
Catalytic dehydrogenative aromatization: an alternative route to functionalized arenes†
Simon A.
Girard‡
a,
Huawen
Huang‡
b,
Feng
Zhou‡
a,
Guo-Jun
Deng
*b and
Chao-Jun
Li
*a
aDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, Montreal, QC H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
bKey Laboratory of Environmentally Friendly Chemistry and Application of Ministry of Education, College of Chemistry, Xiangtan University, Xiangtan 411105, China. E-mail: gjdeng@xtu.edu.cn
First published on 30th January 2015
Abstract
Catalytic dehydrogenative aromatization has emerged as an efficient and environmentally friendly way to access functionalized arenes in recent years. Typically, the compounds bearing an aliphatic six-membered ring, for instance, cyclohexanones, are used as arylation sources. The transformation process commonly involves a sequential, nucleophilic addition, dehydration and catalytically oxidative dehydrogenation, providing convenient entries to carbon–carbon and carbon–heteroatom bond formations. Compared to previous arylation methods, this strategy eliminates the use of harsh reaction conditions and the production of halide wastes, circumvents the issues of chem- and regioselectivities, and offers a milder/greener means for the synthesis of functionalized arenes. It is worth mentioning that this approach distinguishes itself from other methods by frequently using either oxygen as the sole oxidant or hydrogen transfer strategy. Furthermore, an obvious advantage, in terms of a sustainable chemical process, is that water is generated as the only by-product in most cases. This mini-review mainly focuses on our groups' contributions to the direct formation of C–O, C–N and C–C bonds using substituted cyclohexa(e)nones as flexible arylation sources and the extension of these methodologies to the construction of heterocycles.
 Simon A. Girard | Simon A. Girard was born in Saint-Foy-Les-Lyon (France). He received his Bachelor's and Master's degrees from Université Joseph Fourier, Grenoble 1. He is currently working under the supervision of Prof. Dr Chao-Jun Li for his PhD at McGill University in Montréal (Canada). His research interests focus on the development of environmentally friendly and more efficient catalytic methodologies for organic synthesis. |
 Huawen Huang | Huawen Huang was born in Hunan Province, China in 1987. He received his B.S. from Northwest University (2009), and Ph.D. from South China University of Technology (SCUT) under the supervision of Professor Huanfeng Jiang (2014, June). He then joined the Key Laboratory for Environmentally Friendly Chemistry and Application of the Ministry of Education at College of Chemistry, Xiangtan University. His current research is focused on novel oxidative patterns and processes for catalytic organic synthesis. |
 Feng Zhou | Feng Zhou was born in 1983 in Wuhan, China. He received his B.S. degree from Wuhan University in 2006, and his Ph.D. from the Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences in 2011 under the supervision of Prof. Xiyan Lu. He is currently working as a postdoctoral fellow with Prof. Chao-Jun Li at the McGill University in Canada, focusing on transition metal catalyzed reactions. |
 Guojun Deng | Guojun Deng received his B.S. degree from Xiangtan University and completed his Ph.D. degree in 2004 in chemistry from Institute of Chemistry, Chinese Academy of Sciences with Prof. Qinhua Fan. Following his Ph.D., he conducted post-doctoral studies with Prof. Lukas J. Goossen at Max Planck Institute, with Prof. E. Schoffers at Western Michigan University and with Prof. Chao-jun Li at Tulane University and McGill University. In 2009, he joined the College of Chemistry at Xiangtan University as a full professor. His research interests lie in the development of environmentally benign methodologies for the C–C and C–hetero bond formations. |
 Chao-Jun Li | Chao-Jun Li received his B.Sc. at Zhengzhou University (1983), MS at the Chinese Academy of Sciences (1988) and Ph.D. at McGill University (1992). He spent 1992–94 as an NSERC Postdoctoral Fellow at Stanford University (USA), and was an assistant (1994), associate (1998) and full professor (2000) at Tulane University (USA). Since 2003, he has been a Canada Research Chair in Green Chemistry and an E. B. Eddy Chair Professor of Chemistry at McGill University. He is a Fellow of the Royal Society of Canada (Academy of Sciences) and a Fellow of the American Association for the Advancement of Sciences (AAAS). |
1. Introduction
Aromatic compounds are a ubiquitous class of molecules. Besides their tremendous use as organic solvents, dyes, polymers and precursors for the food industry, aromatic moieties represent the core skeleton involved in countless essential biological processes. Consequently, modern medicinal and pharmaceutical chemists have actively pursued the discovery of novel aromatic molecules.1 Accordingly, the synthesis of aromatic compounds, i.e. arylation reactions, remains to date a hot topic of research. Numerous prominent methods have been developed throughout the history of organic chemistry (Scheme 1). The well established Friedel–Crafts reactions (Scheme 1a) have proved to be a robust strategy to access functionalized arenes; however, they suffer from harsh reaction conditions and issues related to chemo- and regioselectivities.2 Benzyne chemistry provides a flexible alternative procedure for arylation (Scheme 1b).3,4 Despite its high reactivity and synthetic diversity, benzynes are not easily available starting materials, which limits their wide application in synthesis.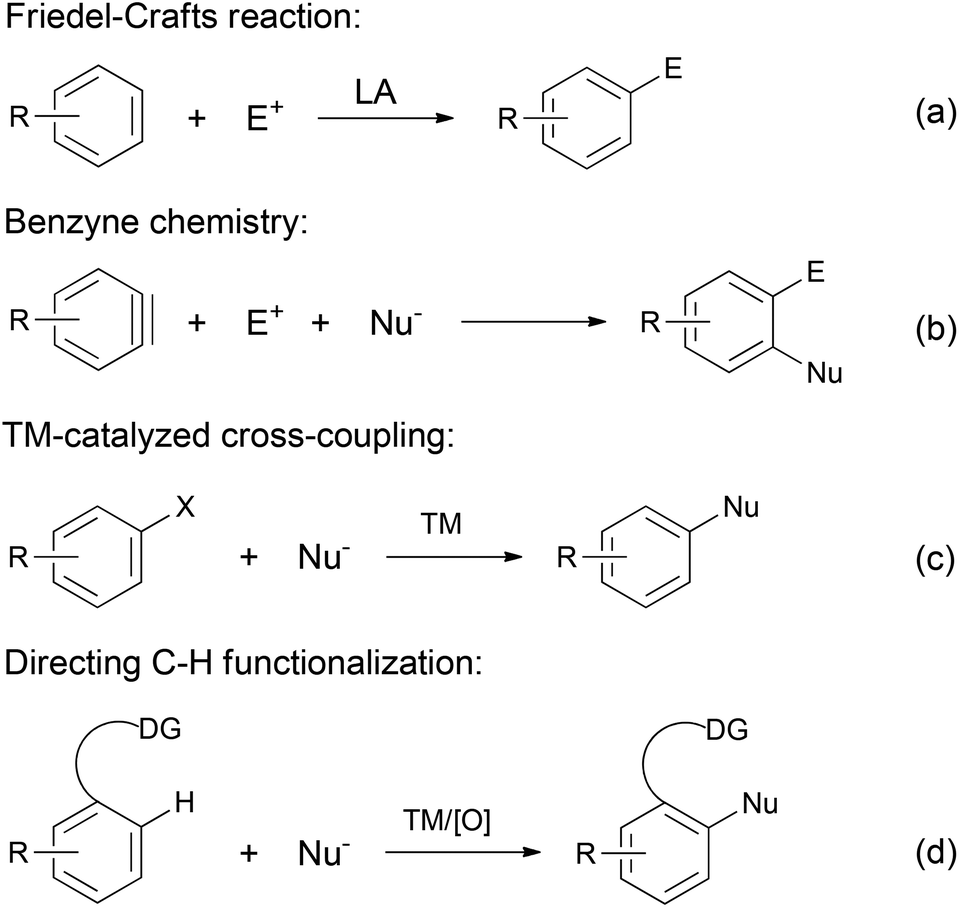 | ||
| Scheme 1 Arylation reactions. LA = Lewis acid; TM = transition metal; DG = directing group. | ||
Over the last few decades, transition metal-catalyzed cross-coupling has emerged as the modern approach for arylation, providing tremendously powerful tools for the versatile construction of aryl C–C and C–heteroatom bonds (Scheme 1c). While various transition metal catalysts have been developed for cross-coupling reactions, palladium-catalyzed reactions are especially effective. Among them, the Heck, the Negishi, the Suzuki–Miyaura, the Stille, and the Hiyama cross-couplings, as well as the more recent Buchwald–Hartwig reaction represent some of the outstanding reactions in this field and have been widely applied in industrial processes.5 Along with the rapid development of organometallic chemistry in the last few decades, a wide range of arylation strategies have been explored and successfully employed, including the use of directing group assisted direct C–H functionalizations (Scheme 1d).6–13 However, a common feature of all the above strategies is the requirement of prefunctionalization of the starting materials. An alternative strategy that obviates such a general requirement would be to go through direct dehydrogenation from substituted cyclohexa(e)nes (Scheme 2).
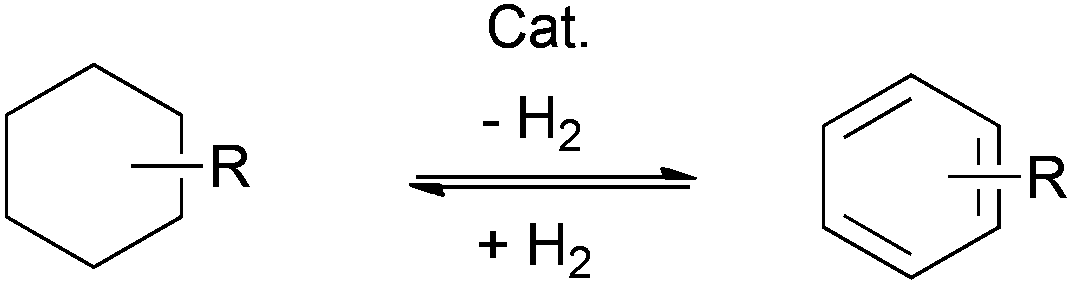 | ||
| Scheme 2 Catalytic dehydrogenation of cyclohexa(e)ne derivatives and hydrogenation of corresponding aryl compounds. | ||
Indeed, the transformation of cyclohexanones to phenols dated back to the early twentieth century, when heterogeneous dehydrogenation was applied to produce phenol.14–16 However, it is only in 2011 that the group of Stahl first presented a palladium-catalyzed homogeneous aerobic dehydrogenation of substituted cyclohexanones to the corresponding phenols.17 This palladium-based catalytic system, via the ingenious combination of two successive β-H eliminations and the aerobic oxidation of palladium, can successfully abstract hydrogen atoms from the six-membered ring to yield the aromatized product. It is conceivable that by introducing an appropriate nucleophile into the catalytic system, through the selective control of nucleophilic addition or conjugate addition followed by dehydrogenation, it would afford substituted arenes or phenols (Scheme 3).
