
Alkyl- and aryl-thioalkylation of olefins with organotrifluoroborates by photoredox catalysis†
Yanjie
Li
,
Kazuki
Miyazawa
,
Takashi
Koike
* and
Munetaka
Akita
*
Chemical Resources Laboratory, Tokyo Institute of Technology, 4259, Nagatsuta, Midori-ku, Yokohama 226-8503, Japan. E-mail: koike.t.ad@m.titech.ac.jp; makita@res.titech.ac.jp; Fax: +81-45-924-5230; Tel: +81-45-924-5230
First published on 11th February 2015
Abstract
A facile and environmentally benign protocol for alkyl- and aryl-thioalkylation of olefins has been developed. Photoredox catalysis with an Ir photocatalyst, [Ir(dF(CF3)ppy)2(bpy)](PF6) (dF(CF3)ppy: 5-trifluoromethyl-2-(2,4-difluorophenyl)pyridine, bpy: 2,2′-bipyridine), induces efficient oxidation of a variety of alkyl- and aryl-thioalkyltrifluoroborates under visible light irradiation at room temperature, leading to the generation of α-thioalkyl radicals via deboronation. The generated α-thioalkyl radicals smoothly react with electron-deficient olefins to afford addition products in good yields. The present photocatalytic method provides us with simple and new access to a range of alkylsulphides under mild reaction conditions.
Introduction
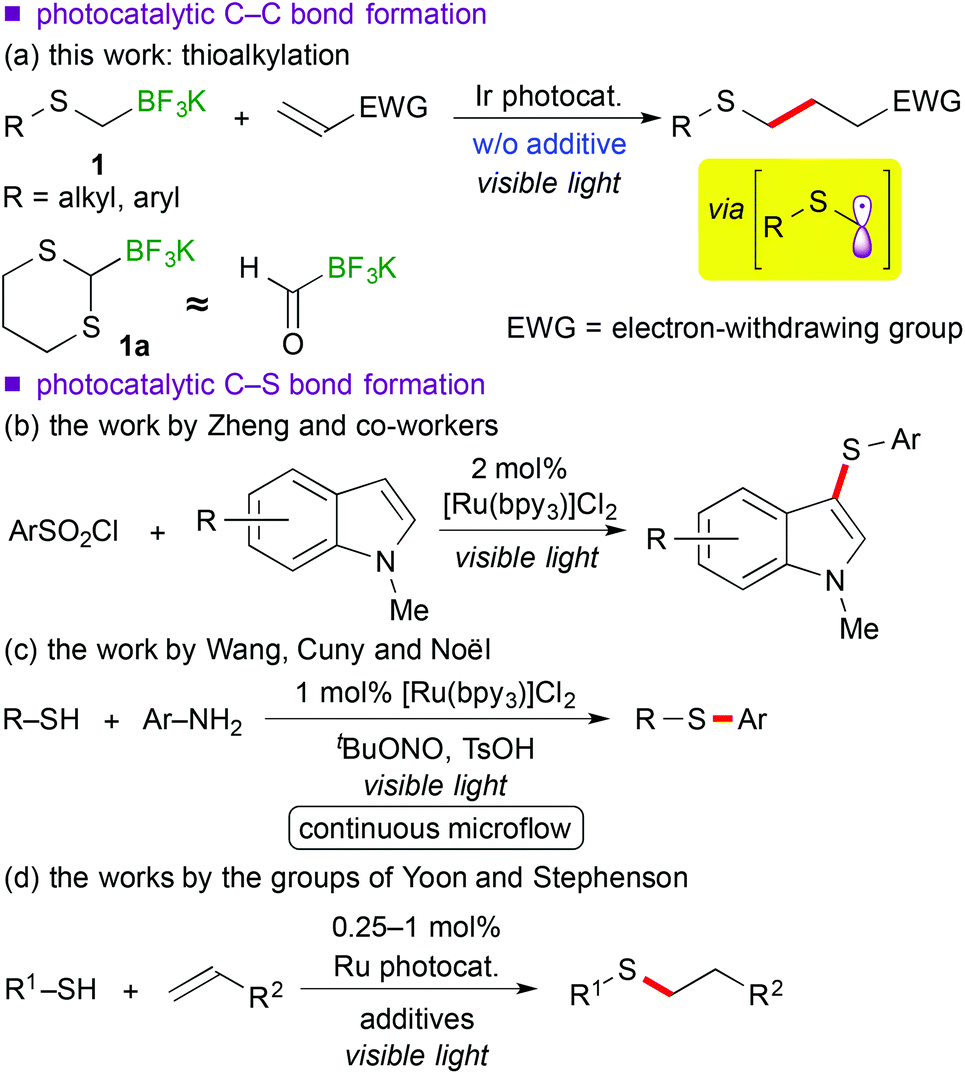
Recently, visible-light-induced single electron transfer (SET) processes mediated by well-defined ruthenium(II) polypyridine complexes and the relevant cyclometalated iridium(III) derivatives have been recognized as a useful redox protocol from the viewpoint of radical chemistry and green chemistry.1 In particular, use of oxidatively generated α-aminoalkyl radicals from various nitrogen-containing compounds by photoredox catalysis has become a powerful method for the synthesis of useful nitrogen-containing building blocks.2,9d The nucleophilic α-aminoalkyl radicals can react with electron-deficient moieties such as unsaturated bonds and arenes to enable functionalization of a carbon atom adjacent to the nitrogen atom. Further expansion into functionalization of ether derivatives advances steadily.3,9b In contrast, reaction of α-sulfur atom substituted alkyl radicals, i.e. α-thioalkyl radicals,4 by photoredox catalysis has not been reported yet, though it can become a new strategy for