
Cu-Mediated 2,2,2-trifluoroethylation of terminal alkynes using 1,1-dichloro-2,2,2-trifluoroethane (HCFC-123)†
En-Jian
Han
a,
Yan
Sun
a,
Qian
Shen
b,
Qing-Yun
Chen
*a,
Yong
Guo
*a and
Yan-Gen
Huang
b
aKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: chenqy@sioc.ac.cn; yguo@sioc.ac.cn
bCollege of Chemistry, Chemical Engineering and Biotechnology, Donghua University, 2999 North Renmin Road, Shanghai 201620, China
First published on 14th August 2015
Abstract
The title reaction provides a novel utilization of ozone-depleting/global-warming HCFCs as a new carbon–carbon cross-coupling model of various terminal alkynes by activating two inert C–Cl bonds successively. This protocol provided trifluoroethylated alkynes efficiently under mild reaction conditions and was compatible with a broad range of functional groups. An example of the synthesis of a terbinafine analogue is shown. Some mechanistic experiments including deuterated reagents and radical/SET inhibitors are described.
Introduction
The strategic incorporation of fluorine or fluorinated moieties into organic substrates imparts useful properties to these molecules.1a Hydrofluorochlorocarbons (HCFCs) are essential materials for the fluorine industry. They are indispensable for the production of a variety of useful fluorinated compounds, although HCFCs cause ozone depletion and global warming.1 An example of the sustainable use of HCFCs involves the synthesis of a very useful polymer polytetrafluoroethylene (PTFE) from the chemical precursor chlorodifluoromethane (HCFC-22) via tetrafluoroethylene. Besides the application in fluorinated materials, HCFCs are also used in the synthesis of agrochemicals, such as pyrethroid insecticides.1dThe challenge in using HCFC molecules in chemical synthesis is activating the inert C–Cl bond without influencing the fluorine moiety. Reported methods which use HCFCs in chemical synthesis typically cause C–F bond cleavage which yields fluoroalkenes. Burton and others have reported the transformation of CF3CH2Cl (HCFC-133a) to a fluoroalkene for further functionalization using dehydrofluorination.2 Chen and Wu have reported reactions between nucleophiles and HCFC-133a under basic and supercritical conditions which gave fluorochlorovinyl derivatives and defluorinated ethers.3 Nucleophilic addition of 1,1-dichloro-2,2,2-trifluoroethane (HCFC-123, 1) to a variety of aldehydes in the presence of Zn has been reported to yield difluoropropenol.4 HCFC-133a can generate either 1,1,1-trifluoroethane or 1,1-difluoroethylene depending on the activation method used.5 Methods to activate the C–Cl bonds of HCFCs which do not affect the fluorine groups via radical intermediates have been developed.5–7 Nevertheless, the reaction partners are limited to alkenes/alkynes,6a,7a phenols/thiophenols,7 and secondary amines.8 To the best of our knowledge, metal-mediated carbon–carbon cross-coupling reactions using HCFCs without defluorination have not been discovered.
Polyfluoro and perfluoroalkyl iodides/bromides (e.g., CF3CH2I, BrCF2P(O)OR, BrCF2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, C4F9I and CF3I) are suitable coupling partners in metal-mediated C–C cross-coupling reactions.9 However, there are few reports of C–C cross-coupling reactions involving fluorinated alkyl chlorides. In addition to the large C–Cl bond energy, the challenges involved in using HCFCs include (Scheme 1a): (1) the cleavage of the C–Cl bond commonly generates a fluoroalkyl radical via a single-electron transfer (SET) process, which can abstract a hydrogen5 or add to unsaturated substrates;6a,7a (2) β-defluorination5,10 is a predominating reaction pathway if an organometallic species is generated after the oxidative addition of a metal to the C–Cl bond.
CH2, C4F9I and CF3I) are suitable coupling partners in metal-mediated C–C cross-coupling reactions.9 However, there are few reports of C–C cross-coupling reactions involving fluorinated alkyl chlorides. In addition to the large C–Cl bond energy, the challenges involved in using HCFCs include (Scheme 1a): (1) the cleavage of the C–Cl bond commonly generates a fluoroalkyl radical via a single-electron transfer (SET) process, which can abstract a hydrogen5 or add to unsaturated substrates;6a,7a (2) β-defluorination5,10 is a predominating reaction pathway if an organometallic species is generated after the oxidative addition of a metal to the C–Cl bond.
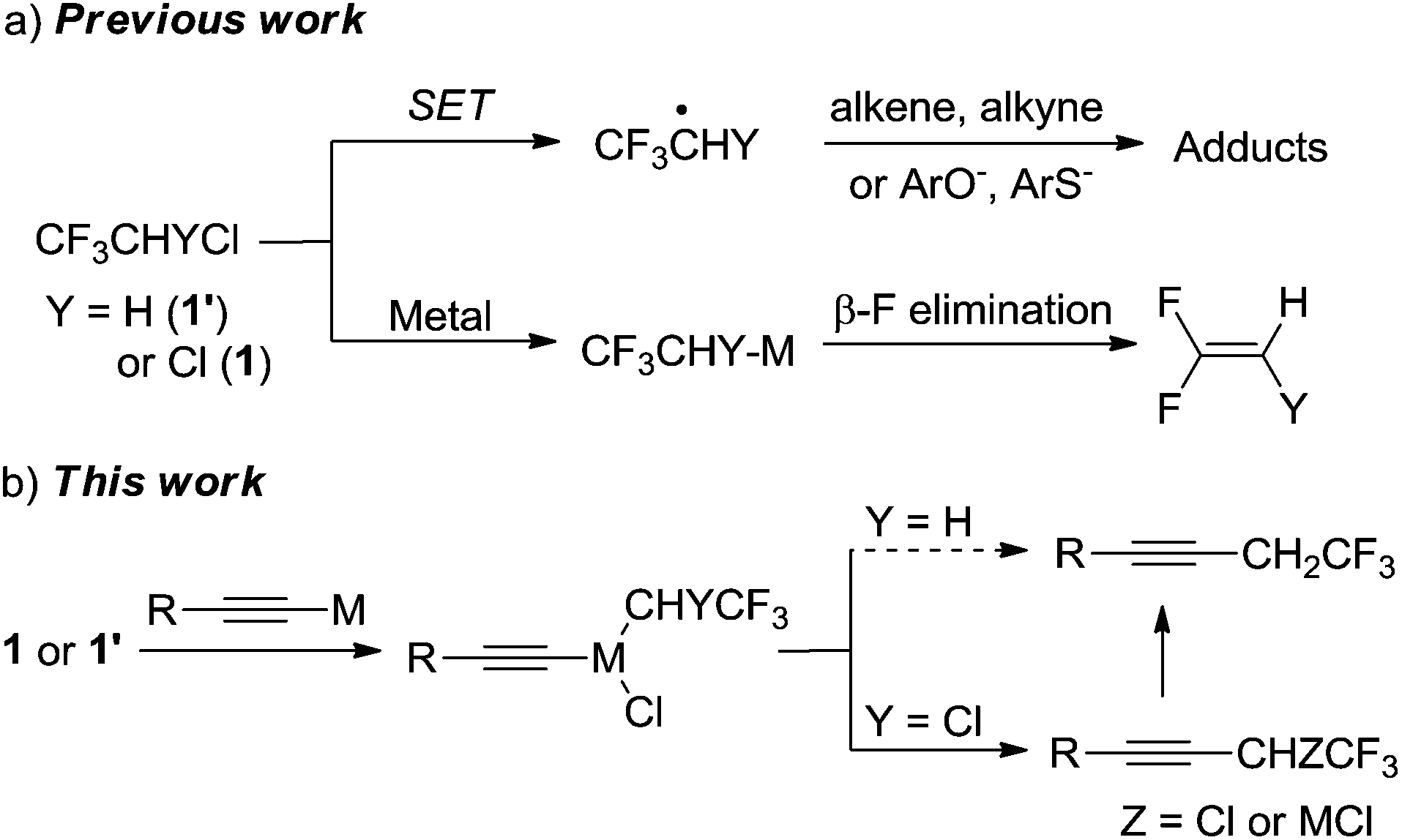 | ||
| Scheme 1 (a) A graphic summary of previous work related to the C–Cl activation of HCFC-133a and HCFC-123. (b) A novel transition metal mediated C–C cross-coupling method using HCFCs. | ||
In 2011, Shibata reported the first example of trifluoromethylation of propargyl halides by a trifluoromethylation of 1-(3-bromoprop-1-ynyl)-4-nitrobenzene using the electrophilic trifluoromethylation reagent [S-(trifluoromethyl)diphenylsulfonium salt].11a In 2012, Szabó reported the trifluoromethylation of propargylic halides and trifluoroacetates using the (Ph3P)3Cu(CF3) reagent to give the selective formation of allenylic or propargylic trifluoromethyl derivatives.11b These methodologies gave trifluoroethylated alkynes with the requirement of a stoichiometric amount of copper metal. In 2013, Cu-catalyzed trifluoromethylation of primary propargylic chlorides with trifluoromethyl trimethylsilane was reported by Nishibayashi.11c A specialized trifluoroethylated alkyne for biological research was synthesized in a good yield by the use of 1,1,1-trifluoro-2-iodoethane and a lithium reagent at an extremely low temperature.11d Some coupling approaches to trifluoromethylated alkynes have also been researched. Ma and his coworkers prepared trifluoromethylated alkynes in copper-catalyzed coupling reactions of terminal alkynes with 2,2,2-trifluorodiazoethane.11e Later, Xu11f and Lee11g independently disclosed the Pd-catalyzed coupling reaction of 1,1,1-trifluoro-2-iodoethane with terminal alkynes or aryl alkynyl carboxylic acids.