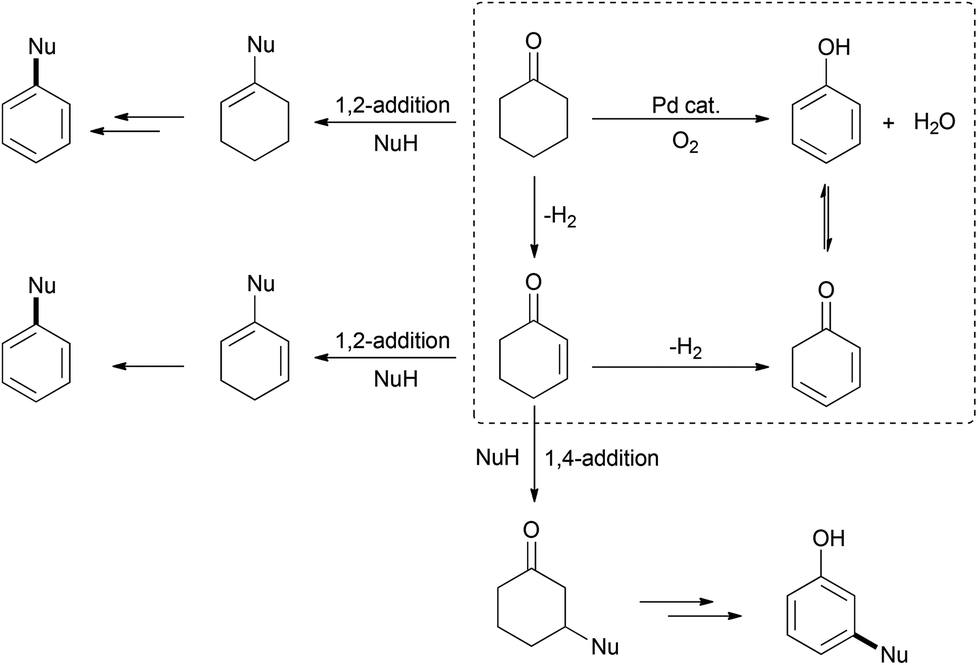 | ||
| Scheme 3 Use of nucleophiles for the synthesis of functionalized arenes. | ||
This mini-review will mainly focus on our groups' contributions toward the development of efficient systems for the formation of substituted arenes through tandem nucleophilic addition and oxidative aromatization. We will first discuss the formation of aryl-ethers from cyclohexanones and alcohols. Then effective procedures for the synthesis of aromatic amines will be presented, which will be followed by the subsequent use of carbon based nucleophiles. Finally, we will illustrate some of the applications of these methodologies in the synthesis of heterocylces.
1.1 C–O bond formation
In 2012, Li's group described a copper-catalyzed aerobic synthesis of aromatic ethers, in which the aromatic moiety stemmed from the substituted cyclohexenone via an oxidative aromatization (Scheme 4).18 While a stoichiometric amount of CuCl2 afforded dehydrogenative coupling under very handy reaction conditions, with the use of a co-catalyst N-hydroxyphthalimide (NHPI, 20 mol%) in the presence of potassium iodide (KI, 1 equiv.) and water (1 equiv.), a catalytic amount of copper triflate (Cu(OTf)2, 10 mol%) provided aryl-ethers in high yields. This reaction tolerated a broad range of alkyl alcohols with various functionalities, including iodo-, nitro-, alkenyl-, and alkynyl- (terminal and internal) groups. As well, natural alcohols such as β-citronellol, (−)-borneol, (−)-menthol, cholesterol and protected serine were successfully functionalized. Diols and diketones were also suitable reactants, and afforded the corresponding diethers in good yields.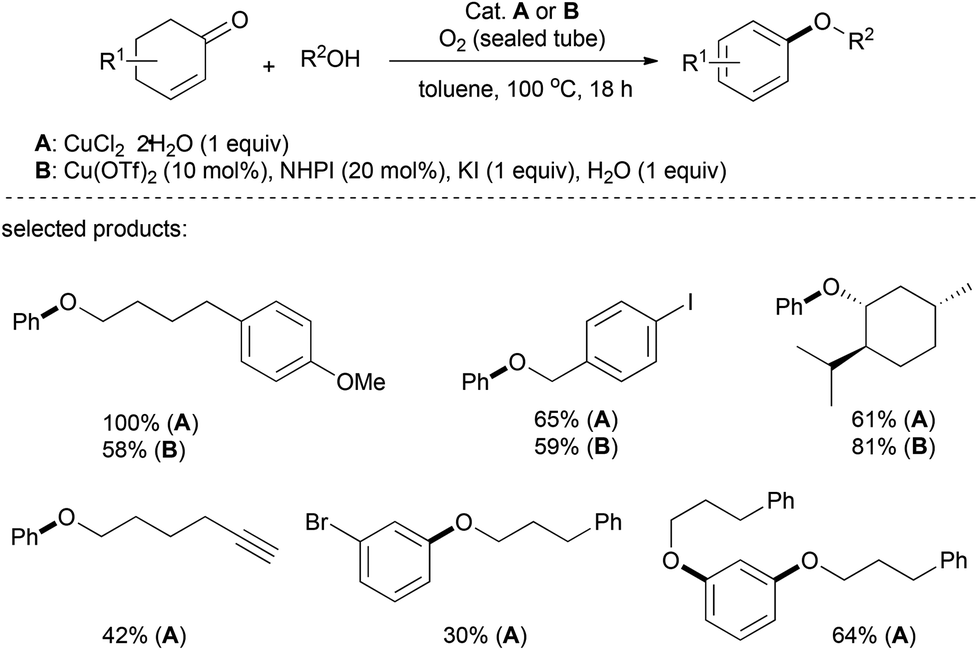 | ||
| Scheme 4 Aryl ethers from phenols/alcohols and cyclohexenones. | ||
Their proposed mechanism is presented in Scheme 5. First, the electrophilic activation of a ketone by the copper complex assists in the formation of the hemiacetal I, and the elimination of water leads to alkoxydiene II. The abstraction of a hydrogen atom by the in situ formed N-oxyl radical leads to diene radical III, which undergoes another hydrogen abstraction to yield the desired product IV.19,20
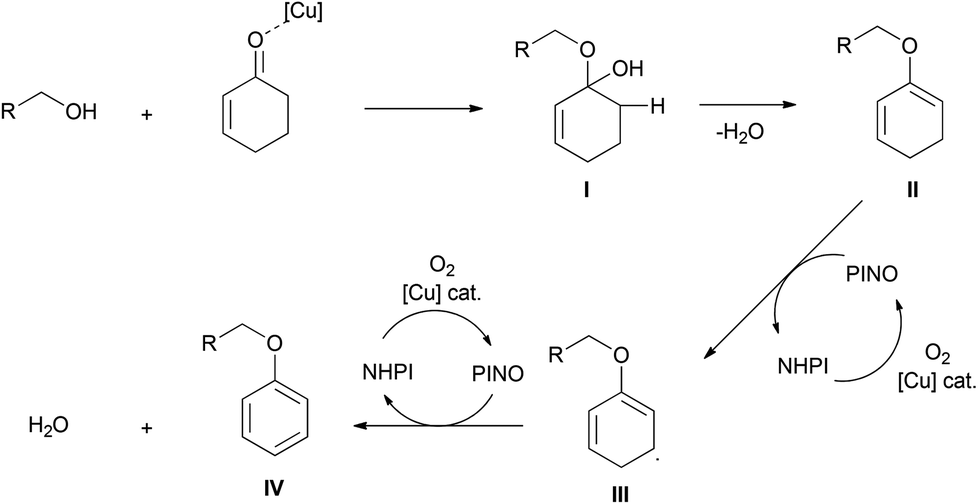 | ||
| Scheme 5 Proposed mechanism for the aryl ether formation. | ||
Shortly afterwards, Lemaire et al. presented a complementary carbon-supported palladium system for the synthesis of aryl ethers (Scheme 6).21,22 Interestingly, cyclohexanones have a higher level of activity than cyclohexenones under the present system and, although requiring an elevated temperature to achieve complete conversion, tetralones were also suitable partners. It is also worth noting that this process was also efficient with 2-methyltetrahydrothiophen-3-one to give the corresponding aryl ether in good yields. Various alkyl alcohols including polyols were suitable for this transformation.
