DOI:
10.1039/C4QI00234B
(Research Article)
Inorg. Chem. Front., 2015,
2, 780-798
DNA/protein binding, DNA cleavage, cytotoxicity, superoxide radical scavenging and molecular docking studies of copper(II) complexes containing N-benzyl-N′-aryl-N′′-benzoylguanidine ligands†
Received
23rd December 2014
, Accepted 24th June 2015
First published on 24th June 2015
Abstract
Five copper(II) complexes containing N,N′,N′′-trisubstituted guanidine ligands were synthesized and characterized by elemental analyses and UV-Visible, FT-IR, EPR and mass spectroscopic techniques. The synthesized copper(II) complexes (1–5) bear the general formula [Cu{C6H5CONC(NR)NHCH2C6H5}2] where R = phenyl (1), 4-methylphenyl (2), 4-ethoxyphenyl (3), 2-methoxyphenyl (4) or 1-naphthyl (5). The four coordinated square planar geometry of the complexes was confirmed by the single crystal X-ray diffraction study. The interaction of the Cu(II) complexes with calf thymus DNA (CT DNA) was explored using absorption and fluorescence spectroscopic methods. The results revealed that the complexes have an affinity constant for DNA in the order of 104 M−1 and the mode of interaction is non-covalent intercalation. The DNA cleavage study showed that the complexes cleaved DNA without any external agent. The interaction of Cu(II) complexes with bovine serum albumin (BSA) was also studied using absorption and fluorescence techniques. The cytotoxic activity of the Cu(II) complexes was probed in vitro against human breast (MCF7) and lung (A549) cancer cell lines. The complexes were also tested against mouse embryonic fibroblast (NIH 3T3) cell lines. Complexes 1 and 3 have good cytotoxic activity which is comparable with the cyclophosphamide drug. The complexes were less cytotoxic towards normal cell lines showing that they affect only cancer cell lines. Superoxide radical scavenging property of the complexes was assessed using the NBT assay. Copper(II) complexes showed appreciable superoxide radical scavenging activity with IC50 values ranging from 1.53 to 5.62 μM. A further molecular docking technique was employed to understand the binding of the complexes toward the molecular target DNA and human DNA topoisomerase I.
1. Introduction
The synthesis of new transition metal complexes possessing the analogous properties of anticancer drugs is a fascinating area of research in bioinorganic chemistry. The electron-rich nature of biomolecules (DNA/protein) and electron-deficient metal ions are believed to have strong interactions, which are thought to be the criteria for the pharmaceutical activities of the complexes. Therefore, the study of the binding properties of metal complexes with DNA and protein is of great significance for the design of new drugs and their application.1
DNA is one of the main molecular targets in the design of anticancer compounds.2 DNA-binding small molecules have attracted interest because of their interference in important mechanisms in the cell, some inducing mutations and cancer, while others have found use as cancer therapeutics. Further, it has been revealed that free radicals can damage proteins, lipids and DNA of bio-tissues, leading to increased rates of cancer, and fortunately antioxidants can prevent this damage due to their free radical scavenging activity.3,4 Therefore much attention has been given to metal complexes that can bind to DNA and also with antioxidant properties.
At the same time, proteins have also attracted enormous research interest as a prime molecular target.5 Serum albumins such as bovine serum albumin (BSA) is the most important protein present in plasma that carries several endo- and exogenous compounds.5 It is essential to explore drug–protein interactions as most of the drugs bound to serum albumins are usually transported as a protein complex.6,7 Attention has also been focused on the proteins that drive and control cell cycle progression.8 Therefore interaction studies of the metal complexes with proteins like BSA become very important for the development of drugs with great potential.
The guanidine molecule, (NH2)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH, is an important ingredient of both organic and inorganic chemistry. Guanidine is noted as a sterically and electronically flexible ligand because of the Y-shaped CN3 unit present in it. The guanidine group is a decisive feature of many biologically active species such as arginine, triazabicyclodecene and saxitoxin. It is found in a growing number of biologically and pharmaceutically relevant compounds. Due to its large spectrum of biological activities the guanidine functional group has been intensively studied as a synthetic goal. Guanidines have a broad spectrum of biological activities like antitumour,9–11 antimalarial, antiinflammatory, urease inhibition,12etc. This is further enhanced by their coordination with metals. A large number of guanidine complexes has been reported with different metals like Cu, Pt, Co, Ni, Ru, Fe, Zn and so on.13,14 But copper has special importance compared to other metals. Copper is an essential element for most aerobic organisms, employed as a structural and catalytic cofactor, and consequently it is involved in many biological pathways.15–17 Also, serum copper levels correlate with tumor incidence, tumor weight, malignant progression, and recurrence in a variety of human cancers supporting the idea that copper could be used as a potential tumor-specific target. Several copper complexes18–31 have now been proposed as potential anticancer substances and cancer inhibiting agents, as they show remarkable anticancer activity and lower general toxicity than platinum compounds. All the above facts have motivated us to develop copper complexes containing N,N′,N′′-trisubstituted guanidine ligands and to evaluate their DNA binding, DNA cleavage, protein binding, cytotoxicity and superoxide radical scavenging properties.
NH, is an important ingredient of both organic and inorganic chemistry. Guanidine is noted as a sterically and electronically flexible ligand because of the Y-shaped CN3 unit present in it. The guanidine group is a decisive feature of many biologically active species such as arginine, triazabicyclodecene and saxitoxin. It is found in a growing number of biologically and pharmaceutically relevant compounds. Due to its large spectrum of biological activities the guanidine functional group has been intensively studied as a synthetic goal. Guanidines have a broad spectrum of biological activities like antitumour,9–11 antimalarial, antiinflammatory, urease inhibition,12etc. This is further enhanced by their coordination with metals. A large number of guanidine complexes has been reported with different metals like Cu, Pt, Co, Ni, Ru, Fe, Zn and so on.13,14 But copper has special importance compared to other metals. Copper is an essential element for most aerobic organisms, employed as a structural and catalytic cofactor, and consequently it is involved in many biological pathways.15–17 Also, serum copper levels correlate with tumor incidence, tumor weight, malignant progression, and recurrence in a variety of human cancers supporting the idea that copper could be used as a potential tumor-specific target. Several copper complexes18–31 have now been proposed as potential anticancer substances and cancer inhibiting agents, as they show remarkable anticancer activity and lower general toxicity than platinum compounds. All the above facts have motivated us to develop copper complexes containing N,N′,N′′-trisubstituted guanidine ligands and to evaluate their DNA binding, DNA cleavage, protein binding, cytotoxicity and superoxide radical scavenging properties.
2. Results and discussion
2.1 Synthesis and characterization
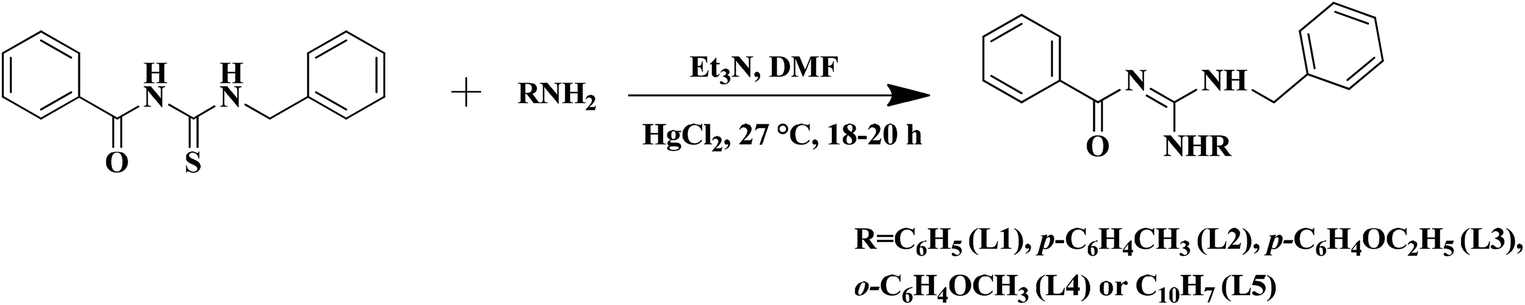
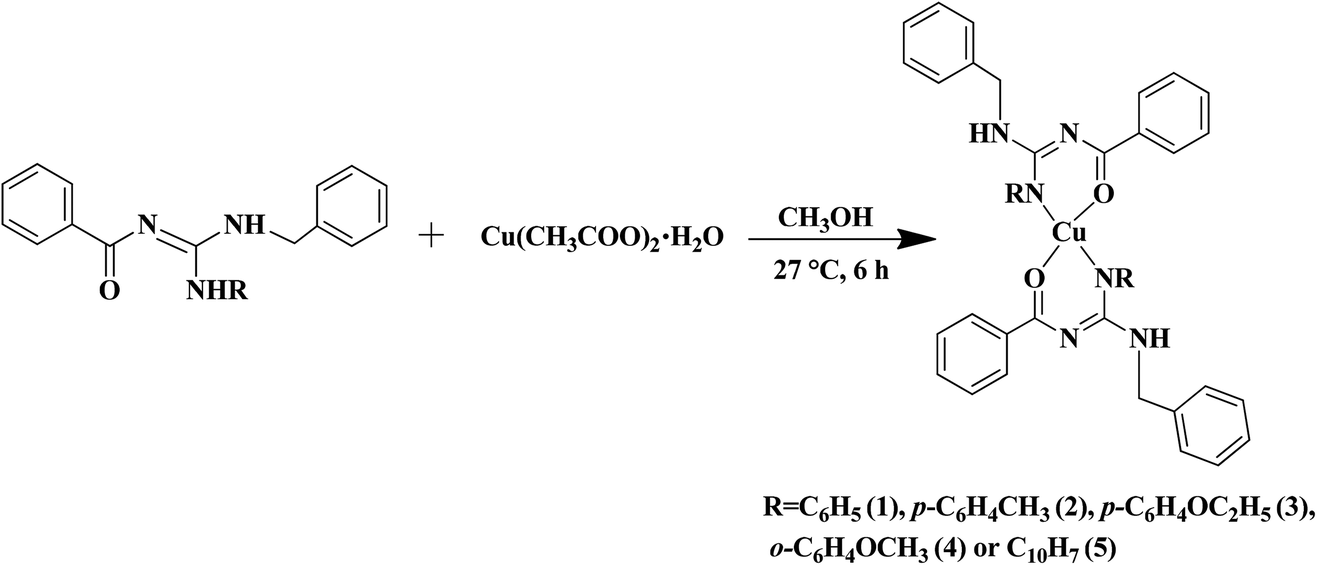
The substituted thiourea was synthesized from benzoyl chloride and benzylamine using the standard procedure.32 The trisubstituted guanidine ligands were obtained from the mercury promoted guanylation reaction between N-benzoyl-N′-benzylthiourea and a suitable amine (Scheme 1).33,34 The syntheses of copper(II) complexes were achieved in good yields by the reaction of guanidine ligands with copper acetate monohydrate (Scheme 2).
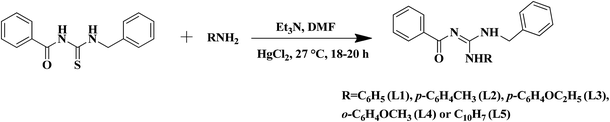 |
| | Scheme 1 Synthesis of N-benzyl-N′-aryl-N′′-benzoylguanidine. | |
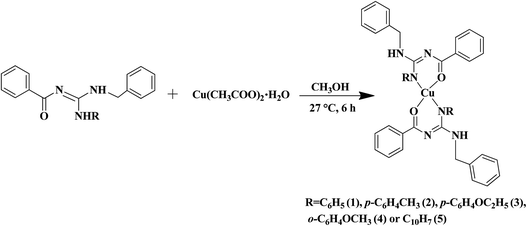 |
| | Scheme 2 Synthesis of copper(II) complexes. | |
The synthesized ligands and complexes were characterized using UV-Visible, FT-IR, NMR/EPR and mass/elemental analysis techniques. The electronic spectra of the ligands showed bands around 272–290 and 235–268 nm regions, which correspond to π → π* and n → π* transitions respectively. In the complexes, a band appeared around 240–271 nm corresponds to intraligand transition. The ligand to metal charge transfer transition (LMCT) is characterized by the appearance of a band around 377–389 nm. There is a broad band around 592–643 nm which is characteristic of the d → d transition.35
In the FT-IR spectra of the ligands there are two N–H bands that appeared around 3165–3419 cm−1. One of the N–H bands is sharp and the other one is weak. The weak N–H band is due to the involvement of hydrogen bonding between the carbonyl oxygen and the N–H hydrogen. The CN absorbs around 1561–1571 cm−1 and CO absorbs around 1591–1602 cm−1.36 On complexation there is a shift in the CO stretching frequency towards the lower value and there is disappearance of the weak N–H band. This clearly confirms that the coordination occurs through carbonyl oxygen and the nitrogen of the N–H after deprotonation.
In the 1H NMR spectra of the ligands (L1–L5), the signal of the NR–H proton appears in the downfield around 12.54–11.93 ppm since it is attached to an aromatic ring. The N–H proton attached to the benzyl moiety appears in the upfield around 5.22–5.10 ppm. The benzyl CH2 proton resonates around 4.74–4.81 ppm. The signals due to all the other aromatic protons are in the expected range. 13C NMR spectra of the ligands confirm the presence of CO (178.1–177.7 ppm) and CN (159.6–158.1 ppm) carbons. Chemical shifts of all the other aromatic and aliphatic carbons were present in the expected regions.