the synthesis of organic sulphides. In addition, sulphides are known as important functional groups in the field of medicinal chemistry and materials chemistry.5Herein we describe the first example of photoredox-catalyzed thioalkylation of olefins through generation of α-thioalkyl radicals via oxidation of alkyl- and aryl-thioalkyltrifluoroborates (Scheme 1(a)). So far, a few examples of synthesis of sulphides by photoredox catalysis have been reported.6 In 1978 and 1979, Kellog et al. presented pioneering studies about reductive formation of sulphides from reaction of organosulfonium salts with Hantzsch ester in the presence of [Ru(bpy)3]Cl2 (bpy = 2,2′-bipyridine).6a,b In 2012, Zheng and co-workers showed coupling reaction of N-methylindoles and arylsulfonyl chlorides via reduction of sulfonyl chlorides (Scheme 1(b)).6c In 2013, Wang, Cuny and Noël described a one-pot Stadler–Ziegler type reaction by photoredox catalysis in a continuous microflow system (Scheme 1(c)).6d Furthermore, radical thiol–ene reactions involved in [Ru(bpz)3]2+ (bpz = 2,2′-bipyrazine) and [Ru(bpy)3]2+ were reported by the groups of Yoon and Stephenson, respectively (Scheme 1(d)).6e,f,g Their proposed reactive intermediates are different from α-thioalkyl radicals.
 | ||
| Scheme 1 Synthesis of sulphides by photoredox catalysis. | ||
Organoborates are widely used in synthetic chemistry because of their easy-to-handle properties and compatibility with a variety of functional groups including sulphides.7 In addition, generation of organic radicals by oxidation of organoborates has been well-documented so far.8 Recently, our group and the groups of Chen and Molander reported on photocatalytic reactions using organoborates.9,10 In particular, we have extensively developed redox-neutral α-heteroatom methylation such as radical aminomethylation9d and radical alkoxymethylation9b of olefins using the corresponding organotrifluoroborates by photoredox catalysis. Although radical thioalkylation is the subject of the present article, the reaction of potassium 1,3-dithian-2-yltrifluoroborate (1a) can be regarded as a synthetic equivalent of formylation (Scheme 1(a)). Because 1,3-dithianes represent protected acyl groups for useful transformations,11 nucleophilic 1,3-dithian-2-yl radicals can be utilized as acyl anion equivalents. Thus, radical dithianylation can be connected to nucleophilic acylation.4d,f,g