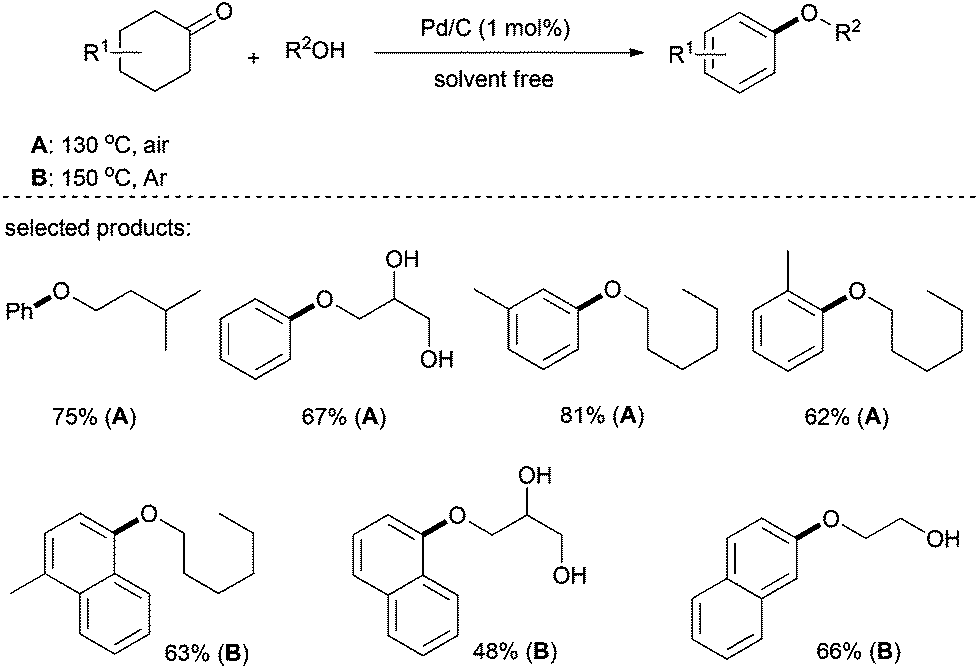 | ||
| Scheme 6 Pd/C catalyzed aryl ether formation. | ||
1.2 C–N bond formation
As exemplified by the formation of aromatic ethers via a copper-catalyzed process, it is conceivable that other nucleophiles such as amines (which are even more nucleophilic) would also undergo analogous nucleophilic addition–dehydrogenative aromatization to deliver the final aromatic products readily (Scheme 7).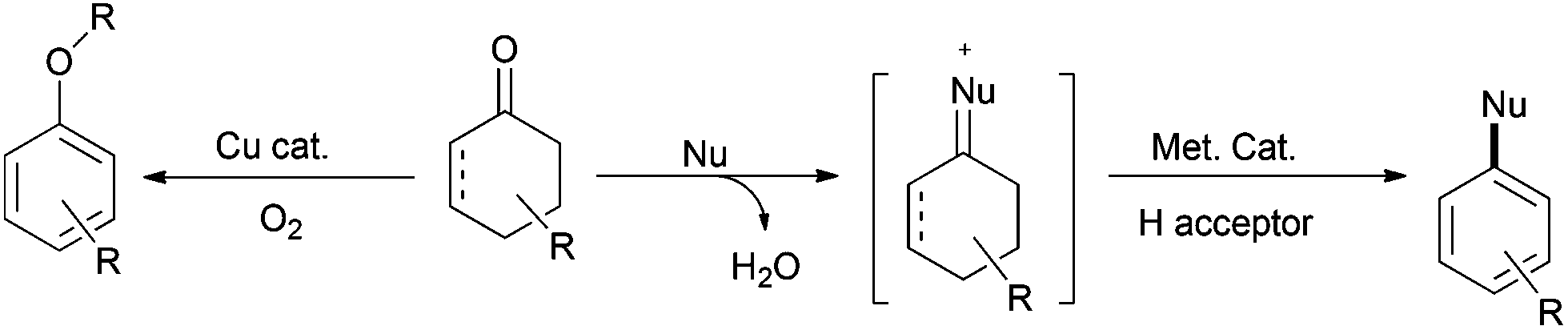 | ||
| Scheme 7 Aromatics via nucleophilic addition–dehydrogenative aromatization. | ||
Indeed, the groups of Li and Deng reported an efficient method for the preparation of aromatic amines. In this reaction, palladium was used as the metal catalyst and O2 as the sole terminal oxidant, generating water as the only by-product (Scheme 8).23 Various alkylamines such as piperidine, morpholine, dibenzylamine, indoline, and even diallylamine reacted smoothly to afford N-arylated tertiary amines. The reactions of anilines and cyclohexanones were achieved as well, under slightly modified reaction conditions (Scheme 8, Condition B).
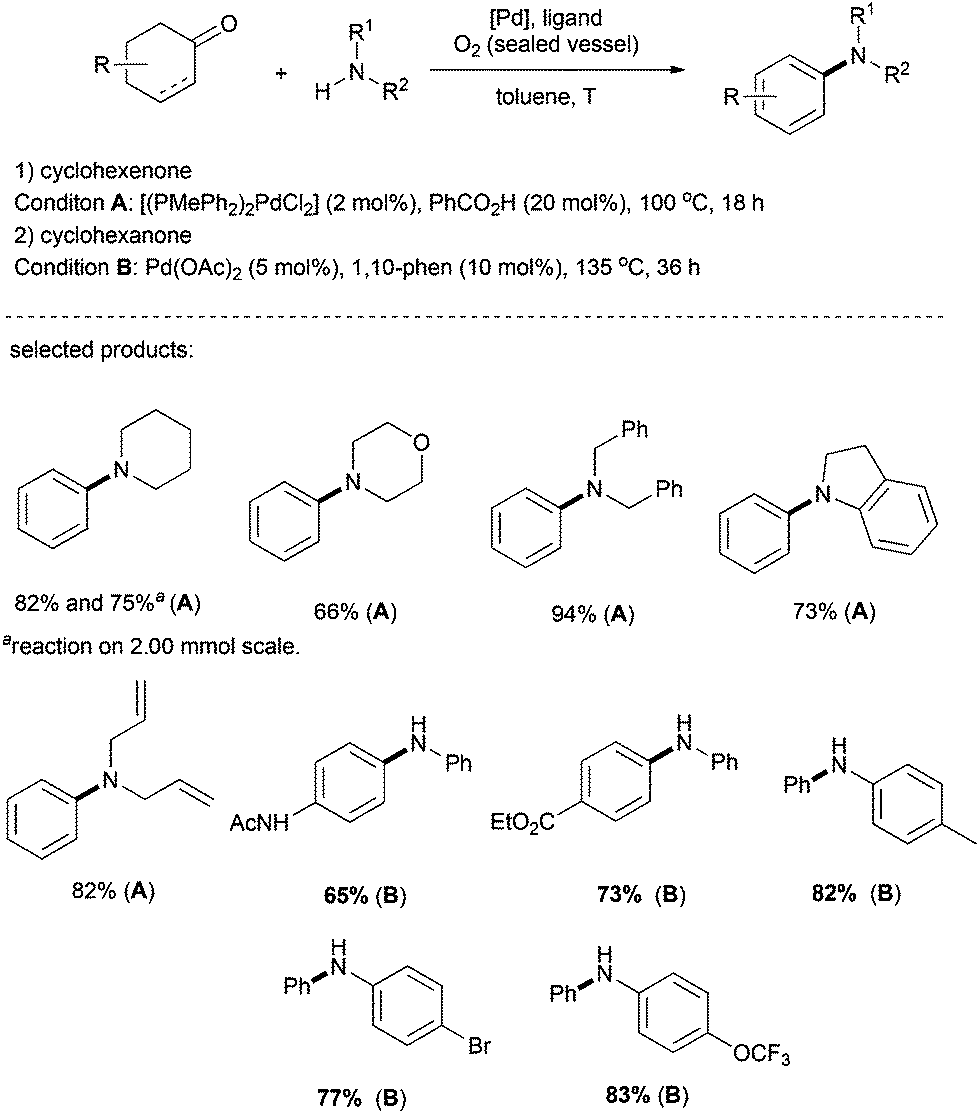 | ||
| Scheme 8 Arylation of amines. | ||
Based on Stahl's work on the palladium-catalyzed aerobic dehydrogenation of substituted cyclohexanones to phenols,17 the authors proposed a plausible mechanism for this transformation (Scheme 9). The reaction starts with the formation of an imine II (via the usual aminol I), which tautomerizes to the more stable enamine III. Palladation of the enamine species III, followed by further tautomerization and β-hydride-elimination, liberates the aryl amine IX or a cyclic diene intermediate VIII and a metal-hydride species VII. The latter leads to the regeneration of the initial catalyst IV in the presence of O2. The product IX will be formed from the diene species VIII in a second catalytic cycle.
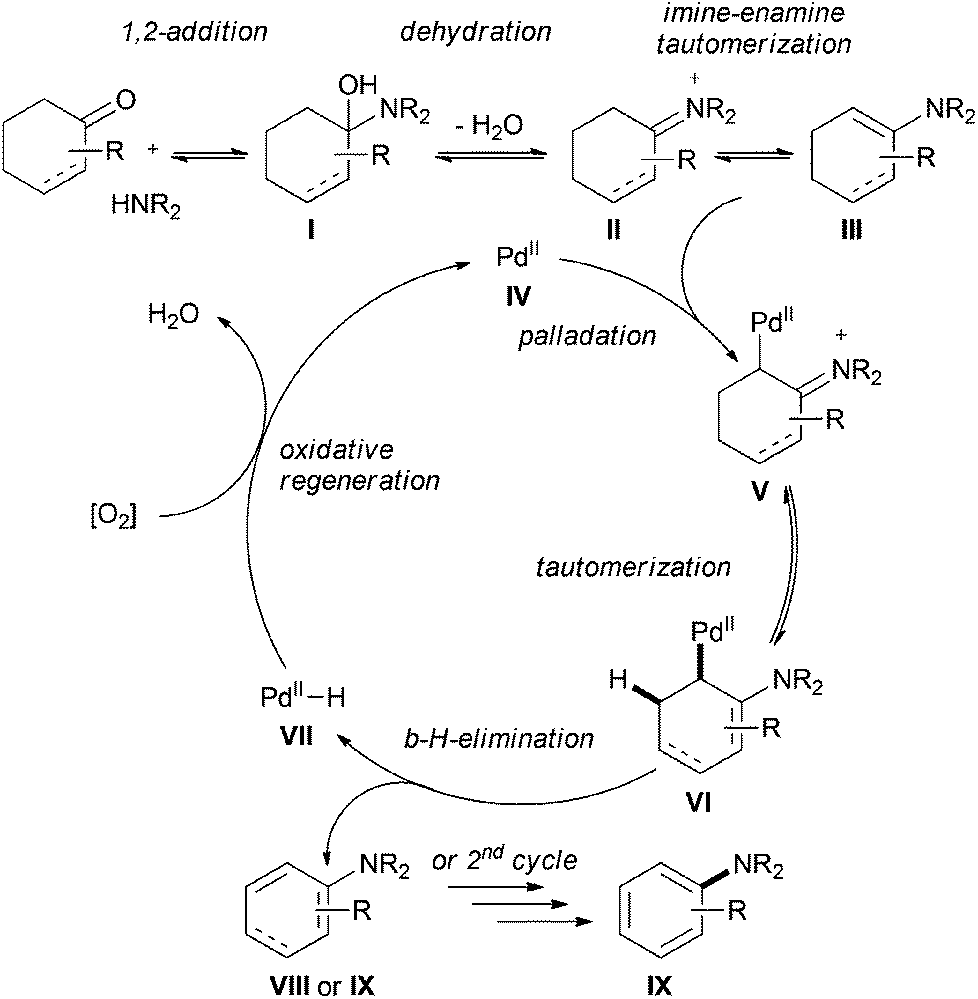 | ||
| Scheme 9 Proposed mechanism for the palladium-catalyzed arylation of amines. | ||
Almost at the same time, Yoshikai's group described an aerobic Pd(OAc)2/Bu4NBr/DMSO catalytic system for the formation of aromatic amines through a palladium-catalyzed dehydrogenation of imines, which were pre-synthesized from the corresponding cyclohexanones and amines (Scheme 10).24 The one-pot fashion of dehydrogenative aromatization from cyclohexanones and amines was then realized under slightly varied reaction conditions. Both alkyl amines and aromatic amines proved to be efficient coupling partners for this transformation.
