
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective syntheses of leuconolam, leuconoxine, and mersicarpine alkaloids from a common intermediate through regiocontrolled cyclizations by Staudinger reactions†
Zining
Li
a,
Qian
Geng
a,
Zhe
Lv
a,
Beau P.
Pritchett
b,
Katsuaki
Baba
b,
Yoshitaka
Numajiri
b,
Brian M.
Stoltz
*b and
Guangxin
Liang
*a
aState Key Laboratory and Institute of Elemento-organic Chemistry, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, China. E-mail: lianggx@nankai.edu.cn
bThe Warren and Katharine Schlinger Laboratory of Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: stoltz@caltech.edu
First published on 20th January 2015
Abstract
Selective syntheses of leuconolam, leuconoxine, and mersicarpine alkaloids bearing distinctive core structures were achieved through Staudinger reactions using a common intermediate. In the key cyclization step, water functioned like a switch to control which core structure to produce. The chemistry allowed for selective syntheses of the group of alkaloids from a simple intermediate through straightforward chemical operations.
Introduction
Leuconolam, leuconoxine, and mersicarpine alkaloids showcase the incredible structural diversity of natural products. These monoterpene indole alkaloid families, though sharing the same biogenetic origin,1 present distinctive skeletons with three completely different polycyclic patterns (1–6, Fig. 1).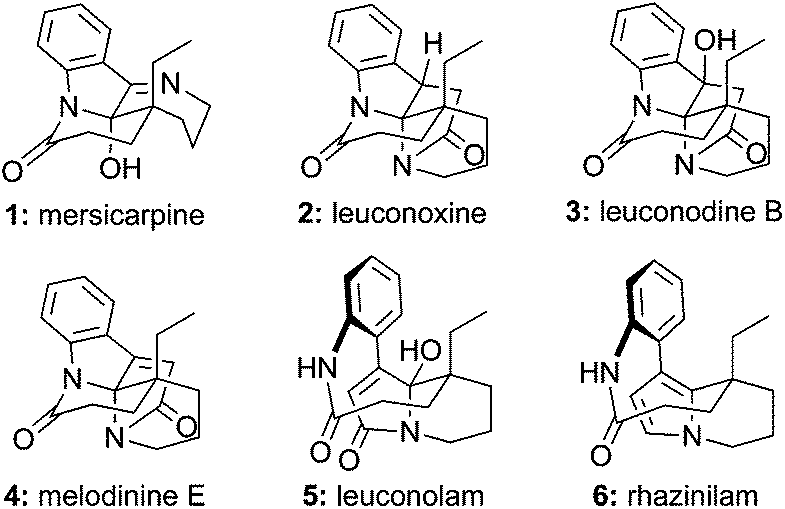 | ||
| Fig. 1 Representative biosynthetically related monoterpene indole alkaloids with distinctive skeleton diversity. | ||
Mersicarpine (1), isolated from both Kopsia and Leuconotis species of plants by Kam and co-workers,2 features a seven-membered cyclic imine, a δ-lactam, and an all-carbon quaternary center around a fully substituted hemiaminal stereogenic center. Although leuconoxine (2),3 leuconodine B (3),4 and melodinine E (4)5 hold the same δ-lactam and indoline moiety as mersicarpine, different bond connections and two additional carbons create an entirely new skeleton distinguished by an aminal functionality, a piperidine ring, and an extra γ-lactam. Leuconolam (5)6 and rhazinilam (6)7 possess an unusual nine-membered lactam and a pyrrole derived-unit. It is proposed that leuconolam is a biosynthetic precusor of melodinine E, which further produces mersicarpine via a skeletal rearrangement and subsequent loss of two carbons in the form of acetic acid.2a The intriguing structural features and biosynthetic connections of these alkaloids make them appealing synthetic targets.8 To date, eight syntheses of mersicarpine9 and three syntheses of leuconoxine-type alkaloids have been reported.9e,g,10 Leuconolam has been accessed through both total synthesis9e,g,11 and oxidative conversion from rhazinilam.12 Rhazinilam has been the focus of numerous synthetic efforts.9g,13
Throughout our efforts toward the total synthesis of mersicarpine,9f,14 we became increasingly interested in its connections with leuconolam and leuconoxine alkaloids. We envisioned rapid access to all three different polycyclic patterns through a versatile intermediate 7 (Scheme 1). Leuconolam (5) could be obtained through disconnection of the C–N bond of melodinine E (4). Melodinine E could be accessed from 11 by an acetylation and aldol condensation sequence. Given that mersicarpine (1) and 11 have the same oxidation state but different bond connections, we conceived that both compounds could be prepared from a common acyclic intermediate 7 through divergent cyclization sequences. We aimed to take advantage of orthogonal protecting groups P1 and P2 on the aniline and amine nitrogens, respectively. Upon the removal of P1, facile hemiaminal formation at the C2 position would afford 8, which could in turn produce compound 9 upon removal of P2. If instead P2 is removed first, a more favourable 6-membered hemiaminal formation would generate intermediate 10, which could produce compound 11 following P1 removal and subsequent lactam formation. It is worth noting that Zhu and co-workers applied a similar strategy in their recent syntheses of these alkaloids, in which they used fine-tuned hydrogenation conditions to control the cyclization sequences.9e Herein, we report a new approach to three different classes of alkaloids using Staudinger reaction as a key ring formation step from a common acyclic intermediate.