Results and discussion
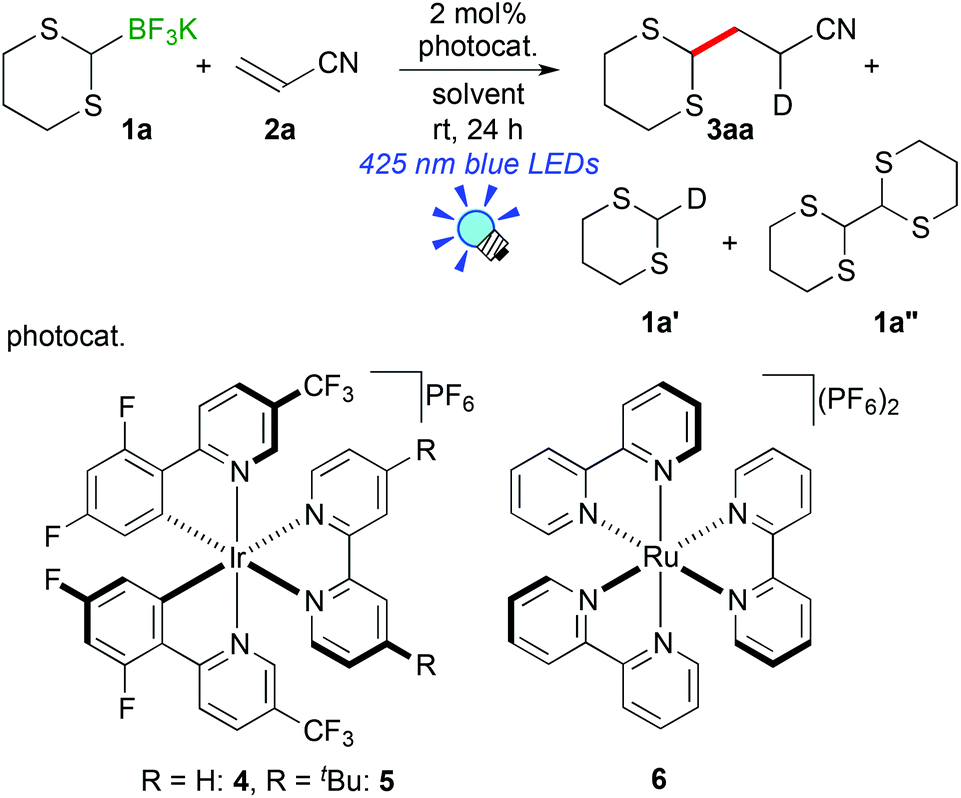
We initially examined the visible-light reaction of potassium 1,3-dithian-2-yltrifluoroborate (1a) with 1 equivalent of acrylonitrile (2a) in the mixed solvent system, acetone-d6 and CD3OD (1/1), in the presence of Ir photocatalyst 4, [Ir(dF(CF3)ppy)2(bpy)](PF6) (dF(CF3)ppy: 5-(trifluoromethyl)-2-(2,4-difluorophenyl)pyridine), which can serve as a strong oxidant (E1/2 = +0.91 V vs. Cp2Fe) when photochemically excited.12As a result, hydrodithianylated product 3aa was obtained in a 56% NMR yield together with the formation of 2-deuterated-1,3-dithiacyclohexane 1a′ (33%) and 1,3-dithianyl dimer 1a″ (10%) (entry 1 in Table 1). The formation of these by-products suggests that the 1,3-dithian-2-yl radical is involved in the present photocatalytic reaction. Next, the use of 2 equivalents of acrylonitrile (2a) dramatically increased the yield of 3aa (90% NMR yield) and suppressed the side reactions (entry 2). Other photocatalysts, [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) (5) (dtbbpy: 4,4′-di-tert-butyl-2,2′-bipyridine) and [Ru(bpy)3](PF6)2 (6),13 resulted in lower yields (entries 3 and 4). CD3OD and the mixed solvent system, acetone-d6 and CD3OD, turned out to be appropriate solvent systems (entries 5–8). Furthermore, the present reaction required both visible-light irradiation and the photocatalyst (entries 9 and 10).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1a : 2a | Photocat. | Solvent | NMR yield of /% | ||
| 3aa | 1a′ | 1a″ | ||||
| a Reaction conditions: a reaction mixture of 1a (0.05 mmol), 2a (0.05 mmol or 0.10 mmol), photocatalyst (1.0 μmol), SiEt4 (an internal standard) and solvent (0.40 mL) was irradiated by using 3 W blue LEDs (λ = 425 ± 15 nm) at room temperature for 24 h. b The reaction was conducted in the dark. | ||||||
| 1 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
4 | Acetone-d6–CD3OD (1/1) | 56 | 33 | 10 |
| 2 | 1:2 |
4 | Acetone-d6–CD3OD (1/1) | 90 | 6 | 3 |
| 3 | 1:2 |