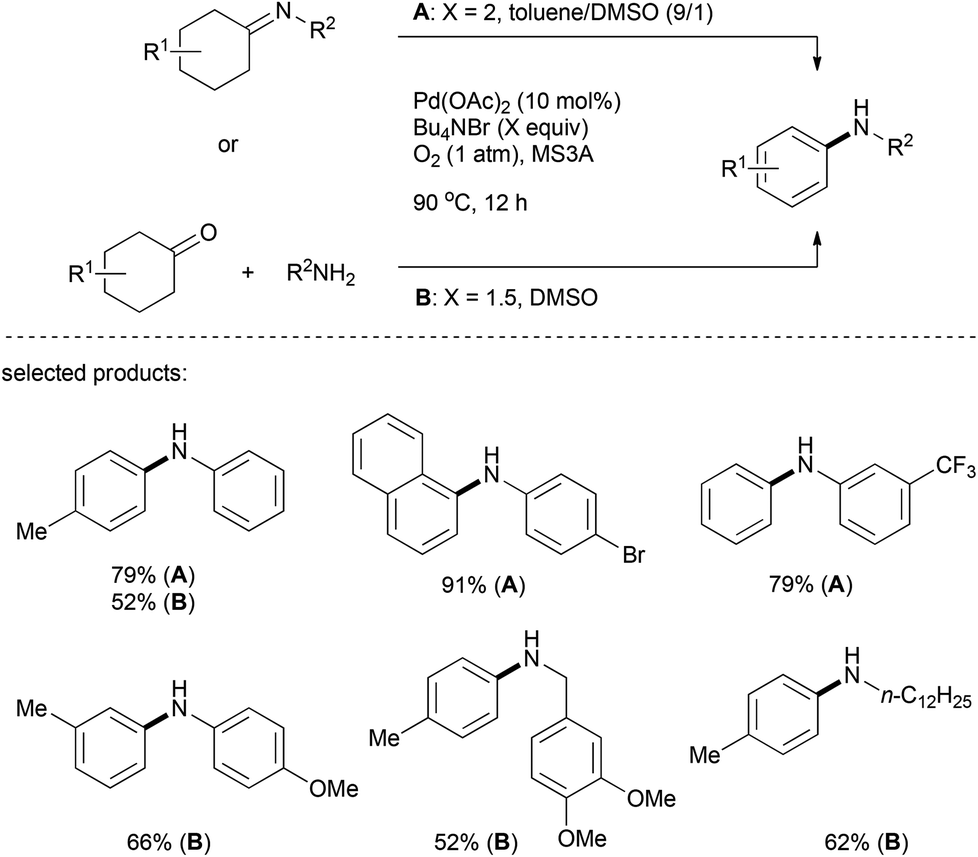 | ||
| Scheme 10 Alternative procedure for the palladium-catalyzed arylation of amines. | ||
It should be noted that prior to this arylation of amines, Deng's group first presented a palladium-catalyzed one-pot diarylamine formation from nitroarenes and cyclohexanones based on the borrowing hydrogen strategy (Scheme 11).25 First, the nitroarene is reduced to aniline by a hydride borrowed from the cyclohexanone via Pd-based β-hydride elimination. Then the ensuing condensation of the aniline and cyclohexanone or cyclohexenone furnishes the imine intermediate, which is followed by a further Pd-catalyzed aerobic dehydrogenation to deliver a diarylamine product.
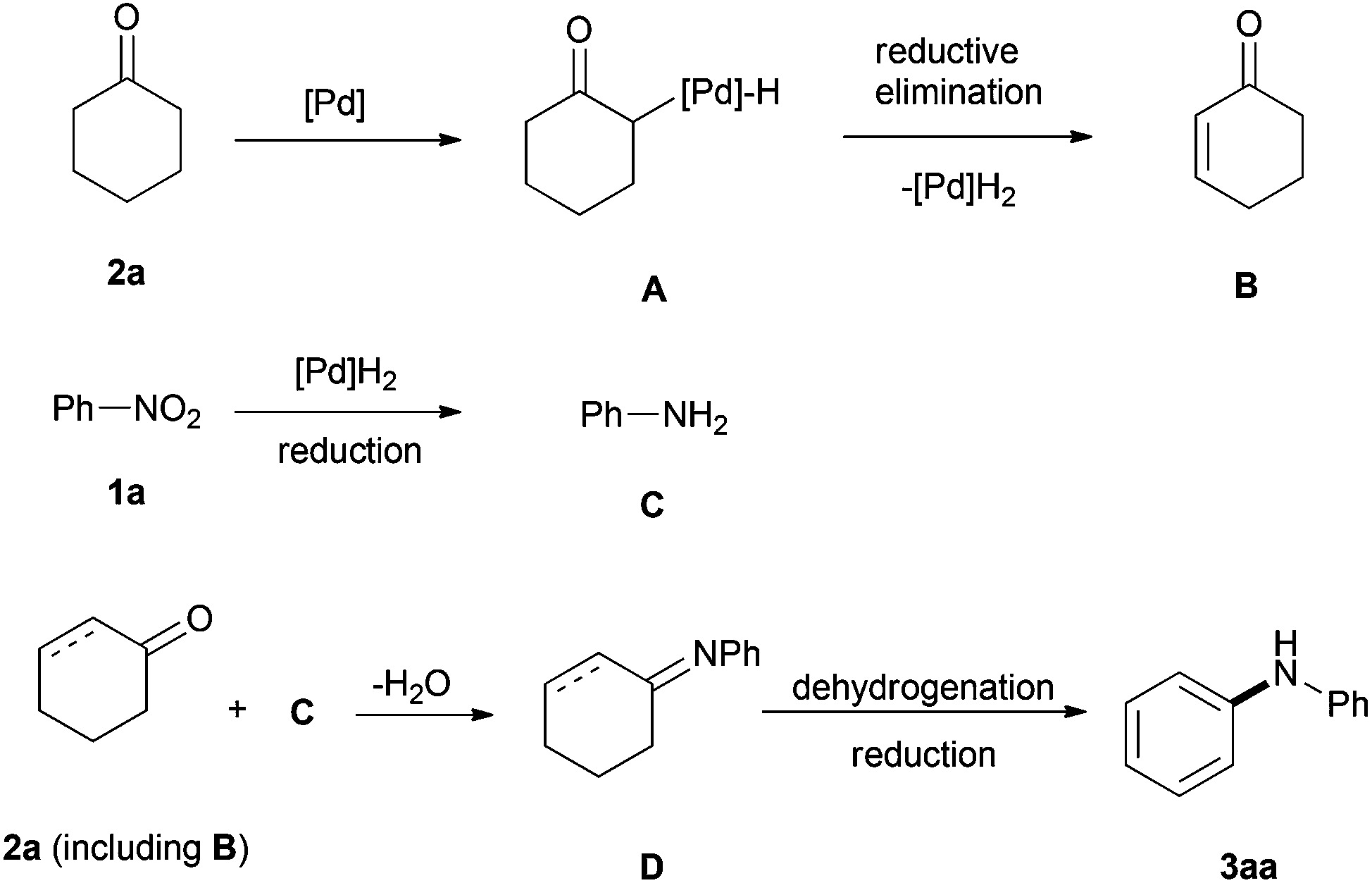 | ||
| Scheme 11 Arylamine formation from nitroarenes by the hydrogen borrowing strategy. | ||
Later on, a metal-free reaction system containing I2/PTSA/DMSO for the preparation of aromatic amines was described by Maycock and co-workers (Scheme 12).26 When 0.5 equiv. of I2 was employed, the reaction proceeded smoothly to afford diarylamine products with ester and halogen (I, Br, and Cl) being tolerated. Interestingly, electrophilic iodination could occur on the relatively electron-rich benzene ring, giving rise to para- or ortho-iodoanilines when 1.1 equiv. of I2 was used.
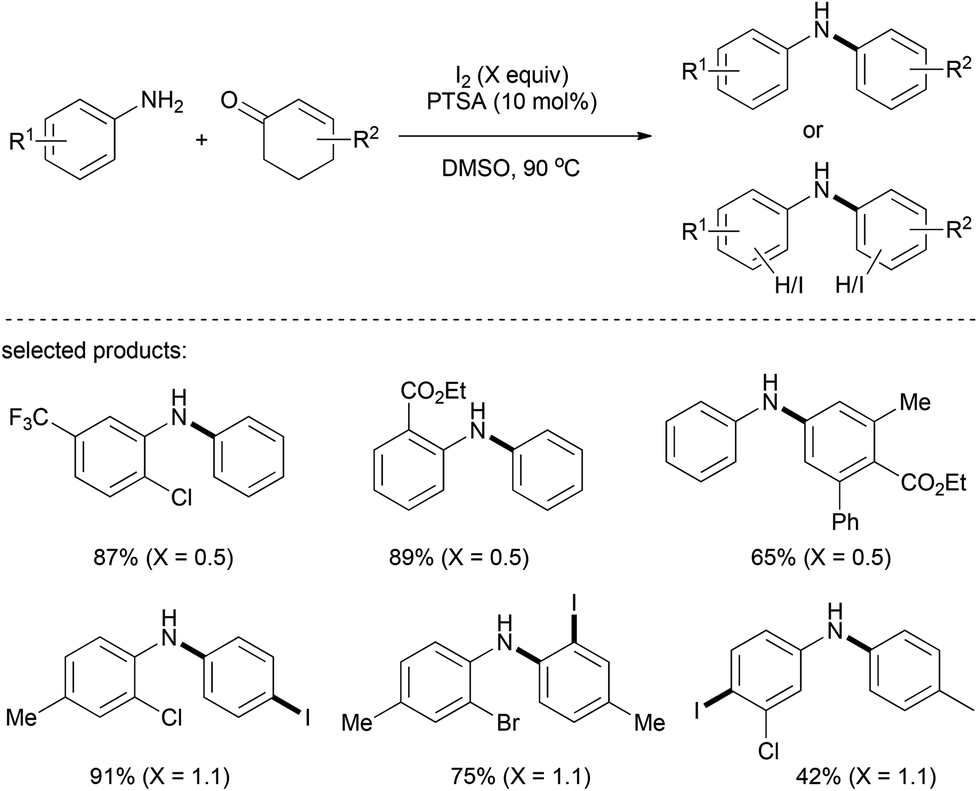 | ||
| Scheme 12 Iodine-catalyzed/mediated arylation of amines. | ||
In 2013, Lemaire et al. developed a carbon-supported palladium system to catalyze the N-arylation of amines using cyclohexanones and tetralones (Scheme 13).27 It is worth noting that the reaction proceeded very efficiently by using only 1–2.5 mol% of Pd/C as the catalyst and 1-octene or nitro-compounds as hydrogen acceptors. In the case of cyclohexanone or β-tetralone, 2 equiv. of 1-octene was required to achieve a high yield. A chemoselective and highly efficient condensation between cyclohexanones and nitroarenes by means of the borrowing hydrogen strategy has also been accomplished under this versatile Pd/C system, albeit using a large excess of cyclohexanones (10 equiv.) and at an elevated reaction temperature (150 °C).
