
Difluoromethyl 2-pyridyl sulfone: a versatile carbonyl gem-difluoroolefination reagent
Bing
Gao
,
Yanchuan
Zhao
,
Jingyu
Hu
and
Jinbo
Hu
*
Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: jinbohu@sioc.ac.cn
First published on 16th December 2014
Abstract
This tutorial account describes a robust carbonyl gem-difluoroolefination reagent, difluoromethyl 2-pyridyl sulfone (2-PySO2CF2H), developed by our group, recently. The guidelines for its laboratory preparation and application are presented in detail for potential users.
 Bing Gao | Bing Gao was born in Sichuan, China in 1986. He received his B.S. degree in chemistry from Jilin University in 2009, and received his Ph.D. degree in 2014 from the Shanghai Institute of Organic Chemistry, Chinese Academy of Science (SIOC, CAS) under the supervision of Professor Jinbo Hu. He is currently a research assistant at the CAS Key Laboratory of Organofluorine Chemistry, SIOC. His doctoral research focused on the preparation and transformation of fluoroalkenes. |
 Yanchuan Zhao | Yanchuan Zhao was born in Jilin, China, in 1983. He received his Ph.D. degree in 2012 from the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences (SIOC, CAS) under the guidance of Professor Jinbo Hu, working in the field of organofluorine chemistry and focusing on the development of novel fluoroalkylation and fluoroolefination reagents. He is currently a postdoctoral research associate with Professor Timothy M. Swager at the Massachusetts Institute of Technology, where he uses 19F NMR to fingerprint various organic molecules. His scientific interests include chemical sensors, molecular recognition, mechanochemistry, and organofluorine chemistry. |
 Jingyu Hu | Jingyu Hu was born in Shandong, China in 1992. After completing his B.S. (East China University of Science and Technology) degree in 2013, he is now pursuing his M.S. at the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences (SIOC, CAS) under the supervision of Professor Jinbo Hu. His current research interests are focussed on fluoroalkylation methodologies. |
 Jinbo Hu | Jinbo Hu was born in Zhejiang, China in 1973. After he completed his B.S. (Hangzhou University) and M.S. (Chinese Academy of Sciences) degrees, he pursued his Ph.D. during 1997 to 2002 at the University of Southern California with Professors G. K. S. Prakash and G. A. Olah. After his postdoctoral work at USC, he joined the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences (SIOC, CAS) as a Research Professor in early 2005, where he is currently the head of the CAS Key Laboratory of Organofluorine Chemistry. He is the recipient of the RSC Fluorine Prize 2009 and the Tan Kah-Kee Young Scientist Award 2012. His current research interests include selective fluorination, defluorination, fluoroalkylation methodologies, and fluorinated materials. |
Introduction
Fluoroalkenes have attracted extensive research interest from the synthetic community for decades owing to their unique properties and important applications.1,2 Recently, exploring new reagents and methods for the synthesis of gem-difluoroolefins with high reliability and broad scope has become particularly active.3,4 Taking into account the cost and availability of the starting materials, the deoxygenative gem-difluoroolefination of carbonyl compounds is particularly attractive.4 We have recently developed a new reagent, difluoromethyl 2-pyridyl sulfone (2-PySO2CF2H), which is capable of converting various aldehydes, ketones, and even some lactones to their corresponding gem-difluoroolefins, efficiently.5 Preliminary mechanistic studies have disclosed that this reaction is a typical Julia–Kocienski olefination.5,6 This reagent has also found broad applications in related synthetic methodologies.7–13 In this tutorial account, we aim to provide a detailed introduction on the preparation and application of 2-PySO2CF2H as a gem-difluoroolefination reagent, which we hope could serve as a useful guideline for potential users.Results and discussion