Herein, we report a novel cross-coupling method in which two distinct steps occur to synthesize trifluoroethylated alkynes (Scheme 1b) inspired by two classical cross-coupling reactions (Cadiot–Chodkiewicz coupling12 and Castro–Stephens coupling13). The first step is a generation of a transition-metal acetylide from the alkyne before the C–Cl activation. The second step is oxidative addition of the organometallic species to the C–Cl bond of a HCFC and then reductive elimination which yields the desired coupling product. This method is a practical and efficient way to trifluoromethylated alkynes by using a cheap metal copper, an ordinary amine and a low-cost chlorofluorohydrocarbon as a fluorinated building block.
Results and discussion
To test our hypothesis, we carried out the reaction between an isolated copper phenyl acetylide (2a′)14 and compound 1 in 1,2-dichloroethane (DCE) at 70 °C, and unfortunately no reaction occurred (Scheme 2, 1). However, on addition of two equivalents of diethylamine to the reaction, which was used to increase the solubility of the copper acetylide, the desired trifluoroethylated acetylene 3a was obtained in 40% yield (Scheme 2, 2). The addition of one equivalent of copper powder increased the yield from 40% to 61% (Scheme 2, 3). Further increasing the equivalents of copper powder and diethylamine did not improve the reaction yield (Scheme 2, 4). Considering that copper acetylide was synthesized from phenylacetylene 2a, we studied a one-step reaction using acetylene 2a as the substrate.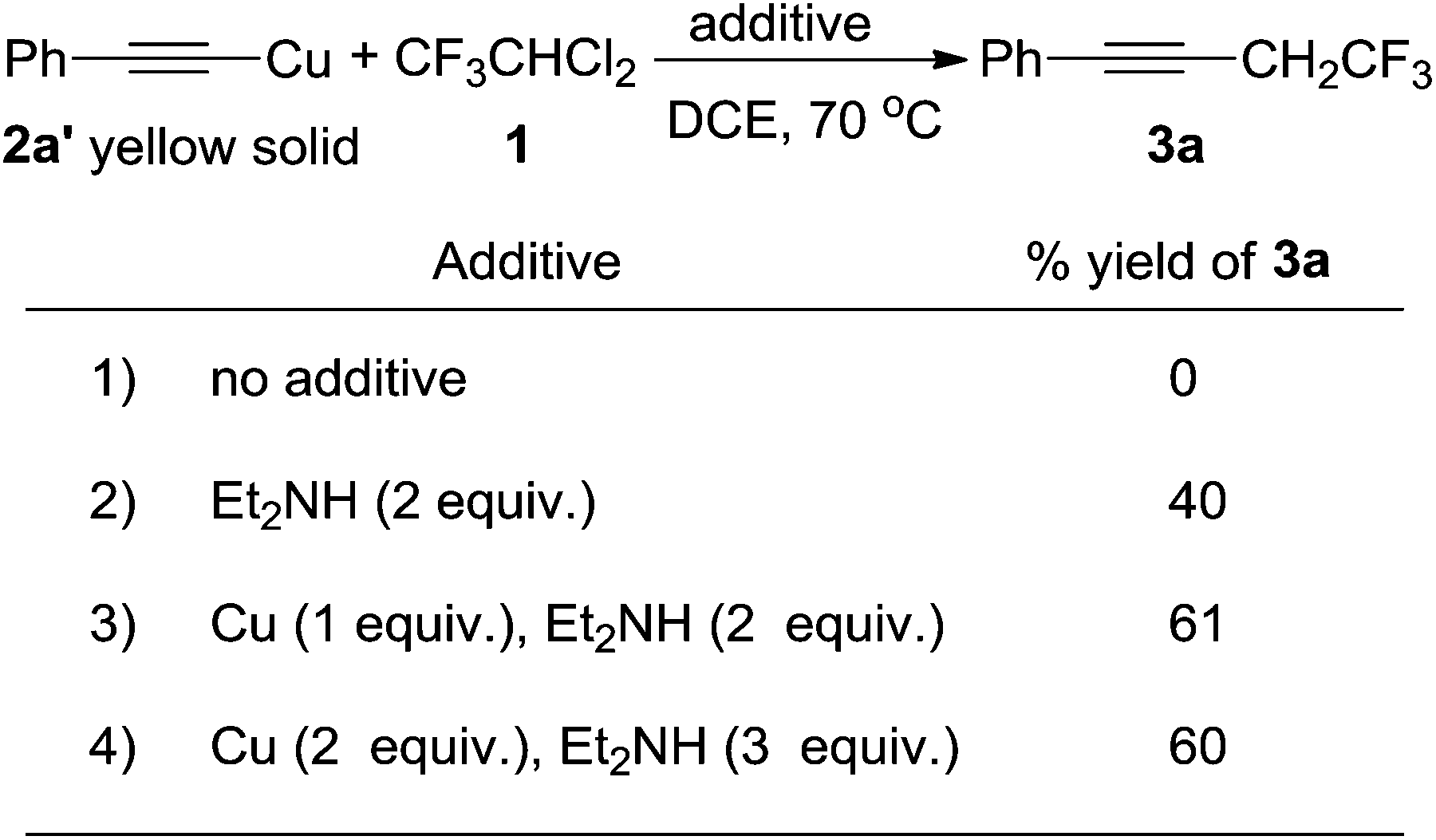 | ||
| Scheme 2 Reactions between the isolated copper acetylide 2a′, compound 1 and varying amounts of copper and diethylamine. | ||
To our delight, a one-pot reaction including acetylene 2a, HCFC-123, copper and diethylamine gave the desired product 3a in 77% yield (Scheme 3, entry 1). To further increase the yield we examined a range of amines. The reaction yields decreased slightly when using di-n-propylamine and di-n-butylamine (Scheme 3, entries 2 and 3). Using diallylamine gave 3a in a 30% yield (Scheme 3, entry 4). The secondary diamine N,N′-dimethyl-1,2-ethanediamine (DMEDA) also gave product 3a in 44% yield, however a side-product (4,4,4-trifluorobuta-1,2-dien-1-yl)benzene was detected (Scheme 3, entry 5). Moreover, the reactions using cyclic or aromatic secondary amines, sterically hindered secondary amines, primary amines, tertiary amines and tertiary diamines, all resulted in low-yielding or no reactions (Scheme 3, entries 6–12). These results suggested that electron-rich secondary linear amines, which are not sterically hindered, facilitated the reaction. What's more, the reaction did not occur when we replaced compound 1 with 2-chloro-1,1,1-trifluoroethane (HCFC-133a, 1′) (Scheme 3, entry 13). Plausibly, the second chlorine atom in compound 1 is required to activate the other C–Cl bond. Therefore, we used the optimal conditions described in Scheme 3 (entry 1) to test the scope of the reaction with a series of alkyne coupling partners (Scheme 4). Other conditions screened can be found in the ESI.† We have examined other solvents, such as DMF, THF, CH3NO2 and CH3CN (Table S1†). Those solvents were not effective.