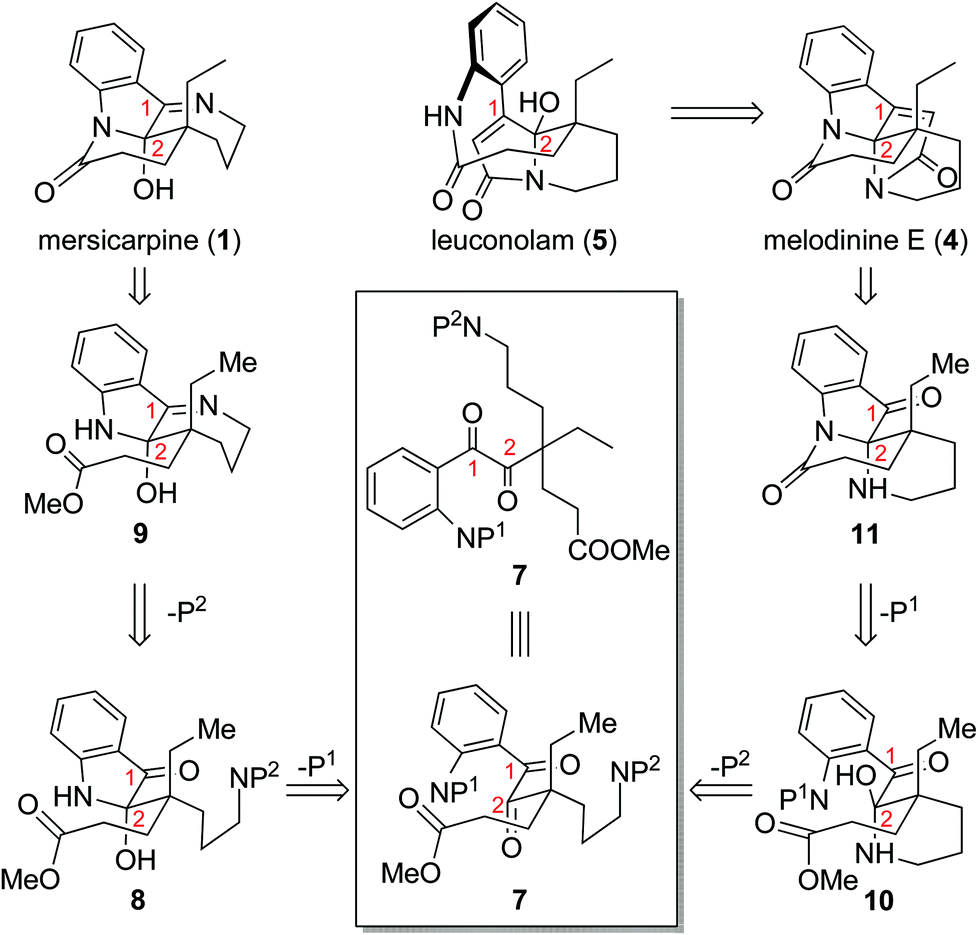 | ||
| Scheme 1 Initial synthetic design of different cyclization sequences leading to distinct molecular skeletons. | ||
Results and discussion
In the forward synthesis, we chose compound 16 (Scheme 2) to be a preferred intermediate with a Boc-protected aniline (NP1) and an azide (NP2) as a masked amine to enable differentiable deprotection. Our quick construction of 16 commenced from a known compound 12.15 Hydroboration-oxidation,16 followed by a Mitsunobu reaction using DPPA17 converted 12 to compound 13 featuring a primary azide. Lactol formation was effected with DIBAL-H, followed by Ohira–Bestmann homologation18 afforded alkyne 14. Oxidation of the primary alcohol, followed by Fischer esterification provided the desired coupling partner for Sonogashira coupling19 with tert-butyl-(2-iodophenyl) carbamate to furnish 15 in good yield. Ruthenium-catalysed oxidation20 of the alkyne afforded 1,2-diketone 16 in 66% yield.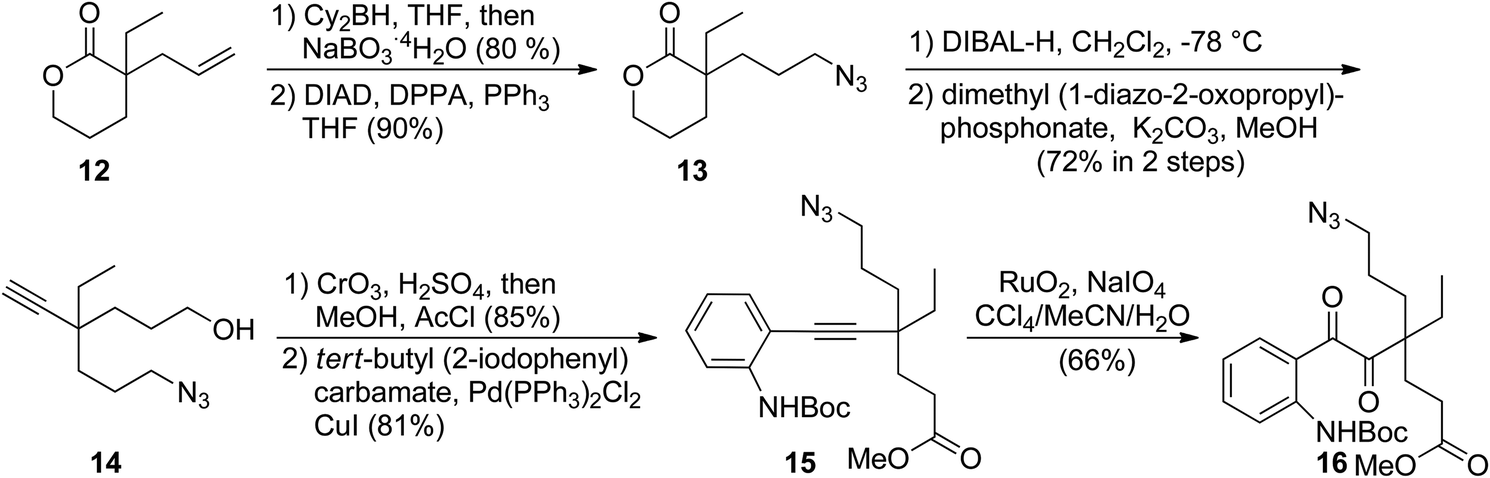 | ||
| Scheme 2 Preparation of the common intermediate 16. | ||
We then turned our attention to exploring divergent cyclization sequences involving 1,2-diketone 16 (Scheme 3). To our surprise, compound 17 didn't undergo spontaneous hemiaminal formation, but was isolated in 69% yield following selective removal of the Boc group in 16 with TMSOTf in the presence of 2,6-lutidine.21 However, treatment of 17 with triphenylphosphine in a mixed solvent of THF and water cleanly furnished mersicarpine in 66% yield. This remarkably simple reaction forms the three remaining rings in mersicarpine under mild conditions. Notably, the oxidation states of diketone in 17 were exploited to rapidly arrive at the target in a redox-free manner. Importantly, a Staudinger reaction in the absence of water gave an inseparable diastereomeric mixture of compound 18, which possesses a totally different polycyclic framework. We hypothesize that an aza-Wittig pathway is operative in the absence of water.22 In the event, the more favourable 6-membered imine is formed, followed by aminal formation with no facial selectivity. Impressively, when a diastereomeric mixture of 18 was treated with sodium hydride in toluene at 50 °C, compound 11 was generated in 85% yield. This finding indicates that an interconversion of the two diastereomeric aminals formed under the reaction conditions funnels the mixture toward a thermodynamically favored product (11).