The Julia–Kocienski olefination has been widely employed as a powerful tool to synthesize alkenes from carbonyl compounds.6 Generally, a carbonyl substrate firstly undergoes a nucleophilic addition with a heteroarenesulfonyl-substituted carbanion, and then the resulting adduct rearranges spontaneously to afford a sulfinate salt, which fragmentizes to give the alkene product. A series of heteroaryl sulfones have been successively developed and investigated since the pioneering discovery by Julia, among which the 1-tert-butyl-1H-tetrazol-5-yl (TBT), 1,3-benzothiazol-2-yl (BT), 1-phenyl-1H-tetrazol-5-yl (PT) sulfones have been found to be most effective. Nevertheless, the 2-pyridyl (2-Py) sulfone derivatives have been less studied and rarely used in conventional Julia–Kocienski reactions. We recently succeeded in the synthesis of monofluorinated alkenes from aldehydes and ketones with TBTSO2CH2F.14 However, its difluoromethyl analogue (TBTSO2CF2H) could not afford the gem-difluoroolefination product under similar conditions, indicating an unusual reactivity as a result of the increased fluorine-substitution. Meanwhile, the BT- and PT-sulfones also provided unsatisfactory results.5b We tried to make minimal modulations on difluoromethyl phenyl sulfone (PhSO2CF2H) that is known as an efficient nucleophilic difluoromethylation reagent.15 The optimization of the aryl group finally led to the new compound 2-PySO2CF2H, which was later found to be a powerful gem-difluoroolefination reagent.Difluoromethyl 2-pyridyl sulfone was prepared according to the following procedure: (1) the difluoromethylation of 2-mercaptopyridine (2-PySH) with difluorochloromethane (HCF2Cl) or diethyl bromodifluoromethanephosphonate [BrCF2P(O)(OEt)2];5,10 (2) the oxidation of 2-PySCF2H under either the NaIO4/RuCl3·xH2O(cat.)/CH3CN/CCl4/H2O conditions or the H2O2/Na2WO4·2H2O(cat.)/MeOH conditions (the later one is preferred; however, caution should be exercised to avoid H2O2 residual during the work-up step).8,9 The raw materials are inexpensive and the reaction can be scaled up to the hectogram scale in the laboratory without the requirement of chromatographic purification (distillation for the first step and crystallization for the second step). Obtained as a colourless crystal, this reagent is fairly stable when exposed to air, moisture, and visible light for more than two years (Fig. 1). It is now commercially available and is also called the “Hu reagent”.10
 | ||
| Fig. 1 Difluoromethyl 2-pyridyl sulfone and its carbonyl gem-difluoroolefination reaction. | ||
In a typical carbonyl gem-difluoroolefination procedure (Fig. 2, condition A),5b an excess amount of base (KOtBu solved in 1.0 mL DMF, 1.8 equiv.) was slowly injected into a DMF solution of a substrate (1.2 equiv.) and 2-PySO2CF2H (0.5 mmol) at −50 °C under a N2 atmosphere, the reaction mixture was subsequently warmed to −40 °C over 15 min and then quenched with 3 M HCl (tips: the temperature during the HCl-quenching step should be maintained at a relatively low level, −40 °C in most cases, so as to avoid side-reactions). Normally, most of the aldehydes and ketones were readily converted to gem-difluoroolefins in high yields. The reaction is reliable, easy-to-operate, and reproducible even on large scales. For instance, both anisic aldehyde (Fig. 2, 80%) and the precursor of thrombin inhibitor SSR182289A16 (Fig. 2, 78%) could be effectively converted to the corresponding olefins on a 5-gram scale.
