
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron(II) complexes of 4-sulfanyl-, 4-sulfinyl- and 4-sulfonyl-2,6-dipyrazolylpyridine ligands. A subtle interplay between spin-crossover and crystallographic phase changes†
Laurence J.
Kershaw Cook
,
Rafal
Kulmaczewski
,
Simon A.
Barrett
and
Malcolm A.
Halcrow
*
School of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT UK. E-mail: m.a.halcrow@leeds.ac.uk; Fax: +44113 343 6565; Tel: +44 113 343 6506
First published on 12th June 2015
Abstract
Oxidation of 4-(methylsulfanyl)-2,6-di(pyrazol-1-yl)pyridine (LSMe) with hydrogen peroxide or mCPBA yields 4-(methylsulfinyl)-2,6-di(pyrazol-1-yl)pyridine (LSOMe) and 4-(methylsulfonyl)-2,6-di(pyrazol-1-yl)pyridine (LSO2Me), respectively. Solid [Fe(LSMe)2][ClO4]2 (1[ClO4]2) is high-spin at room temperature, and exhibits an abrupt spin-transition at T1/2 = 256 K. A shoulder on the cooling side of the χMT vs. T curve is associated with a hysteretic crystallographic phase change, occurring around T↓ = 245 K and T↑ = 258 K. The phase change involves a 180° rotation of around half the methylsulfanyl substituents in the crystal. This contrasts with the previously reported BF4− salt of the same compound, which is isostructural to 1[ClO4]2 at room temperature but transforms to a different crystal phase in its low-spin state. Solid [Fe(LSOMe)2][BF4]2 (2[BF4]2) and [Fe(LSO2Me)2][BF4]2 (3[BF4]2) both exhibit gradual spin-crossover equilibria centred significantly above room temperature. Solution measurements show that the oxidised sulfur centers in 2[BF4]2 and 3[BF4]2 stabilise the low spin states of those complexes.
Introduction
One of the most flexible systems for the study of spin-crossover phenomena,1–3 in bulk materials4–6 and at the nanoscale,7 is the [Fe(bpp)2]2+ family of complexes (bpp is a derivative of 2,6-di(pyrazol-1-yl)pyridine).8–10 A particular advantage of these complexes is that synthetic routes are available to functionalise [Fe(bpp)2]2+ centres at every position of their ligand periphery.11 On one hand, this provides a route to attach functional substituents to [Fe(bpp)2]2+ centres for the production of multifunctional switchable materials.12 On the other hand, it also provides a means to attach tether groups to [Fe(bpp)2]2+, for use in nanostructures or device applications.13,14 No other type of compound that reliably undergoes spin-crossover has such a flexible synthetic and structural chemistry.Through our continued interest in [Fe(bpp)2]2+ chemistry,8,10 we have recently extended the chemistry of these complexes by reporting the first bpp derivatives bearing thiyl and sulfanyl substituents.14–16 Crystals of [Fe(LSMe)2][BF4]2 (1[BF4]2, Scheme 1) proved noteworthy, by undergoing a crystallographic phase change at 205 K that is independent from their abrupt spin-state transition at 265 K.17 Unusually, this phase change was only observed in single crystalline material. Powder samples of the same compound are isostructural with the crystals at room temperature, and exhibit the same spin-transition, but do not transform to the new crystal phase upon further cooling. We report here the corresponding perchlorate salt [Fe(LSMe)2][ClO4]2 (1[ClO4]2), whose structural chemistry differs significantly from 1[BF4]2, as well as new ligands and complexes derived by oxidation of the sulfanyl group in LSMe.
 | ||
| Scheme 1 Ligands referred to in this work. | ||
Results and discussion
The ligand LSMe was prepared by our published method,16 by methylation of 4-mercapto-2,6-dipyrazolylpyridine.14,15 Selective oxidation of LSMe was achieved using aqueous H2O2 or mCPBA, respectively giving LSOMe and LSO2Me in moderate (30–40%) yields after the usual work-up. Although their NMR spectra were similar, the two oxidised ligands were distinguishable by mass spectrometry and by microanalysis. The identity of LSO2Me was also confirmed by an X-ray crystal structure determination (Fig. 1). For comparison with the previously published [Fe(LSMe)2][BF4]2 (1[BF4]2),16 the new salt [Fe(LSMe)2][ClO4]2 (1[ClO4]2) was prepared by complexation of Fe[ClO4]2·6H2O with 2 equiv LSMe in nitromethane solution. Since the oxidised ligands were only available in small quantities, only the BF4− salts of their iron complexes were investigated: [Fe(LSOMe)2][BF4]2·nH2O (2[BF4]2·nH2O; n ≈ 2 by microanalysis) and [Fe(LSO2Me)2][BF4]2 (3[BF4]2).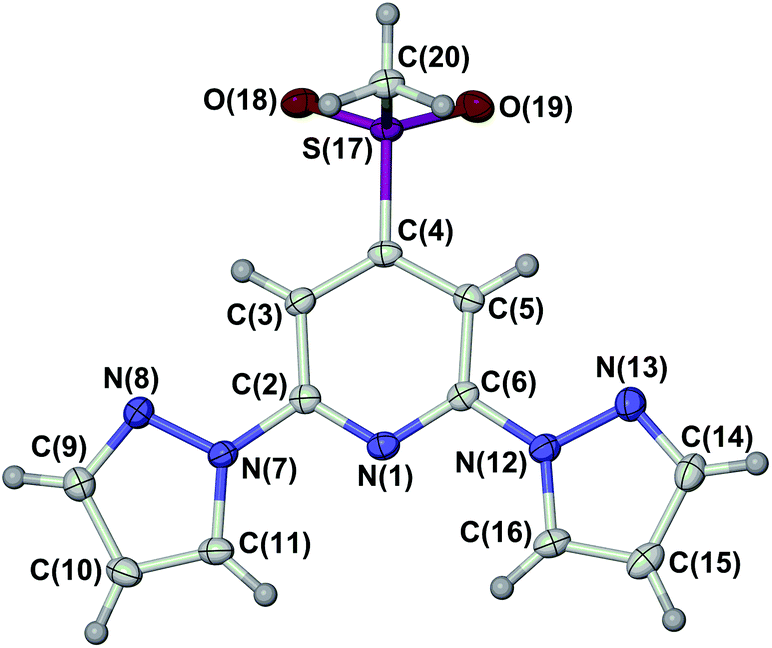 | ||
| Fig. 1 View of the asymmetric unit in the crystal structure of LSO2Me. Atomic displacement ellipsoids are at the 50% probability level. Colour code: C, white; H, pale grey; N, blue; S, purple; O, red. | ||
Slow diffusion of diethyl ether into nitromethane solutions of 1[ClO4]2 yields solvent-free crystals of the complex. Polycrystalline samples are high-spin at 300 K from magnetic susceptibility data (χMT = 3.5 cm3mol−1 K),16 but undergo an abrupt spin-state transition just below room temperature (Fig. 2). The transition exhibits a small thermal hysteresis under the conditions of measurement (T1/2↓ = 255 K, T1/2↑ = 258 K). Unusually, a shoulder is observed on the cooling branch of the transition, but not the warming branch, at approximately two-thirds spin conversion. This is clearly evident in a first-derivative δχMT/δT plot from the same data, which shows an additional maximum at 247 K in cooling mode (Fig. 2). This anomaly in the thermal behaviour of 1[ClO4]2 is also evident by differential scanning calorimetry, which shows endotherm and exotherm peaks corresponding to the spin-transition at T1/2↓ = 255 K and T1/2↑ = 258 K, again with a pronounced shoulder on the cooling curve which is not evident in warming mode (ESI†). Thermodynamic parameters for the spin-transition are ΔH = 10.1 kJ mol−1 and ΔS = 39 J mol−1 K−1.