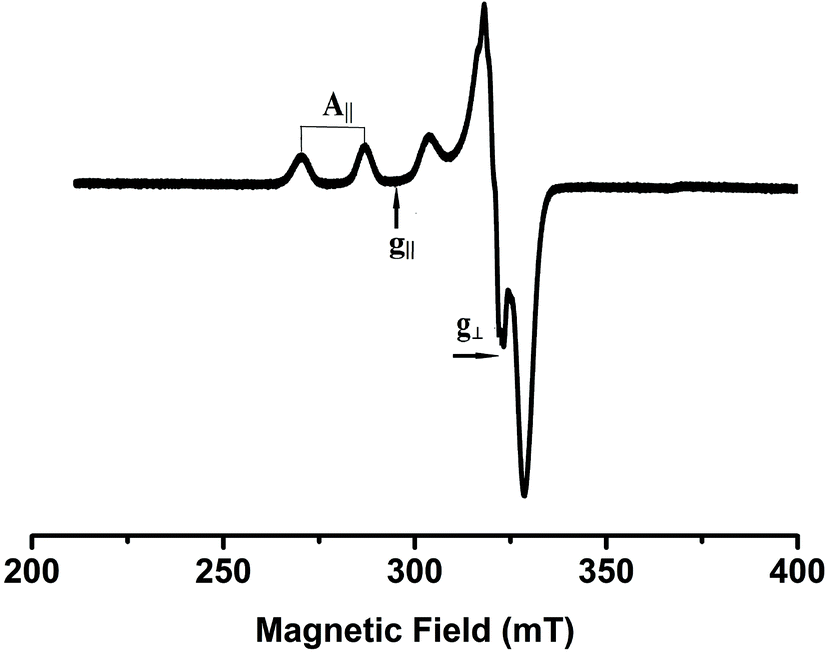
X-Band EPR spectra of the Cu(II) complexes were recorded at liquid nitrogen temperature in DMF solution. All the complexes showed well resolved quartet hyperfine splitting typical of a square planar Cu(II) system (Fig. 1) which was confirmed by the single crystal XRD technique. The frozen solution EPR spectra of the complexes are axial with g|| > g⊥ > 2.00 and the trend in the g value (g|| > g⊥ > 2.00) suggests that the unpaired electron in the Cu(II) ion resides in the dx2−y2 orbital. The observed values (Table 1) of g|| (2.16–2.21) and A|| (∼161–180) × 10−4 cm−1 for 1–5 are consistent with the presence of a square-based geometry as is evident from the crystal structure of 1 and 2. This is also supported by the value of g||/A|| quotient which falls within the range expected for a square planar geometry (121–137 cm).37
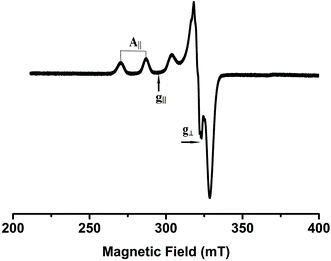 |
| | Fig. 1 EPR spectrum of complex 4 in frozen DMF solution. | |
Table 1 EPR parameters of Cu(II) complexes
| Complex |
Medium & Temp. |
g
||
|
g
⊥
|
A
|| (G) |
|
1
|
Solution state LNT |
2.212 |
2.037 |
174 |
|
2
|
Solution state LNT |
2.162 |
2.024 |
178 |
|
3
|
Solution state LNT |
2.198 |
2.040 |
180 |
|
4
|
Solution state LNT |
2.210 |
2.024 |
161 |
|
5
|
Solution state LNT |
2.213 |
2.015 |
172 |
2.2 Single crystal X-ray crystallographic studies
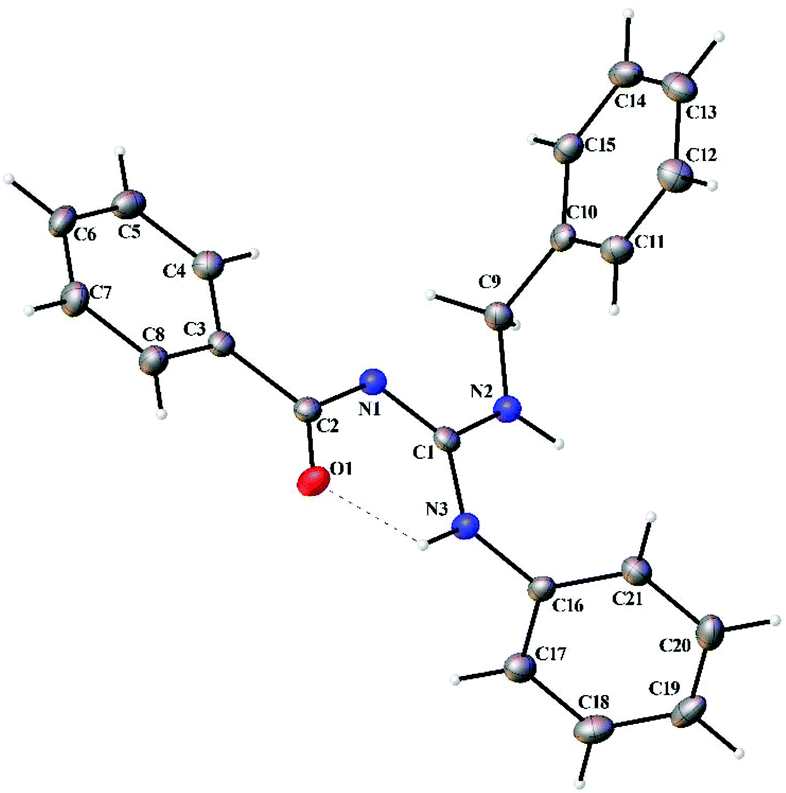
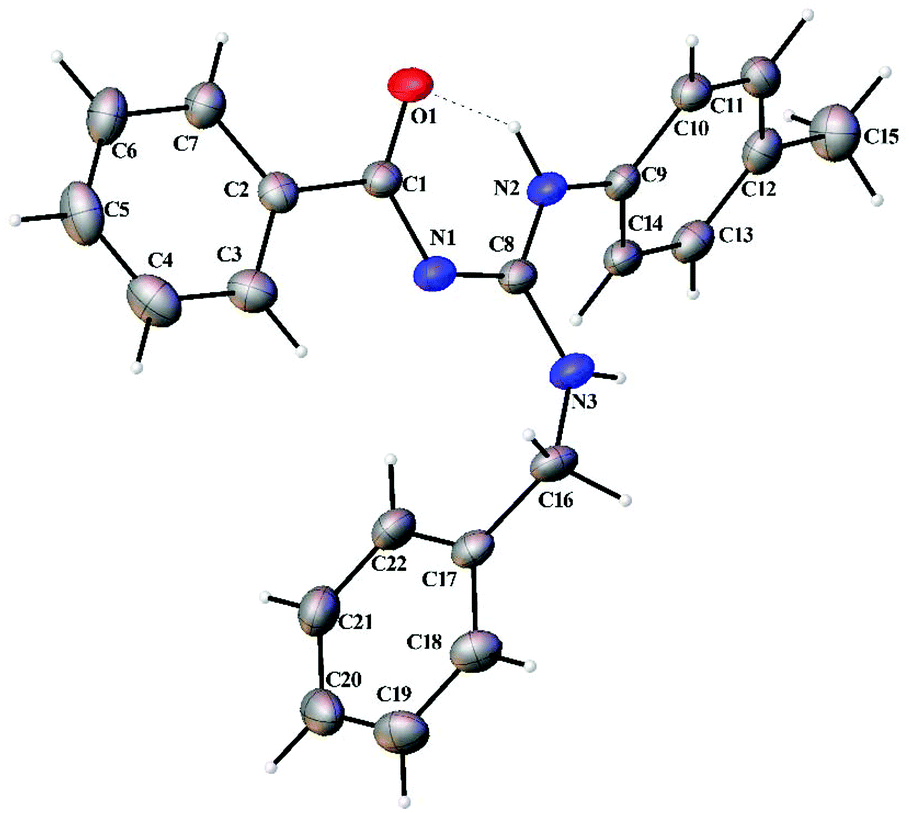
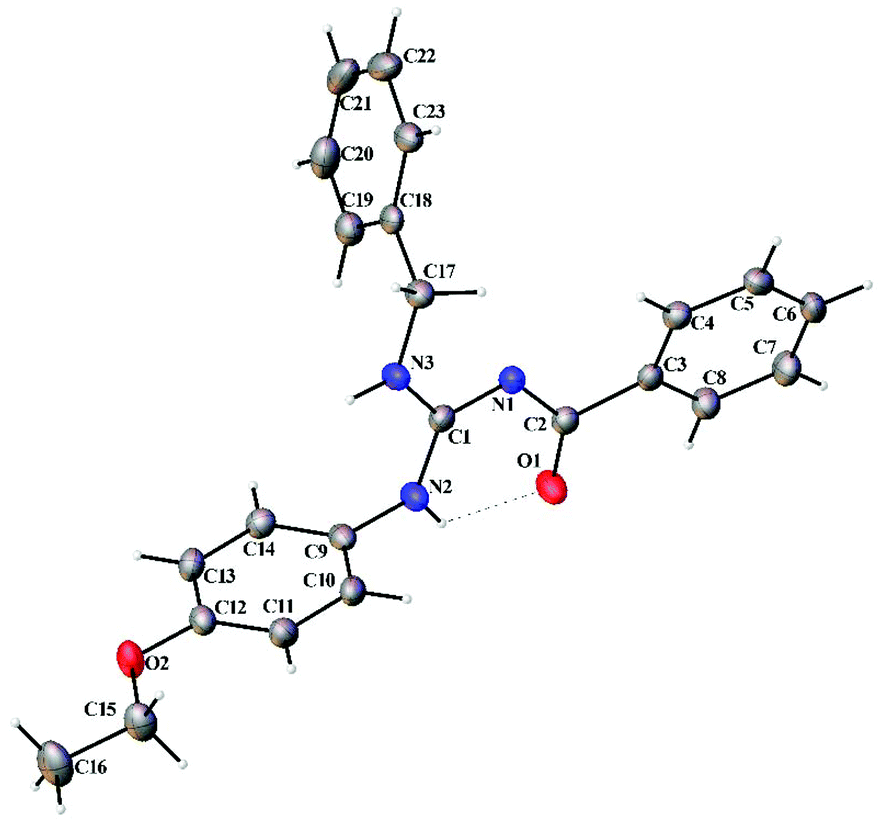
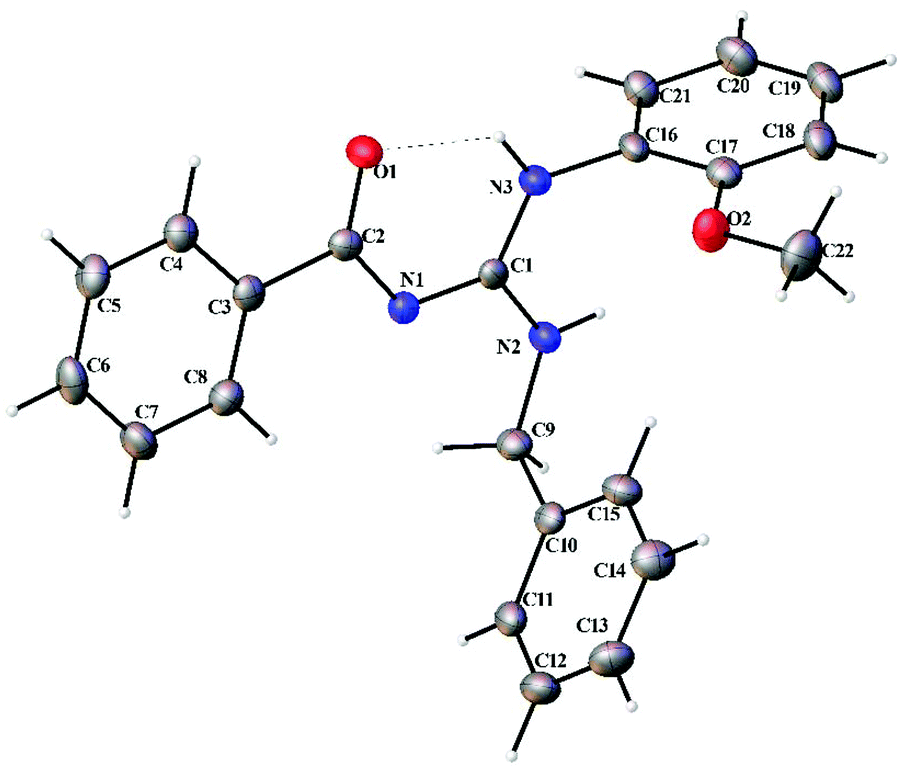
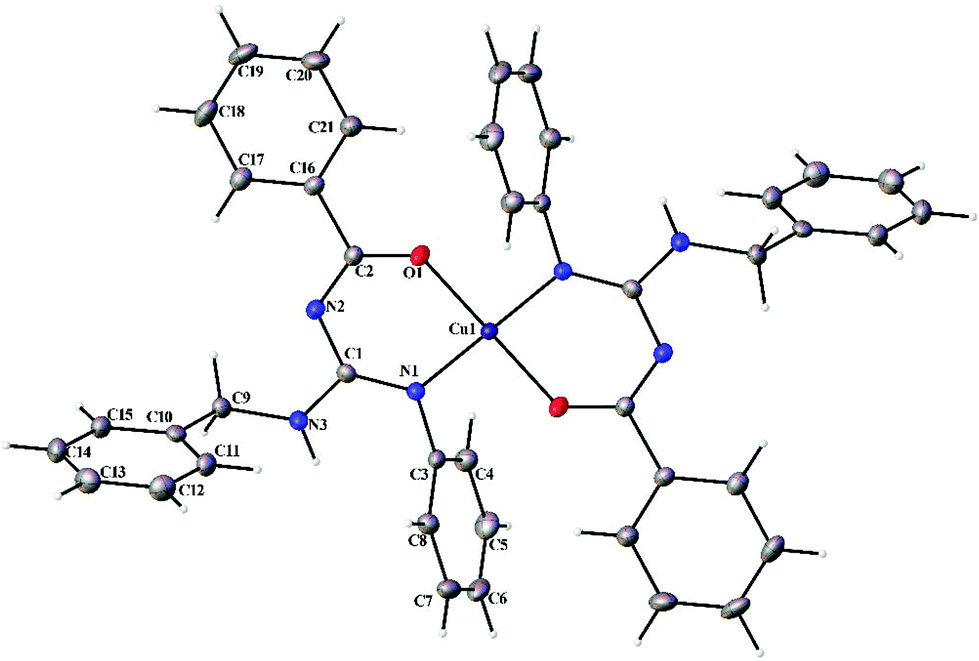
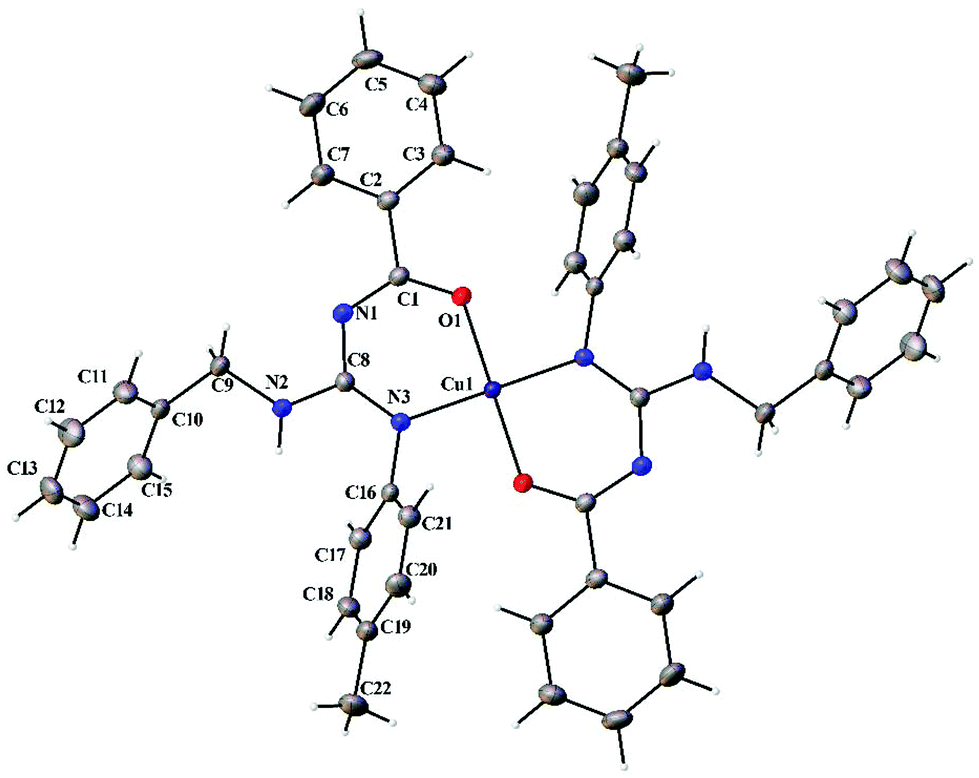
Thermal ellipsoid plots of the ligands (L1–L5) and the complexes (1 and 2) with the atomic labelling scheme are shown in Fig. 2–8. Crystal data and selected interatomic bond lengths and angles are given in Tables 2–5. Suitable crystals of the ligands were grown by slow evaporation of ethanol solutions of the ligands. The crystal structure of the ligands shows the existence of intra and intermolecular hydrogen bonding between carbonyl oxygen and amino nitrogen. There exists a Y-aromaticity in the guanidine moiety as the C–N bond length varies between 1.3281–1.3595 Å (L1), 1.3358–1.3438 Å (L2), 1.3313–1.3526 Å (L3), 1.3387–1.3470 Å (L4) and 1.3352–1.3570 Å (L5).