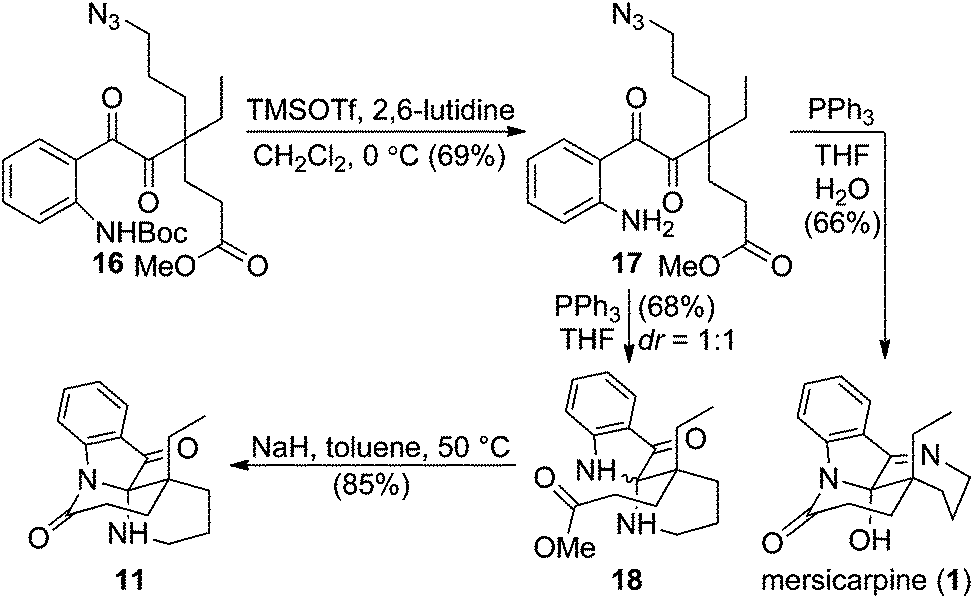 | ||
| Scheme 3 Selective syntheses of mersicarpine and the core structure 11 in leuconoxine-type alkaloids. | ||
Key intermediate 11 facilitated completion of the total syntheses of three leuconoxine-type alkaloids as well as leuconolam (Scheme 4). Acetylation of the free amine in the piperidine ring in 11 proceeded smoothly in neat acetic anhydride at room temperature to afford 19. When treated with LDA at −78 °C, 19 produced leuconodine B readily in 72% yield. The transformation of leuconodine B to melodinine E was fulfilled in 90% yield upon treatment with neat thionyl chloride at room temperature followed by elimination with DBU in THF and subsequent aqueous workup. Initially, we believed that treatment of 20 with DBU would generate melodinine E directly, but surprisingly melodinine E was not detected by 1H NMR spectroscopy in the crude mixture without an aqueous workup. The major product was too sensitive to be isolated and attempted purification of this compound with column chromatography produced melodinine E. High resolution mass spectrometry data suggest that treatment of 20 with DBU yields the proposed structure 21. When the sensitive intermediate 21 was stirred in water at room temperature, melodinine E was produced in 90% yield. Interestingly, when 21 was treated with an aqueous solution of 3 N H2SO4 at 50 °C, leuconolam was generated in 75% yield. Using conditions reported by Zhu and co-workers, leuconolam can also be prepared directly from melodinine E.9e Finally, hydrogenation on melodinine E occurred efficiently to generate leuconoxine in nearly quantitative yield.