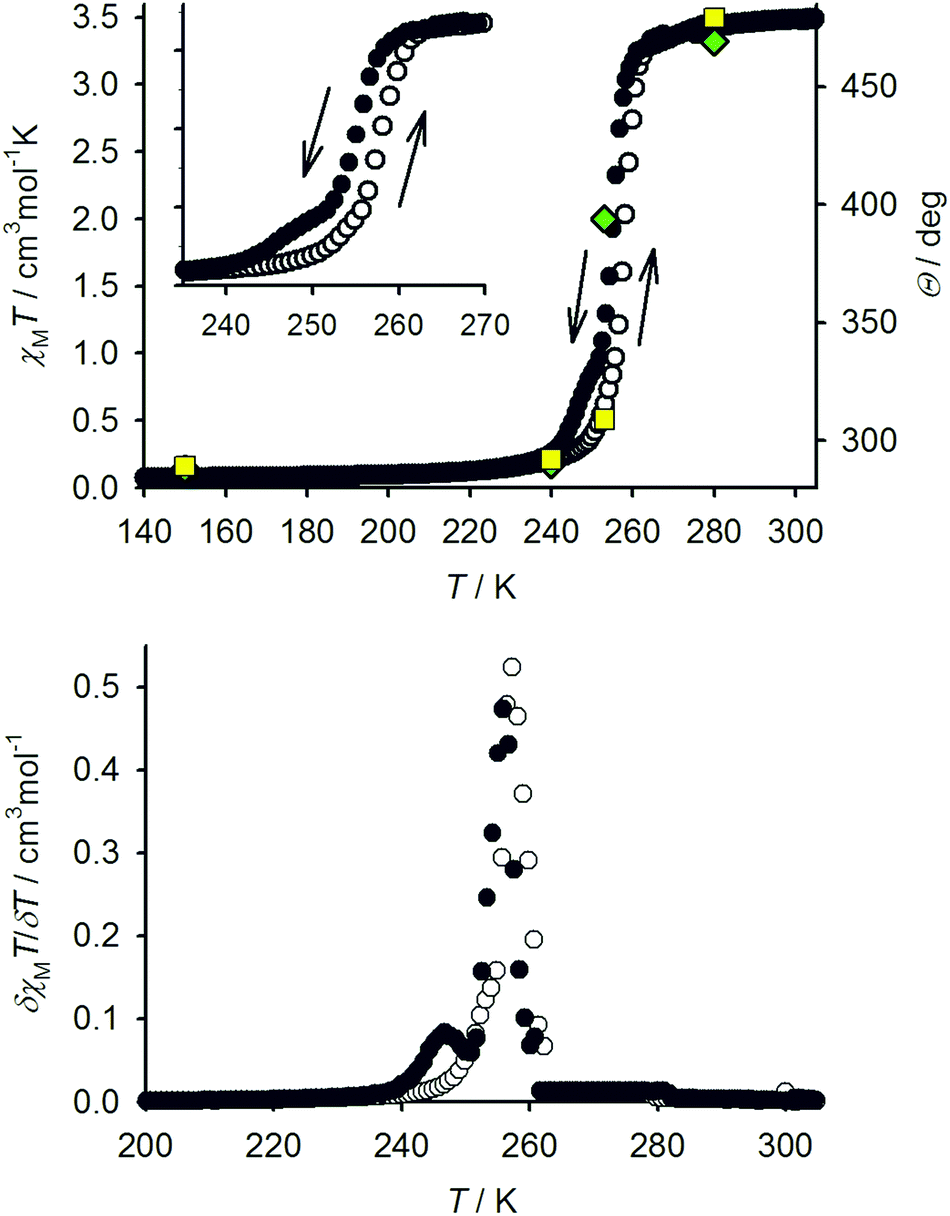 | ||
| Fig. 2 Top: Comparison of the spin-transition in 1[ClO4]2 as measured by variable temperature magnetic susceptibility data with cooling (●) and warming (○) temperature ramps; and, by X-ray crystallography (green diamonds, molecule A; yellow squares, molecule B). An expansion of the susceptibility data near the spin-transition is shown (inset). Bottom: the first derivative of the χMT vs. T data, emphasising the shoulder on the spin-transition in cooling mode (●). A similar, less resolved shoulder may also be present in warming mode (○). | ||
Single crystals of 1[ClO4]2 are isostructural with 1[BF4]2 at room temperature, in the orthorhombic space group Pbcn with Z = 12 (phase 1).16 There are 1.5 formula units in the asymmetric unit, with a half-molecule of the complex lying on a C2 axis (molecule A) and a second whole molecule on a general crystallographic site (molecule B). Cooling the crystal below the spin-transition temperature causes a crystallographic phase change, to the orthorhombic space group P212121 with two unique molecules in the asymmetric unit (Z = 8; phase 2). Although this is a chiral space group, phase 2 of 1[ClO4]2 is racemically twinned. Interestingly, phase 2 is not isostructural with the low-temperature phase of 1[BF4]2, which instead retains the Pbcn space group with a contracted unit cell.16 Unit cell data show that the crystal phase change occurs at a temperature between 240–248 K, just below the onset of the spin-transition which is evident in a contraction of the unit cell a and V parameters below 260 K (Fig. 3). The reverse phase 2 → phase 1 transformation apparently takes place concurrently with the low → high spin state change, at 258 K (Fig. 2 and ESI†). The normalised unit cell volume is unaffected by the crystallographic phase change, within experimental error.
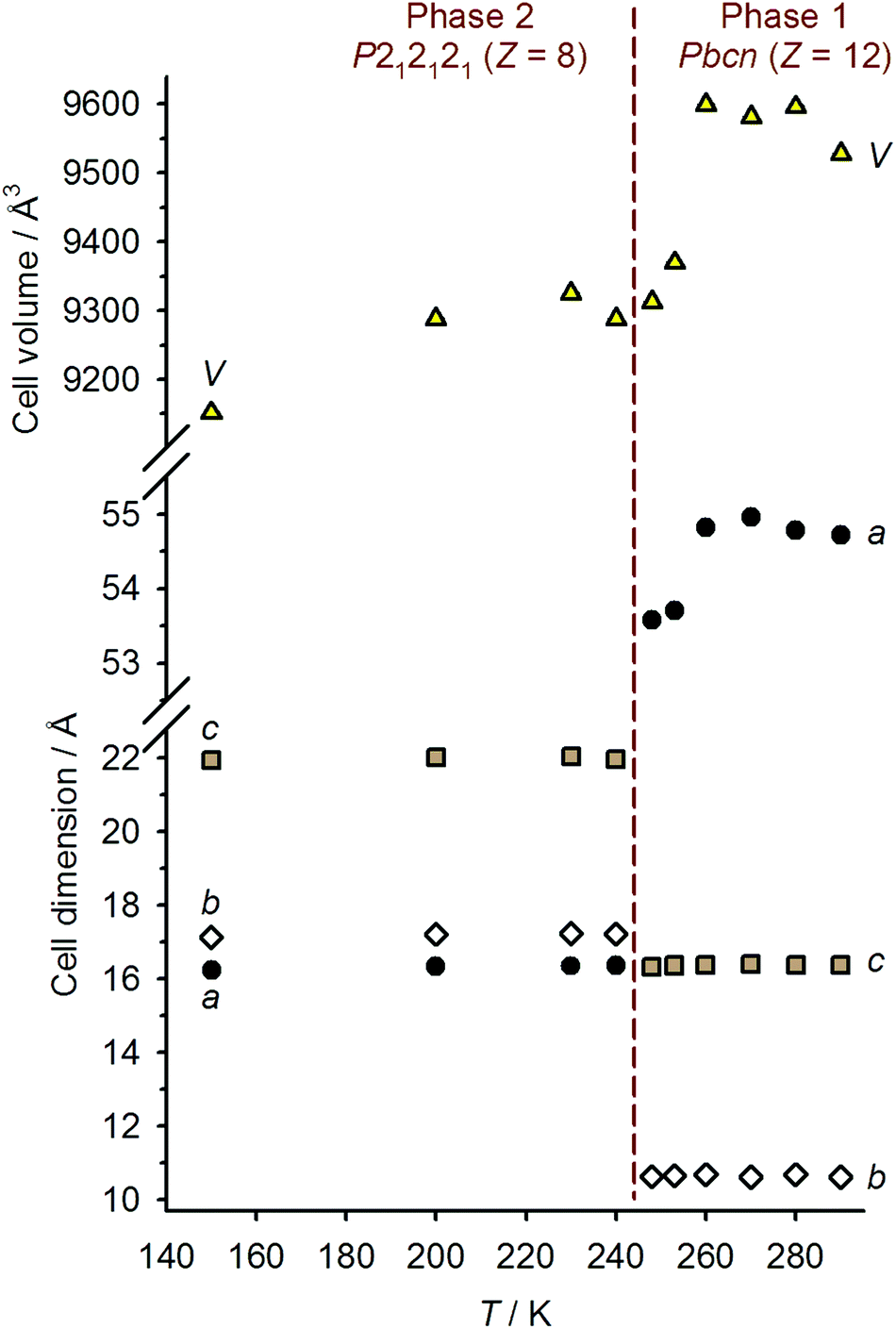 | ||
| Fig. 3 Temperature dependence of the crystallographic unit cell dimensions, and the unit cell volume normalised to Z = 12, for 1[ClO4]2. The dashed line indicates the crystallographic transition temperature. | ||
Full crystallographic refinements of 1[ClO4]2 were achieved at four temperatures (Fig. 4): 280 K (phase 1, when it is high-spin); 253 K (phase 1, just above the phase transition); 240 K (phase 2, just below the phase transition); and 150 K (phase 2). The results are fully consistent with the susceptibility and DSC data, in showing that the bulk of the high → low spin state transition takes place at a slightly higher temperature than the change in crystal symmetry (Fig. 2 and Table 1). At 280 K, the two independent iron centres are both high-spin, with essentially identical Fe–N bond lengths. At 253 K the crystal is still in phase 1 but the Fe–N bond lengths have contracted significantly, indicating that spin-crossover has taken place. While molecule B is essentially low-spin, the metric parameters at molecule A imply an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 high-spin:low-spin population at this temperature (Table 1). The overall spin-state population at 253 K, based on a 1:2 weighted average of the structures of molecules A and B, is in good agreement with the susceptibility data (Fig. 2). This behaviour resembles phase 1 of 1[BF4]2, where half-molecule A also undergoes spin-crossover more gradually than molecule B on cooling.16