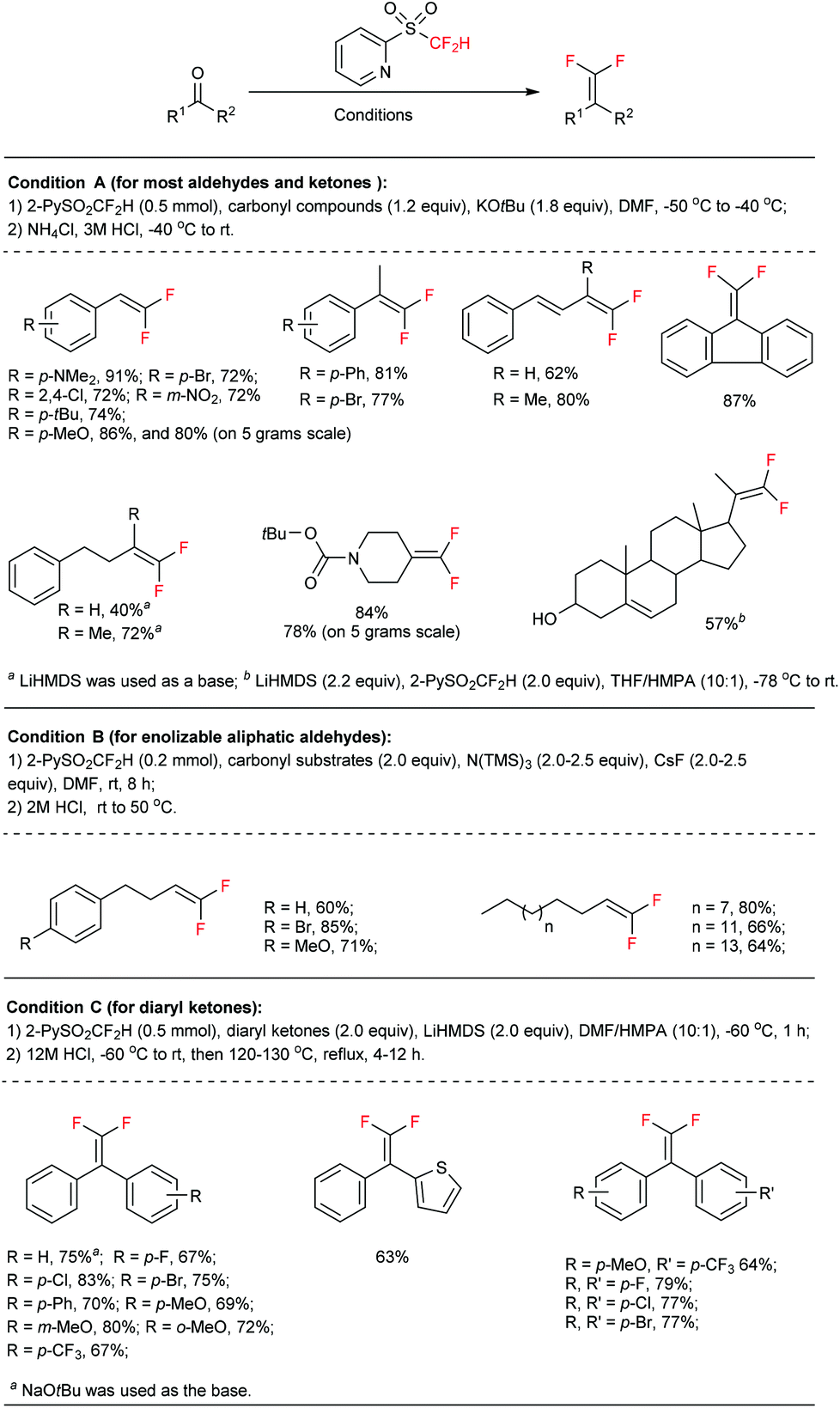 | ||
| Fig. 2 The gem-difluoroolefination of different types of carbonyl compounds with difluoromethyl 2-pyridyl sulfone. | ||
The fluorinated sulfinate salt, a key intermediate after the Smiles rearrangement step in the Julia–Kocienski olefination,17 was found relatively stable under basic conditions and could be captured by CH3I (monitored by 19F NMR).5b These findings clearly suggest a Julia–Kocienski mechanism (Fig. 3, path A). At the onset, the 2-PySO2CF2− anion undergoes nucleophilic addition to the carbonyl group, and then a spontaneous Smiles rearrangement takes place giving the sulfinate intermediate. The latter species undergoes rapid protonolysis to afford the corresponding gem-difluoroolefin when treated with an acid. The understanding of the reaction mechanism allows chemists to find more variations of this important reaction. For instance, a formal nucleophilic iodo- and bromodifluoromethylation of carbonyl compounds has been achieved by changing the reaction pathway from olefination to alkylation.9
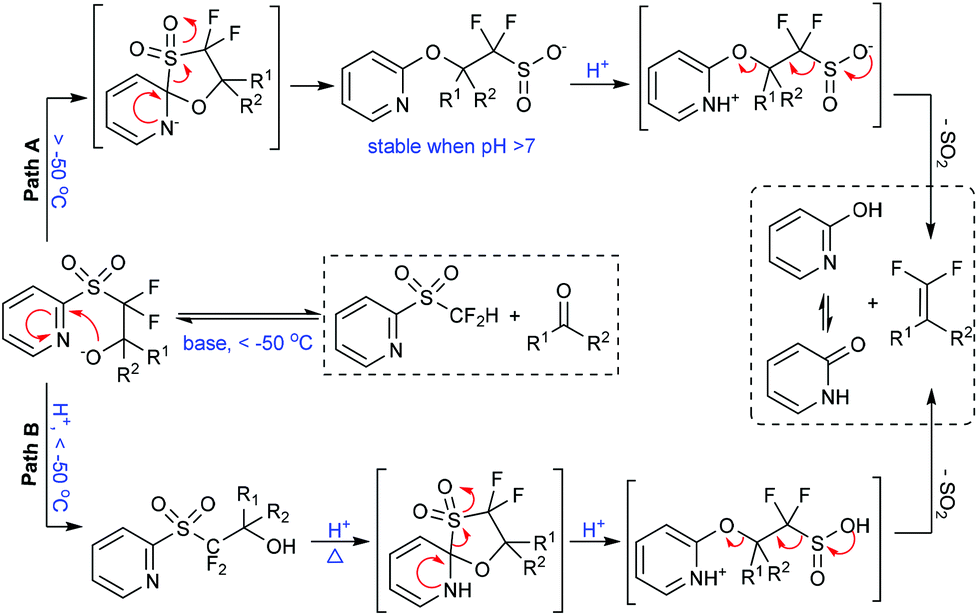 | ||
| Fig. 3 Optional reaction pathways on the basis of mechanistic insights. | ||
In conventional Julia–Kocienski reactions, the non-fluorinated heteroaryl sulfone was firstly deprotonated by using a strong base before its reaction with carbonyl substrates. This pre-generation (of the carbanion) strategy can be used to extend the reaction scope to base-sensitive substrates. However, the Barbier-type in situ generation protocol is essential in our reaction due to the poor stability of the 2-PySO2CF2− anion.18 As a consequence