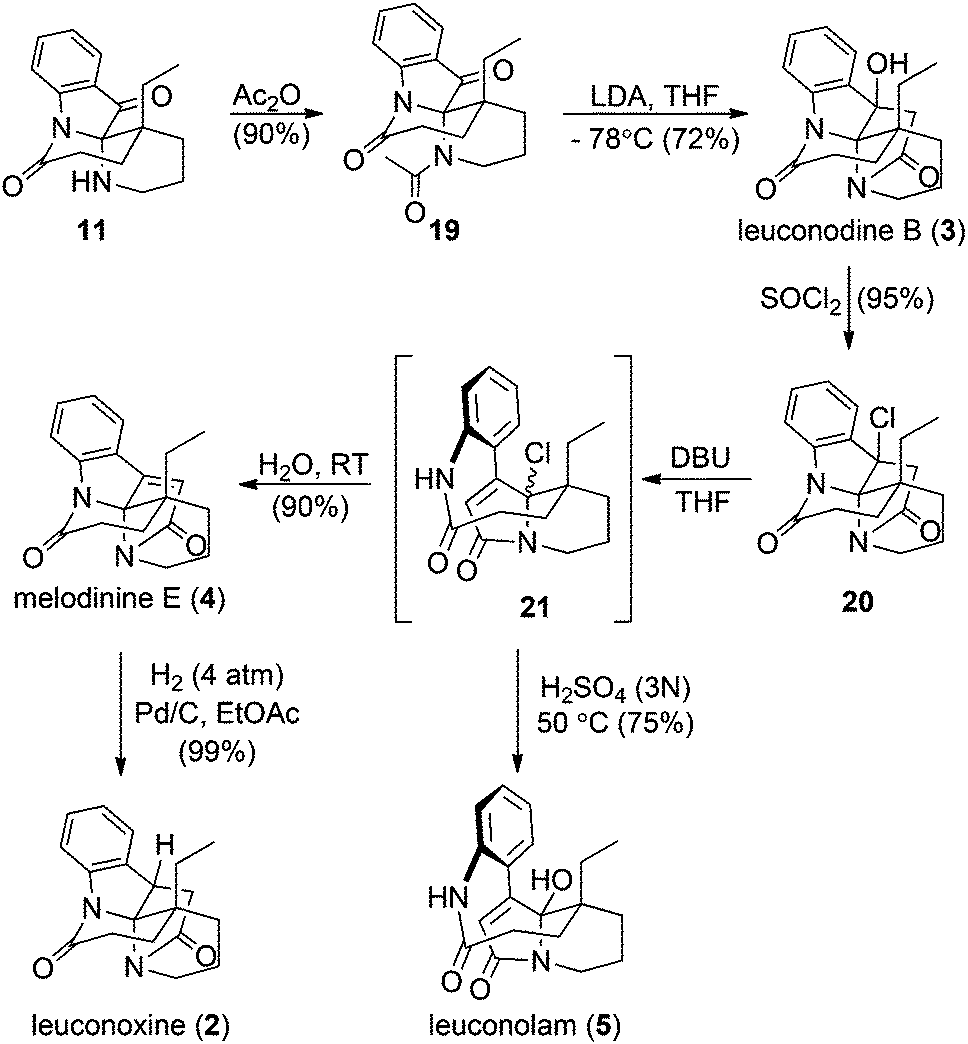 | ||
| Scheme 4 Syntheses of leuconodine B, melodinine E, leuconoxine, and leuconolam. | ||
With efficient racemic syntheses in hand, we took on an effort to produce optically active 12, thereby achieving formal asymmetric syntheses of these alkaloids (Scheme 5). Initially, we hoped diester 22 could undergo an efficient asymmetric allylic alkylation to construct enantioenriched quaternary lactone 23. We found that the reaction with diester 22 proceeded smoothly, but with disappointing enantioselectivity (81% ee). Eventually, we were able to generate optically active 12 from an N-benzyloxy imide 24, which could be readily prepared in 80% yield and 98% ee.23 Reduction of 24 with an excess of NaBH4 formed hydroxamic acid 25 with the desired free primary alcohol. The following acid-induced cyclization of 25 provided the desired lactone (−)-12 in 54% yield over 2 steps.