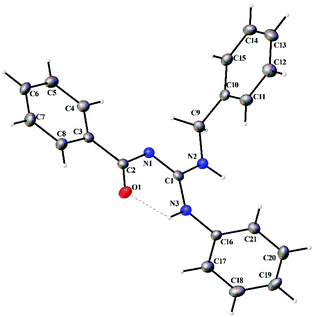 |
| | Fig. 2 Thermal ellipsoid plot of L1. | |
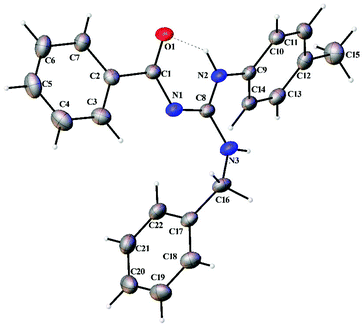 |
| | Fig. 3 Thermal ellipsoid plot of L2. | |
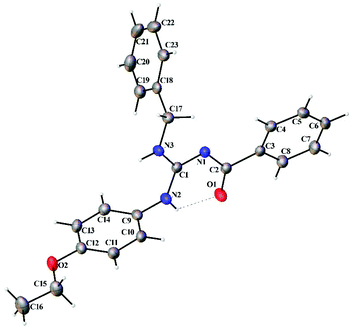 |
| | Fig. 4 Thermal ellipsoid plot of L3. | |
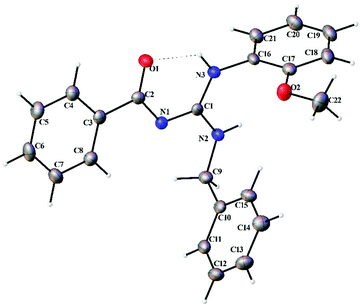 |
| | Fig. 5 Thermal ellipsoid plot of L4. | |
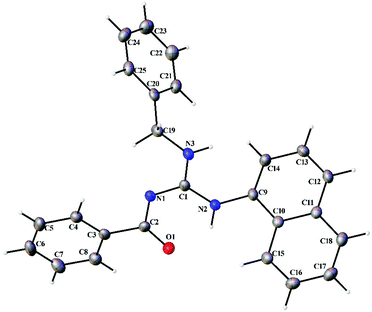 |
| | Fig. 6 Thermal ellipsoid plot of L5. | |
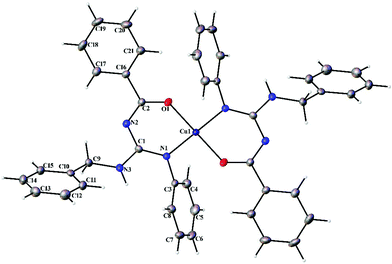 |
| | Fig. 7 Thermal ellipsoid plot of 1. | |
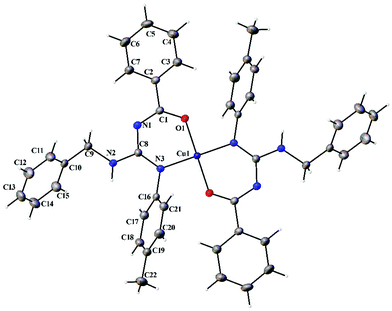 |
| | Fig. 8 Thermal ellipsoid plot of 2. | |
Table 2 Crystallographic data and refinement parameters for ligands L1–L5
| |
L1
|
L2
|
L3
|
L4
|
L5
|
| Empirical formula |
C21H19N3O |
C22H21N3O |
C23H23N3O2 |
C22H21N3O2 |
C25H21N3O |
| Formula weight |
329.39 |
343.42 |
373.44 |
359.42 |
379.45 |
| Temperature (K) |
110(2) |
150(2) |
296.15 |
110(2) |
110(2) |
| Wavelength (Å) |
0.71073 |
0.71073 |
0.71073 |
1.54178 |
1.54178 |
| Crystal system |
Triclinic |
Triclinic |
Monoclinic |
Trigonal |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P121/c1 |
P3(2) |
P |
| Unit cell dimensions |
|
a (Å) |
8.4339(17) |
12.6374(15) |
10.8758(16) |
9.9964(3) |
10.2809(9) |
|
b (Å) |
10.588(2) |
12.7890(15) |
18.848(3) |
9.9964(3) |
10.3681(9) |
|
c (Å) |
10.687(2) |
12.8040(15) |
9.4682(14) |
16.0766(7) |
10.6801(9) |
|
α (°) |
66.082(2) |
73.447(2) |
90 |
90 |
62.458(5) |
|
β (°) |
82.662(2) |
75.957(2) |
92.429(2) |
90 |
79.593(5) |
|
γ (°) |
82.739(2) |
74.303(2) |
90 |
120 |
70.395(6) |
| Volume (Å3) |
862.4(3) |
1878.7(4) |
1939.2(5) |
1391.27(8) |
950.45(14) |
|
Z
|
2 |
4 |
4 |
3 |
2 |
| Density (calculated) Mg m−3 |
1.269 |
1.214 |
1.279 |
1.287 |
1.326 |
| Absorption coefficient (mm−1) |
0.080 |
0.076 |
0.083 |
0.672 |
0.649 |
|
F(000) |
348 |
728 |
792 |
570 |
400 |
| Crystal size (mm3) |
0.56 × 0.52 × 0.25 |
0.65 × 0.61 × 0.53 |
0.519 × 0.512 × 0.206 |
0.16 × 0.04 × 0.04 |
0.12 × 0.06 × 0.04 |
| Theta range for data collection (°) |
2.31 to 27.33 |
1.70 to 27.50 |
1.87 to 28.43 |
5.80 to 59.99 |
4.57 to 59.99 |
| Index ranges |
−10 ≤ h ≤ 10, −13 ≤ k ≤ 13, −13 ≤ l ≤ 13 |
−16 ≤ h ≤ 16, −16 ≤ k ≤ 16, −16 ≤ l ≤ 16 |
−14 ≤ h ≤ 14, −24 ≤ k ≤ 24, −12 ≤ l ≤ 12 |
−11 ≤ h ≤ 11, −11 ≤ k ≤ 11, −17 ≤ l ≤ 17 |
−11 ≤ h ≤ 11, −11 ≤ k ≤ 11, −11 ≤ l ≤ 11 |
| Reflections collected |
9848 |
25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 111 111 |
22873 |
31224 |
16951 |
| Independent reflections [R(int)] |
3835[R(int) = 0.0212] |
8452 [R(int) = 0.0273] |
4741 [R(int) = 0.0327] |
2729 [R(int) = 0.0526] |
2765 [R(int) = 0.0394] |
| Completeness to theta = 27.33°/27.50°/25.24°/59.99°/59.99° |
98.7% |
97.9% |
100.0% |
99.5% |
98% |
| Absorption correction |
Semi-empirical from equivalents |
Semi-empirical from equivalents |
Semi-empirical from equivalents |
Semi-empirical from equivalents |
Semi-empirical from equivalents |
| Max. and min. transmission |
0.9803 and 0.9566 |
0.9608 and 0.9522 |
0.7457 and 0.6776 |
0.9736 and 0.9000 |
0.9745 and 0.9262 |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
3835/0/226 |
8452/0/471 |
4741/0/254 |
2729/1/246 |
2765/0/263 |
| Goodness-of-fit on F2 |
1.065 |
1.045 |
1.052 |
1.111 |
1.084 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.0396, wR2 = 0.0977 |
R
1 = 0.0424, wR2 = 0.1098 |
R
1 = 0.0398, wR2 = 0.0972 |
R
1 = 0.0275, wR2 = 0.0686 |
R
1 = 0.0338, wR2 = 0.0911 |
|
R indices (all data) |
R
1 = 0.0464, wR2 = 0.1023 |
R
1 = 0.0513, wR2 = 0.1153 |
R
1 = 0.0497, wR2 = 0.1036 |
R
1 = 0.0285, wR2 = 0.0692 |
R
1 = 0.0390, wR2 = 0.0939 |
| Largest diff. peak and hole (e Å−3) |
0.261 and −0.228 |
0.282 and −0.267 |
0.254 and −0.221 |
0.125 and −0.127 |
0.158 and −0.222 |
Table 3 Crystallographic data and refinement parameters for complexes 1 and 2
| |
1
|
2
|
| Empirical formula |
C42H36CuN6O2 |
C44H40CuN6O2 |
| Formula weight |
720.31 |
748.36 |
| Temperature (K) |
150(2) |
150(2) |
| Wavelength (Å) |
0.71073 |
0.71073 |
| Crystal system |
Triclinic |
Triclinic |
| Space group |
P |
P |
| Unit cell dimensions |
|
a (Å) |
8.4363(13) |
6.1250(8) |
|
b (Å) |
10.3390(16) |
11.5140(14) |
|
c (Å) |
10.9129(17) |
13.2365(16) |
|
α (°) |
65.535(2) |
81.520(2) |
|
β (°) |
86.346(2) |
87.731(2) |
|
γ (°) |
85.211(2) |
85.984(2) |
| Volume (Å3) |
862.9(2) |
920.6(2) |
|
Z
|
1 |
1 |
| Density (calculated) Mg m−3 |
1.386 |
1.350 |
| Absorption coefficient (mm−1) |
0.680 |
0.640 |
|
F(000) |
375 |
391 |
| Crystal size (mm3) |
0.18 × 0.14 × 0.04 |
0.45 × 0.21 × 0.21 |
| Theta range for data collection (°) |
2.05 to 27.50 |
1.56 to 27.49 |
| Index ranges |
−10 ≤ h ≤ 10, −13 ≤ k ≤ 13, −14 ≤ l ≤ 14 |
−7 ≤ h ≤ 7, −14 ≤ k ≤ 14, −17 ≤ l ≤ 17 |
| Reflections collected |
14794 |
19956 |
| Independent reflections [R(int)] |
3925 [R(int) = 0.0485] |
4161 [R(int) = 0.0214] |
| Completeness to theta = 27.50° |
99.1% |
98.7% |
| Absorption correction |
Semi-empirical from equivalents |
Semi-empirical from equivalents |
| Max. and min. transmission |
0.9733 and 0.8874 |
0.8773 and 0.7616 |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
3925/0/232 |
4161/0/242 |
| Goodness-of-fit on F2 |
1.079 |
1.071 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.0317, wR2 = 0.0846 |
R
1 = 0.0258, wR2 = 0.0677 |
|
R indices (all data) |
R
1 = 0.0326, wR2 = 0.0853 |
R
1 = 0.0266, wR2 = 0.0684 |
| Largest diff. peak and hole (e Å−3) |
0.441 and −0.427 |
0.294 and −0.272 |
Table 4 Selected bond lengths (Å) and angles (°) of ligands
| |
L1
|
L2
|
L3
|
L4
|
L5
|
| O(1)–C(2) |
1.2453(13) |
1.2521(14) |
1.2442(14) |
1.2562(19) |
1.2513(16) |
| N(1)–C(1) |
1.3281(14) |
1.3407(14) |
1.3313(14) |
1.3387(19) |
1.3352(17) |
| N(1)–C(2) |
1.3591(14) |
1.3438(15) |
1.3526(14) |
1.3470(17) |
1.3570(17) |
| N(2)–C(1) |
1.3509(14) |
1.3484(15) |
1.3464(14) |
1.3360(2) |
1.3470(17) |
| N(2)–C(9) |
1.4483(14) |
1.4186(14) |
1.4331(14) |
1.4550(2) |
1.4239(17) |
| N(3)–C(1) |
1.3460(14) |
1.3358(15) |
1.3449(14) |
1.3460(2) |
1.3451(17) |
| N(3)–C(16) |
1.4281(14) |
1.4520(16) |
1.4920(13) |
1.4320(2) |
1.4527(17) |
| N(2)–H(2) |
0.8899 |
0.8800 |
0.8800 |
0.8800 |
0.8800 |
| N(3)–H(3) |
0.8934 |
0.8800 |
0.8800 |
0.8800 |
0.8800 |
| C(1)–N(1)–C(2) |
120.15(9) |
120.55(10) |
120.46(9) |
120.23(13) |
120.51(11) |
| C(1)–N(2)–C(9) |
123.78(9) |
129.58(10) |
123.00(9) |
122.78(12) |
128.41(11) |
| C(1)–N(2)–H(2) |
114.9 |
115.20 |
118.50 |
117.90 |
115.80 |
| C(9)–N(2)–H(2) |
114.9 |
115.20 |
118.50 |
117.90 |
115.80 |
| C(1)–N(3)–C(16) |
123.71(9) |
122.20(10) |
122.95(9) |
124.19(13) |
123.16(11) |
| C(1)–N(3)–H(3) |
121.2 |
118.90 |
118.50 |
118.60 |
118.40 |
| C(16)–N(3)–H(3) |
121.2 |
118.90 |
118.50 |
118.60 |
118.40 |
| N(1)–C(1)–N(3) |
125.53(10) |
122.95(10) |
126.17(10) |
124.43(14) |
123.48(12) |
| N(1)–C(1)–N(2) |
117.36(10) |
117.33(10) |
116.78(10) |
117.82(13) |
117.44(12) |
| N(3)–C(1)–N(2) |
117.08(10) |
119.70(10) |
117.01(10) |
117.74(13) |
119.08(12) |
| O(1)–C(2)–N(1) |
126.88(10) |
126.77(10) |
127.35(10) |
127.06(14) |
127.03(12) |
| O(1)–C(2)–C(3) |
119.33(10) |
118.83(10) |
119.17(9) |
118.02(14) |
118.99(11) |
| N(1)–C(2)–C(3) |
113.79(9) |
114.40(10) |
113.47(9) |
114.92(13) |
113.98(11) |
| N(2)–C(9)–C(10) |
114.44(9) |
115.95(10) |
114.39(9) |
113.88(13) |
114.63(11) |
| N(2)–C(9)–H(9A) |
108.6 |
108.30 |
108.70 |
108.80 |
108.60(11) |
| N(2)–C(9)–H(9B) |
108.6 |
108.30 |
108.70 |
108.80 |
108.60(11) |
Table 5 Selected bond lengths (Å) and angles (°) of complexesa
| |
1
|
2
|
|
Symmetry transformations used to generate equivalent atoms: #1 − x, −y, −z.
|
| Cu(1)–O(1) |
1.9055(11) |
1.9122(9) |
| Cu(1)–O(1)#1 |
1.9055(11) |
1.9122(9) |
| Cu(1)–N(1) |
1.9752(13) |
1.9602(10) |
| Cu(1)–N(1)#1 |
1.9752(13) |
1.9602(10) |
| O(1)–Cu(1)–N(1) |
89.69(5) |
90.03(4) |
| O(1)#1–Cu(1)–N(1)#1 |
89.65(5) |
90.04(4) |
| O(1)#1–Cu(1)–N(1) |
90.31(5) |
89.97(4) |
| O(1)–Cu(1)–N(1)#1 |
90.31(5) |
89.96(4) |
| O(1)#1–Cu(1)–O(1) |
180.0 |
180.0 |
| N(1)–Cu(1)–N(1)#1 |
180.0 |
180.0 |
| C(2)–O(1)–Cu(1) |
128.05(10) |
125.59(8) |
| C(1)–N(1)–Cu(1) |
124.39(9) |
123.25(8) |
| C(3)–N(1)–Cu(1) |
119.49(9) |
120.29(7) |
Complexes 1 and 2 crystallized in the triclinic P space group with a Z of 1. The X-ray diffraction structure of complexes 1 and 2 reveals a perfect square planar geometry with oxygen and nitrogen atoms coordinated to the Cu ion in a trans fashion. The perfect square planar geometry is also reflected by τ4 parameter. τ4 is the parameter used to describe the four coordinate system and is used to determine the tetrahedral/square planar distortion. This term was coined by Dunitz and coworkers,38 and more recently by Keinan and Avnir.39τ4 can be calculated using the formula τ4 = [360° − (α + β)]/141°. The value of τ4 ranges from 1.00 for a perfect tetrahedral geometry, since 360–2 (109.5) = 141, to zero for a perfect square planar geometry, since 360–2 (180) = 0. For the intermediate structures like trigonal pyramidal, τ4 falls in the range of 0–1.00. In complexes 1 and 2, the angle between O(1)–Cu(1)–O(1)#1 and N(1)#1–Cu(1)–N(1) is 180° which shows a perfect square planar geometry. There is an increase in the C–O bond length and a decrease in the C–N bond length (involved in coordination) in 1 and 2 compared to L1 and L2, respectively.
2.3 DNA binding studies
2.3.1 Electronic absorption titration.
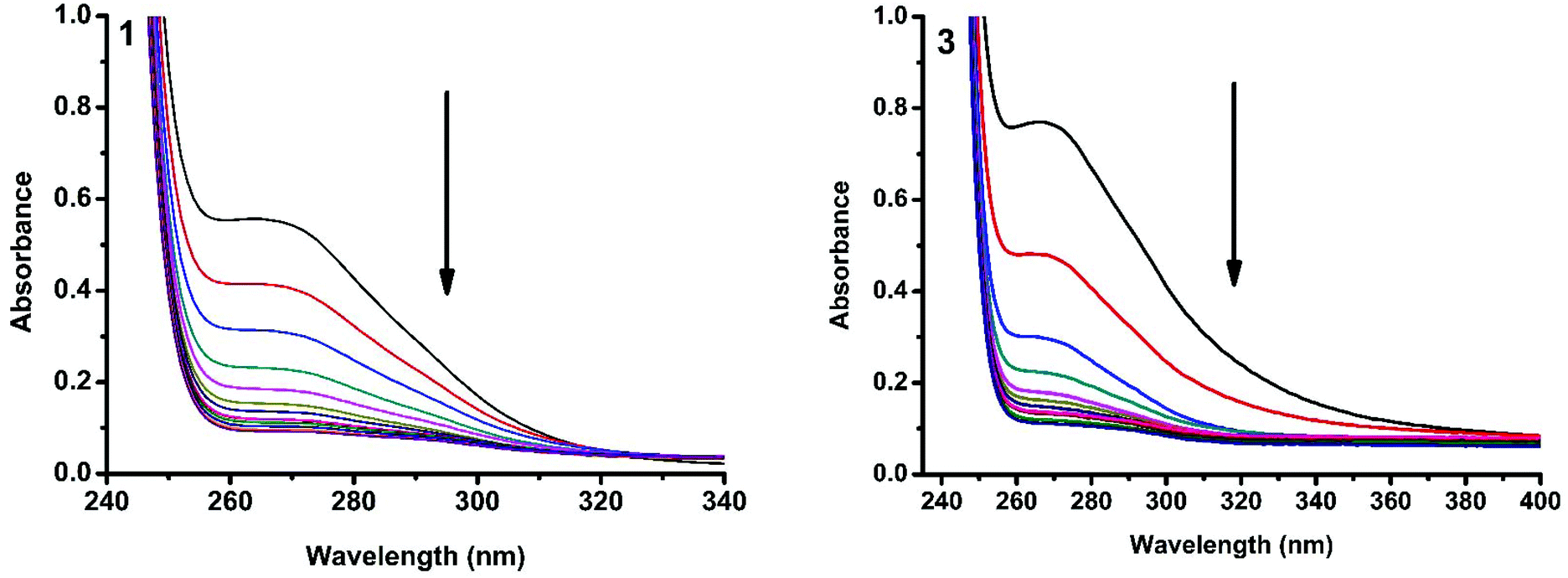
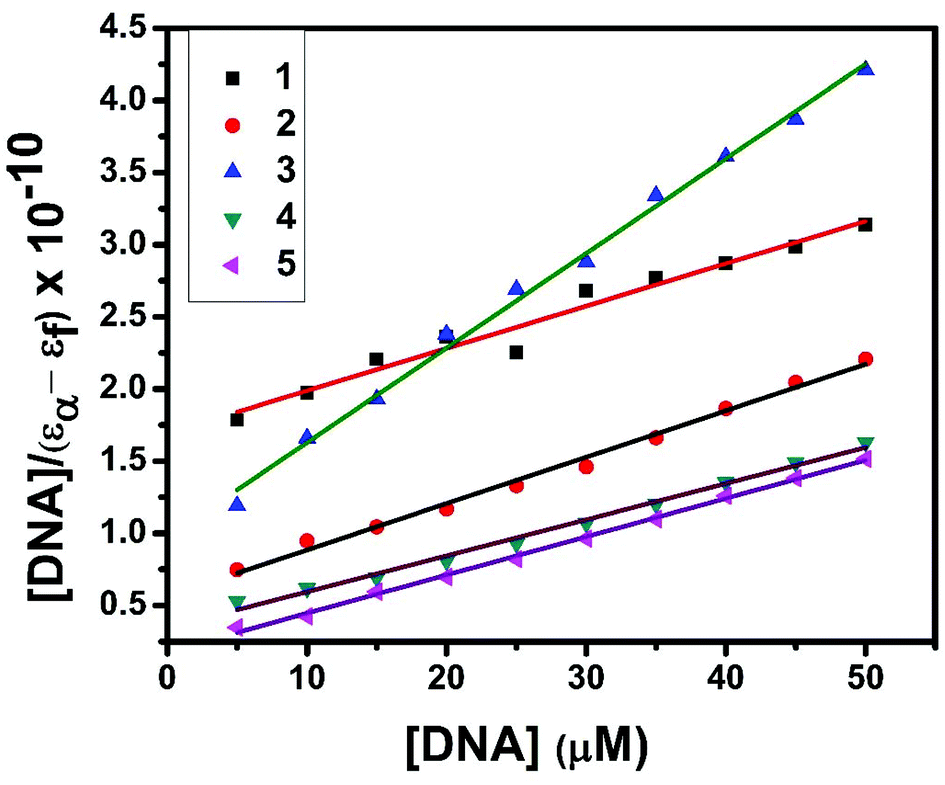
The interaction of copper(II) complexes with CT DNA was investigated using UV-Visible absorption studies. The electronic spectra of Cu(II) complexes (1–5) exhibited a band around 240–271 nm which corresponds to intraligand transition. The observation of the changes in this band upon the incremental addition of DNA was the most widely used method to find out the binding constant. In general, a compound that binds to DNA through intercalation results in hypochromism and bathochromism due to strong stacking interactions between an aromatic chromophore and the base pairs of DNA. The interaction of complexes (1–5) was confirmed by the observed hypochromism with a red shift (Fig. 9 and S1†). The magnitude of hypochromism is in the order 3 > 1 > 2 > 4 > 5, which reflects the DNA binding affinities of the complexes. The intrinsic binding constant Kb was obtained from the ratio of slope to intercept in the plot of [DNA]/(εa − εf) versus [DNA] according to the equation40 [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εb − εf) where [DNA] is the concentration of DNA in base pairs, εa is the apparent extinction coefficient value found by calculating A(observed)/[complex], εf is the extinction coefficient for the free compound, and εb is the extinction coefficient for the compound in the fully bound form. Each set of data, when fitted into the above equation, gave a straight line with a slope of 1/(εb − εf) and a y-intercept of 1/Kb(εb − εf) and the value of Kb was determined from the ratio of slope to intercept (Fig. 11). The magnitudes of intrinsic binding constants (Kb) are given in Table 6. The Kb values were found to be in the range of 1.62 × 104–1.63 × 105 M−1. The high binding ability of complex 3 compared to the other complexes is due to the presence of an ethoxy group in the guanidine ligand. The same is not observed in the case of complex 4 even though a methoxy group is present. The reason for this may be due to the steric effect of the methoxy group which is present at the ortho position.