5 | Acetone-d6–CD3OD (1/1) | 73 | 17 | 2 |
| 4 | 1:2 |
6 | Acetone-d6–CD3OD (1/1) | 79 | 15 | 1 |
| 5 | 1:2 |
4 | CD3OD | 93 | 5 | 1 |
| 6 | 1:2 |
4 | Acetone-d6 | 10 | 12 | 1 |
| 7 | 1:2 |
4 | CD3CN | 17 | 11 | 0 |
| 8 | 1:2 |
4 | DMSO-d6 | 10 | 10 | 5 |
| 9b | 1:2 |
4 | CD3OD | 0 | 0 | 0 |
| 10 | 1:2 |
None | CD3OD | 0 | 0 | 0 |
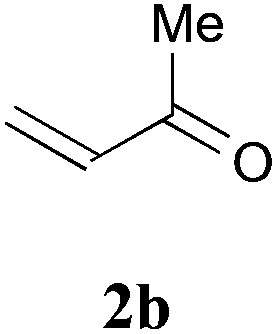
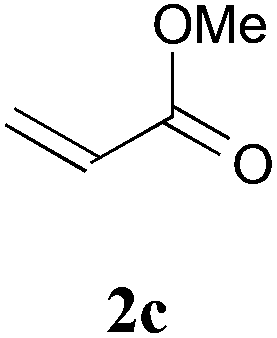
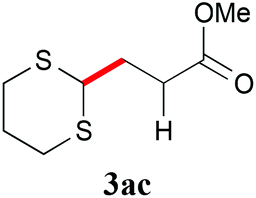
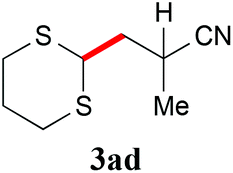
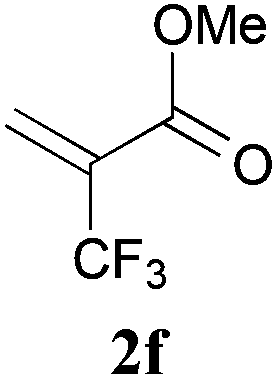
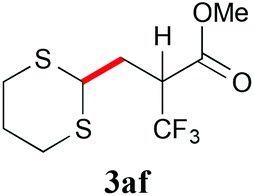
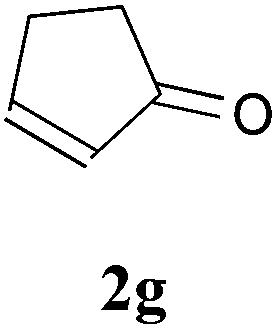
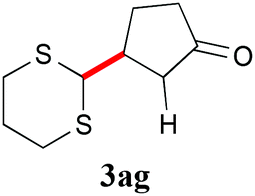
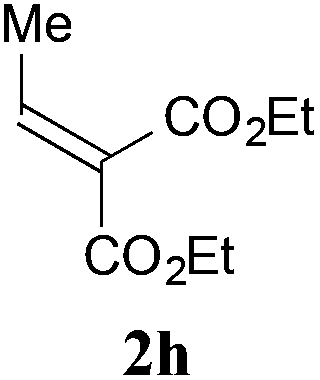
With the optimal reaction conditions in hand, the preparative scale hydrodithianylation of olefins was performed (Table 2). The reactions of typical electron-deficient alkenes, acrylonitrile (2a), 3-buten-2-one (2b) and methyl acrylate (2c), proceeded smoothly to give the corresponding products (3aa–3ac) in good yields (71–80%) (entries 1–3). 1,1-Disubstituted olefins such as methacrylonitrile (2d), α-methylene-γ-butyrolactone (2e) and methyl 2-(trifluoromethyl)acrylate (2f) could also be applied to the present reaction (3ad–3af: 64–80% yields) (entries 4–6). However, olefins with a substituent at the reaction site, 2-cyclopenten-1-one (2g) and diethyl ethylidenemalonate (2h), resulted in low conversions of the olefins and significant formation of deborohydrogenated by-product 1a′ (entries 7 and 8). In contrast, the reaction of fumaronitrile (2i) afforded the hydrodithianylated product (3ai) in an 86% yield (entry 9). These results suggest that the present photocatalytic dithianylation is effective for terminal olefins bearing electron-withdrawing groups and highly electron-deficient internal olefins.
|
|
|||
|---|---|---|---|
| Entry | Olefin 2 | Product 3 | Isolated yield/% |
| a Reaction conditions: a reaction mixture of 1a (0.25 mmol), olefin 2 (0.50 mmol), Ir photocatalyst 4 (5.0 μmol), and degassed MeOH (2.0 mL) was irradiated by using 3 W blue LEDs (λ = 425 ± 15 nm) at room temperature for 24 h. | |||
| 1 |
|
|
80 |
| 2 |
|

|
78 |
| 3 |
|
|
71 |
| 4 |
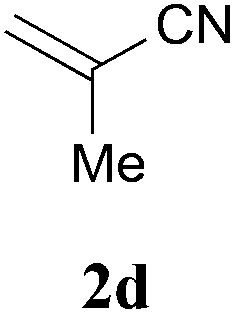
|
|
64 |