:1 high-spin:low-spin population at this temperature (Table 1). The overall spin-state population at 253 K, based on a 1:2 weighted average of the structures of molecules A and B, is in good agreement with the susceptibility data (Fig. 2). This behaviour resembles phase 1 of 1[BF4]2, where half-molecule A also undergoes spin-crossover more gradually than molecule B on cooling.16
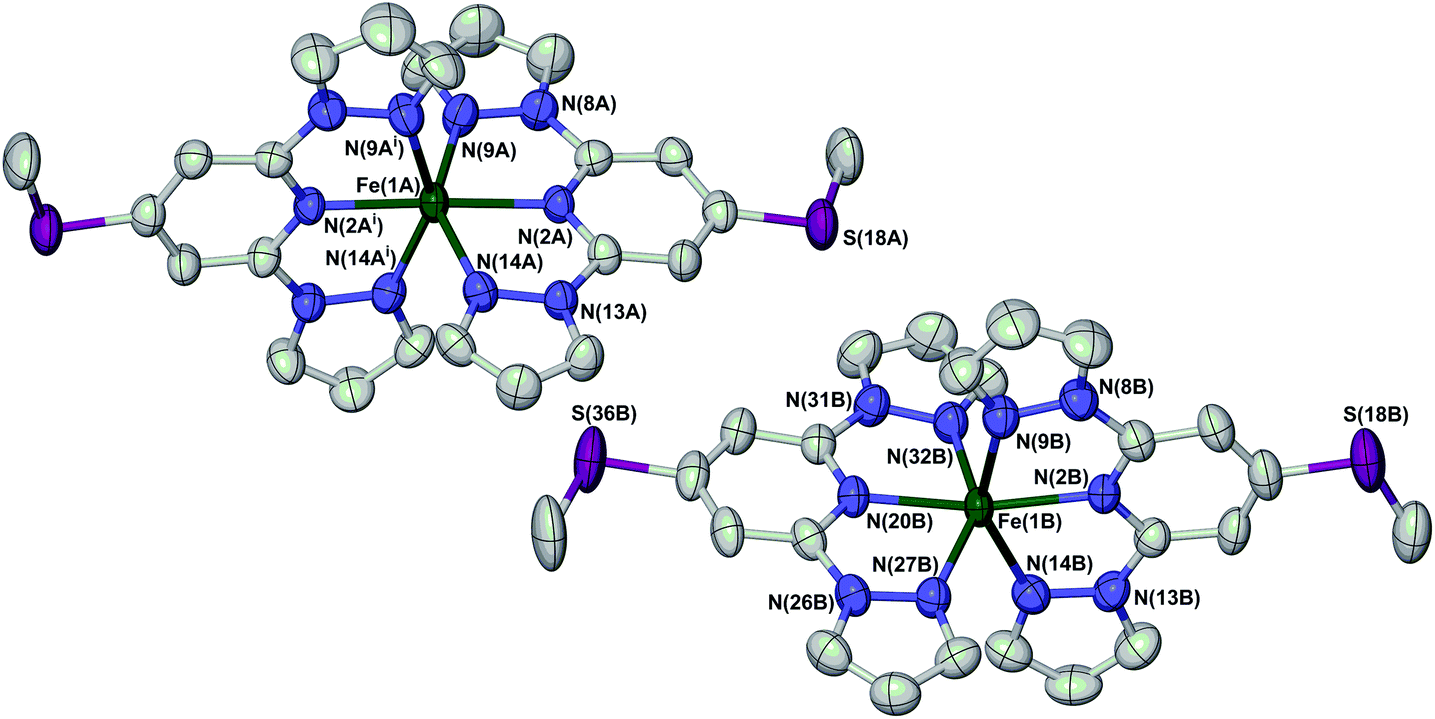 | ||
| Fig. 4 View of the two crystallographically unique [Fe(LSMe)2]2+ cations in phase 1 of 1[ClO4]2 at 280 K. Atomic displacement ellipsoids are at the 50% probability level, and H atoms have been omitted for clarity. The atom numbering scheme in phase 2 is the same as for molecule B of phase 1 (ESI†). Symmetry code: (i) 1 − x, y, 3/2 − z. Colour code: C, white; Fe, green; N, blue; S, purple. | ||
| T (K) | 280 K | 253 K | 240 K | 150 K | |
|---|---|---|---|---|---|
| Phase | Phase 1 | Phase 1 | Phase 2 | Phase 2 | |
| a Symmetry code: (i) 1 − x, y, 3/2 − z. | |||||
| Fe(1A)–N(2A) | 2.115(4) | 2.034(4) | Fe(1A)–N(2A) | 1.902(5) | 1.893(4) |
| Fe(1A)–N(9A) | 2.172(5) | 2.078(5) | Fe(1A)–N(9A) | 1.982(5) | 1.981(4) |
| Fe(1A)–N(14A) | 2.185(5) | 2.092(4) | Fe(1A)–N(14A) | 1.966(6) | 1.967(4) |
| Fe(1A)–N(20A) | 1.908(5) | 1.897(4) | |||
| Fe(1A)–N(27A) | 1.983(6) | 1.978(4) | |||
| Fe(1A)–N(32A) | 1.982(6) | 1.980(4) | |||
| Fe(1B)–N(2B) | 2.126(4) | 1.924(4) | Fe(1B)–N(2B) | 1.904(5) | 1.904(4) |
| Fe(1B)–N(9B) | 2.182(5) | 1.999(5) | Fe(1B)–N(9B) | 1.992(6) | 1.985(4) |
| Fe(1B)–N(14B) | 2.168(5) | 1.988(4) | Fe(1B)–N(14B) | 1.961(6) | 1.960(4) |
| Fe(1B)–N(20B) | 2.120(4) | 1.927(4) | Fe(1B)–N(20B) | 1.898(5) | 1.902(4) |
| Fe(1B)–N(27B) | 2.172(5) | 1.997(4) | Fe(1B)–N(27B) | 1.982(6) | 1.994(4) |
| Fe(1B)–N(32B) | 2.165(5) | 1.995(4) | Fe(1B)–N(32B) | 1.978(6) | 1.971(4) |
| N(2A)–Fe(1A)–N(9A) | 73.61(17) | 76.05(17) | N(2A)–Fe(1A)–N(9A) | 80.1(2) | 79.95(19) |
| N(2A)–Fe(1A)–N(14A) | 73.18(16) | 75.98(15) | N(2A)–Fe(1A)–N(14A) | 79.8(2) | 79.86(18) |
| N(2A)–Fe(1A)–N(2Ai) (ϕ) | 177.7(3) | 176.6(2) | N(2A)–Fe(1A)–N(20A) (ϕ) | 178.4(2) | 178.33(18) |
| N(2A)–Fe(1A)–N(9Ai) | 108.06(17) | 106.39(17) | N(2A)–Fe(1A)–N(27A) | 99.3(2) | 98.59(18) |
| N(2A)–Fe(1A)–N(14Ai) | 105.13(17) | 101.56(16) | N(2A)–Fe(1A)–N(32A) | 101.4(2) | 101.57(17) |
| N(9A)–Fe(1A)–N(14A) | 146.78(16) | 152.03(16) | N(9A)–Fe(1A)–N(14A) | 159.8(2) | 159.68(19) |
| N(9A)–Fe(1A)–N(20A) | 100.8(2) | 100.80(19) | |||
| N(9A)–Fe(1A)–N(9Ai) | 94.7(3) | 92.8(3) | N(9A)–Fe(1A)–N(27A) | 92.7(2) | 92.44(17) |
| N(9A)–Fe(1A)–N(14Ai) | 95.88(18) | 94.81(17) | N(9A)–Fe(1A)–N(32A) | 92.0(2) | 92.03(18) |
| N(14A)–Fe(1A)–N(20A) | 99.3(2) | 99.45(18) | |||
| N(14A)–Fe(1A)–N(27A) | 92.6(2) | 92.77(17) | |||
| N(14A)–Fe(1A)–N(14Ai) | 92.2(3) | 91.0(2) | N(14A)–Fe(1A)–N(32A) | 89.9(2) | 89.81(17) |
| N(20A)–Fe(1A)–N(27A) | 79.3(2) | 79.91(18) | |||
| N(20A)–Fe(1A)–N(32A) | 79.9(2) | 79.92(17) | |||
| N(27A)–Fe(1A)–N(32A) | 159.2(2) | 159.82(18) | |||
| N(2B)–Fe(1B)–N(9B) | 73.44(18) | 79.11(17) | N(2B)–Fe(1B)–N(9B) | 79.4(2) | 79.46(18) |
| N(2B)–Fe(1B)–N(14B) | 72.76(18) | 79.35(17) | N(2B)–Fe(1B)–N(14B) | 79.9(2) | 80.14(17) |
| N(2B)–Fe(1B)–N(20B) (ϕ) | 168.89(19) | 174.88(18) | N(2B)–Fe(1B)–N(20B) (ϕ) | 177.9(2) | 177.72(19) |
| N(2B)–Fe(1B)–N(27B) | 112.82(18) | 102.92(17) | N(2B)–Fe(1B)–N(27B) | 98.6(2) | 98.33(18) |
| N(2B)–Fe(1B)–N(32B) | 101.06(17) | 99.12(17) | N(2B)–Fe(1B)–N(32B) | 101.7(2) | 101.95(18) |