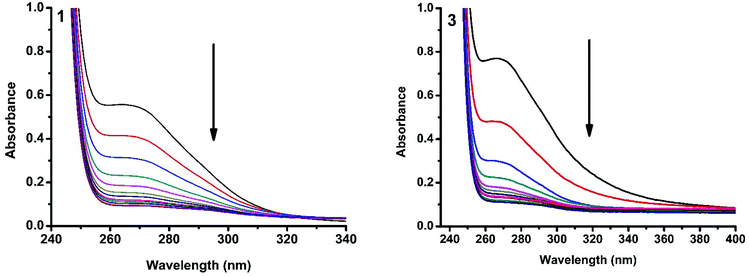 |
| | Fig. 9 Absorption spectra of complexes 1 and 3 in Tris-HCl buffer upon addition of CT DNA. [Complex] = 2.0 × 10−5 M, [DNA] = 0–40 μM. The arrow shows that the absorption intensity decreases upon increasing the DNA concentration. | |
Table 6 DNA binding constant (Kb), quenching constant (Kq) and apparent binding constant (Kapp) values
| Complex |
K
b (M−1) |
K
q (M−1) |
K
app (M−1) |
|
1
|
7.69 × 104 |
8.28 × 104 |
4.14 × 106 |
|
2
|
6.63 × 104 |
8.11 × 104 |
4.05 × 106 |
|
3
|
1.63 × 105 |
8.37 × 104 |
4.18 × 106 |
|
4
|
5.62 × 104 |
7.86 × 104 |
3.93 × 106 |
|
5
|
1.67 × 104 |
7.73 × 104 |
3.86 × 106 |
2.3.2 Ethidium bromide displacement studies.
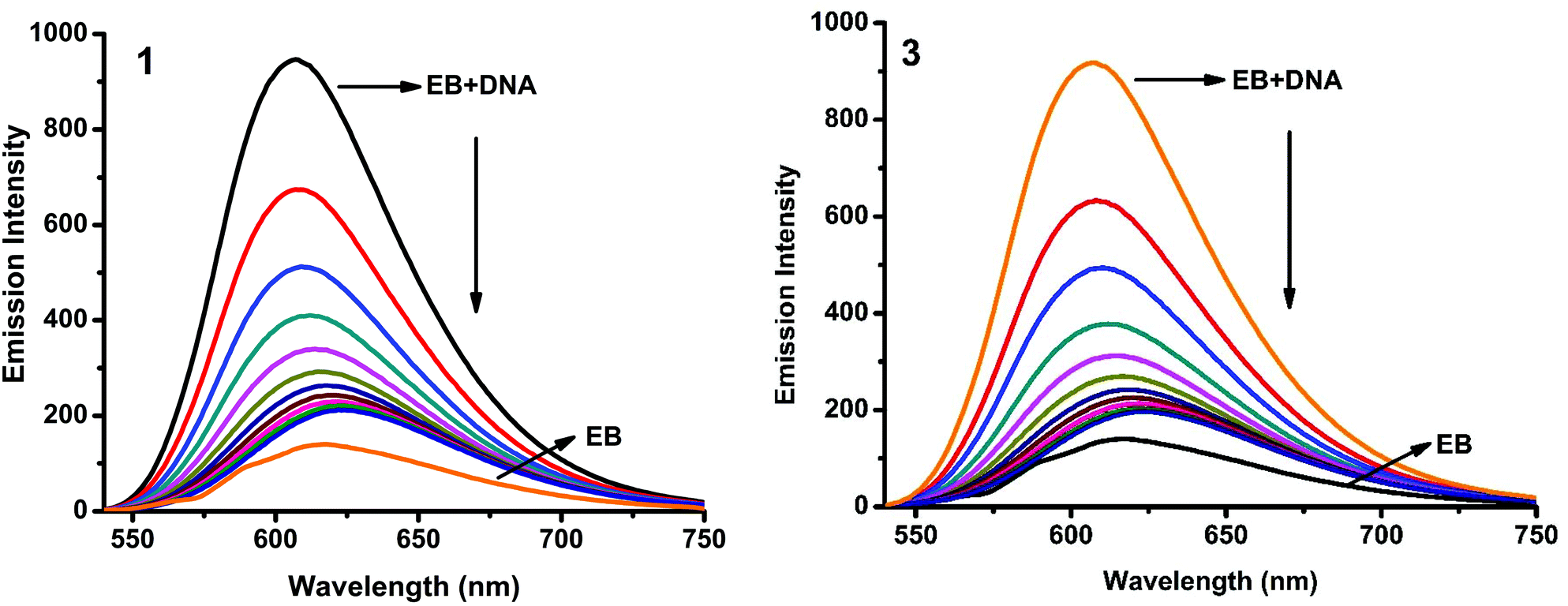
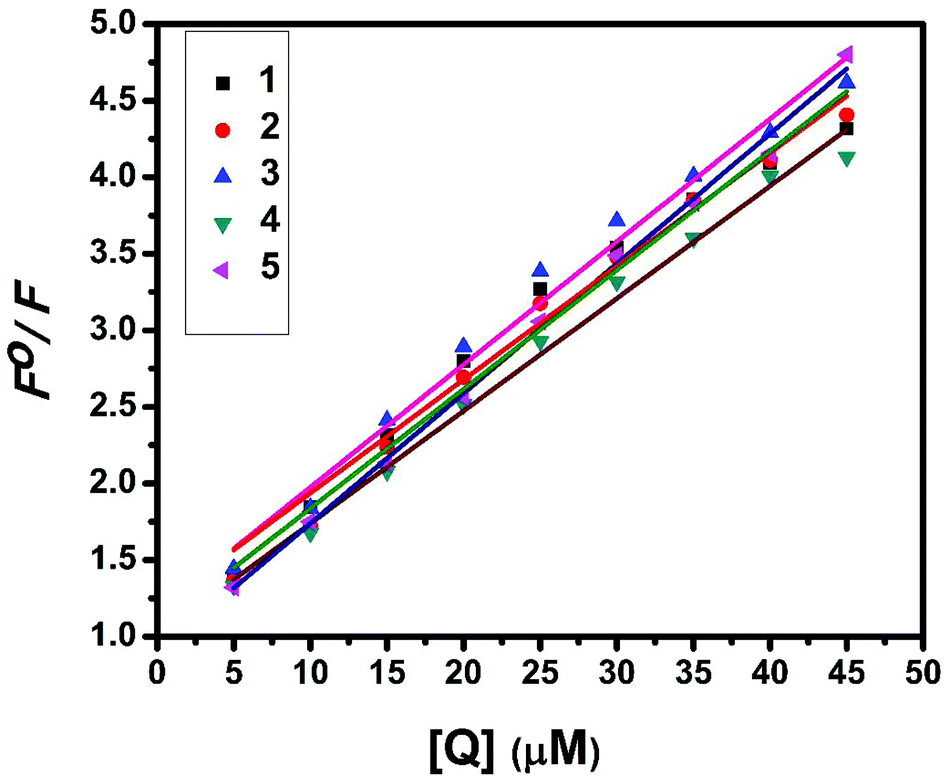
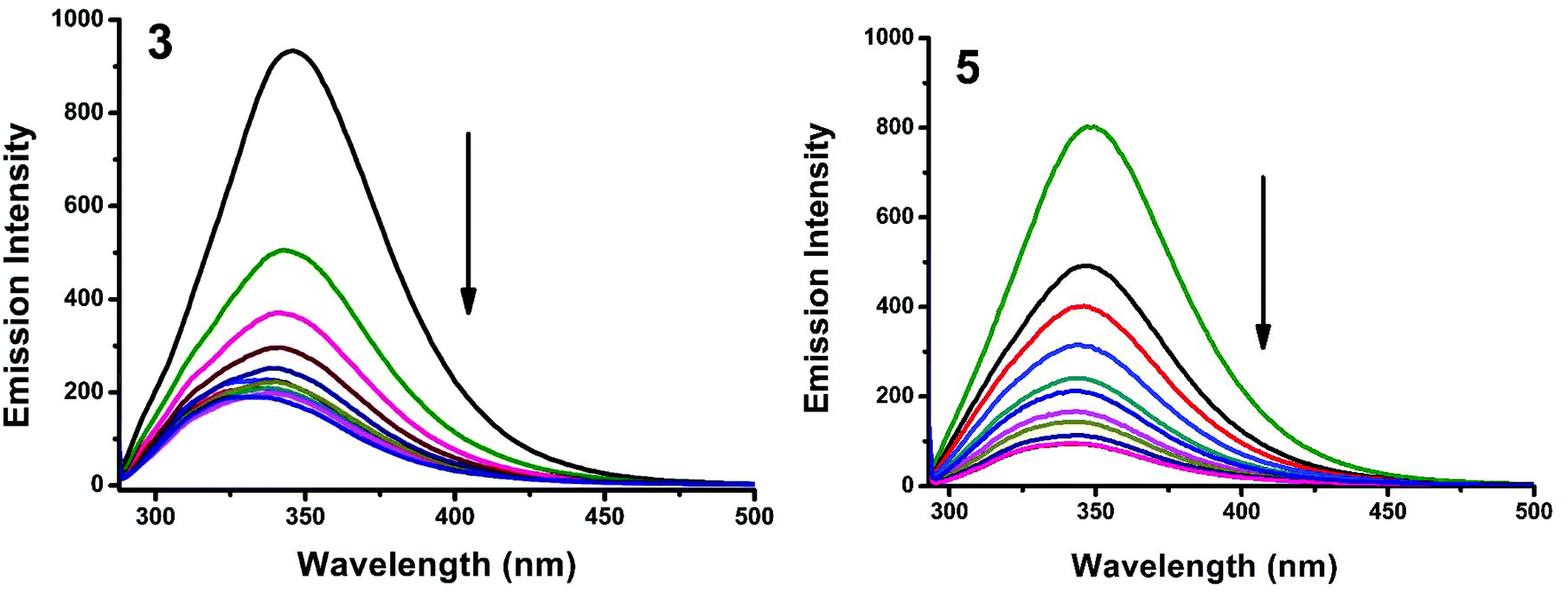
Fluorescence properties have not been observed for the complexes at room temperature in solution or in the presence of CT DNA. So the binding of the complexes with DNA could not be directly predicted through the emission spectra. Hence, a competitive binding study was done to understand the mode of DNA interaction with the complexes.41–43 Ethidium bromide (EB) is one of the most sensitive fluorescent probes that can bind with DNA.44 The fluorescence of EB increases after intercalating into DNA. If the metal complex intercalates into DNA, it leads to a decrease in the binding sites of DNA available for EB, resulting in a decrease in the fluorescence intensity of the CT DNA–EB system.45 The extent of decrease in the fluorescence intensity (quenching) of CT DNA–EB reflects the extent of interaction of the complex with CT DNA. On adding Cu(II) complexes (0–50 μM) to CT DNA–EB, the quenching in the emission of DNA bound EB takes place (Fig. 10 and S2†). Fluorescence quenching is explained by the Stern–Volmer equation46F°/F = 1 + Kq[Q] where F° and F are the fluorescence intensities in the absence and presence of the complex respectively, Kq is a linear Stern–Volmer quenching constant, and [Q] is the concentration of the complex. The slope of the plot of F°/F versus [Q] gave Kq (Fig. 12). The apparent DNA binding constant (Kapp) values were calculated by using the equation KEB[EB] = Kapp [complex] where [complex] is the complex concentration at 50% reduction in the fluorescence intensity of EB, KEB = 1.0 × 107 M−1 and [EB] = 5 μM. Kq and Kapp values are listed in Table 6.
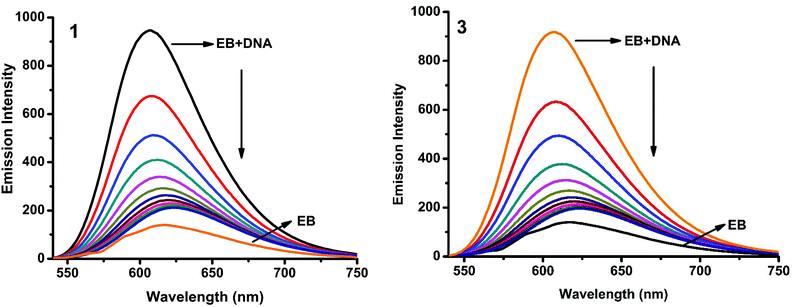 |
| | Fig. 10 Fluorescence quenching curves of EB bound to DNA in the presence of 1 and 3. [DNA] = 5 μM, [EB] = 5 μM and [complex] = 0–50 μM. | |
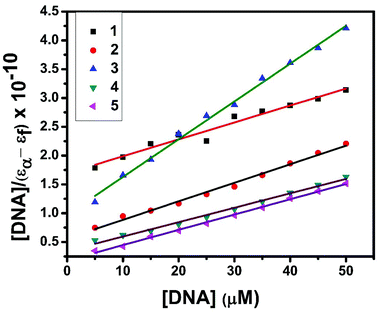 |
| | Fig. 11 Plot of [DNA]/(εa − εf) versus [DNA] for the titration of the complexes with CT DNA. | |
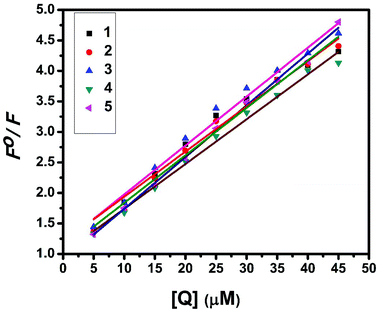 |
| | Fig. 12 Stern–Volmer plot of fluorescence titrations of the complexes with CT DNA. | |
2.4 DNA cleavage
To explore the DNA cleavage ability of complexes 1–5, supercoiled (SC) pBR 322 DNA (40 μM in base pairs) was incubated at 37 °C with the complexes (100 μM) in a 5% DMF/5 mM Tris-HCl/50 mM NaCl buffer at pH 7.2 for 4 h in the absence of an external agent. Complexes 3–5 cleave SC (Form I) DNA into nicked circular (NC) (Form II) DNA [Fig. 13(A)], and the DNA cleavage efficiency follows the order 5 (73.7%) > 3 (50.8%) > 4 (38.2%). Complexes 1 and 2 did not show any appreciable cleavage at the concentration of 100 μM. With increasing the concentration of complexes 1 and 2 (200 μM), DNA cleavage occurs with the percentage of 31.5 and 31.9 respectively [Fig. 13(B)]. The study reveals that 5 cleaves DNA more efficiently than the other complexes, because of the strong partial intercalation of the extended aromatic ring of the naphthyl group.47
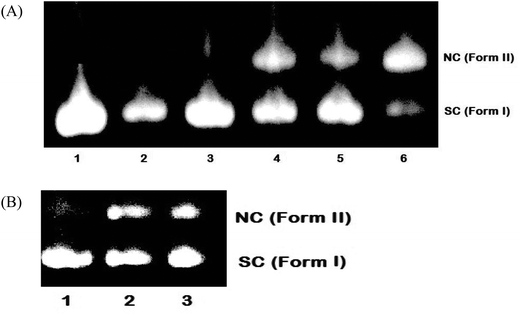 |
| | Fig. 13 Cleavage of supercoiled pBR 322 DNA (40 μM) by complexes 1–5 in a buffer containing 5% DMF: 5 mM Tris-HCl/50 mM NaCl at pH = 7.2 and 37 °C with an incubation time of 4 h. (A) Lane 1, DNA control; lane 2, DNA + 1 (100 μM); lane 3, DNA + 2 (100 μM); lane 4, DNA + 3 (100 μM); lane 5, DNA + 4 (100 μM); lane 6, DNA + 5 (100 μM). (B) Lane 1, DNA control; lane 2, DNA + 1 (200 μM); lane 3, DNA + 2 (200 μM). Forms SC and NC are supercoiled and nicked circular DNA, respectively. | |
2.5 Molecular docking study
2.5.1 Molecular docking with DNA.
Molecular docking is an important in silico computational tool for the rational design of new chemotherapeutic drugs, which predicts non-covalent interactions between the drug molecules and the nucleic acids of DNA. Conformation of docked complexes was analyzed in terms of energy, hydrogen bonding, and hydrophobic interaction between complexes and DNA. From the docking scores, the free energy of binding (FEB) of the complexes was calculated and details are shown in Table 7.