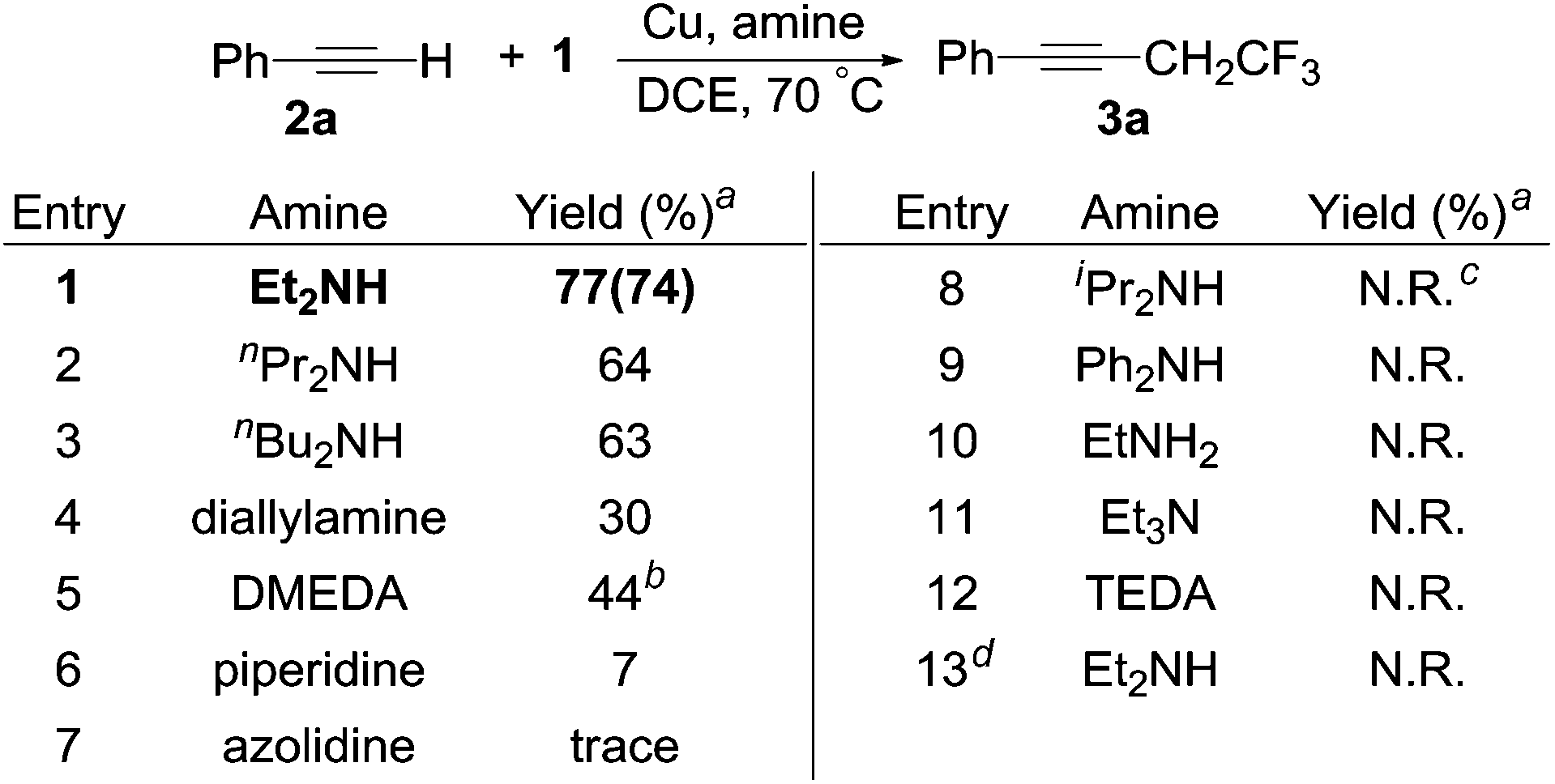 | ||
| Scheme 3 Effect of amines in the reaction between phenylacetylene with 1. The reactions were carried out with 2 (1 mmol) and 1 (2 mmol) in the presence of Cu powder (2 mmol) and amine (3 mmol) in 2 mL DCE at 70 °C in a Schlenk tube under a nitrogen atmosphere for 6 hours. aYields were determined by 19F NMR spectroscopy using benzotrifluoride as an internal standard and an isolated yield is in parentheses. b(4,4,4-Trifluorobuta-1,2-dien-1-yl)benzene (20% yield according to 19F NMR spectroscopy) was found with 3a. cN.R. = no reaction. d1′ was used instead of 1. | ||
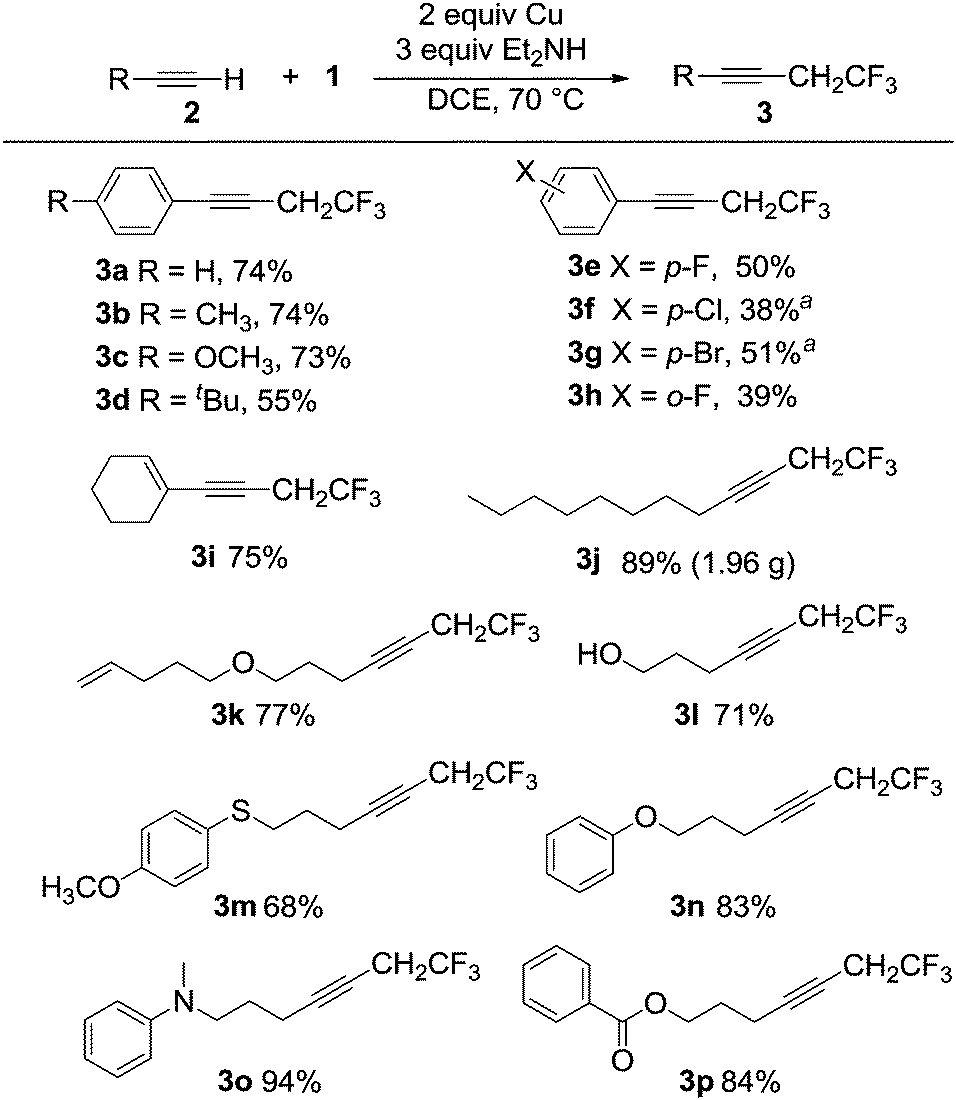 | ||
| Scheme 4 Scope of 2,2,2-trifluoroethylation of various alkynes with 1. Reaction was carried out by using alkyne (1 mmol) and 1 (2 mmol) in the presence of Cu (2 mmol) and diethylamine (3 mmol) in DCE (2 mL) at 70 °C under a nitrogen atmosphere in a Schlenk tube for 6–8 hours and isolated yields are shown. aUsing 1 as the sole solvent. | ||
Reactions involving a series of related electron-rich and electron-poor aromatic alkynes gave products 3a–3h in isolated yields between 38% and 74%, as shown in Scheme 4. The conjugated alkyne 3i and the aliphatic alkyne 3j were obtained in 75% and 89% yields, respectively. The coupling reaction occurred selectively at the alkyne in the presence of a terminal alkene giving product 3k in 77% yield. The reaction was also tolerant of a variety of functional groups, including hydroxyl (3l, 71%), ether and thioether (3m, 68%; 3n, 83% and 3k, 77%), tertiary amine (3o, 94%) and ester groups (3p, 84%). The C–C coupling reaction using the α-bromo carbonyl compound 2q was successful, however the