enolizable aldehydes are not well tolerated under the present highly basic conditions (Fig. 2, condition A). Intriguingly, by using cesium fluoride (CsF) as a desilylating reagent and excess tris(trimethylsilyl)amine [N(TMS)3] as the base precursor,19 the gem-difluoroolefination of enolizable aldehydes was successfully achieved at room temperature in high yields (Fig. 2, condition B).5a These modified reaction conditions avoid the exposure of aldehydes to excess amounts of strong base, and therefore suppress the side reactions.
On the other hand, the gem-difluoroolefination of diaryl ketones had been a long-standing challenge. It was found, on the basis of controlled experiments, that the retro-nucleophilic addition reaction would outstrip the Smiles rearrangement at elevated temperatures under basic conditions.5a This was tentatively ascribed to the steric hindrance of the aryl groups. To solve the problem, we have speculated that an acid-promoted rearrangement could minimize the decomposition of the key nucleophilic addition intermediate (Fig. 3, path B). This hypothesis was finally realized via a one-pot procedure. Particularly, the nucleophilic addition adduct between diaryl ketone and 2-PySO2CF2H was quenched by acid at −60 °C in DMF after 1 h of reaction under basic conditions; the resulting alcohol was further subjected to solvent reflux temperature under acidic conditions for several hours until its full conversion to corresponding gem-difluoroolefin (Fig. 2, condition C). This method gave satisfactory results for various diarylketones and represented the first example of acid-promoted Julia–Kocienski olefination.