Table 7 Molecular docking parameters of complexes 1–5 with B-DNA (PDB ID: 1BNA) dodecamer d(CGCGAATTCGCG)2
| Complex |
Final intermolecular energy, kcal mol−1 |
Final total internal energy (2), kcal mol−1 |
Torsional free energy (3), kcal mol−1 |
Unbound system's energy [= (2)] (4), kcal mol−1 |
Estimated free energy of binding [(1) + (2) + (3) − (4)], kcal mol−1 |
| vdW + Hbond + desolv. energy |
Electrostatic energy |
Total (1) |
|
1
|
−10.24 |
−1.38 |
−11.62 |
−4.24 |
+2.74 |
−4.24 |
−8.88 |
|
2
|
−9.23 |
−0.02 |
−9.25 |
−6.33 |
+2.47 |
−6.33 |
−6.78 |
|
3
|
−9.55 |
−0.00 |
−9.56 |
−6.74 |
+3.84 |
−6.74 |
−5.71 |
|
4
|
−8.90 |
−0.16 |
−9.05 |
−6.13 |
+2.74 |
−6.13 |
−6.31 |
|
5
|
−9.99 |
−0.02 |
−9.96 |
−7.67 |
+2.74 |
−7.67 |
−7.22 |
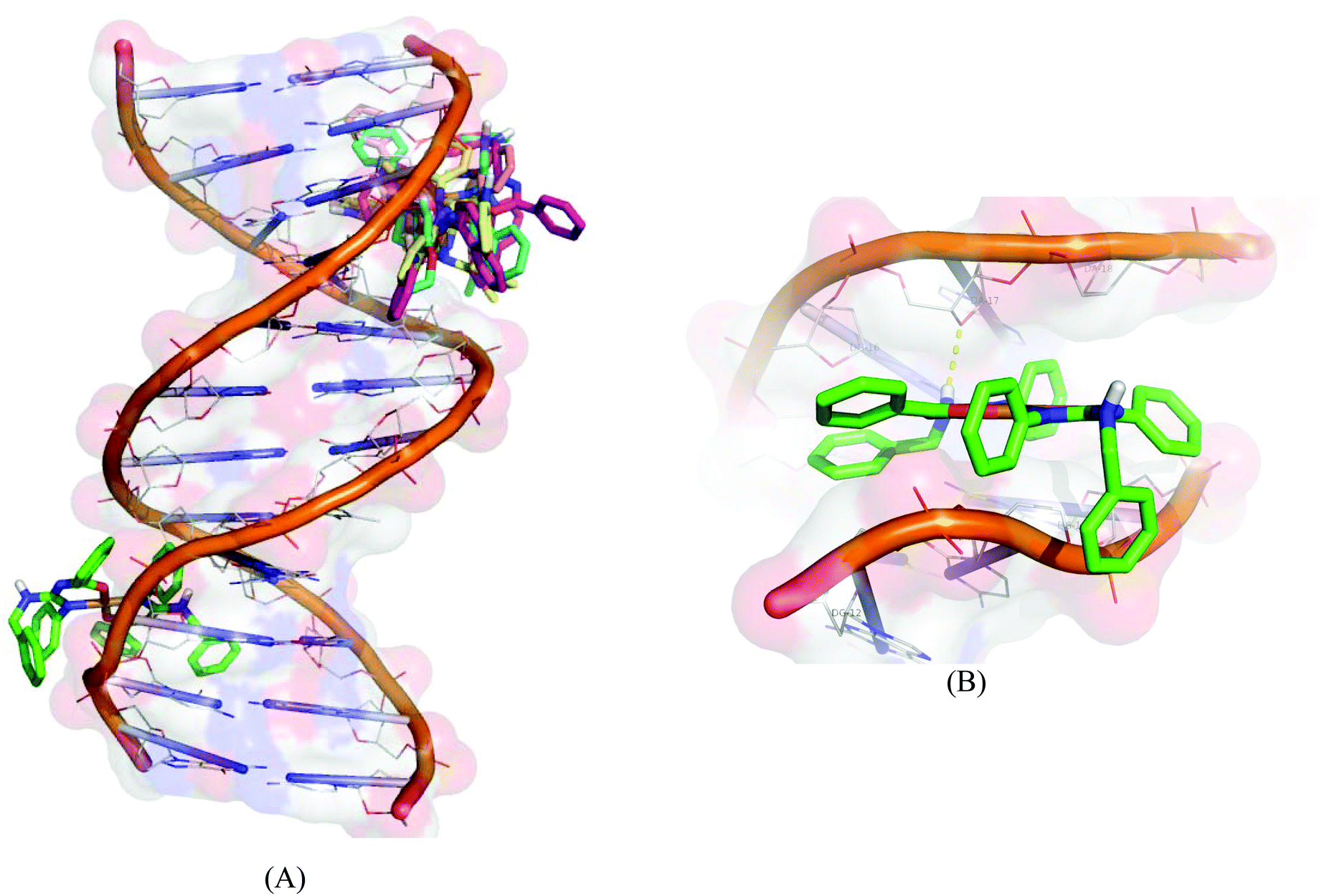
Molecular docking experiments reveal that the docked complexes fit into the DNA comfortably involving van der Waals interaction, hydrophobic and hydrogen bonding contacts with DNA functional groups without disrupting the double helical structure of DNA, which resulted in the binding energy value between −5.71 and −8.88 kcal mol−1. Complex 1 shows a higher binding energy of −8.88 kcal mol−1. Fig. 14 shows the docked models.
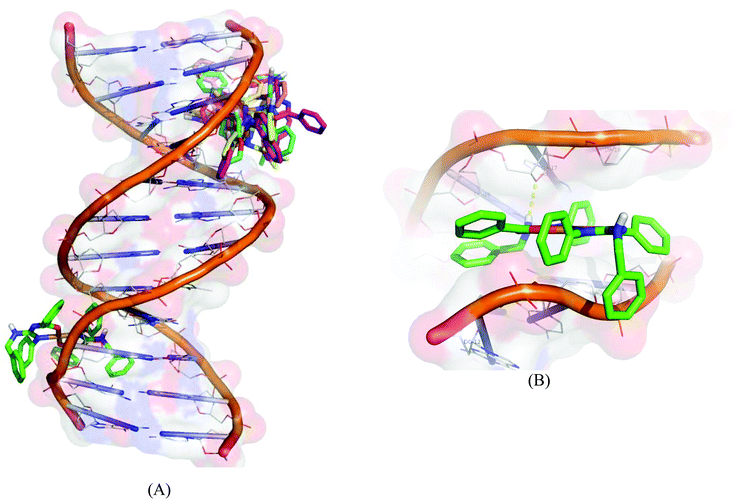 |
| | Fig. 14 Molecular docked model of complexes (1–5) (A) and complex 1 (B) with the DNA dodecamer duplex of sequence d(CGCGAATTCGCG)2 (PDB ID: 1BNA). | |
2.5.2 Molecular docking with human DNA topoisomerase I.
All the copper(II) complexes were subjected to molecular docking with human DNA topoisomerase I using the AutoDock Tools (ADT) version 1.5.6 and AutoDock version 4.2.5.1. The X-ray crystallographic structure of the human DNA topoisomerase I complex (PDB ID: 1SC7) was retrieved from the Protein Data Bank, in which Topo I is bound to the oligonucleotide sequence 50-AAAAAGACTTsX-GAAAATTTTT-30 where ‘s’ is 50-bridging phosphorothiolate of the cleaved strand and ‘X’ represents any of the four bases A, G, C or T. The SH of G11 on the scissile strand was changed to OH and the phosphoester bond of G12 in 1SC7 was rebuilt.48
Docked ligand conformation was analyzed in terms of energy, hydrogen bonding, and hydrophobic interaction between the ligand and DNA topoisomerase I. The free energy of binding (FEB) of the compound was calculated from docking scores and details are given in Table 8. The molecular docking results revealed that all the copper(II) complexes approach towards the DNA cleavage site in the DNA-topoisomerase I forming a stable complex through non-covalent interactions like hydrogen bonding, hydrophobic and van der Waals interactions, resulting in the binding energy value of −10.42 to −12.05 kcal mol−1, subsequently leading to the inhibitory effect on DNA topoisomerase I.
Table 8 Molecular docking parameters of complexes (1–5) with human-DNA-Topo-I complex (PDB ID: 1SC7)
| Complex |
Final intermolecular energy, kcal mol−1 |
Final total internal energy (2), kcal mol−1 |
Torsional free energy (3), kcal mol−1 |
Unbound system's energy [= (2)] (4), kcal mol−1 |
Estimated free energy of binding [(1) + (2) + (3) − (4)], kcal mol−1 |
| vdW + Hbond + desolv. energy |
Electrostatic energy |
Total (1) |
|
1
|
−13.41 |
−0.10 |
−13.51 |
−3.83 |
+2.74 |
−3.83 |
−10.77 |
|
2
|
−12.93 |
+0.04 |
−12.89 |
−5.81 |
+2.47 |
−5.81 |
−10.42 |
|
3
|
−14.50 |
−0.03 |
−14.53 |
−5.79 |
+3.84 |
−5.79 |
−10.69 |
|
4
|
−13.49 |
+0.01 |
−13.48 |
−5.85 |
+2.74 |
−5.85 |
−10.74 |
|
5
|
−14.76 |
−0.04 |
−14.80 |
−5.41 |
+2.74 |
−5.41 |
−12.05 |
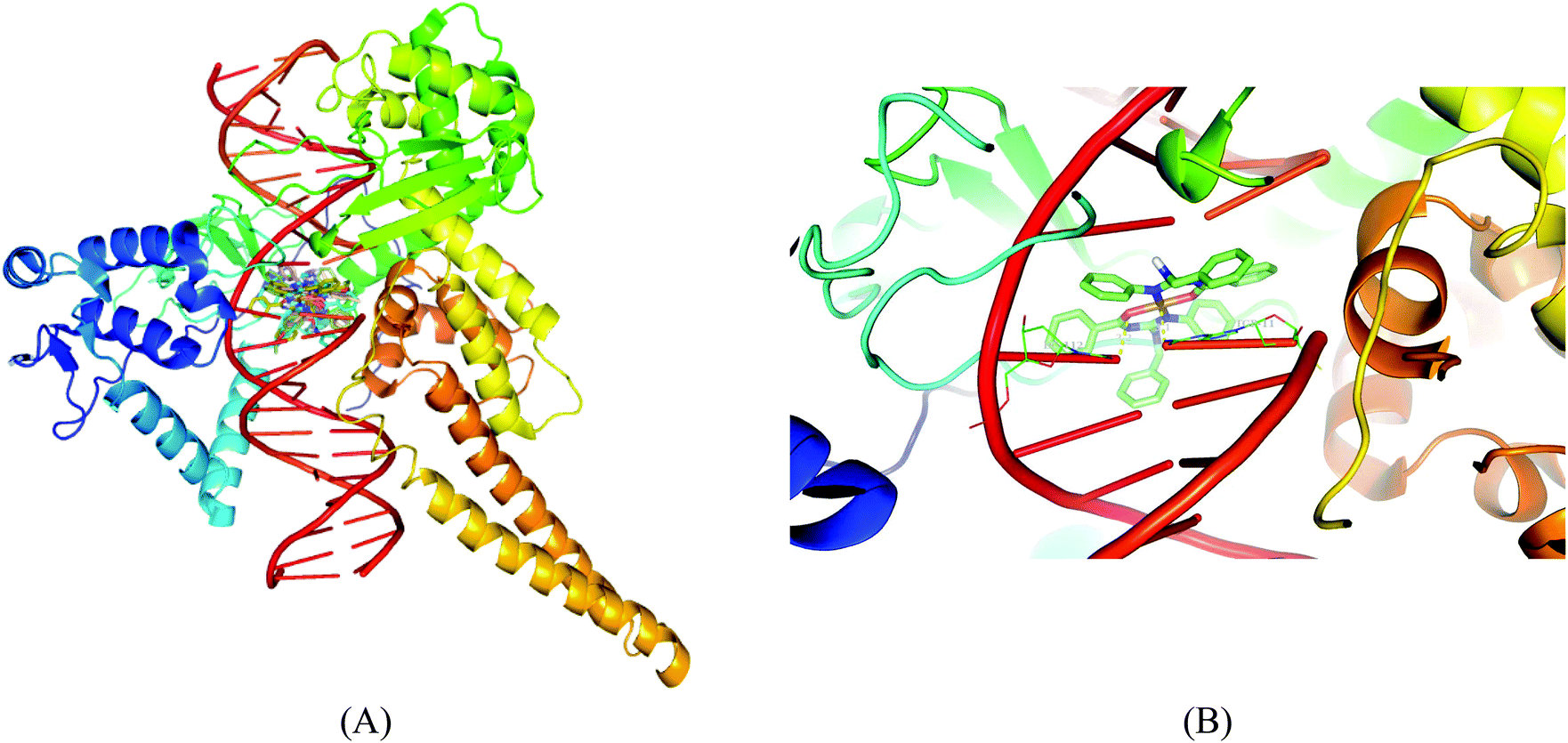
Molecular docking results of DNA-topoisomerase I with the copper complexes show that 5 binds efficiently with the DNA-topoisomerase I receptor and exhibits very high free energy of binding (−12.05 kcal mol−1). The high binding energy of 5 may be due to the presence of the naphthyl moiety. The order of binding affinity of the complexes with the DNA topoisomerase I receptor is 5 > 1 > 4 > 3 > 2. Docking studies reveal that the benzyl N–H and nitrogen attached to the aromatic group exhibit polar interactions with CO of DT-10 residue, and another benzyl N–H and imine nitrogen exhibit polar interactions with CO of TGP-11 residue. Besides these polar interactions, a π–π interaction has been observed between the aromatic ring in the complex and the ring of TGP-11 residue. Interaction of the copper(II) complexes with the DNA topoisomerase I receptor is shown in Fig. 15.
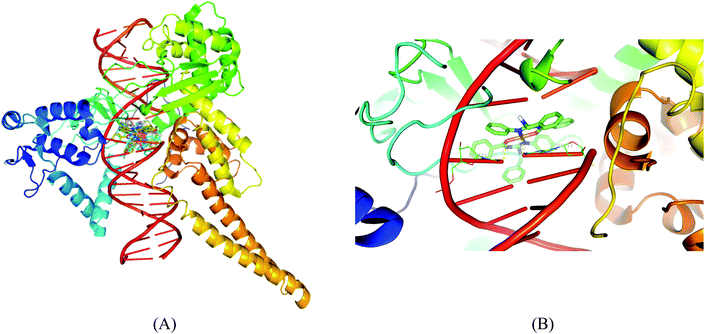 |
| | Fig. 15 Molecular docked model of complexes (1–5) (A) and complex 1 (B) in the cleavage site of human DNA topoisomerase I (PDB ID: 1SC7). | |
Molecular docking studies of the copper(II) complexes with the DNA topoisomerase I receptor revealed that all the docked complexes bind more efficiently suggesting that they can be a potential scaffold to be used for therapeutic purpose. In addition to this, DNA intercalating forces with the active site residues of the protein or to the base pairs of DNA play a vital role in the inhibition of the targeting receptor.49 Interestingly, the complexes occupy the topoisomerase binding site which may suppress the association of topoisomerase with DNA, thus forming a more stable complex with DNA, which results in the topoisomerase inhibition activity and can be the promising DNA targeting anticancer drugs.