amine substituted compound 3q was the major product isolated (Scheme 5). The functional group compatibility of the reaction allowed us to synthesize a fluorinated analogue of terbinafine 3r in 51% yield (Scheme 5). The reaction is not compatible with aryl alkynes bearing electron-deficient functional groups and heteroaryl alkynes bearing nitrogen and sulfur. For example, reactions with 4-acetylphenylacetylene, 2-ethynylpyridine and 2-ethynylthiophene gave no products. In spite of the very small substrate limitation, the reactions are scalable, which makes the reaction practical for further industry application. We were able to synthesize 3a in a 100 mL autoclave, which gave 5 grams of the alkyne product in one pot. In a 20 mL Schlenk tube, 3j could be obtained on a multi-gram scale.
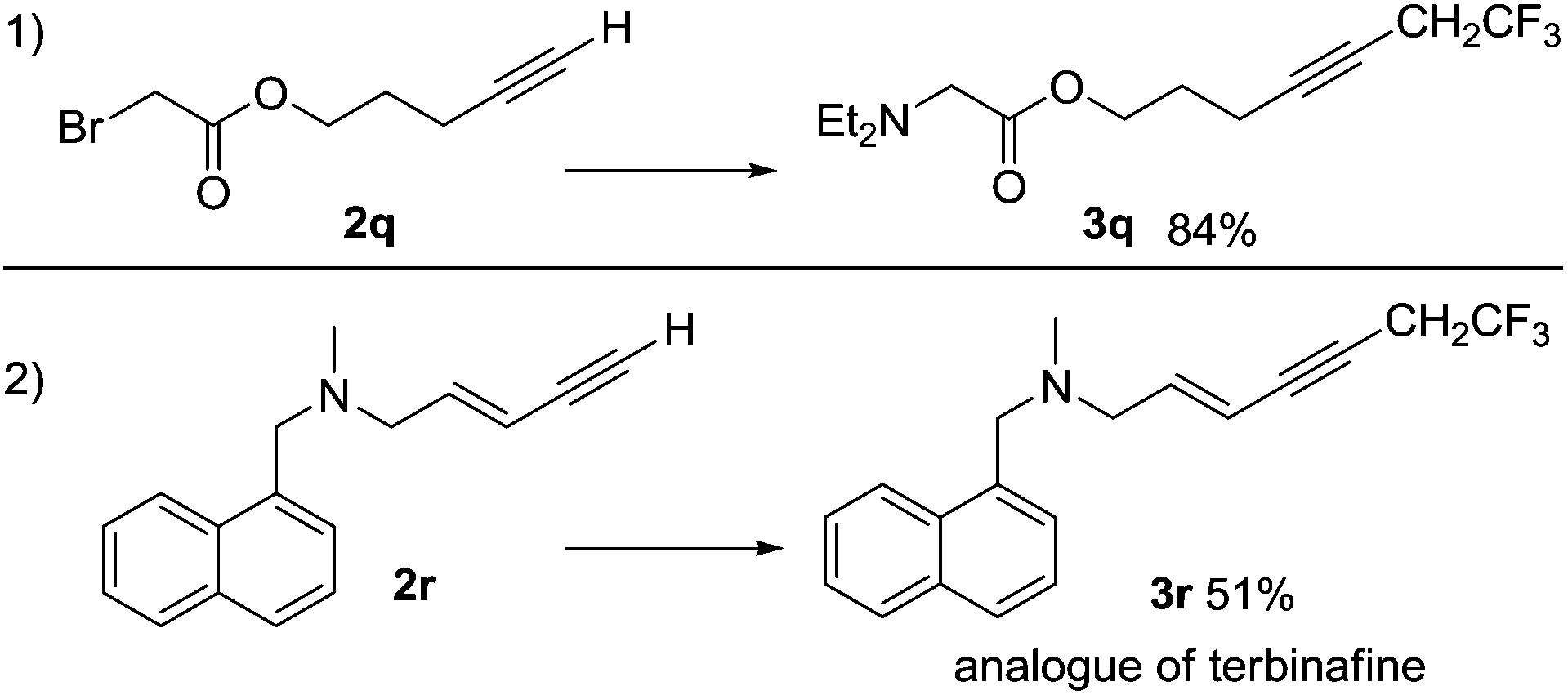 | ||
| Scheme 5 Some special alkyne substrates in the reactions with 1. Reaction conditions as Scheme 4. | ||
To determine the source of protons in the final products we conducted a series of experiments using deuterated materials (Scheme 6). The reaction in which the deuterated alkyne d-2a was used gave the desired product in 64% total yield, of which 17% was the deuterated product. Similar total yields were obtained when using either N-deuterated diethylamine or adding three equivalents of D2O to the reaction mixture, however both gave the deuterated product in an approximately 50% ratio. These results indicated that the protons in the products originated from the acidic protons within the reagents or water in solvents. The reaction in which ethyl-deuterated d10-diethylamine was used did not yield any deuterated products suggesting that redox reactions of