| N(9B)–Fe(1B)–N(14B) | 145.96(18) | 158.42(17) | N(9B)–Fe(1B)–N(14B) | 158.9(2) | 159.36(18) |
| N(9B)–Fe(1B)–N(20B) | 97.03(18) | 96.15(17) | N(9B)–Fe(1B)–N(20B) | 102.2(2) | 102.16(18) |
| N(9B)–Fe(1B)–N(27B) | 96.79(19) | 92.66(18) | N(9B)–Fe(1B)–N(27B) | 93.5(2) | 92.79(17) |
| N(9B)–Fe(1B)–N(32B) | 94.8(2) | 92.72(19) | N(9B)–Fe(1B)–N(32B) | 91.2(2) | 91.87(18) |
| N(14B)–Fe(1B)–N(20B) | 117.01(18) | 105.42(16) | N(14B)–Fe(1B)–N(20B) | 98.7(2) | 98.32(18) |
| N(14B)–Fe(1B)–N(27B) | 92.53(18) | 90.97(17) | N(14B)–Fe(1B)–N(27B) | 93.3(2) | 93.18(17) |
| N(14B)–Fe(1B)–N(32B) | 95.50(19) | 91.85(18) | N(14B)–Fe(1B)–N(32B) | 89.4(2) | 89.34(18) |
| N(20B)–Fe(1B)–N(27B) | 73.36(18) | 79.09(16) | N(20B)–Fe(1B)–N(27B) | 80.0(2) | 80.03(18) |
| N(20B)–Fe(1B)–N(32B) | 73.60(17) | 79.05(16) | N(20B)–Fe(1B)–N(32B) | 79.7(2) | 79.66(18) |
| N(27B)–Fe(1B)–N(32B) | 146.05(17) | 157.93(16) | N(27B)–Fe(1B)–N(32B) | 159.7(2) | 159.69(18) |
| Molecule A: θ | 89.51(4) | 87.88(4) | Molecule A: θ | 88.86(5) | 88.64(3) |
| Σ | 151.5(7) | 125.3(6) | Σ | 89.1(7) | 88.2(6) |
| Θ | 469 | 394 | Θ | 290 | 288 |
| Molecule B: θ | 87.14(4) | 88.48(4) | Molecule B: θ | 89.12(5) | 88.78(3) |
| Σ | 154.4(6) | 95.2(6) | Σ | 90.8(7) | 90.0(6) |
| Θ | 479 | 309 | Θ | 292 | 289 |
At 240 and 150 K the crystal has transformed to phase 2, which now contains two complete, independent formula units in its asymmetric unit. Both molecules of the complex are fully low-spin in this phase, at each temperature. Hence, the phase 1 → phase 2 transformation appears to be required for 1[ClO4]2 to attain its fully low-spin state. As mentioned previously, phase 2 of this material is not isostructural with the low-temperature phase of 1[BF4]2; this is discussed further below.
Phases 1 and 2 of 1[ClO4]2 exhibit similar molecular packing, containing layers of cations. Each cation interacts with two nearest neighbours in the same layer through face-to-face and edge-to-face π–π contacts. The dimensions of these intra-layer interactions in phase 1 are little affected by the partial transition on cooling to 253 K (ESI†). However, the phase 1 → phase 2 transformation leads to a significant slippage and canting of neighbouring molecules within in the cation layers (Fig. 5 and ESI†). The methylsulfanyl groups from each layer protrude into the adjacent layers, occupying cavities bounded by three pyrazolyl groups from two different neighbour molecules. In phase 2, there are a number of intermolecular C–H⋯π contacts from the methyl substituents to these pyrazolyl rings, with C⋯C distances of 3.4–3.6 Å (tabulated in the ESI;† the sum of the van der Waals radii of a methyl group and an aromatic ring is 3.7 Å20). These intermolecular contacts are significantly lengthened in phase 1 at 253 K, when the crystal is still predominantly low-spin. Hence, relief of the intermolecular steric clashes involving the methylsulfanyl substituents is likely to be the driving force for the phase 2 → phase 1 transformation upon warming.16 Importantly, there is no correspondence between the ‘A’ and ‘B’ molecular environments in phases 1 and 2 (ESI†). In phase 1, individual layers are of the ‘A’ and ‘B’ type, which stack parallel to the crystallographic a direction with an ABBABB motif. In contrast, in phase 2, the ‘A’ and ‘B’ molecular environments alternate within each layer.