Functionalities under different chemical circumstances usually exhibit different reactivities. Therefore, one could realize selective transformations via adjusting the reactivity of the reagents or the reaction conditions.20 The 2-PySO2CF2H reagent is capable of chemoselectively gem-difluoroolefinating different carbonyl functionalities under certain conditions (Fig. 4).5a For instance, the aldehyde group could be efficiently converted to the gem-difluorovinyl group under the N(TMS)3/CsF/DMF conditions while the ketone was left untouched. This is probably due to the fact that the terminal carbonyl group (aldehyde) was more reactive toward 2-PySO2CF2− anion than the internal one (ketone). Alternatively, both aldehydes and ketones could be converted to olefins when treated with excess 2-PySO2CF2H under the LiHMDS/DMF conditions at a low temperature. A similar selectivity was also observed for ketoesters under the KHMDS/DMF conditions. Recently, the nucleophilic addition product of 2-PySO2CF2H and sugar lactone was also successfully converted to gem-difluoroolefin under reflux in the TBME solution of NaHCO3.13 It should be noted that we also attempted the gem-difluoroolefination of the carbonyl groups in carboxylic esters and amides with 2-PySO2CF2H reagent, but were not successful.
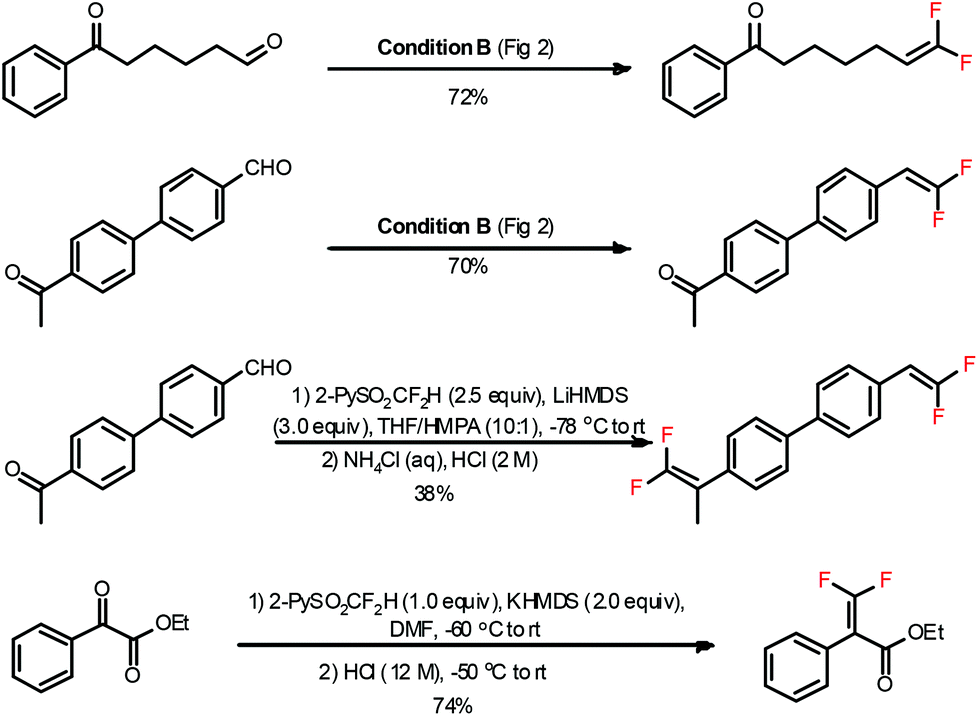 | ||
| Fig. 4 The chemoselective gem-difluoroolefination of carbonyl compounds. | ||
Conclusions
In summary, a new Julia–Kocienski type gem-difluoroolefination reagent, difluoromethyl 2-pyridyl sulfone (2-PySO2CF2H), was recently developed by our group. This reagent can convert diverse carbonyl functionalities to gem-difluorovinyl motifs with high efficacy and good functional group tolerance. For more demanding substrates (such as enolizable aldehydes and diarylketones), modified reaction conditions have been developed to successfully tackle these problems. The chemoselective transformations of aldehydes in the presence of ketone, or ketones in the presence of esters, can be realized under certain reaction conditions. On the other hand, this reagent is currently inapplicable to carboxylic esters and amides, some highly base-sensitive compounds may also not be well tolerated. Nevertheless, with ready availability, ease of operation and excellent performance, the 2-PySO2CF2H reagent promises to find more applications in synthetic fluorine chemistry.Experimental
Preparation of difluoromethyl 2-pyridyl sulfone
Reaction condition A for the gem-difluoroolefination of aromatic aldehydes5b
Under the N2 atmosphere, KOtBu (101 mg, 0.90 mmol) dissolved in DMF (1 mL) was added to a solution of carbonyl substrate (0.6 mmol) and 2-PySO2CF2H (96.6 mg, 0.5 mmol) in 2 mL DMF at −50 °C. The reaction mixture was allowed to warm up to −40 °C within about 15 min (or to room temperature within hours). Then, the reaction is quenched with aqueous saturated NH4Cl (1 mL) at −40 °C followed by 3 N HCl (1 mL). The resulting mixture was then allowed to warm up to room temperature (for electron-deficient aromatic aldehydes, the mixture was heated to 60 °C for 30 min). After completion (monitored by 19F NMR), the reaction mixture was extracted with Et2O (20 mL × 3), the organic phase was then successively washed with saturated aqueous NaHCO3 and brine, and dried over anhydrous MgSO4. After filtration the solvent was evaporated under vacuum and the residue was subjected to flash chromatography to give the pure product.Reaction condition B for the gem-difluoroolefination of aliphatic aldehyde5a