diethylamine might not occur. These experiments don't support a carbenoid species because the ratio of deuterium to hydrogen at the propargyl positions in the corresponding products should be identical to that in the starting alkyne substrates if a carbenoid mechanism is involved.11e
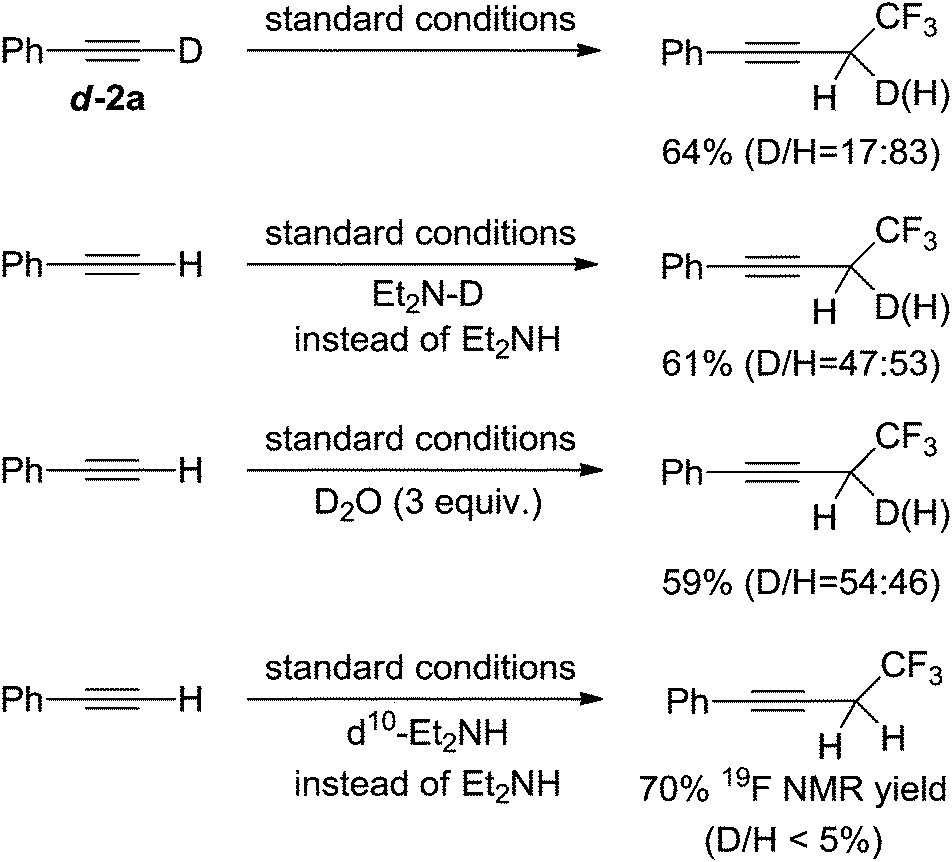 | ||
| Scheme 6 Deuterated experiments for determination of H-sources. | ||
Copper is known to initiate the cleavage of the C–Cl bond in compound 1via a SET in reactions involving phenols, thiophenols and styrene.7a However, we found that the reactions with terminal alkynes were highly tolerant of aromatic rings, conjugated double bonds (3i) and even terminal double bonds (3k) (Scheme 4). Contrast experiments showed that in the absence of an alkyne, styrene (4, Scheme 7) could indeed react with 1via a radical addition.7a However, the reactions between compounds 1, 2a and 4 found that the generation of the radical adduct 5 was much less than that of 3a (Scheme 7). This indicated that the generation of radicals under these reaction conditions was not favorable.
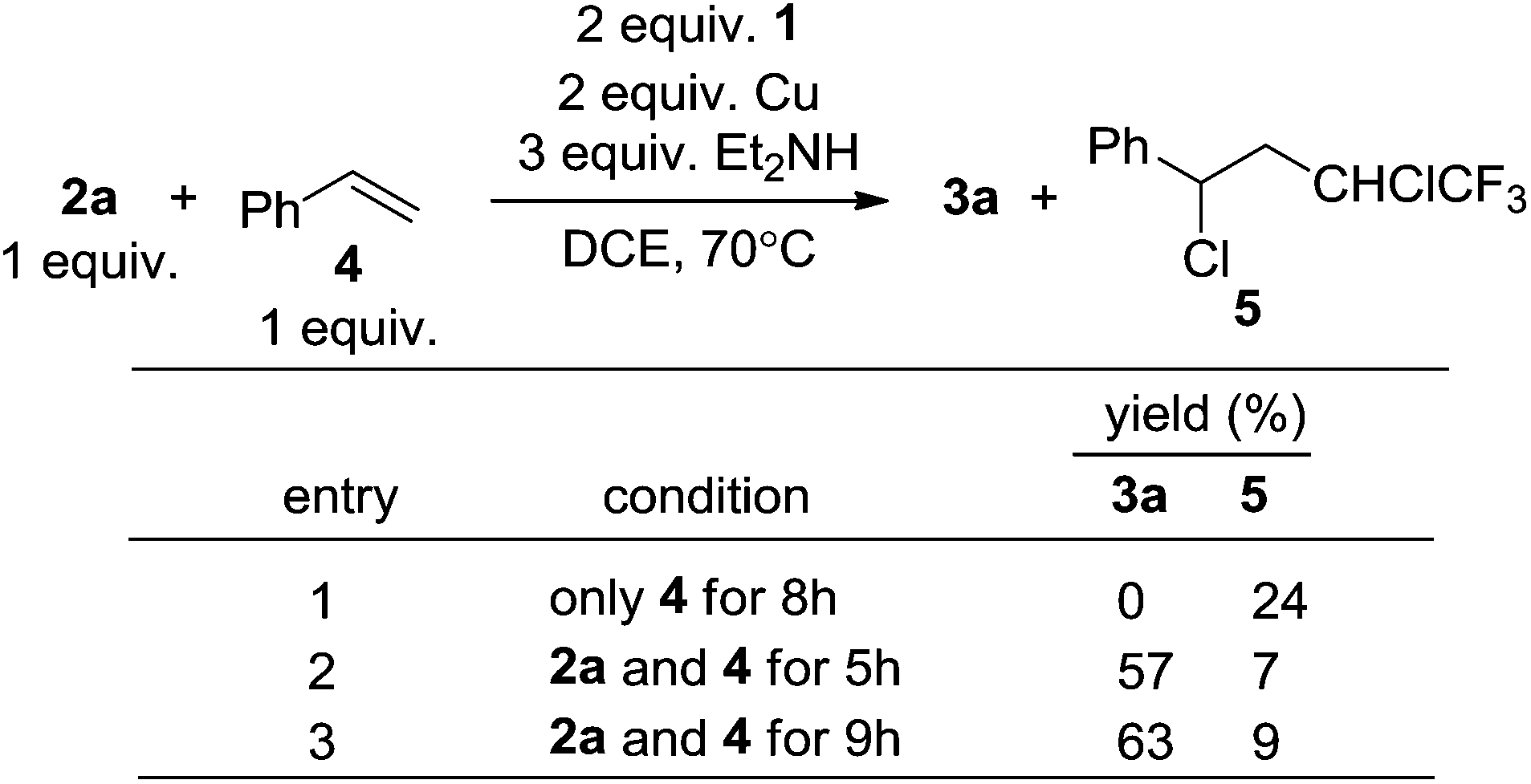 | ||
| Scheme 7 Relative rate between alkyne 2a and alkene 4. | ||
Control experiments including electron transfer and radical inhibitors, including hydroquinone (HQ), 1,4-dinitrobenzene (DNB), 2,6-di-tert-butyl-4-methylphenol (BHT) and 2,2,6,6-tetramethylpiperidin-1-oxy (TEMPO), showed no significant influence on the reaction yields (Scheme 8a/b). Importantly, when the radical trap diallyl ether (DAE) was added to the reaction no radical addition products were detected (Scheme 8), suggesting that fluorinated-alkyl free radicals may not be involved in the reaction.