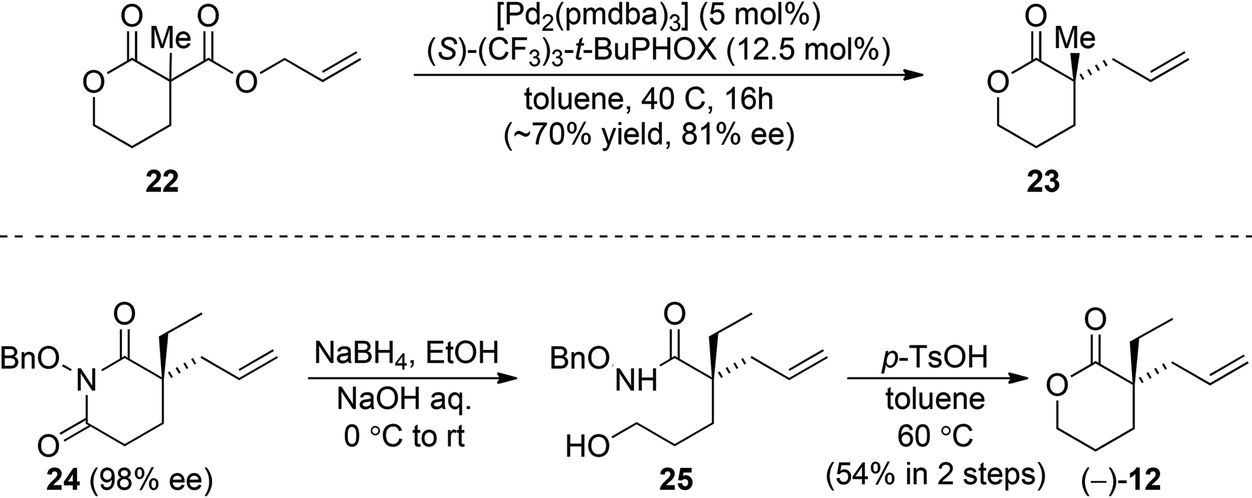 | ||
| Scheme 5 Efforts in preparing optically active 12. | ||
Conclusions
In summary, we have completed total syntheses of mersicarpine (1), leuconoxine (2), leuconodine (3), melodinine E (4), and leuconolam (5) by controlling specific cyclization sequences through a key Staudinger reaction to access different polycyclic frameworks. Additionally, we have achieved enantioselective formal syntheses of these alkaloids by synthesizing enantioenriched lactone 12via an asymmetric allylic alkylation, reduction, and cyclization sequence.Acknowledgements
The NKU authors thank the National Natural Science Foundation of China, Tianjin Natural Science Foundation (grant no. 12JCZDJC26400), the ‘111’ project (B06005) of the Ministry of Education of China, and Program for New Century Excellent Talents in University for financial support. The Caltech authors wish to thank NIH-NIGMS (R01GM080269) for financial support. BPP is grateful to the National Science Foundation for a Graduate Research Fellowship. Toray is thanked for providing YN a postdoctoral opportunity at Caltech.Notes and references
- (a) S. H. Goh and A. R. M. Ali, Tetrahedron Lett., 1986, 27, 2501–2504 CrossRef CAS; (b) J. Hájíček, Collect. Czech. Chem. Commun., 2011, 76, 2023–2083 CrossRef; (c) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694–752 RSC; (d) Y.-Y. Low, F.-J. Hong, K.-H. Lim, N. F. Thomas and T.-S. Kam, J. Nat. Prod., 2014, 77, 327–338 CrossRef CAS PubMed.
- (a) T.-S. Kam, G. Subramaniam, K.-H. Lim and Y.-M. Choo, Tetrahedron Lett., 2004, 45, 5995–5998 CrossRef CAS PubMed; (b) C.-Y. Gan, Y.-Y. Low, N. F. Thomas and T.-S. Kam, J. Nat. Prod., 2013, 76, 957–964 CrossRef CAS PubMed.
- F. Abe and T. Yamauchi, Phytochemistry, 1994, 35, 169–171 CrossRef CAS.
- (a) T. Feng, X.-H. Cai, P.-J. Zhao, Z.-Z. Du, W.-Q. Li and X.-D. Luo, Planta Med., 2009, 75, 1537–1541 CrossRef CAS PubMed; (b) C.-Y. Gan, Y.-Y. Low, N. F. Thomas and T.-S. Kam, J. Nat. Prod., 2013, 76, 957–964 CrossRef CAS PubMed.
