered in the AE and AR reactions of the double bond. Involvement of the triplet reaction substrates can open an additional (triplet) reaction channel resulting in lowering of the general activation barrier. From this viewpoint, the copper or other transition metal surface play the role of catalyst.
Acknowledgements
This work was supported by the Ministry of Education and Science of Ukraine, Research Fund (Grant no. 0113U001694). We thank Professor Hans Ågren (KTH, Stockholm) for the PDC supercomputer use. The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at the Parallel Computer Center (PDC) through the project “Multiphysics Modeling of Molecular Materials”, SNIC 020/11-23.References
-
(a) M. Klessinger and J. Michl, Excited States and Photochemistry of Organic Molecules, VCH Publishers, Inc., New York, 1995 Search PubMed
; (b) J. A. Barltrop and J. D. Coyle, Excited States in Organic Chemistry, Wiley, London, 1975 Search PubMed
-
(a) K. K. Kalninsh and E. F. Panarin, Dokl. Chem., 2014, 456, 103–106 CrossRef CAS
-
(a) S. V. Bondarchuk and B. F. Minaev, J. Mol. Struct.: THEOCHEM, 2010, 952, 1–7 CrossRef CAS PubMed
-
(a) G. J. Hoijtink, Mol. Phys., 1960, 3, 67–70 CrossRef CAS
-
(a) H. Bonin, M. Sauthier and F.-X. Felpin, Adv. Synth. Catal., 2014, 356, 645–671 CrossRef CAS
-
(a) S. V. Bondarchuk, B. F. Minaev and A. Y. Fesak, Int. J. Quantum Chem., 2013, 113, 2580–2588 CrossRef CAS
-
(a) G. Shukri and H. Kasai, Surf. Sci., 2014, 619, 59–66 CrossRef CAS PubMed
-
(a) H. Öström, D. Nordlund, H. Ogasawara, K. Weiss, L. Triguero, L. G. M. Pettersson and A. Nilsson, Surf. Sci., 2004, 565, 206–222 CrossRef PubMed
-
(a) P. Kozyra, E. Broclawik, M. P. Mitoraj and J. Datka, J. Phys. Chem. C, 2013, 117, 7511–7518 CrossRef CAS
-
(a) X. Wang, W. E. Turner II, J. Agarwal and H. F. Schaefer III, J. Phys. Chem. A, 2014, 118, 7560–7597 CrossRef CAS PubMed
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CrossRef CAS
- Materials Studio 5.5, Accelrys, Inc., San Diego, CA, 2008
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS
- C. Ye, S. Hu, W. Yan, J. Duan and C. Jing, J. Phys. Chem. C, 2013, 117, 5785–5791 CAS
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al.Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS
-
(a) W. Kohn and L. J. Sham, Phys. Rev. A, 1965, 140, A1133–A1138 CrossRef
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS PubMed
- C. Peng and H. B. Schlegel, Isr. J. Chem., 1993, 33, 449–454 CrossRef CAS
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, Chem. Phys., 1998, 109, 8218–8224 CAS
- R. F. W. Bader, Atoms in molecules, a quantum theory, Clarendon Press, Oxford, 1990 Search PubMed
- T. A. Keith, AIMAll, Version 10.07.25, TK Gristmill Software, Overland Park KS, USA, http://www.aim.tkgristmill.com, 2010
- T. Lu and F. Chen, J. Comp. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
-
(a) Y. A. Abramov, Acta Crystallogr., Sect. A: Found. Crystallogr., 1997, 53, 264–272 CrossRef
- T. Lu and F. Chen, J. Phys. Chem. A, 2013, 117, 3100–3108 CrossRef CAS PubMed
-
(a) C. Nyberg, C. G. Tengstål and S. Andersson, Chem. Phys. Lett., 1982, 87, 87–91 CrossRef CAS
- D. Cremer and E. A. Kraka, Croat. Chem. Acta, 1984, 57, 1259–1281 Search PubMed
-
(a) E. M. Stuve and R. J. Madix, J. Phys. Chem., 1985, 89, 3183–3185 CrossRef CAS
-
(a) G. Nicolas and J. C. Barthelat, J. Phys. Chem., 1986, 90, 2870–2877 CrossRef CAS
-
(a) A. J. Lee Hanlan, G. A. Ozin and W. J. Power, Inorg. Chem., 1978, 17, 3648–3657 CrossRef
-
(a) P. J. Hay, J. Phys. Chem. A, 2002, 106, 1634–1641 CrossRef CAS
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS
-
(a) B. F. Minaev, S. V. Bondarchuk and M. Girţu, J. Mol. Struct.: THEOCHEM, 2009, 904, 14–20 CrossRef CAS PubMed
-
(a) S. V. Bondarchuk and B. F. Minaev, Chem. Phys., 2011, 389, 68–74 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra12422g |
| This journal is © The Royal Society of Chemistry 2015 |