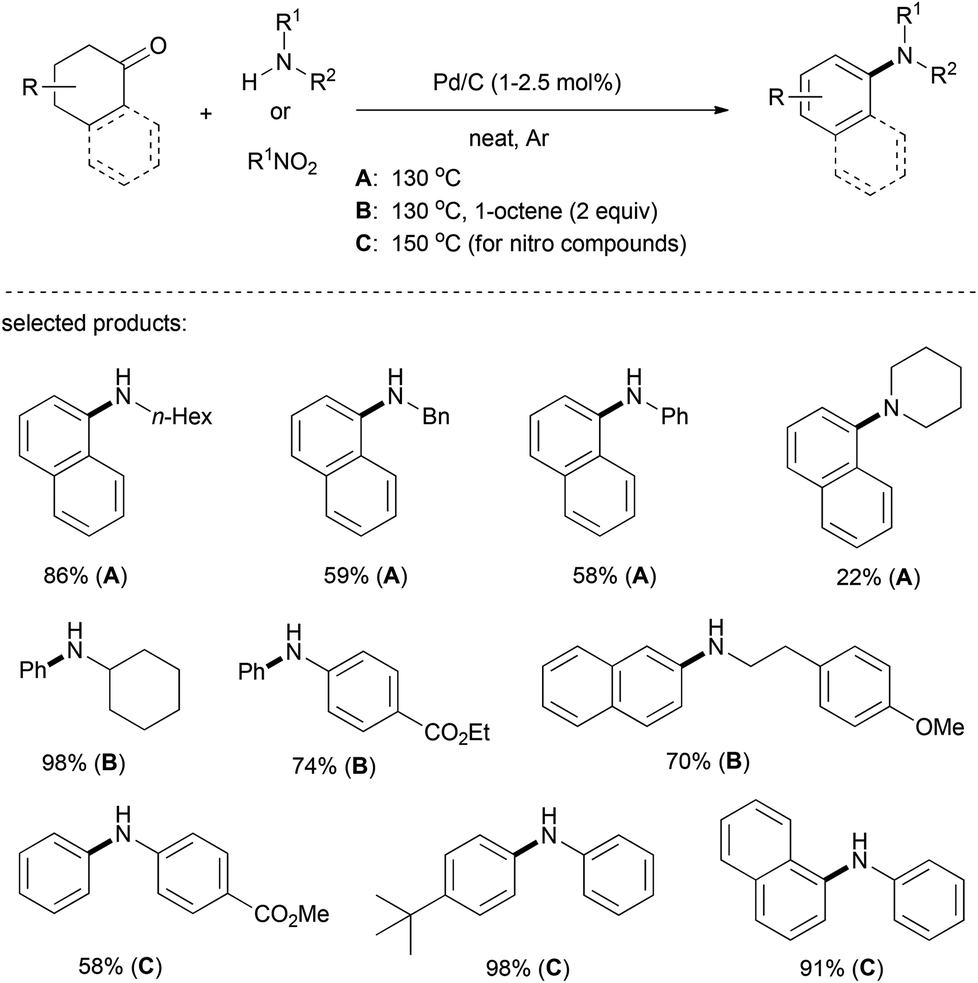 | ||
| Scheme 13 Palladium catalyzed N-arylation of amines. | ||
Recently, the use of aryl sulfonamide in the nucleophilic addition to cyclohexanone and in the ensuing palladium-catalyzed dehydrogenation has been realized by Deng's group.28 Under the optimal catalytic conditions (Pd(TFA)2/1,10-phen/toluene/O2), a range of N-aryl sulfonamides was obtained with a broad functional group tolerance (Scheme 14). Since N-aryl sulfonamides are of great importance as building blocks in pharmaceuticals and bioactive compounds, this palladium-catalyzed aerobic synthesis provides an atom-economical, low-cost, and environmentally friendly method.
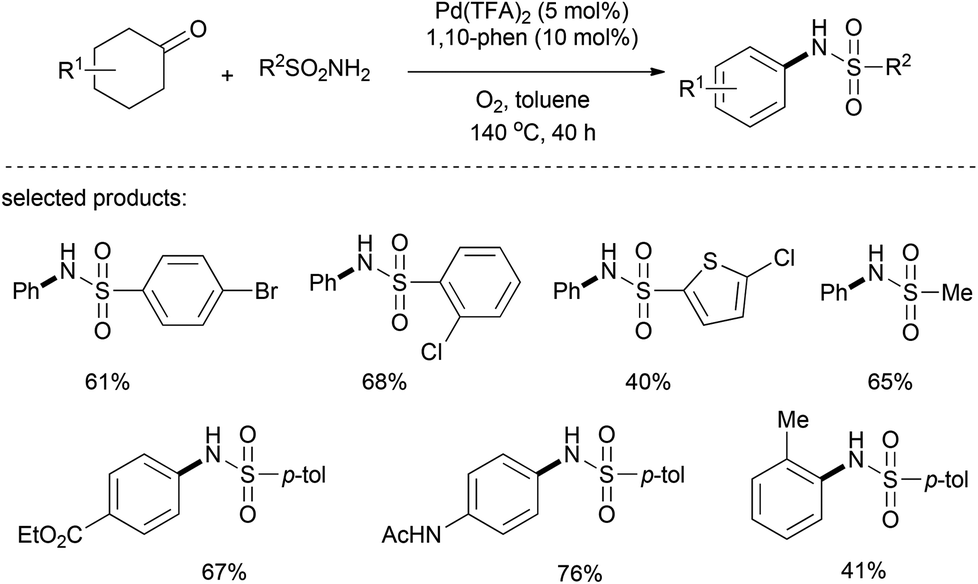 | ||
| Scheme 14 Formation of N-aryl sulfonamides from cyclohexanones and sulfonamides. | ||
In 2013, Stahl et al. reported a protocol for the preparation of primary anilines from tetralones and cyclohexenones on the basis of Pd-catalyzed aerobic dehydrogenation routes to substituted phenols.29 In this catalytic system, a broad range of primary aniline and 1-aminonaphthalene derivatives was generated in moderate to excellent yields (Scheme 15). The reaction of oxime with a stoichiometric amount of Pd(0) via an oxidative addition to the N–O bond was confirmed by the successful isolation of an imino-Pd(II) intermediate. Based on this observation and previous studies on cyclohexanone dehydrogenation, the following Pd-based β-hydride elimination and subsequent tautomerization account for the formation of the desired 1-aminonaphthalenes.