- T. Feng, X.-H. Cai, Y.-P. Liu, Y. Li, Y.-Y. Wang and X.-D. Luo, J. Nat. Prod., 2010, 73, 22–26 CrossRef CAS PubMed.
- S. H. Goh, C. Wei and A. R. M. Ali, Tetrahedron Lett., 1984, 25, 3483–3484 CrossRef CAS.
- (a) H. H. A. Linde, Helv. Chim. Acta, 1965, 48, 1822–1842 CrossRef CAS; (b) A. Banerji, P. L. Majumder and A. Chatterjee, Phytochemistry, 1970, 9, 1491–1493 CrossRef CAS; (c) D. J. Abraham, R. D. Rosenstein, R. L. Lyon and H. H. S. Fong, Tetrahedron Lett., 1972, 13, 909–912 CrossRef.
- For selected syntheses of other alkaloids bearing dearomatized indole moiety, see: (a) S. Han and M. Movassaghi, J. Am. Chem. Soc., 2011, 133, 10768–10771 CrossRef CAS PubMed; (b) X. Zhang, T. Mu, F. Zhan, L. Ma and G. Liang, Angew. Chem., Int. Ed., 2011, 50, 6164–6166 CrossRef CAS PubMed; (c) F. Zhan and G. Liang, Angew. Chem., Int. Ed., 2013, 52, 1266–1269 CrossRef CAS PubMed; (d) S. Li, J. Han and A. Li, Acta Chim. Sin., 2013, 71, 295–298 CrossRef CAS; (e) Y. Sun, R. Li, W. Zhang and A. Li, Angew. Chem., Int. Ed., 2013, 52, 9201–9204 CrossRef CAS PubMed; (f) Y. Sun, P. Chen, D. Zhang, M. Baunach, C. Hertweck and A. Li, Angew. Chem., Int. Ed., 2014, 53, 9012–9016 CrossRef CAS PubMed.
- For total syntheses of mersicarpine, see: (a) J. Magolan, C. A. Carson and M. A. Kerr, Org. Lett., 2008, 10, 1437–1440 CrossRef CAS PubMed; (b) R. Nakajima, T. Ogino, S. Yokoshima and T. Fukuyama, J. Am. Chem. Soc., 2010, 132, 1236–1237 CrossRef CAS PubMed; (c) Y. Iwama, K. Okano, K. Sugimoto and H. Tokuyama, Org. Lett., 2012, 14, 2320–2322 CrossRef CAS PubMed; (d) Y. Iwama, K. Okano, K. Sugimoto and H. Tokuyama, Chem. – Eur. J., 2013, 19, 9325–9334 CrossRef CAS PubMed; (e) Z. Xu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2013, 135, 19127–19130 CrossRef CAS PubMed; (f) Z. Lv, Z. Li and G. Liang, Org. Lett., 2014, 16, 1653–1655 CrossRef PubMed; (g) Y. Yang, Y. Bai, S. Sun and M. Dai, Org. Lett., 2014, 16, 6216–6219 CrossRef CAS PubMed. For the formal total syntheses of mersicarpine, see: (h) A. Biechy and S. Z. Zard, Org. Lett., 2009, 11, 2800–2803 CrossRef CAS PubMed; (i) X. Zhong, Y. Li and F.-S. Han, Chem. – Eur. J., 2012, 18, 9784–9788 CrossRef CAS PubMed.
- A. Umehara, H. Ueda and H. Tokuyama, Org. Lett., 2014, 16, 2526–2529 CrossRef CAS PubMed.
- (a) M. G. Banwell, D. A. S. Beck and A. C. Willis, ARKIVOC, 2006,(no. iii), 163–174 CAS; (b) E. C. Izgu and T. R. Hoye, Chem. Sci., 2013, 4, 2262–2266 RSC.
- A. Décor, D. Bellocq, O. Thoison, N. Lekieffre, A. Chiaroni, J. Ouazzani, T. Cresteil, F. Guéritte and O. Baudoin, Bioorg. Med. Chem., 2006, 14, 1558–1564 CrossRef PubMed.