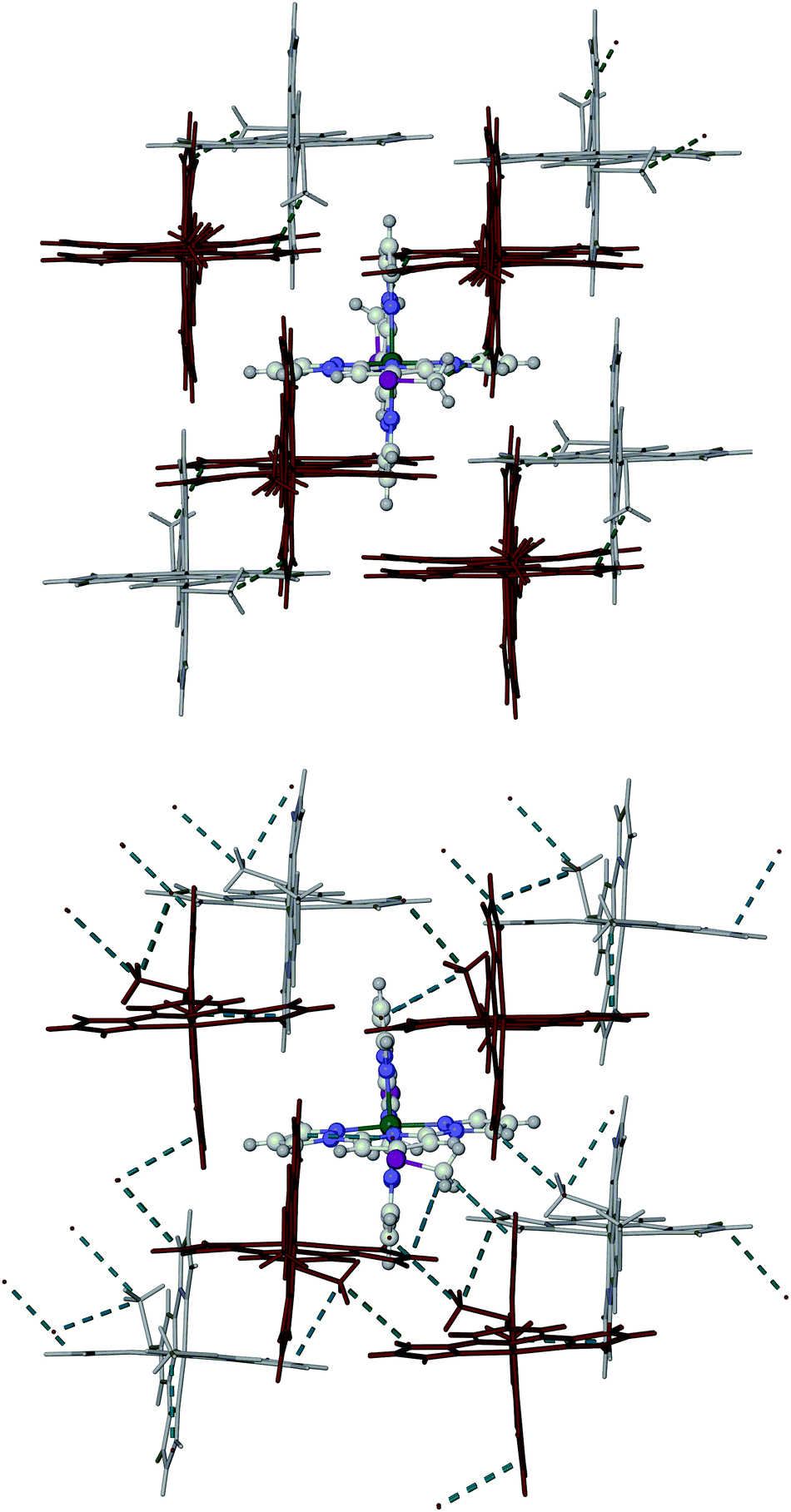 | ||
| Fig. 5 Packing diagrams of 1[ClO4]2 in phase 1 at 253 K (top) and phase 2 at 240 K (bottom). The pale coloured cations are in the same layer as the highlighted molecule, whereas the dark cations are in adjacent layers to the front and back. The close intermolecular C–H⋯π contacts involving the methylsulfanyl groups are also indicated (tabulated in the ESI†). | ||
Interestingly, the orientations of the methylsulfanyl groups in neighbouring molecules are different in the two phases (Fig. 5 and ESI†). In phase 1, the methylsulfanyl groups in each layer are all oriented the same way, while in phase 2 their orientations alternate within the layers. That implies the phase 1 → phase 2 transformation involves a rotation or flipping of around half the methylsulfanyl substituents in the crystal, by ca. 180° (evidence for such a flipping process in the vicinity of its spin transition was also observed in phase 1 of 1[BF4]216). This methylsulfanyl group flipping is the likely origin of the racemic twinning in phase 2 of 1[ClO4]2, while the activation energy associated with the conformational change may also be the cause of the thermal hysteresis. While there is substantial anion disorder in phase 1, this is still present in phase 2 at 240 K (it is frozen out in the 150 K structure). Hence, changes to anion disorder are unlikely to play a role in the phase transition.
The complexes 2[BF4]2·nH2O and 3[BF4]2 were not obtained as single crystals. Powder samples of the compounds are predominantly low-spin at room temperature, but exhibit the onset of gradual thermal spin-crossover as the temperature is raised with T1/2 = 346 ± 2 K (2[BF4]2·nH2O) and >400 K (3[BF4]2; ESI†). The χMT vs. T curve of 2[BF4]2·nH2O is fully reversible upon heating and re-cooling in the range 290 ≤ T ≤ 350 K, showing that its spin-transition is not associated with loss of lattice water over this temperature range.21
The spin-state properties of 1[BF4]2, 2[BF4]2 and 3[BF4]2 in solution were also investigated in solution, which provides a more reliable measure of the effect of the sulfanyl, sulfinyl and sulfonyl substituents on the ligand field of the iron centres. Susceptibility data for 1[BF4]2 and 2[BF4]2 were measured in (CD3)2CO, a weakly associating solvent with a useful liquid range for spin-transitions centred below room temperature. While data for 3[BF4]2 were obtained in CD3NO2, for solubility reasons, the use of these different solvents should have only a minor influence on the complexes’ susceptibility behaviour.22 All three samples exhibit spin-crossover equilibria in these solutions (Fig. 6), with T1/2 = 194 ± 3 K (1[BF4]2), 284 ± 3 K (2[BF4]2) and 294 ± 3 K (3[BF4]2). The trend in T1/2 implies that the more electron-withdrawing sulfone and sulfoxide substituents stabilise the low-spin state of a [Fe(bpp)2]2+ centre, compared to the parent complex 1[BF4]2.