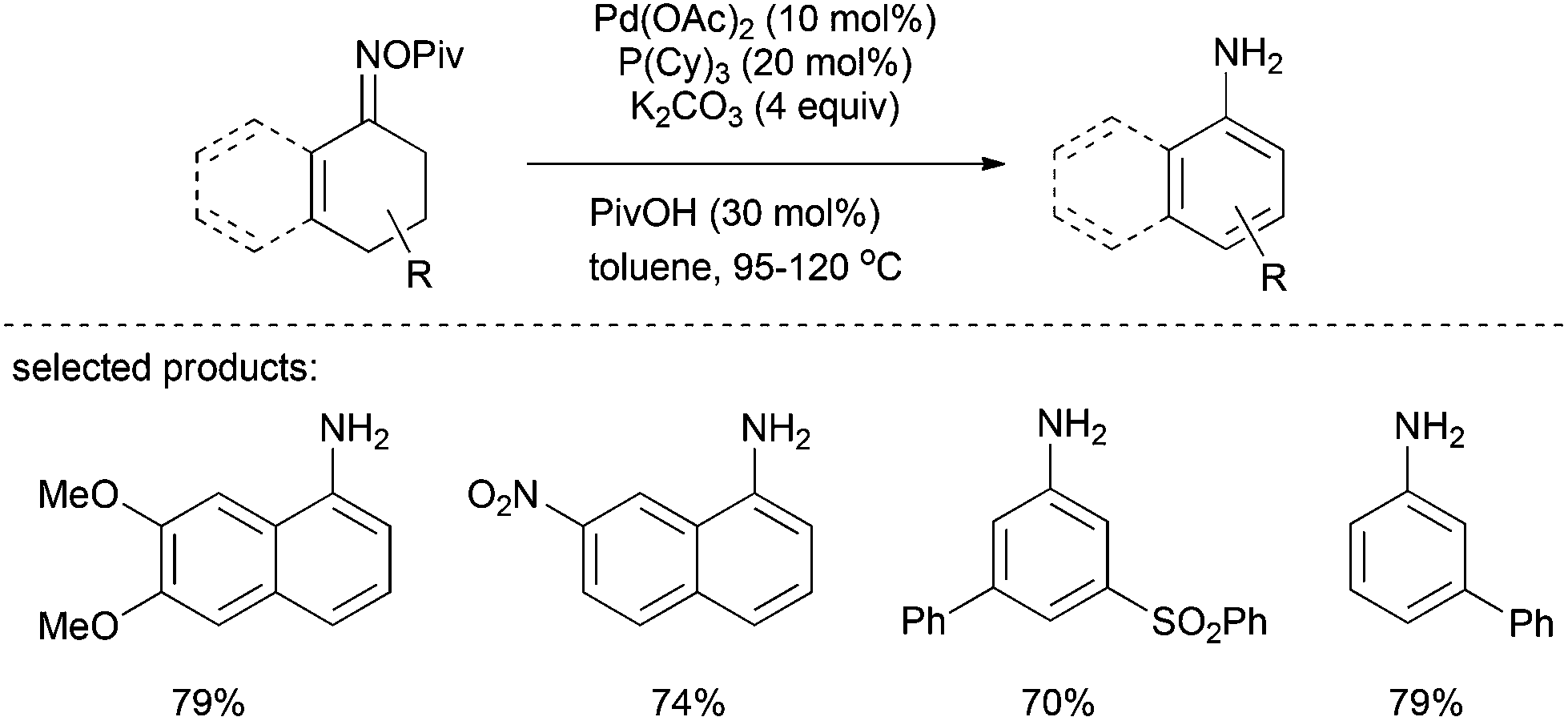 | ||
| Scheme 15 Formation of primary anilines from oxime derivatives. | ||
1.3 C–C bond formation
The same arylation strategy as that for oxygen and nitrogen-based nucleophiles described above can be extended to carbon-based nucleophiles. In 2013, Li's group reported a transition-metal-free one-pot synthesis of biaryls from Grignard reagents and substituted cyclohexanones, in which DDQ functions as the hydrogen acceptor.30 In this one-pot process, Grignard reagent can be conveniently generated in situ from the corresponding aryl iodides; sequential and nucleophilic addition, dehydration and dehydrogenation afford the desired products in up to 99% yield (Scheme 16). Gratifyingly, various functionalities such as halogen, ester and others can be well tolerated in this catalytic system. The heterocycle, 2-iodothiophene, affords 2-phenylthiophene in good yield under optimal conditions. This simple system eliminates the use of transition metals and no additional reagents are required.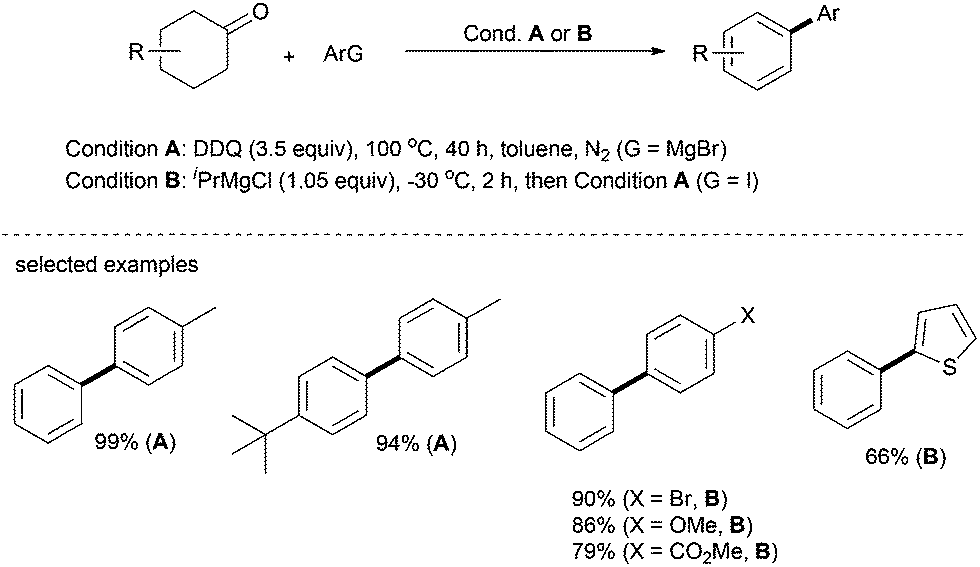 | ||
| Scheme 16 Arylation with C-nucleophiles with Grignard-reagents. | ||
Recently, Deng's group reported a palladium-catalyzed greener approach to access 3-arylindoles from indoles and cyclohexanones, in which molecular oxygen was used as the sole oxidant (Scheme 17).31 This method was applied to various cyclohexanones and showed good regioselectivity as well. Since cyclohexanones are readily available starting materials, this method represents an environmentally benign approach for the preparation of biaryls and arylated heteroarenes.
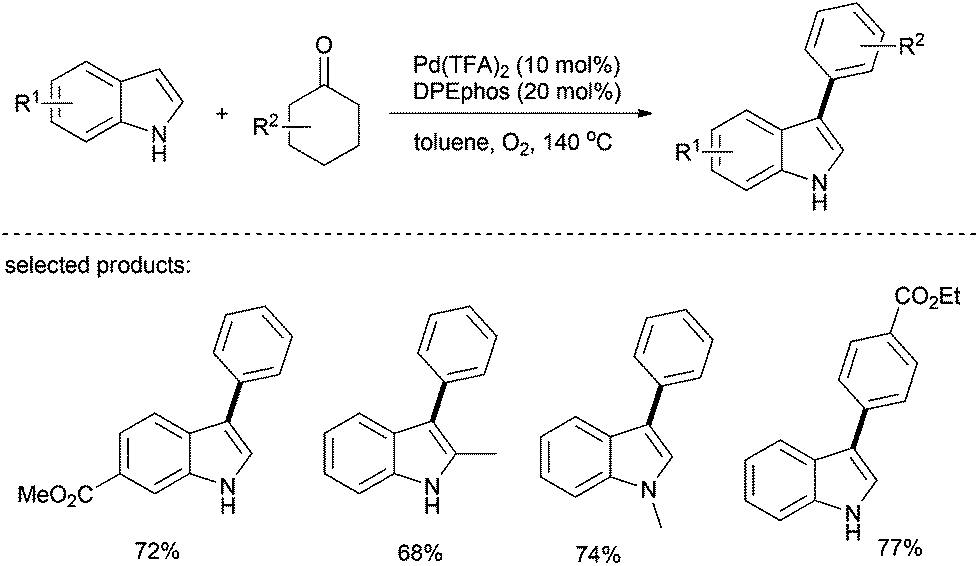 | ||
| Scheme 17 Arylation of indole derivatives. | ||
2. Application to the synthesis of heterocycles
A combination of two couplings at the C1 and C2 positions of a cyclohexanone with its subsequent aromatization would provide new synthetic routes for the construction of heterocycles (Scheme 18).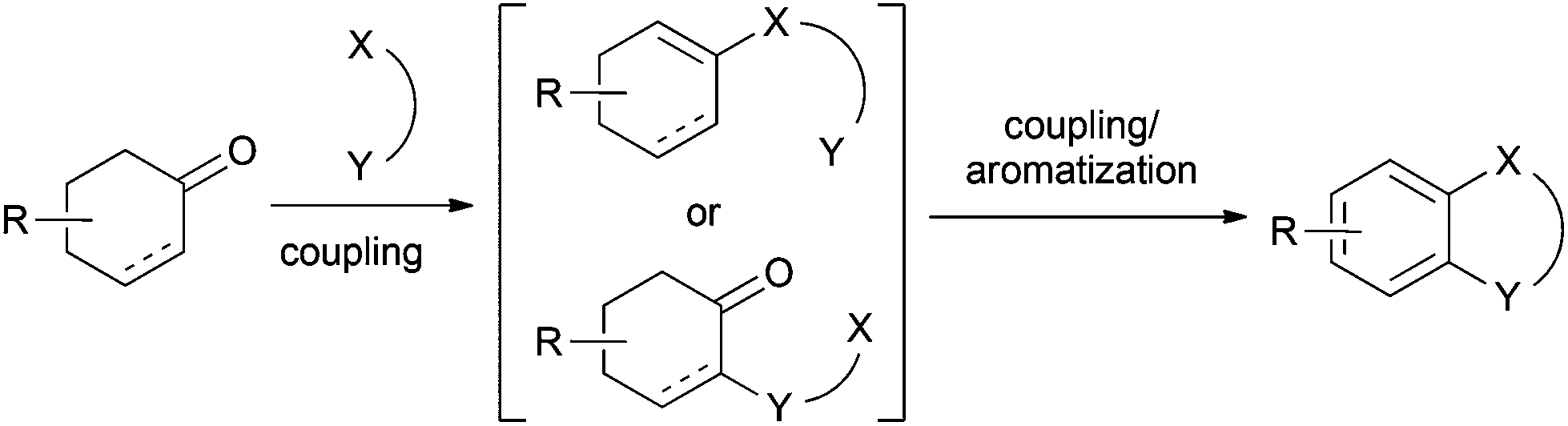 | ||
| Scheme 18 Tandem C1 and C2 couplings of cyclohexanone. | ||
Deng and co-workers presented in 2012 a one-pot synthesis of carbazoles from cyclohexanones and arylhydrazine hydrochlorides (Scheme 19).32 Unlike the widely used Fischer–Borsche reaction, this method could be achieved in a one-pot fashion in the absence of a metal-catalyst by using molecular oxygen as the oxidant. Heating the two substrates in NMP for 24 h under an oxygen atmosphere at 140 °C gave the corresponding carbazole products in moderate to good yields. The use of hydrochloride hydrazines was crucial in this method since it generated, in situ, the stoichiometric amount of an acid required for this transformation.