Under the N2 atmosphere, to an oven-dried Schlenk tube were successively added 2-PySO2CF2H (38.6 mg, 0.2 mmol), N(TMS)3 (93.4 mg, 0.4 mmol), CsF (75.6 mg, 0.4 mmol), DMF (2.0 mL) and aldehyde (0.3 mmol). The reaction mixture was stirred at room temperature until the full consumption of 2-PySO2CF2H (monitored by 19F NMR). After being cooled to 0 °C, saturated aqueous NH4Cl (1.0 mL) and aqueous HCl (2 M, 1.5 mL, 3.0 mmol) were successively added, and the resulting mixture was stirred for another 30 min at 50 °C until full consumption of the sulfinate salt, as determined by 19F NMR. Thereafter, the mixture was poured into ice water (50 mL), and extracted with ether (20 mL × 3). The combined organic layer was washed with brine, dried with anhydrous MgSO4, and concentrated under vacuum. The target product was obtained after the flash chromatography purification with ethyl acetate and petroleum ether.Reaction condition C for the gem-difluoroolefination of diaryl ketones5a
Under the N2 atmosphere, DMF (4.0 mL) and HMPA (0.4 mL) were added to an oven-dried 20 mL Schlenk tube containing 2-PySO2CF2H (96.6 mg, 0.5 mmol) and diaryl ketone (1.0 mmol). The reaction mixture was then cooled to −60 °C in a dry ice-acetone bath. A THF solution of (TMS)2NLi (LiHMDS, 1.0 M, 1.0 mmol) was slowly added within 5 minutes, and then the reaction mixture was stirred at the same temperature for 1 h. Thereafter an aqueous solution of HCl (2.0 M, 1.0 mL) was injected to quench the reaction at −60 °C. After being warmed to room temperature, another portion of HCl (12 M, 1.5 mL) was added, and the reaction mixture was heated to reflux for 4 to 10 hours with an oil bath between 120 and 130 °C. When the reaction was complete, as monitored by 19F NMR, the mixture was poured into ice-water (50 mL) and extracted with ether (20 mL × 3). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The target product was obtained after the flash chromatography purification with ethyl acetate and petroleum ether.The preparation of gem-difluoroolefins on 5-gram scale (e.g. anisic aldehyde)
Under the N2 atmosphere, anisic aldehyde (4.23 g, 31.1 mmol) and 2-PySO2CF2H (5 g, 25.9 mmol) were added to an oven-dried three-necked bottle equipped with magnetic stir bar. 30 mL DMF was injected and the solvent was cooled to −50 °C (dry ice-acetone bath), thereafter 20 mL DMF solution of KOtBu (5.23 g, 46.6 mmol) was slowly added to the reaction mixture within 10 min. The reaction system was stirred at −50 °C for 15 min and then gradually warmed up to −40 °C. Once the starting material was completely converted to sulfinate salt (monitored by 19F NMR), it was quenched with aqueous saturated NH4Cl (20 mL) at −40 °C followed by 3M HCl (20 mL). The resulting mixture was then allowed to warm up to room temperature. After the sulfinate was completely consumed (monitored by 19F NMR), the reaction mixture was diluted with 60 mL water and extracted with Et2O (30 mL × 3), the organic phase was then successively washed with saturated aqueous NaHCO3 and brine, and dried over anhydrous MgSO4. After filtration the solvent was evaporated under vacuum and the residue was subjected to flash chromatography purification to give 3.51 g (80% yield) of a colourless oil.Acknowledgements
We thank the National Basic Research Program of China (2015C931900 and 2012CB215500), the National Natural Science Foundation of China (21372246 and 21421002), Shanghai QMX program (13QH1402400), and the Chinese Academy of Sciences for their financial support.Notes and references
- (a) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2nd edn, 2013 Search PubMed; (b) K. Uneyama, Organofluorine Chemistry, Blackwell, Oxford, 2006 Search PubMed; (c) R. D. Chambers, Organofluorine Chemistry: Fluorinated Alkenes and Reactive Intermediates, Springer, New York, 1997 Search PubMed.