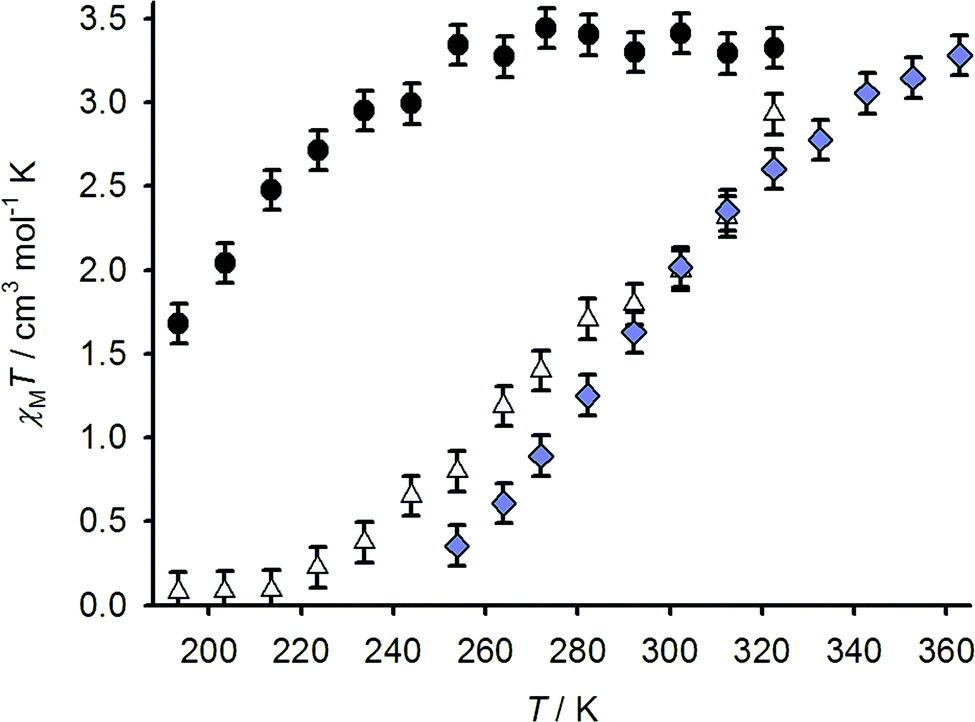 | ||
Fig. 6 Variable temperature magnetic susceptibility data for 1[BF4]2 in (CD3)2CO (●), 2[BF4]2 in (CD3)2CO (△) and 3[BF4]2 in CD3NO2 ( ). ). | ||
Conclusions
The spin-state behaviour and structural chemistry of 1[ClO4]2 and the previously published BF4− salt of the same complex16 are broadly similar, but differ in several respects. The salts are isostructural at room temperature (phase 1, in Pbcn with Z = 12). Moreover, in both phase 1 materials, molecules A and B undergo spin-crossover at a similar temperature but molecule A undergoes the transition less abruptly than molecule B. Finally, both compounds undergo a crystallographic phase change at lower temperature than their spin-crossover T1/2,23 that appears to be triggered by molecule A becoming fully low-spin. However, the low-temperature, fully low-spin phase 2 in 1[ClO4]2 (P212121, Z = 8) is different from 1[BF4]2 (Pbcn, Z = 4). In addition, 1[BF4]2 exhibits the phase 1 → phase 2 transition in single crystalline samples, but not as a bulk powder.16 In contrast, low-spin 1[ClO4]2 adopts phase 2 in both types of sample. The crystallographic phase change in 1[ClO4]2 is hysteretic, occurring at T↓ = 245 ± 3 K and T↑ = 258 ± 1 K (the same temperature as the low → high spin-state transition). That may reflect the activation energy associated with the flipping of the methylsulfanyl groups that takes places during the structure change.Solid 1[BF4]2 and 1[ClO4]2 belong to a growing family of [Fe(bpp)2]2+ derivatives, that adopt this mode of crystal packing but exhibit different, spin-state dependent phase behaviour (ESI†). Other examples include iron(II) complex salts of 4-bromo-2,6-di(pyrazol-1-yl)pyridine (LBr) and 4-iodo-2,6-di(pyrazol-1-yl)pyridine (LI), which undergo gradual spin-crossover above room temperature and retain the same crystal symmetry in both spin states (Pbcn, Z = 4);16,24 and, 4-ethynyl-2,6-di(pyrazol-1-yl)pyridine (LCCH), which exhibits a more complicated interplay between three different crystal phases (ESI†).25
While characterisation of 2[BF4]2·nH2O and 3[BF4]2 has been limited by their poor crystallinity, it is clear that oxidation of the sulfanyl substituents in [Fe(LSMe)2]2+ results in stabilisation of its low-spin state, thus increasing T1/2 for spin-crossover. That is counter-intuitive at first glance, since more electron-withdrawing sulfinyl and sulfonyl substituents would be expected to reduce the basicity of the bpp moiety, and thus weaken the ligand field in its complexes. We are currently undertaking a more detailed investigation of this question, for future publication.
Experimental
4-(Methylsulfanyl)-2,6-di(pyrazol-1-yl)pyridine (LSMe) and the salt [Fe(LSMe)2][BF4]2 (1[BF4]2) were prepared by our previously reported methods.16 All other reagents were purchased commercially and used as supplied.Synthesis of 4-(methylsulfinyl)-2,6-di(pyrazol-1-yl)pyridine (LSOMe)
Solid LSMe (0.17 g, 0.61 mmol) was dissolved in glacial acetic acid (10 cm3), with stirring. Aqueous hydrogen peroxide (30% w/v; 0.35 cm3, 3.57 mmol) was then carefully added to the solution, and the mixture was stirred at room temperature for 5 h. The resultant yellow solution was neutralised with aqueous NaOH, leading to a cloudy precipitate which was extracted with dichloromethane (2 × 50 cm3). The extracts were dried over MgSO4 and filtered, then evaporated to dryness. The brown residue was eluted through a silica gel column (eluent 99:1 dichloromethane:methanol) yielding LSOMe as a pale brown microcrystalline solid. Yield 52 mg, 31%. Mp 142–144 °C. Found C, 52.3; H, 4.00; N, 25.4. Calcd for C12H11N5OS C, 52.7; H, 4.06; N, 25.6%. ES-MS m/z 296.1 [Na(LSOMe)]+. 1H NMR (CDCl3) δ 2.89 (s, 3H, SOCH3), 6.54 (dd, 1.9 and 2.7 Hz, 2H, Pz H4), 7.80 (d, 1.9 Hz, 2H, Pz H3), 8.11 (s, 2H, Py H3/5), 8.58 (d, 2.7 Hz, 2H, Pz H5). 13C NMR (CDCl3) δ 43.3 (SOCH3), 103.7 (Py C3/5), 108.7 (Pz C4), 127.3 (Pz C5), 143.1 (Pz C3), 150.8 (Py C2/6), 162.6 (Py C4).
Synthesis of 4-(methylsulfonyl)-2,6-di(pyrazol-1-yl)pyridine (LSO2Me)
A solution of LSMe (0.14 g, 0.52 mmol) in dichloromethane (15 cm3) was cooled to 0 °C. 3-Chloroperbenzoic acid (0.25 g, 1.13 mmol) was then added carefully, and the mixture was stirred at this temperature for 1 h, before being warmed to room temperature and stirred for a further 5 h. The solution was diluted to 35 cm3 with chloroform, and washed sequentially with aqueous NaHCO3 (20 cm3), NaOH (20 cm3) and H2O (20 cm3). The organic solution was dried with MgSO4, filtered and evaporated to dryness to yield a crude yellow solid. Recrystallisation from a minimum volume of n-hexane at −20 °C afforded small yellow needle crystals. Yield 63 mg, 42%. Mp 203–205 °C. Found C, 49.5; H, 3.80; N, 24.1. Calcd for C12H11N5O2S C, 49.8; H, 3.83; N, 24.2%. ES-MS m/z 290.1 [H(LSO2Me)]+, 312.1 [Na(LSO2Me)]+, 601.1 [Na(LSO2Me)2]+. 1H NMR (CDCl3) δ 3.19 (s, 3H, SO2CH3), 6.56 (dd, 1.7 and 2.6 Hz, 2H, Pz H4), 7.82 (d, 1.7 Hz, 2H, Pz H3), 8.35 (s, 2H, Py H3/5), 8.57 (d, 2.6 Hz, 2H, Pz H5). 13C NMR (CDCl3) δ 43.6 (SO2CH3), 106.7 (Py C3/5), 109.0 (Pz C4), 127.4 (Pz C5), 143.5 (Pz C3), 151.4 (Py C2/6), 154.0 (Py C4).Synthesis of [Fe(LSMe)2][ClO4]2 (1[ClO4]2)
A solution of LSMe (31 mg, 0.12 mmol) and Fe[ClO4]2·6H2O (18 mg, 0.06 mmol) in nitromethane (10 cm3) was stirred at room temperature for 1 h. The solution was filtered and concentrated to ca. 3 cm3, then the product was precipitated as a bright yellow powder by addition of diethyl ether. Yield 30 mg, 65%. Found C, 37.1; H, 2.80; N, 17.9%. Calcd for C24H22Cl2FeN10O8S2 C, 37.5; H, 2.88; N, 18.2%.CAUTION! Although we have experienced no problems in handling this compound, metal–organic perchlorates are potentially explosive and should be handled with due care in small quantities.
