
Passive optical mapping of structural evolution in complex fluids
Abstract
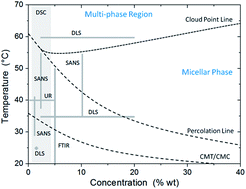
Self-assembling complex systems exhibit properties that involve a broad spectrum of thermal, structural, morphological, and optical transitions. Various techniques have been used to assess different aspects of the phase transitions in these complex systems. However, because of inherent technical constraints, structural information is usually provided only within narrow ranges of concentrations and temperatures. We show here that by effectively suppressing multiple scattering, low-coherence dynamic light scattering permits assessing the aggregation dynamics of self-assembling systems in a completely passive manner and over ranges of concentration and temperatures well beyond the limits of traditional approaches. The power spectral analysis of scattered intensity fluctuations permits a reliable characterization of multiple relaxation times. We demonstrate that the entire phase diagram can be covered in a consistent way and structural phase transitions can be mapped over a broad optical regime from weak to strong scattering.
 Please wait while we load your content...
Please wait while we load your content...