2.6 Protein binding studies
2.6.1 Absorbance and fluorescence studies.
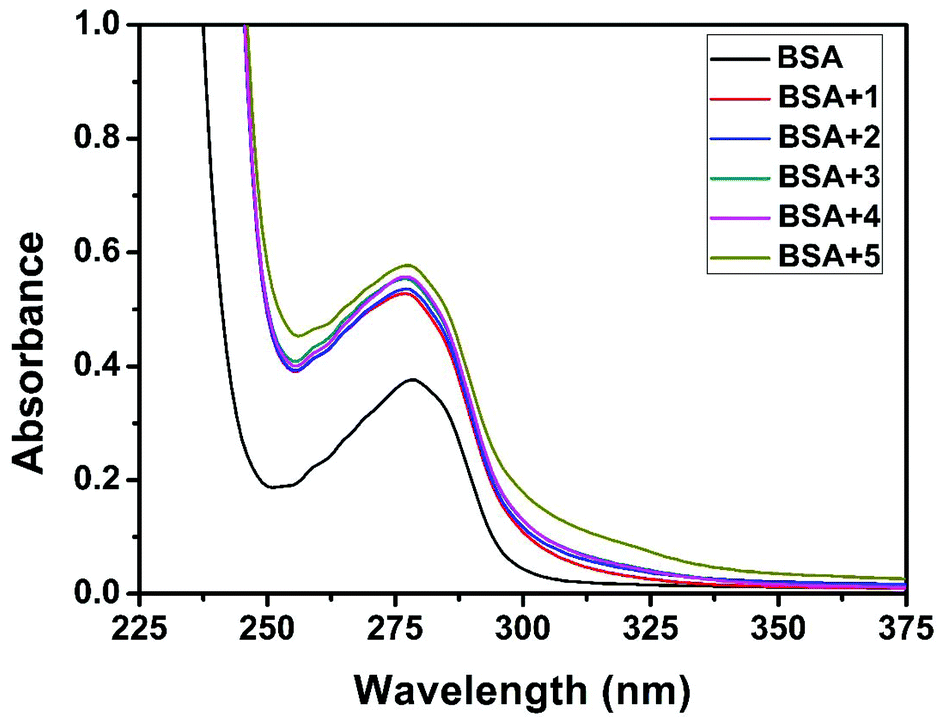
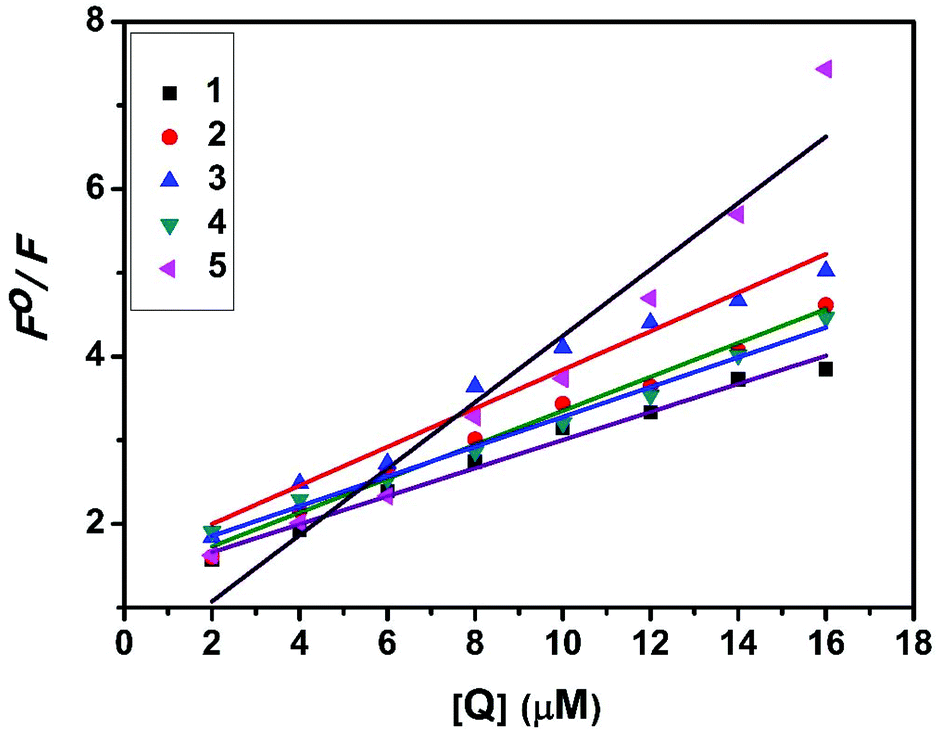
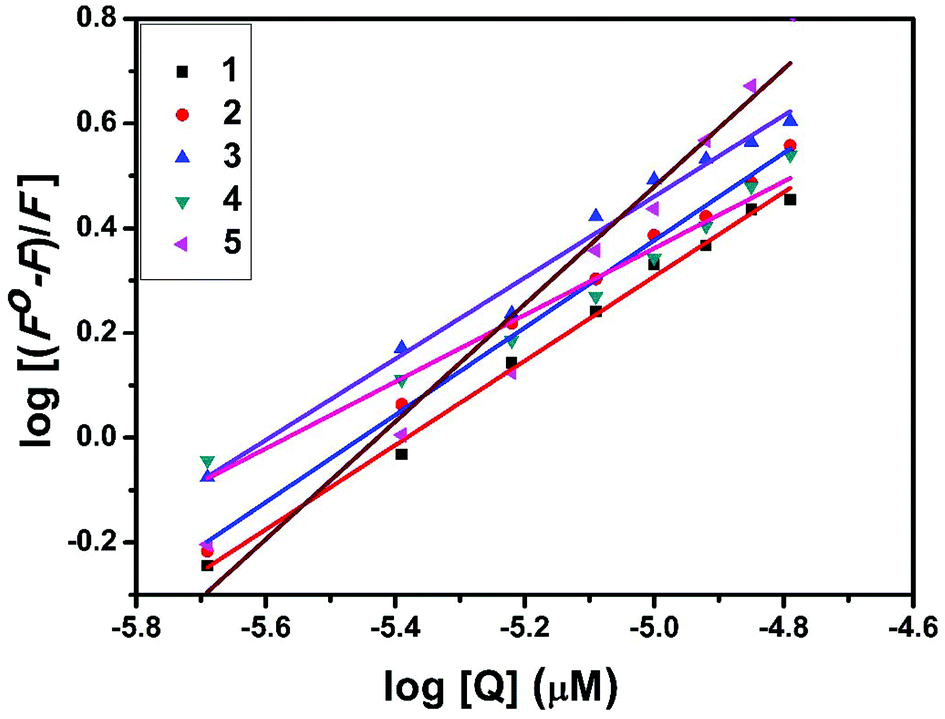
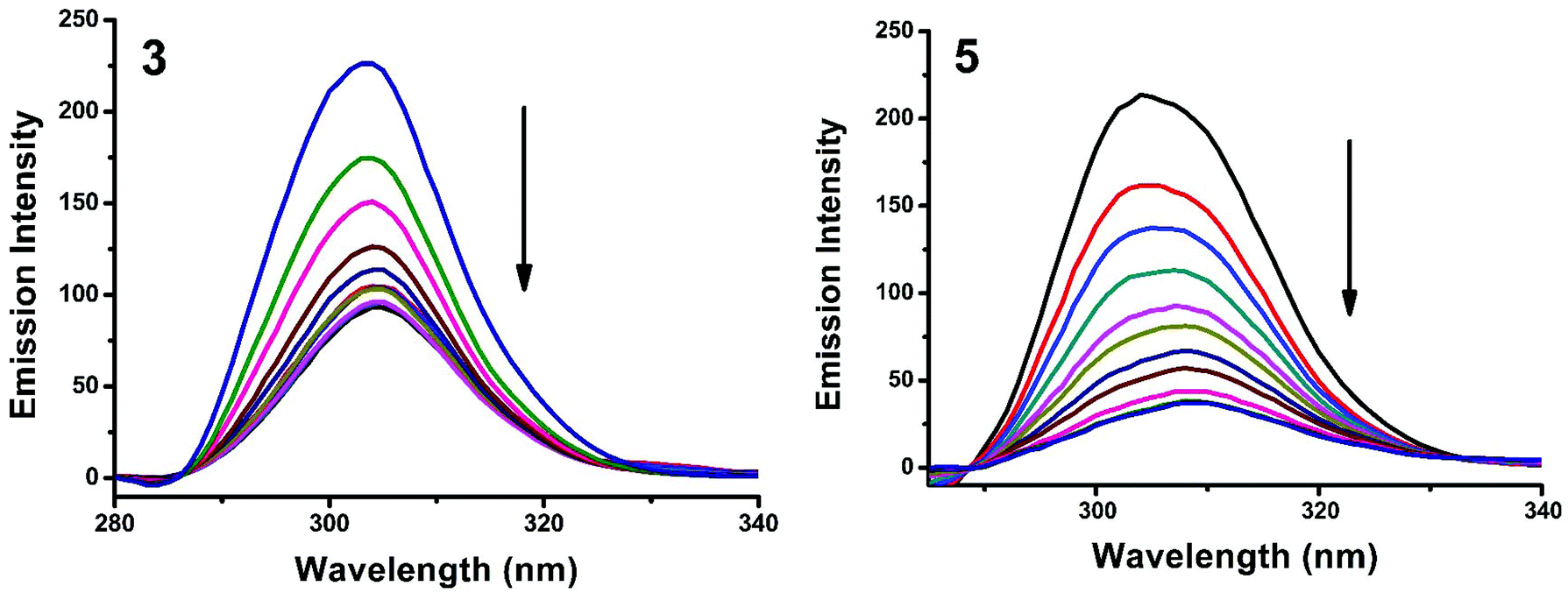
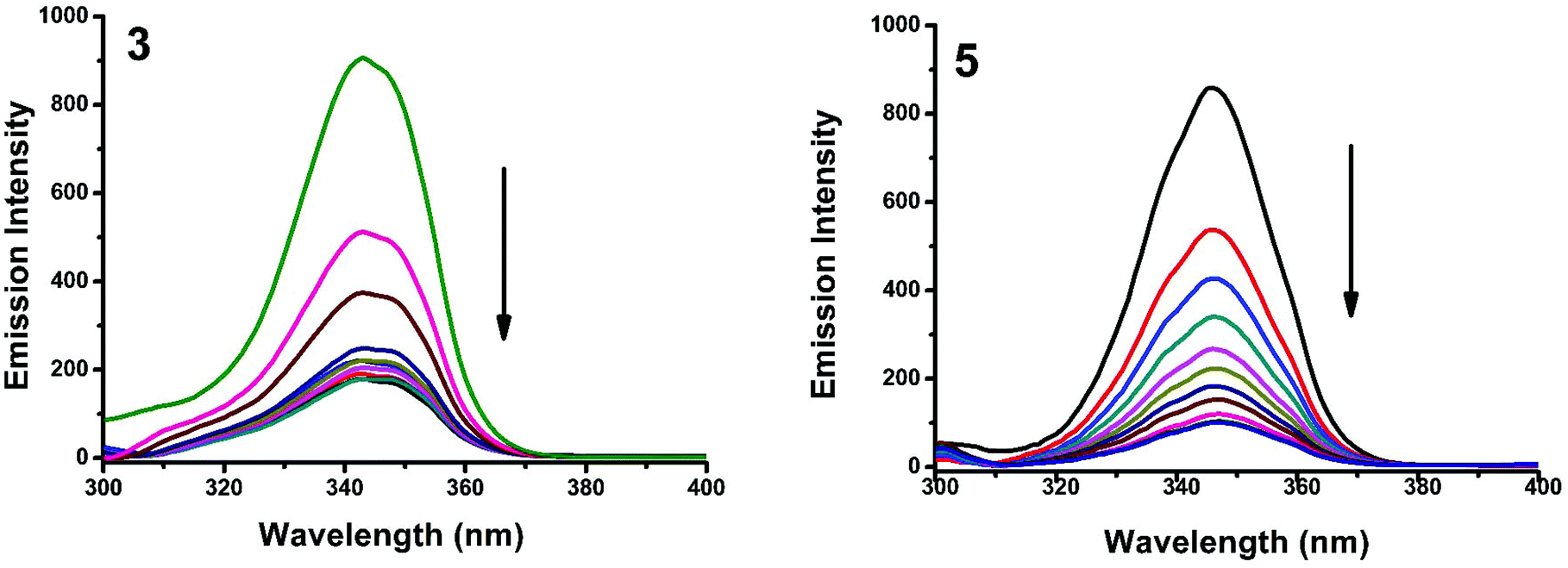
The interaction of complexes with BSA was investigated using fluorescence studies. BSA (1 μM) was titrated with various concentrations of the complexes (0–20 μM). Fluorescence spectra were recorded in the range of 290–500 nm upon excitation at 280 nm. The changes observed in the fluorescence emission spectra of BSA on the addition of increasing concentrations of the copper(II) complexes are shown in Fig. 16 and S3.† On the addition of complexes 1–5 to BSA, there is a significant decrease in the fluorescence intensity of BSA at 346 nm up to 77.19, 83.22, 81.78, 80.08 and 88.41% with a blue shift of 11, 11, 14, 8 and 8 nm respectively. The decrease in fluorescence intensity with the blue shift shows the interaction between the complexes and BSA.50,51 The fluorescence quenching is described by the Stern–Volmer relationship F°/F = 1 + Kq[Q] where F° and F demonstrate the fluorescence intensities in the absence and presence of a quencher, respectively, Kq is a linear Stern–Volmer quenching constant, and [Q] is the quencher concentration. The quenching constant (Kq) can be calculated from the plot of F°/F versus [Q] (Fig. 18). When small molecules bind independently to a set of equivalent sites, on a macromolecule, the equilibrium between free and bound molecules is represented by the Scatchard equation52,53 log[(F° − F)/F] = logKb + nlog[Q] where Kb is the binding constant of the complex with BSA and n is the number of binding sites. From the plot of log[(F° − F)/F] versus log[Q] (Fig. 19), the number of binding sites (n) and the binding constant (Kb) values have been obtained. Kq, Kb and n values for the interaction of the copper(II) complexes with BSA are provided in Table 9. The calculated value of n is around 0.7–1 for the complexes, proving the existence of a single binding site in BSA for all of the complexes. From the values of Kq and Kb, it is proved that complex 5 interacts with BSA more strongly than the rest of the complexes. Surprisingly the complex which interacts weakly with CT DNA binds strongly with BSA.54
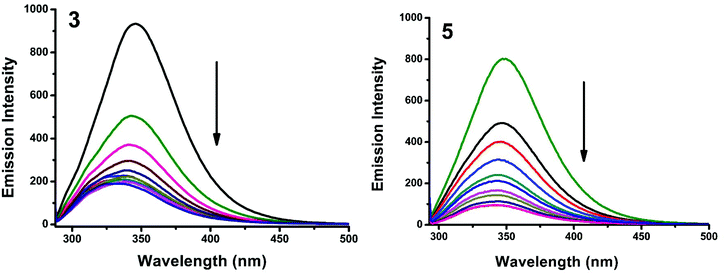 |
| | Fig. 16 Fluorescence quenching curves of BSA in the absence and presence of 3 and 5. [BSA] = 1 μM and [complex] = 0–20 μM. | |
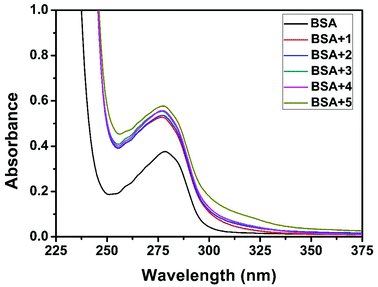 |
| | Fig. 17 The absorption spectra of BSA (10 μM) and BSA with 1–5 (4 μM). | |
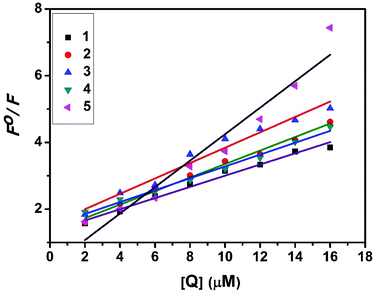 |
| | Fig. 18 Stern–Volmer plot of the fluorescence titrations of the complexes with BSA. | |
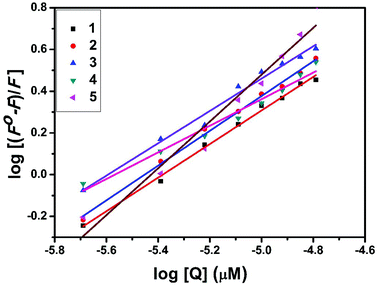 |
| | Fig. 19 Scatchard plot of the fluorescence titrations of the complexes with BSA. | |
Table 9 Protein binding constant (Kb), quenching constant (Kq) and number of binding sites (n) values
| Complex |
K
b (M−1) |
K
q (M−1) |
n
|
|
1
|
7.09 × 103 |
1.52 × 105 |
0.6942 |
|
2
|
5.42 × 104 |
2.25 × 105 |
0.8693 |
|
3
|
2.15 × 104 |
2.30 × 105 |
0.7747 |
|
4
|
1.60 × 104 |
1.94 × 105 |
0.7809 |
|
5
|
2.49 × 106 |
4.33 × 105 |
1.1799 |
Quenching usually occurs either by dynamic or static mode. Dynamic quenching is a process in which the fluorophore and the quencher come into contact during the transient existence of the excited state. On the other hand, static quenching refers to the formation of a fluorophore–quencher complex in the ground state.55 UV-Visible absorption spectroscopy is the tool to determine the type of quenching involved. Addition of the complexes to BSA leads to an increase in BSA absorption intensity without affecting the position of the absorption band (Fig. 17). It showed the existence of static interactions between BSA and the complexes.
