
Modifying the carbon fiber–epoxy matrix interphase with silicon dioxide nanoparticles
Abstract
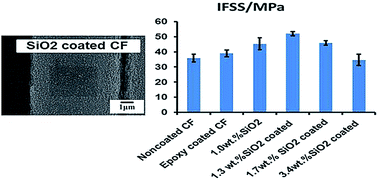
Nanoparticle modification of the carbon fiber–epoxy interface was investigated by depositing silicon dioxide (SiO2) nanoparticles on the surface of carbon fibers (CFs) using a conventional sizing process by immersing CFs tows in a SiO2 nanoparticle suspension. The surface density of the nanoparticles and the uniformity of their dispersion were characterized by scanning electron microscopy. CF–epoxy composites were fabricated to study the effect of the SiO2 nanoparticle coating on the CF–epoxy composite interfacial properties. Interfacial adhesion was assessed by measuring the interfacial shear strength (IFSS) with the single fiber fragmentation test. A 44% improvement of the IFSS was obtained with a SiO2 relative concentration of 1.3 wt%, compared with non-coated CFs. This increase was the result of an increase of the shear modulus of the matrix in the interphase region caused by the presence of the SiO2 nanoparticles. Coating CFs with SiO2 nanoparticles has been shown to be a simple, scalable, and cost effective method to increase the interfacial mechanical properties in CF–epoxy composites.
 Please wait while we load your content...
Please wait while we load your content...