| 5 |
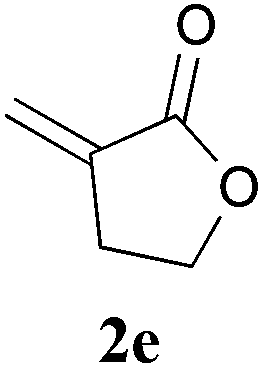
|
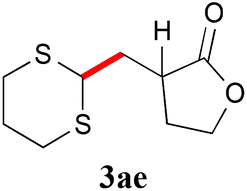
|
78 |
| 6 |
|
|
80 |
| 7 |
|
|
— |
| 8 |
|
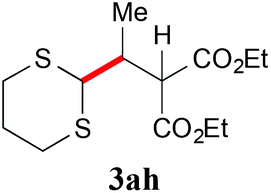
|
— |
| 9 |
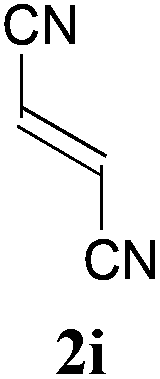
|
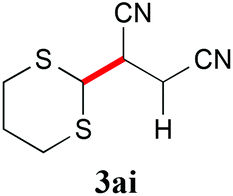
|
86 |
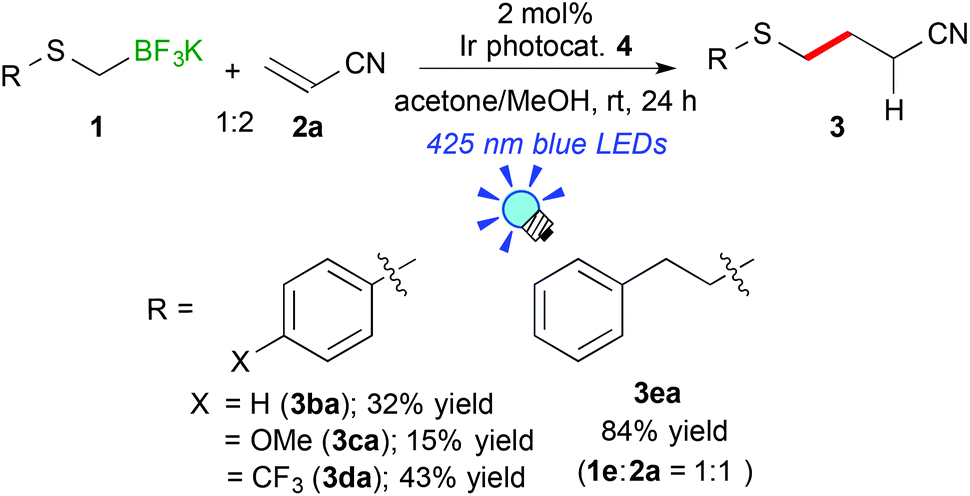
Next, the scope of aryl- and alkyl-thiomethyltrifluoroborates was examined (Scheme 2). The reactions of potassium phenylthiomethyltrifluoroborate (1b), potassium 4-methoxyphenylthiomethyltrifluoroborate (1c), and potassium 4-trifluoromethylphenylthiomethyltrifluoroborate (1d) with acrylonitrile (2a) afforded the corresponding arylthiomethylated products (3ba–da) in moderate yields (15–43%) due to the formation of a considerable amount of arylthiomethanes 1′ as a by-product. Notably, the electron-withdrawing CF3 group hinders the formation of the byproduct. These results suggest that the electron-withdrawing group stabilizes the generated arylthiomethyl radical under these reaction conditions. In contrast, potassium phenethylthiomethyltrifluroborate (1e) gave the alkylthiomethylated product 3ea in an excellent yield (84% yield). In addition, the reaction of 1e did not require an excess amount of olefin.
 | ||
| Scheme 2 Examination of substituents on the sulphur atom in thiomethyltrifluoroborates. | ||
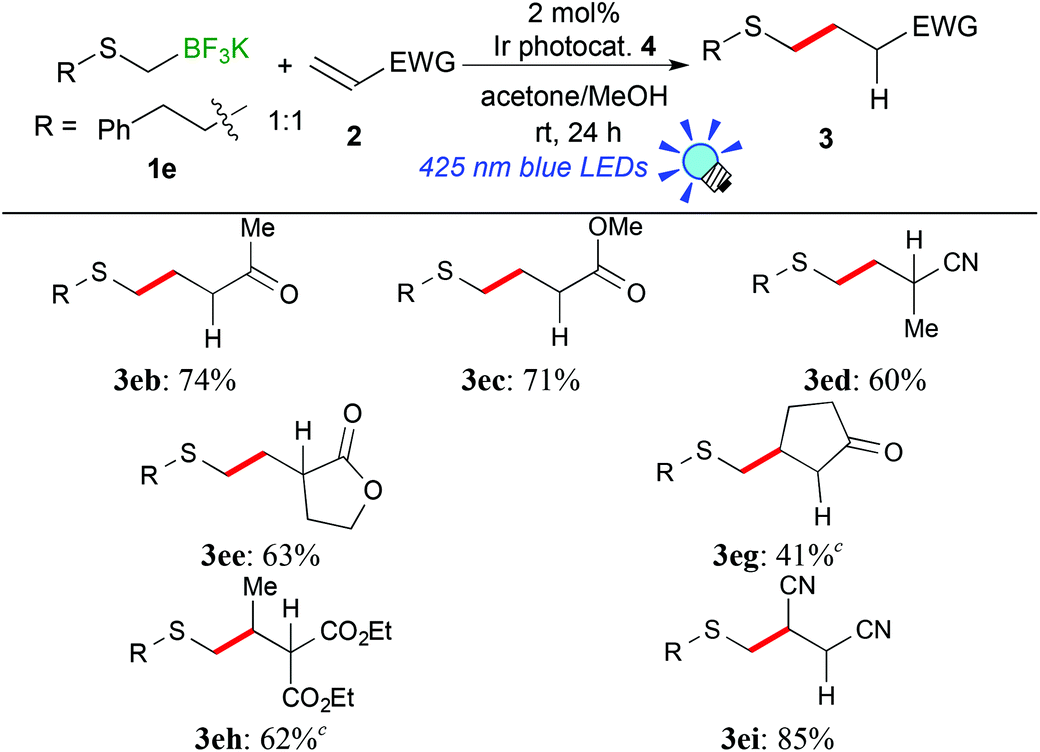
In the light of high reactivity and selectivity of potassium phenethylthiomethyltrifluoroborate (1e), the scope of olefins was investigated (Table 3). Electron-deficient olefins (2b–e, 2i) were also applicable to the present reaction. It should be noted that the reaction with 1e enabled alkylthiomethylation of olefins with a substituent at the reaction site, 2-cyclopenten-1-one (2g) and diethyl ethylidenemalonate (2h), which could not be applied to the reaction with 1avide supra. The alkylthiomethylated products were obtained in 41% (3eg) and 62% yields (3eh) with the use of an excess amount of borate and 5 mol% of Ir catalyst 4. These results suggest that the phenethylthiomethyl radical serves as a species more nucleophilic than the 1,3-dithian-2-yl radical and arylthiomethyl radicals presumably due to steric and electronic reasons.