- For recent applications of gem-difluoroolefins, see: (a) J. Ichikawa, M. Yokota, T. Kudo and S. Umezaki, Angew. Chem., Int. Ed., 2008, 47, 4870 CrossRef CAS PubMed; (b) M. Takachi, Y. Kita, M. Tobisu, Y. Fukumoto and N. Chatani, Angew. Chem., Int. Ed., 2010, 49, 8717 CrossRef CAS PubMed; (c) P. J. Riss and F. I. Aigbirhio, Chem. Commun., 2011, 47, 11873 RSC; (d) M. Ohashi, H. Saijo, M. Shibata and S. Ogoshi, Eur. J. Org. Chem., 2013, 443 CrossRef CAS; (e) K. Fuchibe, Y. Mayumi, N. Zhao, S. Watanabe, M. Yokota and J. Ichikawa, Angew. Chem., Int. Ed., 2013, 52, 7825 CrossRef CAS PubMed; (f) Y. Xiong, X. Zhang, T. Huang and S. Cao, J. Org. Chem., 2014, 79, 6395 CrossRef CAS PubMed; (g) B. Gao, Y. Zhao, C. Ni and J. Hu, Org. Lett., 2014, 16, 102 CrossRef CAS PubMed; (h) B. Gao, Y. Zhao and J. Hu, Angew. Chem., Int. Ed., 2014 DOI:10.1002/anie.201409705.
- (a) M. Hu, Z. He, B. Gao, L. Li, C. Ni and J. Hu, J. Am. Chem. Soc., 2013, 135, 17302 CrossRef CAS PubMed; (b) M.-O. Turcotte-Savard and J.-F. Paquin, Org. Biomol. Chem., 2013, 11, 1367 RSC; (c) S. Y. Han and I. H. Jeong, Org. Lett., 2010, 12, 5518 CrossRef CAS PubMed; (d) G. K. S. Prakash, J. Hu, Y. Wang and G. A. Olah, Angew. Chem., Int. Ed., 2004, 43, 5203 CrossRef CAS PubMed; (e) J. Ichikawa, H. Fukui and Y. Ishibashi, J. Org. Chem., 2003, 68, 7800 CrossRef CAS PubMed.
- (a) F. Wang, L. Li, C. Ni and J. Hu, Beilstein J. Org. Chem., 2014, 10, 344 CrossRef PubMed; (b) J. Zheng, J. Cai, J.-H. Lin, Y. Guo and J.-C. Xiao, Chem. Commun., 2013, 49, 7513 RSC; (c) C. S. Thomoson, H. Martinez and W. R. Dolbier, J. Fluorine Chem., 2013, 150, 53 CrossRef CAS PubMed; (d) D. J. Burton, Z.-Y. Yang and W. Qiu, Chem. Rev., 1996, 96, 1641 CrossRef CAS PubMed, and references therein; (e) I. Nowak and M. J. Robins, Org. Lett., 2005, 7, 721 CrossRef CAS PubMed; (f) S. A. Fuqua, W. G. Duncan and R. M. Silverstein, J. Org. Chem., 1965, 30, 2543 CrossRef CAS.