2.6.2 Characteristics of synchronous fluorescence spectra.
Synchronous fluorescence spectroscopy can give valuable information on the microenvironment near different fluorophores, and the difference between the emission and excitation wavelengths (Δλ) reflects the spectra of different fluorophores.56 Tyrosine, tryptophan and phenylalanine residues are responsible for the fluorescence properties of BSA. In this experiment, synchronous fluorescence spectra of BSA in the absence and presence of increasing concentrations (2–20 μM) of 1–5 were obtained at different Δλ. At Δλ = 15 nm, the synchronous spectrum of BSA is characteristic of tyrosine while the corresponding spectrum at Δλ = 60 nm is characteristic of tryptophan.56–59 On the addition of the complexes, the fluorescence intensity of tyrosine residue at 300 nm decreased in the magnitude of 57.6, 68.1, 66.8, 59.7 and 82.6% with 2, 3, 1, 5, 5 nm red shifts for complexes 1–5 respectively (Fig. 20 and S4†). Similarly, there was also a decrease in the intensity of tryptophan residue at 340 nm. The magnitude of decrease was 77.8, 83.5, 81.19, 80.9 and 88.1% with 3, 4, 1, 2, 3 nm red shifts for complexes 1–5 respectively (Fig. 21 and S5†). The synchronous fluorescence spectra clearly suggested that the fluorescence intensity of both tryptophan and tyrosine was affected with increasing concentration of the complexes. The results indicate that the interaction of complexes with BSA affects the conformation of both tryptophan and tyrosine micro-regions.
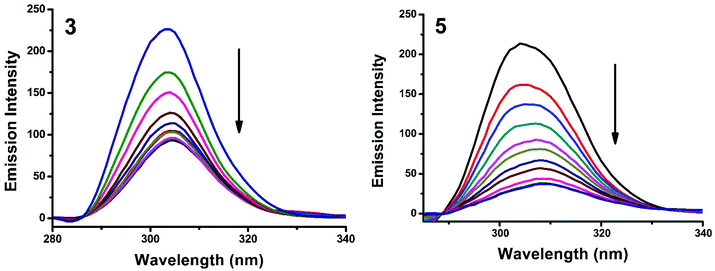 |
| | Fig. 20 Synchronous spectra of BSA (1 μM) as a function of concentration of 3 and 5 (0–20 μM) with Δλ = 15 nm. | |
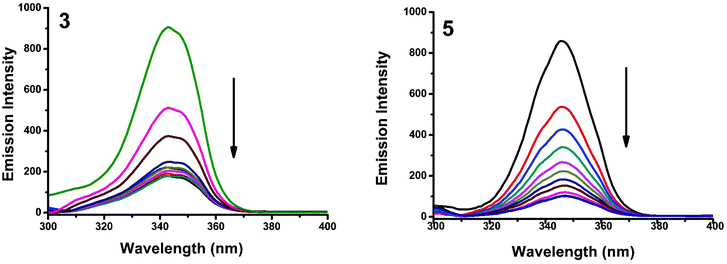 |
| | Fig. 21 Synchronous spectra of BSA (1 μM) as a function of concentration of 3 and 5 (0–20 μM) with Δλ = 60 nm. | |
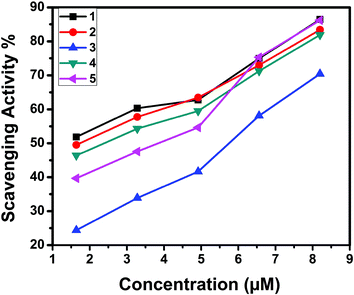
2.7 Superoxide scavenging study
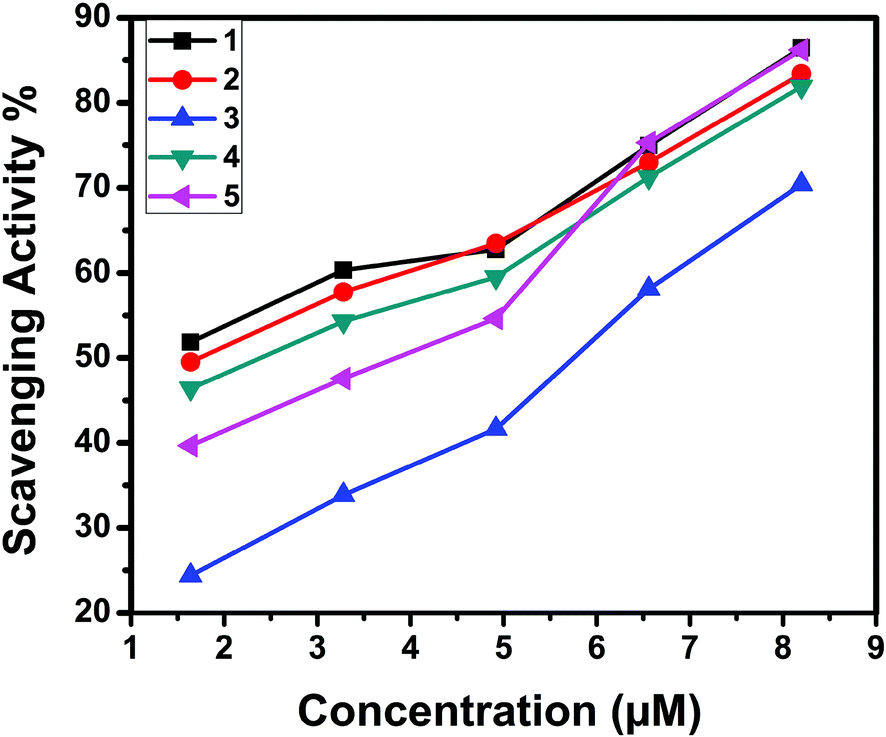
The superoxide dismutase (SOD) activity of the complexes was investigated by the NBT assay. Several complexes containing transition metals60 including copper are known to mimic SOD, although their structures are totally unrelated to the native enzyme.61 The percentage inhibition of formazan formation at various concentrations of complexes was measured by measuring the absorbance at 560 nm and plotted to a straight line. As the concentration of the tested complexes was increased, the slope (m) was decreased. Percentage inhibition of the reduction of NBT was plotted against the concentration of the complexes (Fig. 22). The complexes exhibited SOD-like activity at biological pH with the IC50 values in the range of 1.53–5.62 μM. The superoxide scavenging data are shown in Table 10. Even though the SOD-mimetic complexes obtained were not as effective as the enzyme (bovine erythrocyte SOD, IC50 = 2.1 × 10−7 M; horseradish SOD, IC50 = 7.0 × 10−8 M),62,63 their experimental IC50 values were below 20.0 × 10−6 M, so our complexes can be considered as active SOD mimetics.64,65
 |
| | Fig. 22 Inhibition of superoxide radicals by complexes 1–5. | |
Table 10 IC50 values (μM) calculated from SOD assays of Cu(II) complexes (1–5)
| Complex |
IC50 (μM) |
|
1
|
1.53 |
|
2
|
1.78 |
|
3
|
5.62 |
|
4
|
2.66 |
|
5
|
3.58 |
2.8 Cytotoxicity
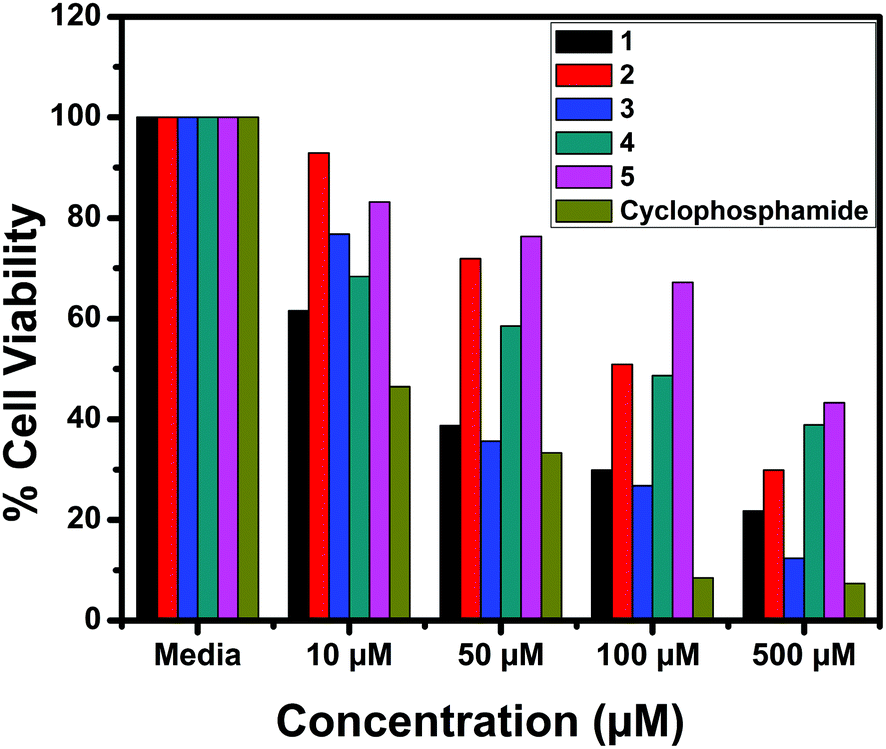
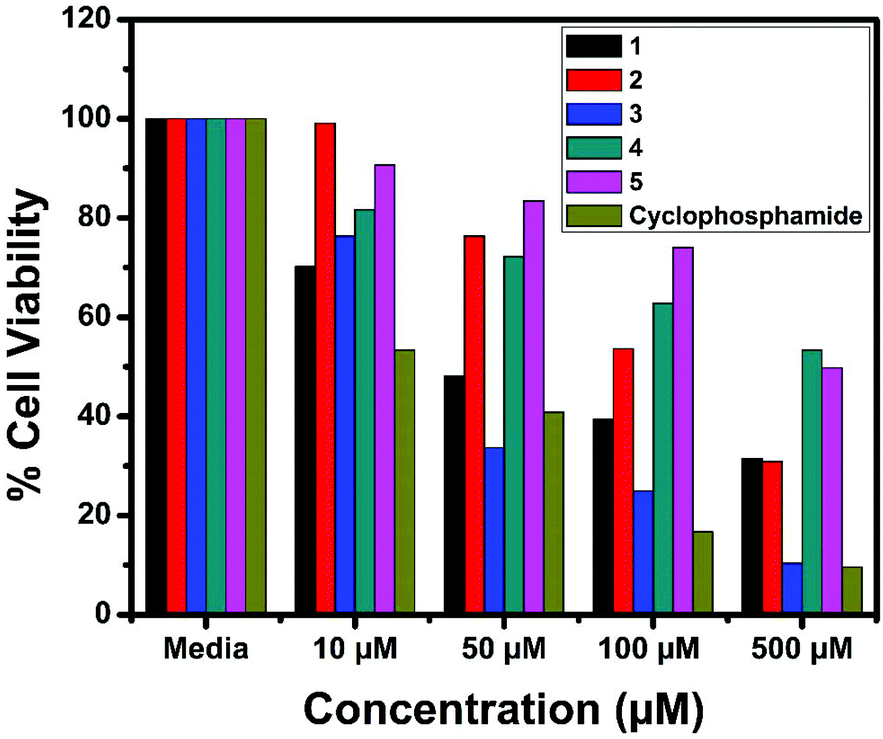
The in vitro cytotoxic activity of the complexes (1–5) was determined against human breast (MCF7), lung (A549) cancer and mouse embryonic fibroblast (NIH 3T3) cell lines using the MTT assay.66Fig. 23, 24 and S6† show the cytotoxicity of the complexes (1–5) after 24 h incubation on MCF7, A549 and NIH 3T3 cell lines respectively. Complexes were dissolved in DMSO and a blank sample containing the same volume of DMSO was taken as the control to identify the activity of the solvent in this experiment. Cyclophosphamide was used as a positive control to assess the cytotoxicity of the tested complexes. The results were analyzed by means of cell inhibition expressed as IC50 values and are shown in Table 11. The IC50 values show that complex 3 exhibited a higher inhibitory effect than the other complexes and the IC50 is very close to the value of cyclophosphamide. Complex 1 also shows lower IC50 values with a good cytotoxic effect. The rest of the complexes 2, 4 and 5 show only moderate cytotoxicity. Fortunately all the complexes were less toxic towards the normal cell as was evident from the higher IC50 values (above 550 μM). The cytotoxicity results are in good agreement with the DNA binding ability of the complexes.
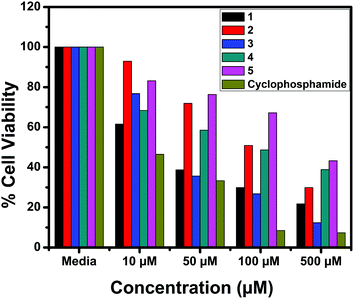 |
| | Fig. 23 Cytotoxicity of complexes 1–5 after 24 h incubation on MCF7 cell lines. | |
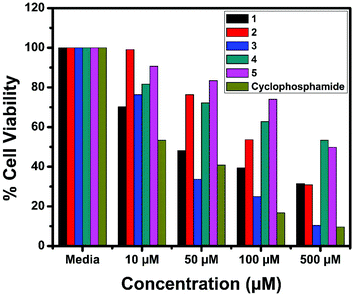 |
| | Fig. 24 Cytotoxicity of complexes 1–5 after 24 h incubation on A549 cell lines. | |
Table 11
In vitro cytotoxicity of the Cu(II) complexes in MCF7, A549 and NIH 3T3 cell lines
| Compound |
IC50 (μM) |
| MCF7 |
A549 |
NIH 3T3 |
|
1
|
42.71 |
52.98 |
900.34 |
|
2
|
107.02 |
122.2 |
723.40 |
|
3
|
37.1 |
45.71 |
600.30 |
|
4
|
204.52 |
256.32 |
572.81 |
|
5
|
198.6 |
232.6 |
670.22 |
| Cyclophosphamide |
11.89 |
41.81 |
— |
3. Experimental section
3.1 Materials and methods
Analytical grade reagents and chemicals (>95% purity) were purchased from Sigma Aldrich/Merck and used as received. Solvents were purified according to the standard procedures. The melting points were determined on a Lab India instrument and are uncorrected. The elemental analyses were performed using a Vario EL–III CHNS analyzer. FT-IR spectra were obtained as KBr pellets using a Nicolet-iS5 spectrophotometer. UV-Visible spectra were recorded using a Shimadzu-2600 spectrophotometer. Emission spectra were recorded on a Jasco V-630 spectrofluorometer using 5% DMF in buffer as the solvent. NMR spectra were recorded in CDCl3 by using TMS as an internal standard on a Bruker 500 MHz spectrometer. EPR spectra were recorded on a JEOL EPR spectrometer at liquid nitrogen temperature operating at X-band frequency (9.1 GHz).
3.2 Synthesis of N,N′,N′′-trisubstituted guanidines
The guanidine ligands were synthesized from N-benzoyl-N′-benzylthiourea by a guanylation method. The thiourea was mixed with the desired substituted amine in DMF in an equimolar ratio with two equivalents of triethylamine. The temperature was maintained below 5 °C using an ice bath and one equivalent of mercuric chloride was added to the reaction mixture with vigorous stirring. The ice bath was removed after 30 minutes, while the stirring continued overnight. The progress of the reaction was monitored using TLC until all the thiourea was consumed. 10 mL of chloroform was added to the reaction mixture and the suspension was filtered through a celite bed to remove the HgS residue. The solvent was evaporated under reduced pressure and the solid residue was dissolved in 10 mL of CH2Cl2, then washed with water and the organic phase was dried over anhydrous Na2SO4. The residue obtained after evaporation of the solvent was recrystallized from ethanol to obtain crystals of the title compounds.
3.2.1
N-Benzyl-N′-phenyl-N′′-benzoylguanidine (L1).
N-Benzoyl-N′-benzylthiourea (1.3517 g, 5 mmol), aniline (0.456 mL, 5 mmol), triethylamine (1 mL, 10 mmol) and HgCl2 (1.3576 g, 5 mmol) were used. Yield: 82%. Colourless solid. M.p.: 101 °C. UV–Vis (CHCl3): λmax, nm (ε, dm3 mol−1 cm−1) 245 (19100), 273 (26900). FT-IR (KBr, ν cm−1): 3414, 3174 (N–H), 1599 (CO), 1568 (CN). 1H NMR (500 MHz, CDCl3): δ, ppm 4.78 (s, 2H), 5.25 (s, 1H), 7.24–7.46 (m, 13H), 8.26–8.27 (d, J = 7 Hz, 2H), 12.19 (s, 1H). 13C NMR (125 MHz, CDCl3): δ, ppm 45.2 (aliphatic CH2), 125.5, 126.9, 127.5, 127.6, 127.8, 128.8, 129.1, 130.0, 131.2, 136.0, 138.5 (aromatic C), 158.7 (CN), 177.8 (CO). HRMS Calcd for C21H19N3O: 329.1528. Found: 329.1523.
3.2.2
N-Benzyl-N′-(4-methylphenyl)-N′′-benzoylguanidine (L2).