 | (1) |
|
a Reaction conditions: a reaction mixture of 1a (0.25 mmol), olefin 2 (0.25 mmol), Ir photocatalyst 4 (5.0 μmol), degassed acetone (1.0 mL) and MeOH (1.0 mL) was irradiated by using 3 W blue LEDs (λ = 425 ± 15 nm) at room temperature for 24 h.
b Isolated yields.
c
1e:2 = 2:1, 5 mol% of Ir catalyst 4 was used.
|
|---|
|
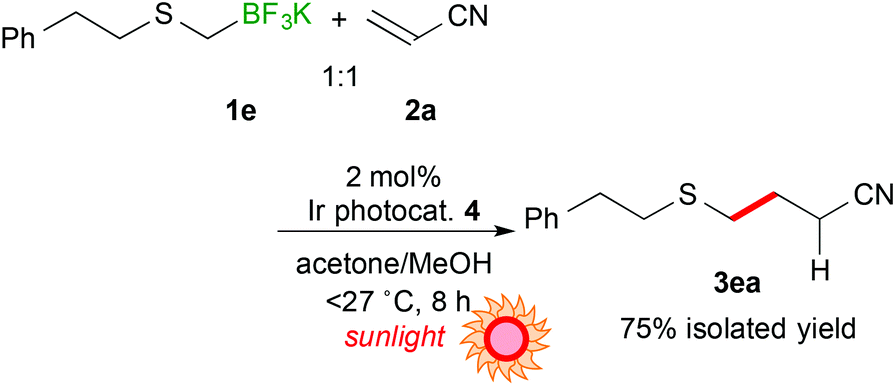
Utilization of sunlight as a light source for photoreaction is promising in terms of green and sustainable chemistry. Thus, sunlight-driven photocatalytic alkylthiomethylation of 2a with 1e was performed. As a result, the reaction proceeded more efficiently (8 h) than the reaction irradiated by using blue LEDs (eqn (1)).
To gain an insight into the reaction mechanism, electrochemical analysis of organoborates and luminescence quenching experiments were carried out (see the ESI†). Oxidation potentials for all the above-mentioned α-thioalkylborates (1a–1e) were less than +0.72 V vs. Cp2Fe, indicating that they can be easily oxidized by the photoactivated Ir photocatalyst 4 (E1/2 = +0.91 V vs. Cp2Fe). In addition, potassium phenethylthiomethyltrifluoroborate (1e) strongly quenched luminescence from the triplet excited state of 4, supporting that the first SET event occurs between the excited Ir species and 1.
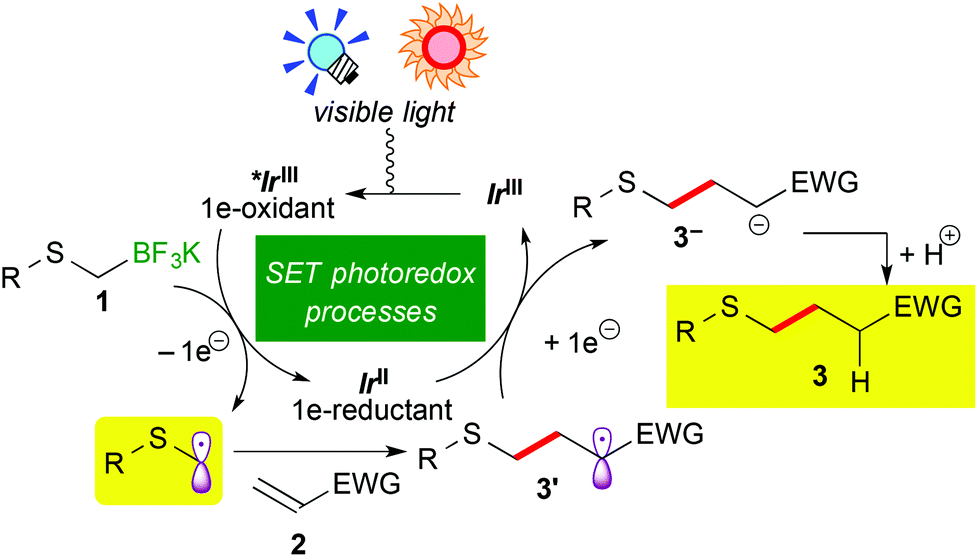
A plausible reaction mechanism is illustrated in Scheme 3. Visible light irradiation induces the excitation of the Ir photocatalyst (IrIII) into the photoactivated state (*IrIII), which serves as a strong oxidant. One-electron oxidation of alkyl- or aryl-thioalkylborate 1 by *IrIII generates an α-thioalkyl radical via deboronation accompanying the formation of the highly reduced Ir species, IrII. Subsequent addition of the α-thioalkyl radical to electron-deficient olefins 2 affords the radical intermediate 3′, which undergoes a second SET event from IrII to be converted into carboanionic intermediate 3−. Finally, protonation by the solvent produces alkyl- or aryl-thioalkylated product 3. A control experiment in CD3OH revealed that the H atom in the product 3 does not result from hydrogen abstraction from the CD3 group but from protonation with the OH group (see the ESI†). At present, we cannot rule out the radical propagation mechanism at all. But the present reaction required continuous irradiation of visible light to proceed steadily (see the ESI†). Therefore, the radical propagation process is not the major reaction pathway, if any.