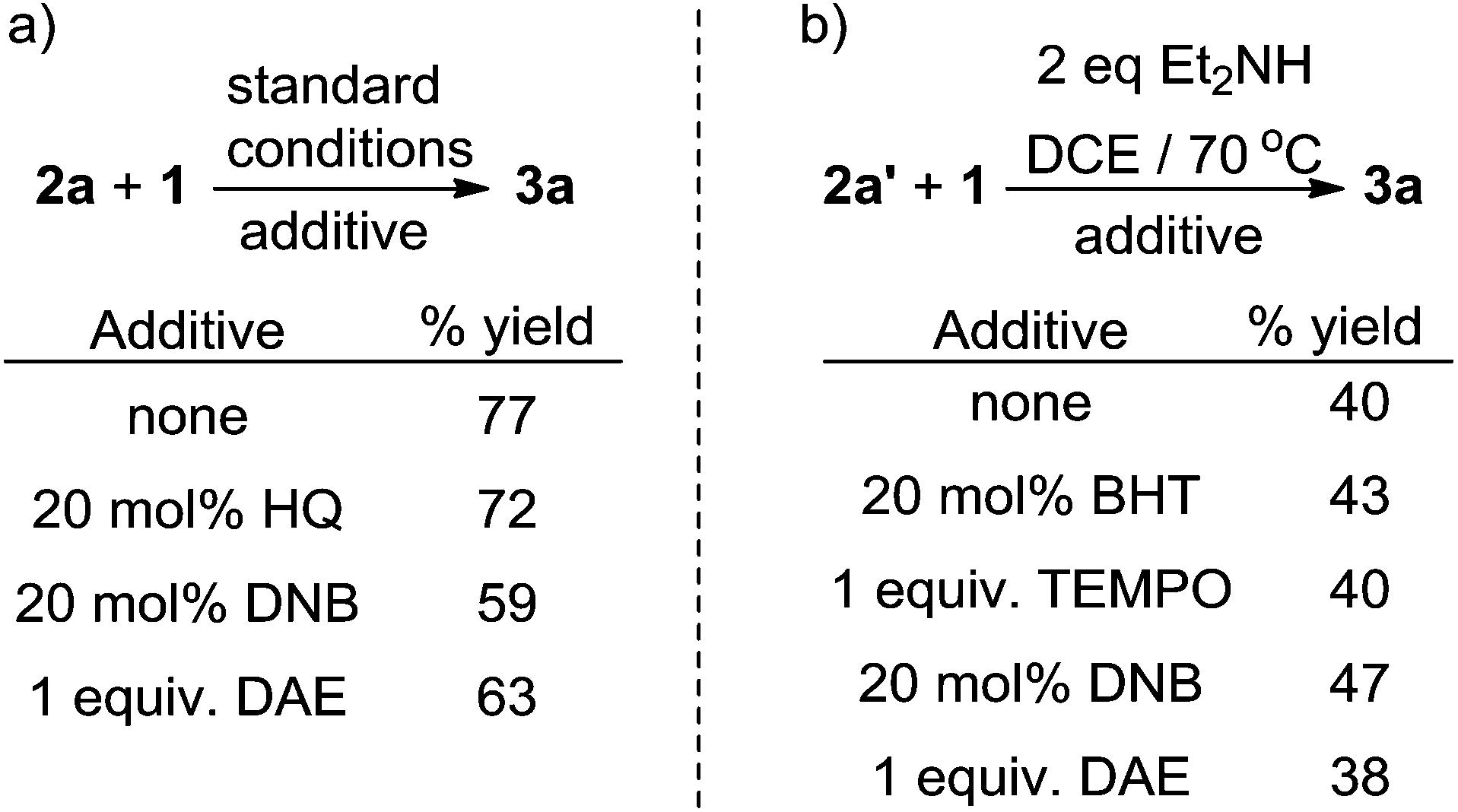 | ||
| Scheme 8 Mechanism experiments with radical scavengers. | ||
Therefore, an outer-sphere radical process is unlikely to be the predominating pathway and an oxidative addition mechanism may be involved.15 However, rigorous investigations are necessary to unambiguously elucidate the detailed mechanism.
Conclusions
We have successfully developed a Cu-mediated 2,2,2-trifluoroethylation of a series of terminal alkynes using HCFC-123. These reactions are compatible with a range of functional groups and can be performed on a gram scale in an efficient and economic manner. Mechanistic experiments indicate that an outer-sphere radical process is unlikely to be the predominating pathway. This study has revealed the first Csp3–Csp cross-coupling reaction using a commonly available HCFC, providing a profitable way to utilize the ozone-depleting and globe-warming chemicals to generate valuable fluorinated molecules. Detailed mechanistic studies and further applications of this reaction are in progress in our laboratory.Experimental section
General information
NMR spectra were obtained on 300 or 400 MHz spectrometers and recorded at 25 °C. Chemical shifts for 1H NMR spectra are reported in ppm downfield from TMS, chemical shifts for 13C NMR spectra are recorded in ppm relative to internal chloroform (δ 77.0 ppm for 13C), and chemical shifts for 19F NMR are reported in ppm downfield from fluorotrichloromethane (CFCl3). Coupling constants (J) are reported in hertz. The terms m, s, d, t, q and br refer to multiplet, singlet, doublet, triplet, quartet and broad, respectively. 13C NMR was broad-band decoupled from hydrogen nuclei.1 Absorbance frequencies in infrared spectra (IR) are given at maximum intensity in cm−1. The mass analyzer type used for HRMS is time-of-flight mass spectrometer (TOF-MS) or Fourier transform ion cyclotron resonance mass spectrometer (FTICR-MS). Column chromatography was performed using silica gel (mesh 300–400).All reagents were used as received from commercial sources or prepared as described in references. All reagents were weighed and handled in air. Substrates 1 and 1′ were purchased from commercial sources and used as received. Substrates 2k,162m,172n,172o,182p,192q19 and 2r20 were prepared according to literature procedures.