- (a) B. Gao, Y. Zhao, M. Hu, C. Ni and J. Hu, Chem. – Eur. J., 2014, 20, 7803 CrossRef CAS PubMed; (b) Y. Zhao, W. Huang, L. Zhu and J. Hu, Org. Lett., 2010, 12, 1444 CrossRef CAS PubMed.
- (a) J. B. Baudin, G. Hareau, S. A. Julia and O. Ruel, Tetrahedron Lett., 1991, 32, 1175 CrossRef CAS; (b) J. B. Baudin, G. Hareau, S. A. Julia and O. Ruel, Bull. Soc. Chim. Fr., 1993, 130, 856 CAS; (c) P. R. Blakemore, W. J. Cole, P. J. Kocieński and A. Morley, Synlett, 1998, 26 CrossRef CAS PubMed; (d) P. J. Kocienski, A. Bell and P. R. Blakemore, Synlett, 2000, 365 CAS; (e) B. Zajc and R. Kumar, Synthesis, 2010, 1822 CrossRef CAS PubMed.
- Y. Zhao, L. Zhang, G. Xu, J. Zheng and J. Hu, Sci. Sin.: Chim., 2011, 41, 1833 CrossRef.
- G. K. S. Prakash, C. Ni, F. Wang, J. Hu and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 2559 CrossRef CAS PubMed.
- Y. Zhao, B. Gao and J. Hu, J. Am. Chem. Soc., 2012, 134, 5790 CrossRef CAS PubMed.
- Q. Zhou, A. Ruffoni, R. Gianatassio, Y. Fujiwara, E. Sella, D. Shabat and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 3949 CrossRef CAS PubMed.
- G. K. S. Prakash, C. Ni, F. Wang, Z. Zhang, R. Haiges and G. A. Olah, Angew. Chem., Int. Ed., 2013, 52, 10835 CrossRef CAS PubMed.
- W. Miao, C. Ni, Y. Zhao and J. Hu, J. Fluorine Chem., 2014, 167, 231 CrossRef CAS PubMed.
- X. Liu, Q. Yin, J. Yin, G. Chen, X. Wang, Q.-D. You, Y.-L. Chen, B. Xiong and J. Shen, Eur. J. Org. Chem., 2014, 6150 CrossRef CAS.
- L. Zhu, C. Ni, Y. Zhao and J. Hu, Tetrahedron, 2010, 66, 5089 CrossRef CAS PubMed.
- (a) C. Ni and J. Hu, Synlett, 2011, 770 CAS; (b) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465 RSC; (c) G. K. S. Prakash and J. Hu, Acc. Chem. Res., 2007, 40, 921 CrossRef CAS PubMed; (d) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015 DOI:10.1021/cr5002386.
- J.-M. Altenburger, G. Y. Lassalle, M. Matrougui, D. Galtier, J.-C. Jetha, Z. Bocskei, C. N. Berry, C. Lunven, J. Lorrain, J.-P. Herault, P. Schaeffer, S. E. O'Connor and J.-M. Herbert, Bioorg. Med. Chem., 2004, 12, 1713 CrossRef CAS PubMed.
- (a) L. A. Warren and S. Smiles, J. Chem. Soc., 1930, 1327 RSC; (b) J. F. Burnett and R. E. Zahler, Chem. Rev., 1951, 49, 273 CrossRef.
- C. Blomberg, The Barbier Reaction and Related One-Step Processes, Springer-Verlag, Berlin, 1993 Search PubMed.
- (a) T. Billard, S. Bruns and B. R. Langlois, Org. Lett., 2000, 2, 2101 CrossRef CAS PubMed; (b) S. Large, N. Roques and B. R. Langlois, J. Org. Chem., 2000, 65, 8848 CrossRef CAS PubMed; (c) M. Hu, B. Gao, C. Ni, L. Zhang and J. Hu, J. Fluorine Chem., 2013, 155, 52 CrossRef CAS PubMed.
- (a) R. A. Shenvi, D. P. O'Malley and P. S. Baran, Acc. Chem. Res., 2009, 42, 530 CrossRef CAS PubMed; (b) N. A. Afagh and A. K. Yudin, Angew. Chem., Int. Ed., 2010, 49, 262 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2015 |