 | ||
| Scheme 3 A plausible reaction mechanism. | ||
Conclusions
We have developed alkyl- and aryl-thioalkylation of olefins using the corresponding organotrifluoroborates by visible-light-induced photoredox catalysis. The present photocatalytic protocol enables facile generation of α-thioalkyl radicals and access to a new range of alkylsulphides without any additives under mild reaction conditions: room temperature and visible light irradiation including natural sunlight. Now our effort is directed to the synthesis of new useful compounds through photoredox processes.Acknowledgements
The financial support from the Japanese government (Grants-in-Aid for Scientific Research: no. 26288045) is gratefully acknowledged. Y.L. also gratefully acknowledges financial support from the Japanese Government (Monbukagakusho: MEXT) Scholarship (no. 121540).Notes and references
- For selected reviews on photoredox catalysis, see: (a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (c) F. Teplý, Collect. Czech. Chem. Commun., 2011, 76, 859 CrossRef; (d) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (e) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC; (f) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (g) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734 CrossRef CAS PubMed; (h) M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727 CrossRef; (i) J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977 CrossRef PubMed; (j) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (k) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem. – Eur. J., 2014, 20, 3874 CrossRef CAS PubMed; (l) T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562 RSC; (m) T. Koike and M. Akita, Top. Catal., 2014, 57, 967 CrossRef CAS.
- For selected reports on the reaction of aminoalkyl radicals by photoredox catalysis, see: (a) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114 CrossRef CAS PubMed; (b) P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672 CrossRef CAS PubMed; (c) Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338 CrossRef CAS PubMed; (d) S. Cai, X. Zhao, X. Wang, Q. Liu, Z. Li and D. Z. Wang, Angew. Chem., Int. Ed., 2012, 51, 8050 CrossRef CAS PubMed; (e) X. Ju, D. Li, W. Li, W. Yu and F. Bian, Adv. Synth. Catal., 2012, 354, 3561 CrossRef CAS; (f) S. Zhu, A. Das, L. Bui, H. Zhou, D. P. Curran and M. Rueping, J. Am. Chem. Soc., 2013, 135, 1823 CrossRef CAS PubMed; (g) H. Zhou, P. Lu, X. Gu and P. Li, Org. Lett., 2013, 15, 5646 CrossRef CAS PubMed; (h) A. Singh, A. Arora and J. D. Weaver, Org. Lett., 2013, 15, 5390 CrossRef CAS PubMed; (i) P. Zhang, T. Xiao, S. Xiong, X. Dong and L. Zhou, Org. Lett., 2014, 16, 3264 CrossRef CAS PubMed; (j) Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257 CrossRef CAS PubMed; (k) C. K. Prier and D. W. C. MacMillan, Chem. Sci., 2014, 5, 4173 RSC; (l) A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602 CrossRef CAS PubMed.
- (a) R. S. Andrews, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2010, 49, 7274 CrossRef CAS PubMed; (b) K. Qvortrup, D. A. Rankic and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 626 CrossRef CAS PubMed.
- (a) U.-C. Yoon, H.-J. Kim and P. S. Mariano, Heterocycles, 1989, 29, 1041 CrossRef; (b) E. Hasegawa, M. A. Brumfield, P. S. Mariano and U.-C. Yoon, J. Org. Chem., 1988, 53, 5435 CrossRef CAS; (c) U. C. Yoon, Y. C. Kim, J. J. Choi and D. U. Kim, J. Org. Chem., 1992, 57, 1422 CrossRef CAS; (d) J. H. Byers, C. C. Whitehead and M. E. Duff, Tetrahedron Lett., 1996, 37, 2743 CrossRef CAS; (e) T. Ikeno, M. Harada, N. Arai and K. Narasaka, Chem. Lett., 1997, 26, 169 CrossRef; (f) M. de Greef and S. Z. Zard, Tetrahedron, 2004, 60, 7781 CrossRef CAS; (g) W. Du, L. Tian, J. Lai, X. Huo, X. Xie, X. She and S. Tang, Org. Lett., 2014, 16, 2470 CrossRef CAS PubMed.
- For selected reviews, see: (a) T. Kondo and T. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef CAS PubMed; (b) D. J. Procter, J. Chem. Soc., Perkin Trans. 1, 2001, 335 RSC; (c) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS PubMed; (d) C.-F. Lee, Y.-C. Liu and S. S. Badsara, Chem. – Asian J., 2004, 9, 706 CrossRef PubMed; (e) I. P. Beletskaya and V. P. Ananikov, Eur. J. Org. Chem., 2007, 3431 CrossRef CAS; (f) J. Clayden and P. MacLellan, Beilstein J. Org. Chem., 2011, 7, 582 CrossRef CAS PubMed; (g) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed; (h) C. C. Eichman and J. P. Stambuli, Molecules, 2011, 16, 590 CrossRef CAS PubMed; (i) H. Liu and X. Jiang, Chem. – Asian J., 2013, 8, 2546 CrossRef CAS PubMed; (j) P. Chauhan, S. Mahajan and D. Enders, Chem. Rev., 2014, 114, 8807 CrossRef CAS PubMed.