Preparation of (phenylethynyl)copper 2a′14
CuI (2.02 g, 10.5 mmol) was dissolved in NH3·H2O (28% NH3 solution, 25 mL) and EtOH (15 mL) to form a blue solution. While stirring, phenylacetylene (1.02 g, 10.0 mmol) was added dropwise to the solution. The system was allowed to stand for 2 h to form a yellow precipitate. The precipitate was filtered out and successively washed with NH3·H2O (10% NH3 solution, 3 × 25 mL), H2O (3 × 25 mL), EtOH (3 × 25 mL), and Et2O (3 × 25 mL). The bright yellow solid was then dried under high vacuum to afford the desired polymeric copper acetylide 2a′, which was used without further purification.General procedure for the reaction of (phenylethynyl)copper 2a′ with 1,1-dichloro-2,2,2-trifluoroethane (HCFC-123)
To a 5 mL Schlenk tube were added (phenylethynyl)copper 2a′ (165 mg, 1 mmol) and copper powder (0 to 2 mmol). The tube was then evacuated and backfilled with N2 (3 times). HCFC-123 (2 mmol), Et2NH (0 to 3 mmol) and DCE (2 mL) were added into this Schlenk tube subsequently under N2. The tube was sealed and heated to 70 °C (oil bath). After stirring for 8 h, the reaction mixture was cooled to room temperature and benzotrifluoride was added. The yield was determined by 19F NMR spectroscopy using benzotrifluoride as an internal standard before working up the reaction. If necessary, the reaction mixture was diluted with petroleum ether (PE, 10 mL) and the precipitate was removed by filtration. The filtrate was concentrated and the residue was purified with silica gel chromatography (petroleum ether as the eluent) to give the pure product 3a.Screening conditions for reactions of phenylacetylene 2a with 1,1-dichloro-2,2,2-trifluoroethane (HCFC-123) in the presence of various metals and salts (Tables 1S and 2S†)
To a 5 mL Schlenk tube was added metal powder (x mmol). The tube was then evacuated and backfilled with N2 (3 times). HCFC-123 (y mmol), phenylacetylene 2a (102 mg, 1 mmol), amine (z mmol) and solvent (2 mL) were added into this Schlenk tube subsequently under N2. The Schlenk tube was sealed and heated to 60–80 °C (oil bath). After stirring for 8 h, the reaction mixture was cooled to room temperature and benzotrifluoride was added. The yield was determined by 19F NMR spectroscopy using benzotrifluoride as an internal standard before working up the reaction. If necessary, the reaction mixture was diluted with PE (10 mL) and the precipitate was removed by filtration. The filtrate was concentrated in a vacuum and the residue was purified with silica gel chromatography (petroleum ether as the eluent) and concentrated in a vacuum to give the pure product 3a.General procedure for the copper-mediated 2,2,2-trifluoroethylation of 2,2-dichloro-1,1,1-trifluoroethane (HCFC-123) with various alkynes
To a 5 mL Schlenk tube was added Cu powder (128 mg, 2 mmol). The tube was then evacuated and backfilled with N2 (3 times). HCFC-123 (306 mg, 2 mmol), alkyne (1 mmol), Et2NH (219 mg, 3 mmol) and DCE (2 mL) were added into this Schlenk tube subsequently under N2. The Schlenk tube was sealed and heated to 70 °C (oil bath). After stirring for 8 h, the reaction mixture was cooled to room temperature and diluted with PE (10 mL). The precipitate was removed by filtration. The solvent was removed in a vacuum and the residue was purified with silica gel chromatography (petroleum ether as the eluent) to provide the pure product.Autoclave procedure: Cu powder (3.8 g, 60 mmol), phenylacetylene (4.1 g, 40 mmol), diethylamine (8.8 g, 120 mmol), 1,1-dichloro-2,2,2-trifluoroethane (12.2 g, 80 mmol) and 1,2-dichloroethane (40 mL) were added to an autoclave (0.1 L). After removal of air in the autoclave under vacuum, the reaction was run at 70 °C for 7 h. The reaction was stopped and allowed to stand overnight. The solid was removed by filtration and rinsed with ethyl acetate. The organic phase was washed with deionized water (50 mL) and a saturated solution of sodium chloride (50 mL). The organic phase was dried over anhydrous sodium sulfate. After removal of the solvent, the residue was distilled under vacuum to give the product as a colorless liquid (oil bath 120 °C/2 mmHg, 5.2 g, 28 mmol, 70%).
Yield: 89%; 1.96 g; yellow oil. 1H NMR (300 MHz, CDCl3) δ 3.00 (qm, J = 9.0 Hz, 2H), 2.13–2.19 (m, 2H), 1.47–1.52 (m, 2H), 1.27–1.35 (m, 10H), 0.88 (t, J = 9.0 Hz, 3H); 19F NMR (282 MHz, CDCl3) δ −67.5 (t, J = 9.8 Hz); 13C NMR (100 MHz, CDCl3) δ 124.5 (q, J = 275 Hz), 85.0, 68.1 (q, J = 5 Hz), 29.2, 29.1, 28.8, 28.4, 26.1 (q, J = 34 Hz), 22.7, 18.6, 14.1; IR (neat) ν/cm−1: 2930.6, 2858.4, 2119.9, 1642.8, 1493.9, 1468.4, 1422.0, 1357.2, 1257.8, 1157.9, 1139.6, 1112.9, 908.8, 834.4, 656.3. Anal. calcd for C12H19F3: C 65.43, H 8.69; found: C 65.50, H 8.70.
Isotopic labeling experiments
(1) To a 5 mL Schlenk tube was added Cu powder (128 mg, 2 mmol). The tube was then evacuated and backfilled with N2 (3 times). HCFC-123 (306 mg, 2 mmol), terminal deuterated alkyne (103 mg, 1 mmol), Et2NH (219 mg, 3 mmol) and DCE (2 mL) were added into this Schlenk tube subsequently under N2. The Schlenk tube was sealed and heated to 70 °C (oil bath). After stirring for 8 h, the reaction mixture was cooled to room temperature and diluted with PE (10 mL). The precipitate was removed by filtration. The solvent was removed and the residue was purified with silica gel chromatography (PE) and concentrated in a vacuum to provide the pure product.(2) To a 5 mL Schlenk tube was added Cu powder (128 mg, 2 mmol). The tube was then evacuated and backfilled with N2 (3 times). HCFC-123 (306 mg, 2 mmol), alkyne (102 mg, 1 mmol), N-deuterated diethylamine (222 mg, 3 mmol) and DCE (2 mL) were added into this Schlenk tube subsequently under N2. The Schlenk tube was sealed and heated to 70 °C (oil bath). After stirring for 8 h, the reaction mixture was cooled to room temperature and diluted with PE (10 mL). The precipitate was removed by filtration. The solvent was removed and the residue was purified with silica gel chromatography (PE) to provide the pure product.
(3) To a 5 mL Schlenk tube was added Cu powder (128 mg, 2 mmol). The tube was then evacuated and backfilled with N2 (3 times). HCFC-123 (306 mg, 2 mmol), alkyne (102 mg, 1 mmol), Et2NH (219 mg, 3 mmol), D2O (57 mg, 3 mmol) and DCE (2 mL) were added into this Schlenk tube subsequently under N2. The Schlenk tube was sealed and heated to 70 °C (oil bath). After stirring for 8 h, the reaction mixture was cooled to room temperature and diluted with PE (10 mL). The precipitate was removed by filtration. The solvent was removed and the residue was purified with silica gel chromatography (PE) to provide the pure product.
(4) To a 5 mL Schlenk tube was added Cu powder (128 mg, 2 mmol). The tube was then evacuated and backfilled with N2 (3 times). HCFC-123 (306 mg, 2 mmol), alkyne (102 mg, 1 mmol), d10-Et2NH (249 mg, 3 mmol) and DCE (2 mL) were added into this Schlenk tube subsequently under N2. The Schlenk tube was sealed and heated to 70 °C (oil bath). After stirring for 8 h, the reaction mixture was cooled to room temperature and diluted with PE (10 mL). The precipitate was removed by filtration. The solvent was removed and the residue was purified with silica gel chromatography (PE) and concentrated in a vacuum to provide the pure product.