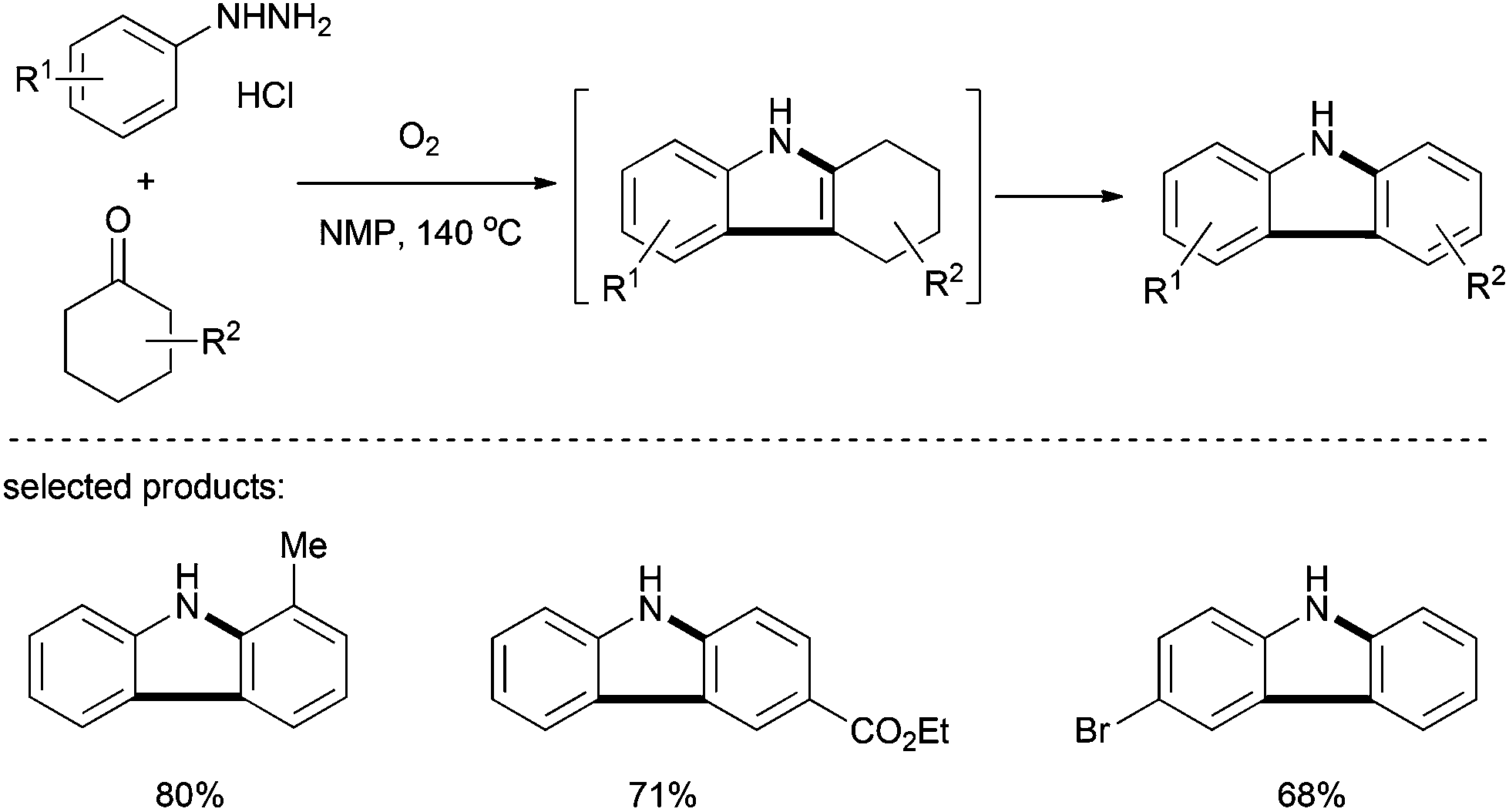 | ||
| Scheme 19 One-pot synthesis of carbazoles from cyclohexanones and arylhydrazine hydrochlorides. | ||
One year later, the same group employed 2-aminobenzenethiols as double-site coupling reagents for the metal free synthesis of phenothiazines (Scheme 20).33 Various functionalized phenothiazines were successfully synthesised using a combination of benzyl aryl sulfone and potassium iodide under an oxygen atmosphere. Cyclohexanones bearing electron-donating groups at the para position gave the corresponding phenothiazines in good yields with several 2-aminobenzenethiols. However the reaction is strongly influenced by steric hindrance.
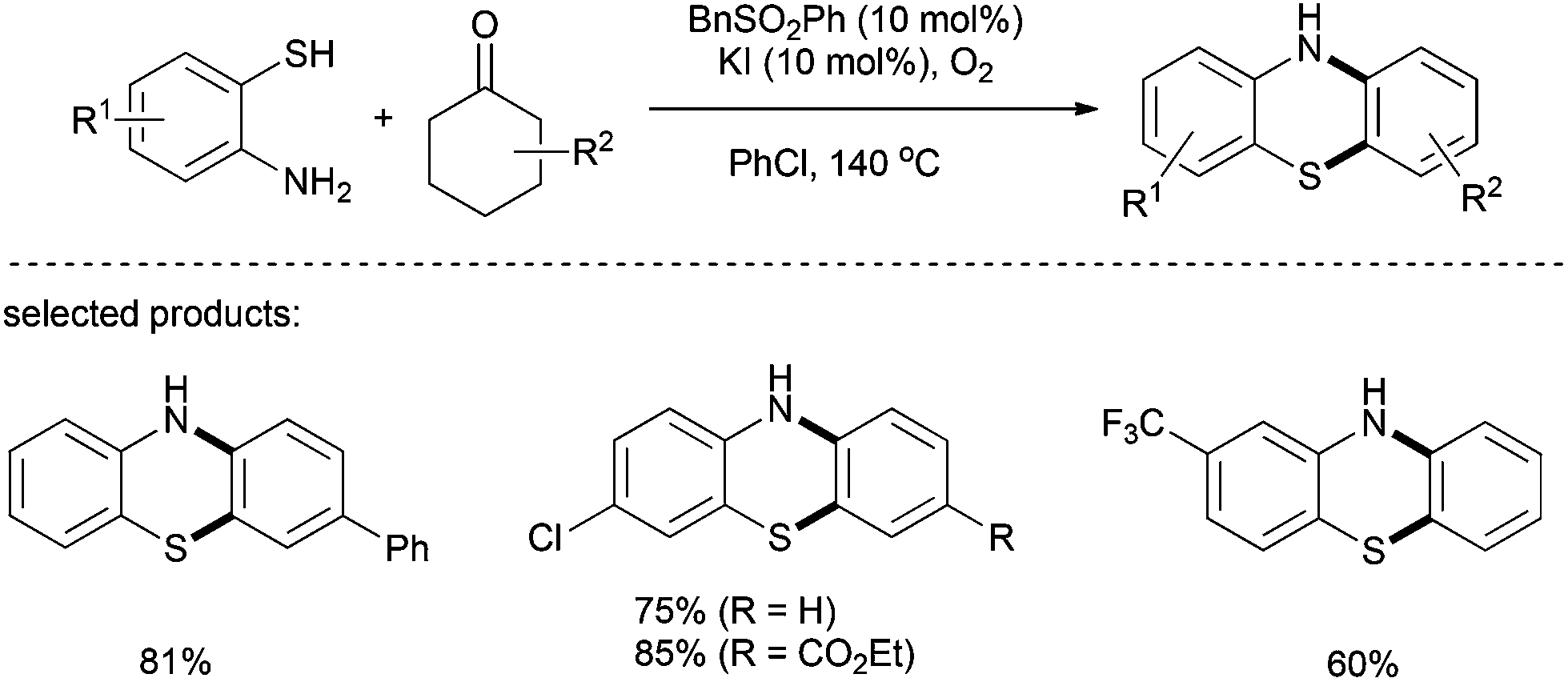 | ||
| Scheme 20 Metal-free one-pot synthesis of phenothiazines from cyclohexanones and 2-aminobenzenethiols. | ||
The group of Hong described, in 2013, an interesting approach to the synthesis of coumarins (Scheme 21).34 The reaction proceeded via a Pd(II)-catalyzed dehydrogenation–oxidative Heck-cyclization. Using Pd(TFA)2 in combination with copper salt and molecular oxygen as oxidants in pivalic acid, a broad range of highly functionalized coumarins were prepared in moderate to good yields. Based on the proposed mechanistic pathway, a further functionalization of coumarins was possible.
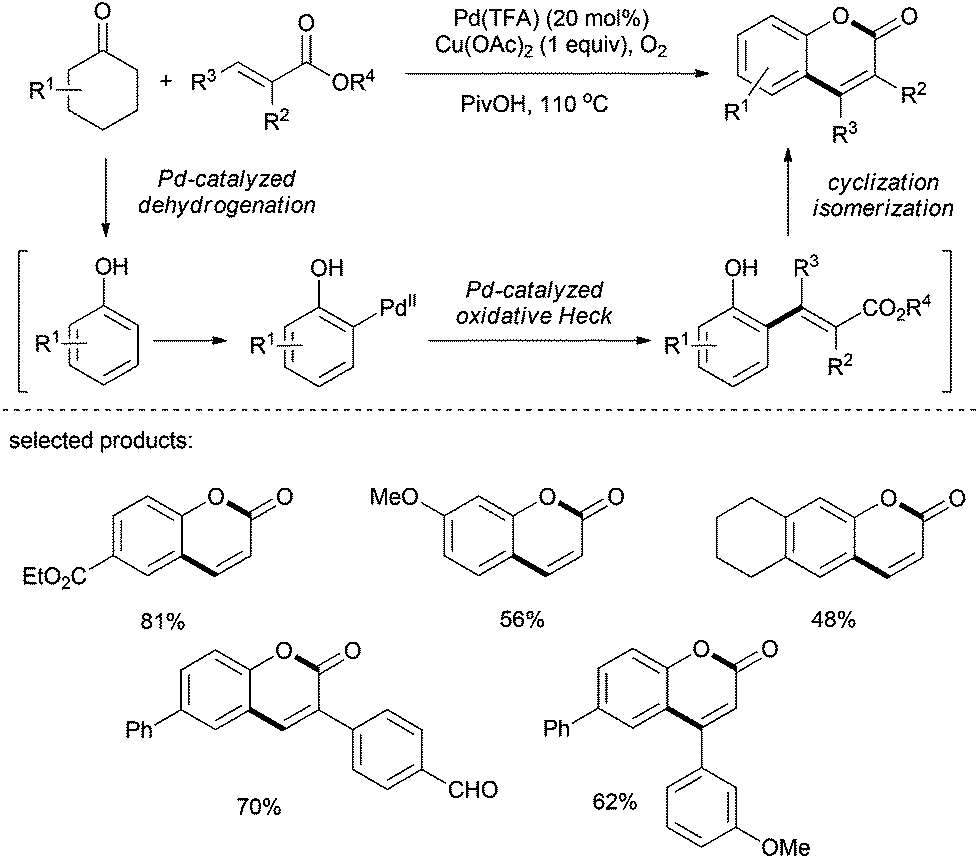 | ||
| Scheme 21 Synthesis of coumarins. | ||
Jiang and co-workers introduced the catalytic I2/PTSA/O2 system for the construction of 2-aminobenzothiazoles from thioureas and cyclohexanones through a sulfanylphenol intermediate via the generation of an α-C–S bond.35 This reaction was performed at a relatively low temperature compared to the previous dehydrogenation, albeit a strong organic acid, PTSA, was required. While substituted cyclohexanones afforded 2-aminobenzothiazoles with generally moderate yields, the treatment of α- or β-tetralones with thioureas delivered naphtho[2,1-d]thiazoles in excellent yields. Notably, apart from free thiourea, N-alkyl, N-aryl, and even disubstituted thioureas showed high reactivities (Scheme 22).