- For representative total syntheses of rhazinilam, see: (a) A. H. Ratcliffe, G. F. Smith and G. N. Smith, Tetrahedron Lett., 1973, 14, 5179–5184 CrossRef; (b) P. Magnus and T. Rainey, Tetrahedron, 2001, 57, 8647–8651 CrossRef CAS; (c) J. A. Johnson and D. Sames, J. Am. Chem. Soc., 2000, 122, 6321–6322 CrossRef CAS; (d) J. A. Johnson, N. Li and D. Sames, J. Am. Chem. Soc., 2002, 124, 6900–6903 CrossRef CAS PubMed; (e) A. L. Bowie, C. C. Hughes and D. Trauner, Org. Lett., 2005, 7, 5207–5209 CrossRef CAS PubMed; (f) Z. Liu, A. S. Wasmuth and S. G. Nelson, J. Am. Chem. Soc., 2006, 128, 10352–10353 CrossRef CAS PubMed; (g) A. L. Bowie and D. Trauner, J. Org. Chem., 2009, 74, 1581–1586 CrossRef CAS PubMed; (h) Z. Gu and A. Zakarian, Org. Lett., 2010, 12, 4224–4227 CrossRef CAS PubMed; (i) L. McMurray, E. M. Beck and M. Gaunt, Angew. Chem., Int. Ed., 2012, 51, 9288–9291 CrossRef CAS PubMed; (j) J.-B. Gualtierotti, D. Pasche, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2014, 53, 9926–9930 CrossRef CAS PubMed.
- Z. Li and G. Liang, Tetrahedron Lett., 2013, 54, 242–244 CrossRef CAS PubMed.
- J. L. Herrmann and R. H. Schlessinger, J. Chem. Soc., Chem. Commun., 1973, 711–712 RSC.
- H. C. Brown and B. C. S. Rao, J. Am. Chem. Soc., 1956, 78, 5694–5695 CrossRef CAS.
- (a) B. Lal, B. N. Pramanik, M. S. Manhas and A. K. Bose, Tetrahedron Lett., 1977, 18, 1977–1980 CrossRef; (b) Z. Xu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2013, 52, 3272–3276 CrossRef CAS PubMed.
- (a) S. Müller, B. Liepold, G. J. Roth and H. J. Bestmann, Synlett, 1996, 521–522 CrossRef PubMed; (b) G. J. Roth, B. Liepold, S. G. Müller and H. J. Bestmann, Synthesis, 2004, 59–62 CrossRef CAS PubMed.
- (a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef; (b) C. C. Li, Z. X. Xie, Y. D. Zhang, J. H. Chen and Z. Yang, J. Org. Chem., 2003, 68, 8500–8504 CrossRef CAS PubMed.
- (a) R. Zibuck and D. Seebach, Helv. Chim. Acta, 1988, 71, 237–240 CrossRef CAS; (b) M. F. Semmelhack, S. R. Campagna, M. J. Federle and B. L. Bassler, Org. Lett., 2005, 7, 569–572 CrossRef CAS PubMed.
- H. M. M. Bastiaans, J. L. van der Baan and H. C. J. Ottenheijm, J. Org. Chem., 1997, 62, 3880–3889 CrossRef CAS.
- For recent reviews on Aza-Wittig reactions, see: (a) S. Eguchi, Y. Matsushita and K. Yamashita, Org. Prep. Proced. Int., 1992, 24, 209–243 CrossRef CAS; (b) P. M. Fresneda and P. Molina, Synlett, 2004, 1–17 CrossRef CAS PubMed.
- (a) D. C. Behenna, Y. Liu, T. Yurino, J. Kim, D. E. White, S. C. Virgil and B. M. Stoltz, Nat. Chem., 2012, 4, 130–133 CrossRef CAS PubMed; (b) N. B. Bennett, D. C. Duquette, J. Kim, W.-B. Liu, D. C. Behenna, S. C. Virgil and B. M. Stoltz, Chem. – Eur. J., 2013, 19, 4414–4418 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and procedures, compound characterization data, copies of 1H and 13C NMR spectra for new compounds. See DOI: 10.1039/c4qo00312h |
| This journal is © the Partner Organisations 2015 |