- (a) D. M. Hedstrand, W. H. Kruizinga and R. M. Kellog, Tetrahedron Lett., 1978, 19, 1255 CrossRef; (b) T. J. van Bergen, D. M. Hedstrand, W. H. Kruizinga and R. M. Kellog, J. Org. Chem., 1979, 44, 4953 CrossRef CAS; (c) M. Chen, Z.-T. Huang and Q.-Y. Zheng, Chem. Commun., 2012, 48, 11686 RSC; (d) X. Wang, G. D. Cuny and T. Noël, Angew. Chem., Int. Ed., 2013, 52, 7860 CrossRef CAS PubMed; (e) E. L. Tyson, M. S. Ament and T. P. Yoon, J. Org. Chem., 2013, 78, 2046 CrossRef CAS PubMed; (f) E. L. Tyson, Z. L. Niemeyer and T. P. Yoon, J. Org. Chem., 2014, 79, 1427 CrossRef CAS PubMed; (g) M. H. Keylor, J. E. Park, C.-J. Wallentin and C. R. J. Stephenson, Tetrahedron, 2014, 70, 4264 CrossRef CAS.
- (a) G. A. Molander and J. Ham, Org. Lett., 2006, 8, 2031 CrossRef CAS PubMed; (b) H. Stefani, R. Cella and A. S. Vieira, Tetrahedron, 2007, 63, 3623 CrossRef CAS; (c) G. A. Molander and N. Ellis, Acc. Chem. Res., 2007, 40, 275 CrossRef CAS PubMed; (d) S. Darses and J.-P. Genet, Chem. Rev., 2008, 108, 288 CrossRef CAS PubMed; (e) A. S. Vieira, P. F. Fiorante, J. Zukerman-Schpector, D. Alves, G. V. Botteselle and H. A. Stefani, Tetrahedron, 2008, 64, 7234 CrossRef CAS.
- (a) G. B. Schuster, Pure Appl. Chem., 1990, 62, 1565 CrossRef CAS; (b) Y. Nishigaichi, T. Orimi and A. Takuwa, J. Organomet. Chem., 2009, 694, 3837 CrossRef CAS; (c) G. Sorin, R. M. Mallorquin, Y. Contie, A. Baralle, M. Malacria, J.-P. Goddard and L. Fensterbank, Angew. Chem., Int. Ed., 2010, 49, 8721 CrossRef CAS PubMed; (d) G. A. Molander, V. Colombel and V. A. Braz, Org. Lett., 2011, 13, 1852 CrossRef CAS PubMed; (e) J. W. Lockner, D. D. Dixon, R. Risgaard and P. S. Baran, Org. Lett., 2011, 13, 5628 CrossRef CAS PubMed; (f) Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, M. D. Bel and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292 CrossRef CAS PubMed; (g) T. W. Liwosz and S. R. Chemler, Org. Lett., 2013, 15, 3034 CrossRef CAS PubMed; (h) K. Komeyama, T. Kashihara and K. Takaki, Tetrahedron Lett., 2013, 54, 1084 CrossRef CAS; (i) S. R. Neufeldt, C. K. Seigerman and M. S. Sanford, Org. Lett., 2013, 15, 2302 CrossRef CAS PubMed.
- (a) Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414 CrossRef CAS; (b) K. Miyazawa, Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2013, 49, 7249 RSC; (c) T. Koike and M. Akita, Synlett, 2013, 24, 2492 CrossRef CAS; (d) K. Miyazawa, T. Koike and M. Akita, Adv. Synth. Catal., 2014, 356, 2749 CrossRef CAS.
- (a) H. Huang, G. Zhang, L. Gong, S. Zhang and Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280 CrossRef CAS PubMed; (b) J. C. Tellis, D. N. Primer and G. A. Molandar, Science, 2014, 345, 443 CrossRef PubMed.
- (a) E. J. Corey and D. Seebach, Angew. Chem., Int. Ed. Engl., 1965, 4, 1075 CrossRef CAS; (b) E. J. Corey and D. Seebach, Angew. Chem., Int. Ed. Engl., 1965, 4, 1077 CrossRef CAS; (c) D. Seebach, Angew. Chem., Int. Ed. Engl., 1979, 18, 239 CrossRef.
- The photoexcited state of [Ir(dF(CF3)ppy)2(bpy)](PF6) (4) is a strong oxidant (E1/2 = +0.91 V vs. Cp2Fe); see: (a) M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef CAS; (b) D. Hanss, J. C. Freys, G. Bernardinelli and O. S. Wenger, Eur. J. Inorg. Chem., 2009, 4850 CrossRef CAS.
- The photoexcited state of [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) (5) and [Ru(bpy)3](PF6)2 (6) are weaker oxidants (E1/2 = +0.80 V and +0.36 V vs. Cp2Fe, respectively) than that of 4; see: ref. 11a and K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data. See DOI: 10.1039/c4qo00352g |
| This journal is © the Partner Organisations 2015 |