Relative rate between alkyne 2a and alkene 4: to a 5 mL Schlenk tube was added Cu powder (102 mg, 2 mmol). The tube was then evacuated and backfilled with N2 (3 times). HCFC-123 (382 mg, 2.5 mmol), alkyne 2a (102 mg, 1 mmol), styrene 4 (104 mg, 1 mmol), Et2NH (219 mg, 3 mmol) and DCE (2 mL) were added into this Schlenk tube subsequently under N2. The Schlenk tube was sealed and heated to 70 °C (oil bath). After stirring for 5–9 h, the reaction mixture was cooled to room temperature and benzotrifluoride was added. The yield was determined by 19F NMR before working up. If necessary, the reaction mixture was diluted with PE (10 mL) and the precipitate was removed by filtration. The filtrate was concentrated and the residue was purified with silica gel chromatography (PE) and concentrated in a vacuum to give the pure product. Product 5 is a known compound.7a
Mechanism experiments with radical scavengers
Acknowledgements
Support of our work by the National Basic Research Program of China (973 Program) (no. 2012CB821600), the National Natural Science Foundation of China (no. 21421002, 21032006, 21172241) and the Chinese Academy of Sciences is gratefully acknowledged.Notes and references
- (a) P. Kirsch, Modern Fluoroorganic Chemistry. Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2nd edn, 2013 Search PubMed; (b) D. A. Good and J. S. Francisco, Chem. Rev., 2003, 103, 4999 CrossRef CAS PubMed; (c) V. G. Nenajdenko, V. M. Muzalevskiy and A. V. Shastin, Chem. Rev., 2015, 115, 973 CrossRef CAS PubMed; (d) P. L. Johnson, J. Labelled Compd. Radiopharm., 2007, 50, 47 CrossRef CAS PubMed.
- (a) R. Anilkumar and D. J. Burton, Tetrahedron Lett., 2002, 43, 6979 CrossRef CAS; (b) R. Anilkumar and D. J. Burton, J. Fluorine Chem., 2005, 126, 835 CrossRef CAS PubMed; (c) J. M. Bainbridge, S. J. Brown, P. N. Ewing, R. R. Gibson and J. M. Percy, J. Chem. Soc., Perkin Trans. 1, 1998, 2541 RSC.
- (a) K. Wu and Q.-Y. Chen, Tetrahedron, 2002, 58, 4077 CrossRef CAS; (b) K. Wu and Q.-Y. Chen, J. Fluorine Chem., 2002, 113, 79 CrossRef CAS.
- M. Tamura and A. Sekiya, J. Fluorine Chem., 1995, 71, 119 CrossRef CAS.
- X.-J. Tang and Q.-Y. Chen, Chem. Sci., 2012, 3, 1694 RSC.
- (a) Z.-Y. Long and Q.-Y. Chen, J. Org. Chem., 1999, 64, 4775 CrossRef CAS PubMed; (b) X.-T. Huang and Q.-Y. Chen, J. Org. Chem., 2001, 66, 4651 CrossRef CAS PubMed; (c) X.-T. Huang, Z.-Y. Long and Q.-Y. Chen, J. Fluorine Chem., 2001, 111, 107 CrossRef CAS; (d) K. Wu and Q.-Y. Chen, Chin. J. Chem., 2004, 22, 371 CrossRef CAS PubMed. For a review, see: (e) C.-P. Zhang, Q.-Y. Chen, Y. Guo, J.-C. Xiao and Y.-C. Gu, Chem. Soc. Rev., 2012, 41, 4536 RSC.
- (a) X.-J. Tang and Q.-Y. Chen, Org. Lett., 2012, 14, 6214 CrossRef CAS PubMed; (b) X.-J. Tang and Q.-Y. Chen, J. Fluorine Chem., 2015, 169, 1 CrossRef CAS PubMed.
- Y. Xu, W. R. Dolbier Jr. and X. X. Rong, J. Org. Chem., 1997, 62, 1576 CrossRef CAS.
- For selected recent publications for the metal mediated coupling reaction of fluorinated alkyl iodides/bromides, see: (a) Y. Zhao and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 1033 CrossRef CAS PubMed; (b) Z. Feng, Q.-Q. Min, Y.-L. Xiao, B. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1669 CrossRef CAS PubMed; (c) Q.-Q. Min, Z. Yin, Z. Feng, W.-H. Guo and X. Zhang, J. Am. Chem. Soc., 2014, 136, 1230 CrossRef CAS PubMed; (d) Q. Qi, Q. Shen and L. Lu, J. Am. Chem. Soc., 2012, 134, 6548 CrossRef CAS PubMed; (e) Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034 CrossRef CAS PubMed; (f) Y.-L. Xiao, W.-H. Guo, G.-Z. He, Q. Pan and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 9909 CrossRef CAS PubMed.
- K. Uneyama, T. Katagiri and H. Amii, Acc. Chem. Res., 2008, 41, 817 CrossRef CAS PubMed.
- (a) H. Kawai, T. Furukawa, Y. Nomura, E. Tokunaga and N. Shibata, Org. Lett., 2011, 13, 3596 CrossRef CAS PubMed; (b) T. S. N. Zhao and K. J. Szabó, Org. Lett., 2012, 14, 3966 CrossRef CAS PubMed; (c) Y. Miyake, S. Ota, M. Shibata, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2013, 49, 7809 RSC; (d) Y. Matsuya, D. Ihara, M. Fukuchi, D. Honma, K. Itoh, A. Tabuchi, H. Nemoto and M. Tsuda, Bioorg. Med. Chem., 2012, 20, 2564 CrossRef CAS PubMed; (e) C.-B. Liu, W. Meng, F. Li, S. Wang, J. Nie and J.-A. Ma, Angew. Chem., Int. Ed., 2012, 51, 6227 CrossRef CAS PubMed; (f) Y.-S. Feng, C.-Q. Xie, W.-L. Qiao and H.-J. Xu, Org. Lett., 2013, 15, 936 CrossRef CAS PubMed; (g) J. Hwang, K. Park, J. Choe, H. Min, K. H. Song and S. Lee, J. Org. Chem., 2014, 79, 3267 CrossRef CAS PubMed.
- J.-L. Philippe, W. Chodkiewicz and P. Cadiot, Tetrahedron Lett., 1970, 11, 1795 CrossRef.
- R. D. Stephens and C. E. Castro, J. Org. Chem., 1963, 28, 3313 CrossRef CAS.
- For synthesis of copper phenyl acetylide, see: (a) W. Shi, Y. Luo, X. Luo, L. Chao, H. Zhang, J. Wang and A. Lei, J. Am. Chem. Soc., 2008, 130, 14713 CrossRef CAS; (b) C. Theunissen, M. Lecomte, K. Jouvin, A. Laouiti, C. Guissart, J. Heimburger, E. Loire and G. Evano, Synthesis, 2014, 1157 Search PubMed.
- It is reasonable to propose a concerted oxidation addition mechanism because “metal complexes that do not readily undergo one-electron processes tend to react by two-electron mechanisms”. (a) J. F. Hartwig, Organotransition Metal Chemistry: from Bonding to Catalysis, University Science Books, Sausalito, 2010, pp. 301–320 Search PubMed; (b) A. I. Konovalov, A. Lishchynskyi and V. V. Grushin, J. Am. Chem. Soc., 2014, 136, 13410 CrossRef CAS PubMed; (c) D.-H. Yu, J.-N. Shao, R.-X. He and M. Li, Chin. Chem. Lett., 2015, 26, 564 CrossRef CAS PubMed.
- J. F. Reichwein, M. C. Patel and B. L. Pagenkopf, Org. Lett., 2001, 3, 4303 CrossRef CAS PubMed.
- M. Rivara, M. K. Patel, L. Amori and V. Zuliani, Bioorg. Med. Chem. Lett., 2012, 22, 6401 CrossRef CAS PubMed.
- A. Chevalier, C. Massif, P.-Y. Renard and A. Romieu, Chem. – Eur. J., 2013, 19, 1686 CrossRef CAS PubMed.
- B. D. Stevens and S. G. Nelson, J. Org. Chem., 2005, 70, 4375 CrossRef CAS PubMed.
- M. Alami, F. Ferri and Y. Gaslain, Tetrahedron Lett., 1996, 37, 57 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details for experimental conditions, copies of 19F NMR, 1H NMR and 13C NMR spectra for isolated compounds. See DOI: 10.1039/c5qo00210a |
| This journal is © the Partner Organisations 2015 |