 | ||
| Scheme 22 Iodine-catalyzed synthesis of 2-aminobenzothiazoles from thioureas and cyclohexanones. | ||
In addition to benzothiazole synthesis, recently Deng and co-workers have effectively utilized aryl amides as coupling partners for the preparation of benzoxazoles.36 An elevated temperature (160 °C) was essential for the success of this metal-free transformation, and a stoichiometric amount of DMSO was employed as a co-oxidant. A series of substituted 2-arylbenzoxazoles with alkyl, halogen, hydroxyl, and ester was prepared in moderate to good yields (Scheme 23). Remarkably, the non-dehydrogenated intermediate product (i.e. tetrahydrobenzoxazole) could successfully be isolated by shortening the reaction time to 2 h.
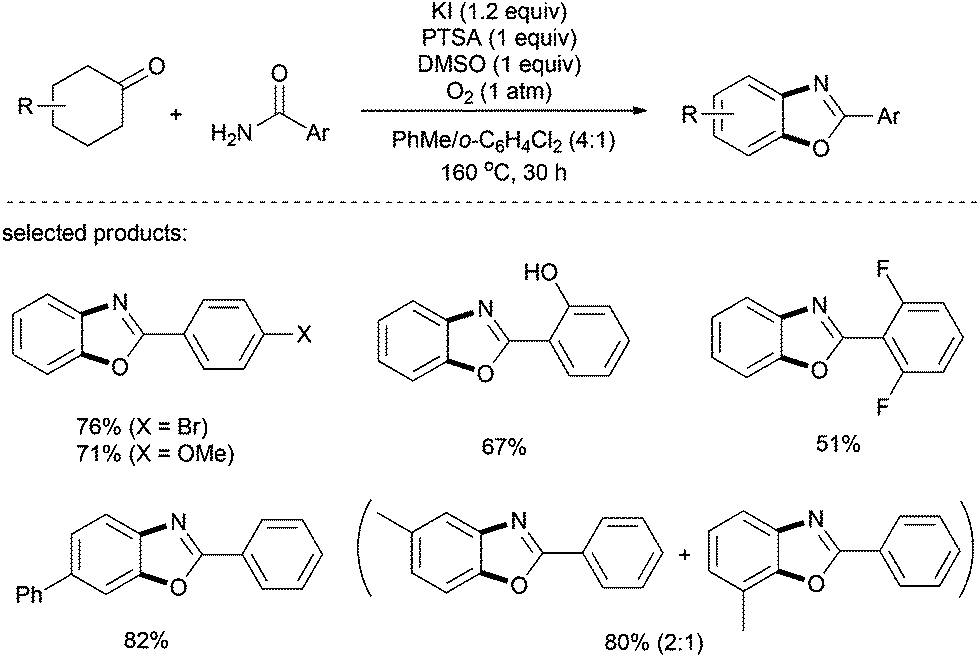 | ||
| Scheme 23 Preparation of benzoxazoles from cyclohexanones and aryl amides. | ||
3. Conclusions
In the past few years, catalytic dehydrogenative aromatization has evolved into a current of intense investigations. In this context, many impressive, synthetically useful reactions have been reported via this non-cross-coupling strategy. As an efficient route to functionalized arenes in organic synthesis, the direct formation of C–O, C–N and C–C bonds has been realized by our groups. Some representative synthetic applications for the construction of heterocycles have also been incorporated into this mini-review. Throughout all these catalytic oxidative processes, the distinctive feature is that molecular oxygen is used as a terminal oxidant, thus delivering water as the main byproduct in most cases. Low-cost and easily available starting materials, highly efficient atom economy and product diversity make this strategy particularly practical and fascinating. For further prospect in this field, a broader range of coupling partners and more synthetic applications under simpler, milder conditions will be expected. On the other hand, the even more challenging aspect is to develop a general method for the synthesis of functionalized arenes via a modified non-cross-coupling strategy, specifically, through the temporary dearomatization–introduction of coupling partners–rearomatization sequences, as exemplified by the formation of aniline from phenol.Acknowledgements
We are grateful to the Canada Research Chair (Tier 1), the FQRNT, NSERC and CFI for their support (to CJL), G.-J. Deng acknowledges financial support from the National Natural Science Foundation of China (21172185, 21372187), the New Century Excellent Talents in University from Ministry of Education of China (NCET-11-0974), and the Scientific Research Foundation for Returned Scholars, Ministry of Education of China (2011-1568).Notes and references
- L. A. Thompson and J. A. Ellman, Chem. Rev., 1996, 96, 555–600 CrossRef CAS PubMed.
- G. Sartori and R. Maggi, Advances in Friedel-Crafts Acylation Reactions: Catalytic and Green Processes, Taylor & Francis, 2009 Search PubMed.
- W. Sander, Acc. Chem. Res., 1999, 32, 669–676 CrossRef CAS.
- M. R. Bryce and M. Vernon, in Advances in Heterocyclic Chemistry, ed. A. R. Katritzky and A. J. Boulton, Academic Press, 1981, vol. 28, pp. 183–229 Search PubMed.
- E.-I. Negishi and A. de Meijere, Handbook of Organopalladium Chemistry for Organic Synthesis, Wiley-Interscience, New York, 2002 Search PubMed.
- R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef CAS PubMed.
- T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212–11222 CrossRef CAS PubMed.
- K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2011, 45, 788–802 CrossRef PubMed.
- L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC.
- J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC.
- L. Ackermann, Acc. Chem. Res., 2013, 47, 281–295 CrossRef PubMed.
- S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef CAS.
- X.-S. Zhang, K. Chen and Z.-J. Shi, Chem. Sci., 2014, 5, 2146–2159 RSC.
- P. Sabatier, Ber. Dtsch. Chem. Ges., 1911, 44, 1984–2001 CrossRef CAS.
- A. Skita and H. Ritter, Ber. Dtsch. Chem. Ges., 1911, 44, 668–676 CrossRef CAS.
- H. C. Chitwood, J. T. Fitzpatrick, G. W. Fowler and B. T. Freure, Ind. Eng. Chem., 1952, 44, 1696–1698 CrossRef CAS.
- Y. Izawa, D. Pun and S. S. Stahl, Science, 2011, 333, 209–213 CrossRef CAS PubMed.
- M.-O. Simon, S. A. Girard and C.-J. Li, Angew. Chem., Int. Ed., 2012, 51, 7537–7540 CrossRef CAS PubMed.
- C. Galli, P. Gentili and O. Lanzalunga, Angew. Chem., Int. Ed., 2008, 47, 4790–4796 CrossRef CAS PubMed.
- F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800–3842 CrossRef CAS PubMed.
- M. Sutter, R. Lafon, Y. Raoul, E. Métay and M. Lemaire, Eur. J. Org. Chem., 2013, 5902–5916 CrossRef CAS.
- M. Sutter, N. Sotto, Y. Raoul, E. Métay and M. Lemaire, Green Chem., 2013, 15, 347–352 RSC.
- S. A. Girard, X. Hu, T. Knauber, F. Zhou, M.-O. Simon, G.-J. Deng and C.-J. Li, Org. Lett., 2012, 14, 5606–5609 CrossRef CAS PubMed.
- A. Hajra, Y. Wei and N. Yoshikai, Org. Lett., 2012, 14, 5488–5491 CrossRef CAS PubMed.
- Y. Xie, S. Liu, Y. Liu, Y. Wen and G.-J. Deng, Org. Lett., 2012, 14, 1692–1695 CrossRef CAS PubMed.
- M. T. Barros, S. S. Dey, C. D. Maycock and P. Rodrigues, Chem. Commun., 2012, 48, 10901–10903 RSC.
- M. Sutter, M.-C. Duclos, B. Guicheret, Y. Raoul, E. Métay and M. Lemaire, ACS Sustainable Chem. Eng., 2013, 1, 1463–1473 CrossRef CAS.
- X. Cao, Y. Bai, Y. Xie and G.-J. Deng, J. Mol. Catal. A: Chem., 2014, 383–384, 94–100 CrossRef CAS PubMed.
- W. P. Hong, A. V. Iosub and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 13664–13667 CrossRef CAS PubMed.
- F. Zhou, M.-O. Simon and C.-J. Li, Chem. – Eur. J., 2013, 19, 7151–7155 CrossRef CAS PubMed.
- S. Chen, Y. Liao, F. Zhao, H. Qi, S. Liu and G.-J. Deng, Org. Lett., 2014, 16, 1618–1621 CrossRef CAS PubMed.
- F. Xiao, Y. Liao, M. Wu and G.-J. Deng, Green Chem., 2012, 14, 3277–3280 RSC.
- Y. Liao, P. Jiang, S. Chen, F. Xiao and G.-J. Deng, RSC Adv., 2013, 3, 18605–18608 RSC.
- D. Kim, M. Min and S. Hong, Chem. Commun., 2013, 49, 4021–4023 RSC.
- J. Zhao, H. Huang, W. Wu, H. Chen and H. Jiang, Org. Lett., 2013, 15, 2604–2607 CrossRef CAS PubMed.
- X. Cao, X. Cheng, Y. Bai, S. Liu and G.-J. Deng, Green Chem., 2014, 16, 4644–4648 RSC.
Footnotes |
| † This article is an invited contribution to celebrate Prof. Ei-ichi Negishi's 80th birthday with Organic Chemistry Frontiers. |
| ‡ S. A. Girard, H. Huang and F. Zhou contributed equally. |
| This journal is © the Partner Organisations 2015 |