Synthesis of [Fe(LSOMe)2][BF4]2·nH2O (2[BF4]2·nH2O, n ≈ 2)
Method as above, using LSOMe (40 mg, 0.15 mmol) and Fe[BF4]2·6H2O (24 mg, 0.07 mmol). The product was obtained as a bright orange powder. Yield 46 mg, 85%. Found C, 35.4; H, 2.80; N, 16.8%. Calcd for C24H22B2F8FeN10O2S2·2H2O C, 35.5; H, 3.23; N, 17.2%. 1H NMR (CD3NO2) δ 2.9 (SOCH3), 29.2 (Py H3/5), 31.7 (Pz H5), 47.8 (Pz H4), 52.8 (Pz H3).Synthesis of [Fe(LSO2Me)2][BF4]2 (3[BF4]2)
Method as above, using LSO2Me (37 mg, 0.13 mmol) and Fe[BF4]2·6H2O (20 mg, 0.06 mmol). The product is a dark red powder. Yield 39 mg, 80%. Found C, 35.4; H, 2.80; N, 16.9%. Calcd for C24H22B2F8FeN10O4S2 C, 35.7; H, 2.74; N, 17.3%. 1H NMR (CD3NO2) δ 3.3 (SO2CH3), 19.9 (Py H3/5), 23.4 (Pz H5), 34.3 and 35.5 (Pz H3 and H4).Single crystal X-ray structure determinations
Diffraction data for were measured using an Agilent Supernova dual-source diffractometer, using monochromated Cu-Kα radiation (λ = 1.54184 Å). The diffractometer is fitted with an Oxford Cryostream low-temperature device. Experimental details of the structure determinations are given in Table 2. All the structures were solved by direct methods (SHELXS9726), and developed by full least-squares refinement on F2 (SHELXL9726). Crystallographic figures were prepared using XSEED,27 which incorporates POVRAY.28| LSO2Me | 1[ClO4]2 | ||||
|---|---|---|---|---|---|
| a R = ∑[|Fo| − |Fc|]/∑|Fo|. b wR = [∑w(Fo2 − Fc2)/∑wFo4]1/2. | |||||
| T (K) | 100(2) | 280(2) | 253(2) | 240(2) | 150(2) |
| Molecular formula | C12H11N5O2S | C24H22Cl2FeN10O8S2 | C24H22Cl2FeN10O8S2 | C24H22Cl2FeN10O8S2 | C24H22Cl2FeN10O8S2 |
| M r | 289.32 | 769.39 | 769.39 | 769.39 | 769.39 |
| Crystal class | Monoclinic | Orthorhombic | Orthorhombic | Orthorhombic | Orthorhombic |
| Space group | P21/n | Pbcn | Pbcn | P212121 | P212121 |
| a (Å) | 5.63390(10) | 54.8011(19) | 53.7002(12) | 16.3532(4) | 16.2202(4) |
| b (Å) | 14.1865(4) | 10.6074(3) | 10.6448(2) | 17.2208(5) | 17.1650(4) |
| c (Å) | 16.1192(4) | 16.3787(5) | 16.3472(3) | 21.9658(7) | 21.9195(6) |
| β (°) | 91.402(2) | — | — | — | — |
| V (Å3) | 1287.95(5) | 9520.9(5) | 9344.5(3) | 6185.9(3) | 6102.8(3) |
| Z | 4 | 12 | 12 | 8 | 8 |
| μ (mm−1) | 2.338 | 7.153 | 7.288 | 7.340 | 7.440 |
| Measured reflections | 4740 | 22818 |
18968 |
17806 |
17530 |
| Independent reflections | 2515 | 9301 | 9050 | 10969 |
10533 |
| R int | 0.028 | 0.033 | 0.028 | 0.038 | 0.046 |
| R 1, I > 2σ(I)a | 0.036 | 0.078 | 0.080 | 0.058 | 0.049 |
| wR2, all datab | 0.093 | 0.222 | 0.231 | 0.163 | 0.120 |
| Goodness of fit | 1.058 | 1.046 | 1.035 | 1.032 | 1.037 |
| Flack parameter | — | — | — | 0.462(7) | 0.546(5) |
See http://dx.doi.org/10.1039/b000000x/ for crystallographic files in .cif format.
X-ray structure refinements
At 253 and 280 K, the compound adopts the space group Pbcn with 1.5 formula units in its asymmetric unit (Z′ = 1.5). Half a complex cation spans the C2 axis 1/2, y, 3/4 (molecule A), and a whole complex dication (molecule B) and three ClO4− anions lie on general crystallographic positions. All three anions are disordered over two or three sites, whose relative occupancies are slightly different at the two temperatures. The refined restraints Cl–O = 1.42(2) and O⋯O = 2.32(2) Å were applied to the disordered anions. All wholly occupied non-H atoms, plus partial Cl atoms with occupancy >0.5, were refined anisotropically and all H atoms were placed in calculated positions and refined using a riding model.
In contrast, at 240 and 150 K the crystals exhibit the space group P212121 with two complex dications and four ClO4− anions on general crystallographic sites in the asymmetric unit (Z′ = 2). The crystals are racemic twins in this space group, with Flack parameters refining close to 0.5 at both temperatures. ADSYMM and NEWSYMM analyses detected no missed higher symmetry, however, and were both consistent with this choice of space group.29 At 240 K, all four unique anions are disordered over two sites, which were modelled with refined distance restraints as above. No disorder is present in the 150 K model, and no restraints were applied at that temperature. All crystallographically ordered non-H atoms were refined anisotropically in these structures, while H atoms were placed in calculated positions and refined using a riding model.
Other measurements
Elemental microanalyses were performed by the University of Leeds School of Chemistry microanalytical service. Electrospray mass spectra (ESMS) were obtained on a Bruker MicroTOF spectrometer, from MeCN feed solutions. All mass peaks have the correct isotopic distributions for the proposed assignments. The differential scanning calorimetry measurement used a TA Instruments DSC Q20 calorimeter, heating at a rate of 10 K min−1.Magnetic susceptibility measurements were performed on a Quantum Design VSM SQUID magnetometer, in an applied field of 5000 G. Data were acquired in cooling and warming modes, on a 5 K min−1 temperature ramp. A diamagnetic correction for the sample was estimated from Pascal's constants;16 a diamagnetic correction for the sample holder was also measured separately, and applied to the data. Magnetic susceptibility measurements in solution were obtained by Evans method using a Bruker Avance500 spectrometer operating at 500.13 MHz.30 Tetramethylsilane was added to all the solutions as an internal standard. A diamagnetic correction for the sample,17 and a correction for the variation of the density of the solvent with temperature,31 were applied to these data.
Acknowledgements
This work was funded by the EPSRC grants EP/I014039/1, EP/K012568/1 and EP/K00512X/1. The authors thank Dr Oscar Cespedes (School of Physics and Astronomy, University of Leeds) for help with the magnetic susceptibility measurements.Notes and references
- P. Gütlich and H. A. Goodwin, Spin Crossover in Transition Metal Compounds I–III, Top. Curr. Chem., 2004, pp. 233–235 Search PubMed.
- M. A. Halcrow, Spin-crossover materials - properties and applications, John Wiley & Sons, Chichester, 2013, p. 568 Search PubMed.
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313 RSC; P. Gütlich, Eur. J. Inorg. Chem., 2013, 581 CrossRef CAS; P. Gütlich, A. B. Gaspar and Y. Garcia, Beilstein J. Org. Chem., 2013, 9, 342 CrossRef PubMed.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119 RSC.
- J. Tao, R.-J. Wei, R.-B. Huang and L.-S. Zheng, Chem. Soc. Rev., 2012, 41, 703 RSC; P. Guionneau, Dalton Trans., 2014, 43, 382 RSC.
- M. Shatruk, H. Phan, B. A. Chrisostomo and A. Suleimenova, Coord. Chem. Rev., 2015, 289–290, 62 CrossRef CAS.
- M. Cavallini, Phys. Chem. Chem. Phys., 2012, 14, 11867 RSC; H. J. Shepherd, G. Molnár, W. Nicolazzi, L. Salmon and A. Bousseksou, Eur. J. Inorg. Chem., 2013, 653 CrossRef CAS.
- M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493 CrossRef CAS.
- J. Olguín and S. Brooker, Coord. Chem. Rev., 2011, 255, 203 CrossRef.
- L. J. Kershaw Cook, R. Mohammed, G. Sherborne, T. D. Roberts, S. Alvarez and M. A. Halcrow, Coord. Chem. Rev., 2015, 289–290, 2 CrossRef CAS.
- M. A. Halcrow, New J. Chem., 2014, 38, 1868 RSC.
- M. Nihei, L. Han and H. Oshio, J. Am. Chem. Soc., 2007, 129, 5312 CrossRef CAS PubMed; M. Nihei, N. Takahashi, H. Nishikawa and H. Oshio, Dalton Trans., 2011, 40, 2154 RSC; R. González-Prieto, B. Fleury, F. Schramm, G. Zoppellaro, R. Chandrasekar, O. Fuhr, S. Lebedkin, M. Kappes and M. Ruben, Dalton Trans., 2011, 40, 7564 RSC; K. Takahashi, Y. Hasegawa, R. Sakamoto, M. Nishikawa, S. Kume, E. Nishibori and H. Nishihara, Inorg. Chem., 2012, 51, 5188 CrossRef PubMed.
- M. Matsuda and H. Tajima, Chem. Lett., 2007, 36, 700 CrossRef CAS; M. S. Alam, M. Stocker, K. Gieb, P. Müller, M. Haryono, K. Student and A. Grohmann, Angew. Chem., Int. Ed., 2010, 49, 1159 CrossRef PubMed; M. Cavallini, I. Bergenti, S. Milita, J. C. Kengne, D. Gentili, G. Ruani, I. Salitros, V. Meded and M. Ruben, Langmuir, 2011, 27, 4076 CrossRef PubMed; D. Secker, S. Wagner, S. Ballmann, R. Härtle, M. Thoss and H. B. Weber, Phys. Rev. Lett., 2011, 106, 136807 CrossRef PubMed; V. Meded, A. Bagrets, K. Fink, R. Chandrasekar, M. Ruben, F. Evers, A. Bernand-Mantel, J. S. Seldenthuis, A. Beukman and H. S. J. van der Zant, Phys. Rev. B: Condens. Matter, 2011, 83, 245415 CrossRef; A. K. Botcha, S. Basak and R. Chandrasekar, RSC Adv., 2014, 4, 34760 RSC.
- L. Pukenas, F. Benn, E. Lovell, A. Santoro, L. J. Kershaw Cook, M. A. Halcrow and S. D. Evans, J. Mater. Chem. C 10.1039/c5tc01233c.
- L. J. Kershaw Cook, J. Fisher, L. P. Harding and M. A. Halcrow, Dalton Trans., 2015, 44, 9417 RSC.
- L. J. Kershaw Cook, H. J. Shepherd, T. P. Comyn, C. Baldé, O. Cespedes, G. Chastanet and M. A. Halcrow, Chem. – Eur. J., 2015, 21, 4805 CrossRef CAS PubMed.
- C. J. O'Connor, Prog. Inorg. Chem., 1982, 29, 203 CrossRef.
- J. K. McCusker, A. L. Rheingold and D. N. Hendrickson, Inorg. Chem., 1996, 35, 2100 CrossRef CAS; P. Guionneau, M. Marchivie, G. Bravic, J.-F. Létard and D. Chasseau, Top. Curr. Chem., 2004, 234, 97 CrossRef.
- J. M. Holland, J. A. McAllister, C. A. Kilner, M. Thornton-Pett, A. J. Bridgeman and M. A. Halcrow, J. Chem. Soc., Dalton Trans., 2002, 548 RSC; J. Elhaïk, D. J. Evans, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2005, 1693 RSC; M. Haryono, F. W. Heinemann, K. Petukhov, K. Gieb, P. Müller and A. Grohmann, Eur. J. Inorg. Chem., 2009, 2136 CrossRef CAS; S. Vela, J. J. Novoa and J. Ribas-Arino, Phys. Chem. Chem. Phys., 2014, 16, 27012 RSC.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaca, New York, USA, 3rd edn, 1960, pp. 257–264 Search PubMed.
- See e.g. M. Sorai, J. Ensling, R. M. Hasselbach and P. Gütlich, Chem. Phys., 1977, 20, 197 CrossRef CAS; K. H. Sugiyarto, D. C. Craig, A. D. Rae and H. A. Goodwin, Aust. J. Chem., 1994, 47, 869 CrossRef; K. H. Sugiyarto, K. Weitzner, A. D. Rae and H. A. Goodwin, Aust. J. Chem., 1997, 50, 869 CrossRef; M. Clemente-León, E. Coronado, M. C. Giménez-López and F. M. Romero, Inorg. Chem., 2007, 46, 11266 CrossRef PubMed; T. D. Roberts, F. Tuna, T. L. Malkin, C. A. Kilner and M. A. Halcrow, Chem. Sci., 2012, 3, 349 RSC; M. B. Bushuev, V. A. Daletsky, D. P. Pishchur, Y. V. Gatilov, I. V. Korolkov, E. B. Nikolaenkova and V. P. Krivopalov, Dalton Trans., 2014, 43, 3906 RSC.
- S. A. Barrett, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2011, 40, 12021 RSC.
- Crystallographic phase transitions at the spin-crossover T1/2 temperature are much more common.6.
- N. T. Madhu, I. Salitros, F. Schramm, S. Klyatskaya, O. Fuhr and M. Ruben, C. R. Chim., 2008, 11, 1166 CrossRef CAS.
- I. Šalitroš, O. Fuhr, A. Eichhöfer, R. Kruk, J. Pavlik, L. Dlháň, R. Boča and M. Ruben, Dalton Trans., 2012, 41, 5163 RSC; I. Šalitroš, L. Pogány, M. Ruben, R. Boča and W. Linert, Dalton Trans., 2014, 43, 16584 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 CrossRef CAS.
- POVRAY, v. 3.5, Persistence of Vision Raytracer Pty. Ltd., Williamstown, Victoria, Australia, 2002. http://www.povray.org Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 2003 RSC; E. M. Schubert, J. Chem. Educ., 1992, 69, 62 CrossRef CAS.
- J. C. Philip and H. B. Oakley, J. Chem. Soc. Trans., 1924, 125, 1189 RSC; W. A. Felsing and S. A. Durban, J. Am. Chem. Soc., 1926, 48, 2885 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Definitions of the structural indices used in Table 1, additional crystallographic Figures and Tables; DSC data; magnetic susceptibility data for 2[BF4]2 and 3[BF4]2; and a comparison of 1[ClO4]2 with other [Fe(bpp)2]2+ derivatives that adopt this mode of crystal packing. CCDC 1060840 (L3), 1060841 (1[ClO4]2 at 280 K), 1060842 (1[ClO4]2 at 253 K), 1060843 (1[ClO4]2 at 240 K) and 1060844 (1[ClO4]2 at 150 K). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00083a |
| This journal is © the Partner Organisations 2015 |