N-Benzoyl-N′-benzylthiourea (1.3517 g, 5 mmol), 4-methylaniline (0.5357 g, 5 mmol), triethylamine (1 mL, 10 mmol) and HgCl2 (1.3576 g, 5 mmol) were used. Yield: 81%. Colourless solid. M.p.: 94 °C. UV–Vis (CHCl3): λmax, nm (ε, dm3 mol−1 cm−1) 235 (13500), 272 (22600). FT-IR (KBr): ν, cm−1 3299, 3145 (N–H), 1594 (CO), 1571 (CN). 1H NMR (500 MHz, CDCl3) δ, ppm: 2.07 (s, 3H), 4.81–4.80 (d, J = 5.5 Hz, 2H), 5.22 (s, 1H), 7.17–7.50 (m, 12H), 8.29–8.30 (d, J = 7.5 Hz, 2H), 12.07 (s, 1H). 13C NMR (125 MHz, CDCl3): δ, ppm 20.9 (aliphatic CH3), 45.2 (aliphatic CH2), 125.7, 127.5, 127.8, 128.7, 129.1, 130.6, 131.1, 133.1, 137.0, 138.6 (aromatic C), 158.9 (CN), 177.7 (CO). HRMS Calcd for C22H21N3O: 343.1685. Found: 343.1685.
3.2.3
N-Benzyl-N′-(4-ethoxyphenyl)-N′′-benzoylguanidine (L3).
N-Benzoyl-N′-benzylthiourea (1.3517 g, 5 mmol), 4-ethoxyaniline (0.644 mL, 5 mmol), triethylamine (1 mL, 10 mmol) and HgCl2 (1.3576 g, 5 mmol) were used. Yield: 79%. Colourless solid. M.p.: 141 °C. UV–Vis (CHCl3): λmax, nm (ε, dm3 mol−1 cm−1) 240 (35400), 271 (39600). FT-IR (KBr): ν, cm−1 3419, 3192 (N–H), 1594 (CO), 1567 (CN). 1H NMR (500 MHz, CDCl3): δ, ppm 1.42–1.45 (t, J = 7 Hz, 3H), 4.02–4.06 (q, J = 7 Hz, 2H), 4.78–4.80 (d, J = 5.5 Hz, 2H), 5.10 (s, 1H), 6.92–6.94 (dd, J = 2 Hz, 2 Hz, 2H), 7.21–7.48 (m, 10 H), 8.28–8.30 (d, J = 7.5 Hz, 2H), 11.93 (s, 1H). 13C NMR (125 MHz, CDCl3): δ, ppm 14.7 (aliphatic CH3), 45.1 (aliphatic CH2), 63.7 (aliphatic CH2–O), 115.7, 127.5, 127.6, 127.8, 128.7, 129.1, 131.1, 138.6, 158.0 (aromatic C), 159.4 (CN), 177.7 (CO). HRMS Calcd for C23H23N3O2: 373.1790. Found: 373.1790.
3.2.4
N-Benzyl-N′-(2-methoxyphenyl)-N′′-benzoylguanidine (L4).
N-Benzoyl-N′-benzylthiourea (1.3517 g, 5 mmol), 2-methoxyaniline (0.564 mL, 5 mmol), triethylamine (1 mL, 10 mmol) and HgCl2 (1.3576 g, 5 mmol) were used. Yield: 84%. Colourless solid. M.p.: 134 °C. UV–Vis (CHCl3): λmax, nm (ε, dm3 mol−1 cm−1) 237 (7900), 272 (12500), 290 (10800). FT-IR (KBr): ν, cm−1 3285, 3165 (N–H), 1602 (CO), 1561 (CN). 1H NMR (500 MHz, CDCl3): δ, ppm 3.77 (s, 3H), 4.79 (s, 2H), 5.26 (s, 1H), 6.91–6.92 (d, J = 6 Hz, 1H), 6.95–6.98 (t, J = 7.5 Hz, 1H), 7.17–7.46 (m, 10H), 8.26–8.28 (d, J = 7.5 Hz, 2H), 11.96 (s, 1H). 13C NMR (125 MHz, CDCl3): δ, ppm 45.3 (aliphatic CH2), 55.7 (aliphatic CH3–O), 111.8, 121.0, 127.5, 127.8, 128.8, 129.2, 131.1, 138.7 (aromatic C), 158.7 (CN), 177.7 (CO). HRMS Calcd for C22H21N3O2: 359.1634. Found: 359.1639.
3.2.5
N-Benzyl-N′-(1-naphthyl)-N′′-benzoylguanidine (L5).
N-Benzoyl-N′-benzylthiourea (1.3517 g, 5 mmol), 1-naphthylamine (0.7159 g, 5 mmol), triethylamine (1 mL, 10 mmol) and HgCl2 (1.3576 g, 5 mmol) were used. Yield: 70%. Colourless solid. M.p.: 116 °C. UV–Vis (CHCl3): λmax, nm (ε, dm3 mol−1 cm−1) 240 (28700), 273 (27700), 290 (26400). FT-IR (KBr): ν, cm−1 3418, 3207 (N–H), 1591 (CO), 1567 (CN). 1H NMR (500 MHz, CDCl3): δ, ppm 4.79–4.80 (d, J = 5.5 Hz, 2H), 5.12 (s, 1H), 7.27–7.93 (m, 14 H), 8.15 (s, 1H), 8.36–8.37 (d, J = 7.5 Hz, 2H), 12.54 (s, 1H). 13C NMR (125 MHz, CDCl3): δ, ppm 45.1 (aliphatic CH2), 122.8, 124.1, 125.6, 127.0, 127.3, 127.4, 127.9, 128.2, 128.4, 128.6, 129.2, 129.7, 131.2, 131.9, 134.7, 138.5 (aromatic C), 159.6 (CN), 178.1 (CO); HRMS Calcd for C25H21N3O: 379.1685. Found: 379.1690.
3.3 Synthesis of copper(II) complexes (1–5)
A methanolic solution of copper acetate monohydrate was added into the methanolic solution of the appropriate guanidine ligand. The reaction mixture was stirred for 6 h at room temperature. The solid product formed was filtered and washed with methanol. Suitable crystals of complexes (1 and 2) were grown by the vapour diffusion method using CHCl3/n-hexane.
3.3.1 Bis(N-benzyl-N′-phenyl-N′′-benzoylguanidinato)copper(II) (1).
L1 (0.3294 g, 1 mmol) and Cu(CH3COO)2·H2O (0.0998 g, 0.5 mmol) were used. Yield: 79%. Blue solid. M.p.: 210 °C. Anal. Calcd for C42H36CuN6O2 (720.32): C, 70.03; H, 5.04; N, 11.67. Found: C, 70.27; H, 5.12; N, 11.71. UV-Vis (CHCl3): λmax, nm (ε, dm3 mol−1 cm−1) 219 (24100), 271 (45200), 388 (2400), 596 (72). FT-IR (KBr): ν, cm−1 3421 (N–H), 1589 (CO), 1552 (CN). EPR (LNT): ‘g’ values 2.21, 2.04.
3.3.2 Bis(N-benzyl-N′-(4-methylphenyl)-N′′-benzoylguanidinato)copper(II) (2).
L2 (0.3434 g, 1 mmol) and Cu(CH3COO)2·H2O (0.0998 g, 0.5 mmol) were used. Yield: 81%. Blue solid. M.p.: 211 °C. Anal. Calcd for C44H40CuN6O2 (748.37): C, 70.62; H, 5.39; N, 11.23. Found: C, 69.69; H, 5.16; N, 11.19. UV-Vis (CHCl3): λmax, nm (ε, dm3 mol−1 cm−1) 271 (46800), 389 (3000), 615 (92). FT-IR (KBr): ν, cm−1 3425 (N–H), 1590 (CO), 1553 (CN). EPR (LNT): ‘g’ values 2.16, 2.02.
3.3.3 Bis(N-benzyl-N′-(4-ethoxyphenyl)-N′′-benzoylguanidinato)copper(II) (3).
L3 (0.3734 g, 1 mmol) and Cu(CH3COO)2·H2O (0.0998 g, 0.5 mmol) were used. Yield: 83%. Green solid. M.p.: 194 °C. Anal. Calcd for C46H44CuN6O4 (808.27): C, 68.34; H, 5.49; N, 10.40. Found: C, 69.04; H, 5.61; N, 10.40. UV-Vis (CHCl3): λmax, nm (ε, dm3 mol−1 cm−1) 240 (65300), 271 (65800), 377 (3800), 601 (92). FT-IR (KBr): ν, cm−1 3427 (N–H), 1590 (CO), 1545 (CN). EPR (LNT): ‘g’ values 2.20, 2.01.
3.3.4 Bis(N-benzyl-N′-(2-methoxyphenyl)-N′′-benzoylguanidinato)copper(II) (4).
L4 (0.3594 g, 1 mmol) and Cu(CH3COO)2·H2O (0.0998 g, 0.5 mmol) were used. Yield: 79%. Light blue solid. M.p.: 178 °C. Anal. Calcd for C44H40CuN6O4 (780.37): C, 67.72; H, 5.17; N, 10.77. Found: C, 67.50; H, 5.08; N, 11.19. UV-Vis (CHCl3): λmax, nm (ε, dm3 mol−1 cm−1) 271 (46800), 389 (3000), 615 (92). FT-IR (KBr): ν, cm−1 3419 (N–H), 1589 (CO), 1552 (CN). EPR (LNT): ‘g’ values 2.2105, 2.024.
3.3.5 Bis(N-benzyl-N′-(1-naphthyl)-N′′-benzoylguanidinato)copper(II) (5).
L5 (0.3794 g, 1 mmol) and Cu(CH3COO)2·H2O (0.0998 g, 0.5 mmol) were used. Yield: 81%. Green solid. M.p.: 224 °C. Anal. Calcd for C50H40CuN6O2 (820.43): C, 73.20; H, 4.91; N, 10.24. Found: C, 72.94; H, 4.89; N, 10.17. UV-Vis (CHCl3): λmax, nm (ε, dm3 mol−1 cm−1) 240 (37000), 271 (37700), 592 (104). FT-IR (KBr): ν, cm−1 3429 (N–H), 1588 (CO), 1547 (CN). EPR (LNT): ‘g’ values 2.21, 2.02.
3.4 Single crystal X-ray diffraction studies
A Bruker APEX2 (Mo Kα) or Bruker GADDS (Cu Kα) X-ray (three-circle) diffractometer was employed for crystal screening, unit cell determination, and data collection. Integrated intensity information for each reflection was obtained by reduction of the data frames with the program APEX2.67 The integration method employed a three dimensional profiling algorithm and all data were corrected for Lorentz and polarization factors, as well as for crystal decay effects. Finally, the data were merged and scaled to produce a suitable data set. The absorption correction program SADABS68 was employed to correct the data for absorption effects. Systematic reflection conditions and statistical tests of the data suggested the space group (Tables 2 and 3). Solutions were obtained readily using SHELXTL (XS).69 Hydrogen atoms were placed in idealized positions and were set riding on the respective parent atoms. All non-hydrogen atoms were refined with anisotropic thermal parameters. The structures were refined (weighted least squares refinement on F2) to convergence.69,70 Olex2 was employed for the final data presentation and structure plots.70
3.5 DNA binding studies
The DNA binding experiments were performed in Tris-HCl/NaCl buffer (50 mM Tris-HCl/1 mM NaCl buffer, pH 7.2) using DMF (10%) solutions of the complexes. The concentration of CT DNA was determined from the absorption intensity at 260 nm with an ε value71 of 6600 M−1 cm−1. Absorption titration experiments were performed using different concentrations of CT DNA, while keeping the complex concentration constant. Samples were equilibrated before recording each spectrum. The concentration of the complex used was 20 μM; CT DNA of varying concentrations (5–40 μM) was added each time and the significant absorbance change was noted.
The competitive binding of each complex with EB has been investigated by the fluorescence spectroscopic technique in order to examine whether the complex can displace EB from its CT DNA–EB complex. Ethidium bromide solution was prepared using Tris-HCl/NaCl buffer (pH 7.2). The test solution was added in aliquots of 5 μM concentration to DNA–EB and the change in fluorescence intensity at 596 nm (450 nm excitation) was noted down.
3.6 DNA cleavage studies
A mixture of Tris buffer (5 mM Tris-HCl/50 mM NaCl buffer, pH 7.2), pBR 322 plasmid DNA (350 μg mL−1) and complexes 1–5 was incubated for 4 h at 37 °C. A dye solution (0.05% bromophenol blue and 5% glycerol) was added to the reaction mixture prior to electrophoresis. The samples were then analyzed by 1.5% agarose gel electrophoresis [Tris-HCl/boric acid/EDTA (TBE) buffer, pH 8.0] for 2 h at 60 mV. The gel was stained with 0.5 μg mL−1 ethidium bromide, visualized by UV light and photographed. The extent of cleavage of the pBR 322 DNA was determined by measuring the intensities of the bands using an AlphaImager HP instrument.
3.7 Molecular docking studies
The X-ray crystal structure of B-DNA (PDB ID: 1BNA) dodecamer d(CGCGAATTCGCG)2 and human DNA Topo-I complex (PDB ID: 1SC7) were obtained from the Protein Data Bank (http://www.rcsb.org/pdb). The 2D structure of copper complexes was drawn using ChemDraw Ultra 12.0 (ChemOffice 2010). Chem3D Ultra 12.0 was used to convert the 2D structure into 3D and the energy was minimized using the semi-empirical AM1 method. Molecular docking studies have been done using the AutoDock Tools (ADT) version 1.5.6 and the AutoDock version 4.2.5.1 docking program.72 The energy calculations were performed using genetic algorithms. The outputs were exported to PyMol for visual inspection of the binding modes and for possible polar and hydrophobic interactions of the complexes with DNA.
3.8 Protein binding studies
The binding of copper(II) complexes (1–5) with BSA was studied using fluorescence spectra recorded at a fixed excitation wavelength of 280 nm and the emission was monitored at 335 nm. The excitation and emission slit widths and scan rates were constantly maintained for all the experiments. Stock solution of BSA was prepared in 50 mM phosphate buffer (pH 7.2) and stored in the dark at 4 °C for further use. Concentrated stock solutions of each test compound were prepared by dissolving them in DMF–phosphate buffer (5:95) and diluted with phosphate buffer to obtain required concentrations. 2.5 mL of BSA solution was titrated by successive additions of a 10−6 M stock solution of the complexes using a micropipette. For synchronous fluorescence spectra measurements, the same concentrations of BSA and the complexes were used and the spectra were recorded at two different Δλ values of 15 and 60 nm.
3.9 Superoxide scavenging study
The superoxide (O2−) radical scavenging assay was done based on the ability of the complexes to inhibit formazan formation by scavenging the superoxide radicals generated in the riboflavin–light–NBT system.73 Each 3 mL reaction mixture contained 50 mM sodium phosphate buffer (pH 7.6), 20 mg riboflavin, 10 mM methionine and 0.1 mg NBT. The reaction was started by illuminating the reaction mixture with different concentrations of the test complexes (2–10 μM) for 90 s. After illumination, the absorbance was measured at 560 nm immediately. The entire reaction assembly was enclosed in a box lined with aluminium foil. The above reaction mixture without the test sample was used as the control. All the tests were run in triplicate and various concentrations of the complexes were used to fix the concentration range at which complexes showed around 50% activity. The percentage activity was calculated using the formula [(A0 − AC)/A0] × 100, where A0 and AC are the absorbance in the absence and presence of the tested complex respectively.
3.10
In vitro cytotoxicity evaluation by the MTT assay
Cytotoxicity of the copper(II) complexes was evaluated on human breast (MCF7), lung cancer (A549) and mouse embryonic fibroblast (NIH 3T3) cell lines which were obtained from the National Centre for Cell Science, Pune, India. Cell viability was determined using the MTT assay method. The MCF7 and A549 cells were grown in Eagle's minimum essential medium containing 10% fetal bovine serum (FBS) while NIH 3T3 fibroblasts were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS. For screening experiments, the cells were seeded into 96-well plates in 100 mL of the respective medium containing 10% FBS, at a plating density of 10000 cells per well and incubated at 37 °C for 24 h prior to the addition of the complexes. Complexes 1–5 dissolved in DMSO (10–500 μM) were seeded to the wells. Triplication was maintained, and the medium without the complexes served as the control. After 24 h, the wells were treated with 20 μL MTT [5 mg mL−1 phosphate buffered saline (PBS)] and incubated at 37 °C for 4 h. The medium with MTT was then removed separately and the formed formazan crystals were dissolved in 100 mL DMSO. The absorbance at 570 nm was measured using an ELISA plate reader. The graph was plotted between the percentage of cell inhibition and the concentration of the complexes. IC50 values were calculated from the percentage of inhibition. The percentage of cell inhibition was determined using the formula, % inhibition = [mean OD of untreated cells (control)/mean OD of treated cells (control)] × 100.
4. Conclusions
Copper(II) complexes containing N,N′,N′′-trisubstituted benzyl based guanidine ligands were synthesized and characterized by analytical, spectroscopic and single crystal X-ray diffraction studies with a view to evaluate their biological applications. The DNA and protein binding of the complexes were investigated using absorbance and fluorescence spectroscopic techniques. The complexes bind reasonably well to DNA in the order of 104 M−1 through intercalation. The DNA cleavage ability shows that complex 5 has a better cleaving ability compared to the other complexes. The protein binding ability of the complexes was examined by fluorescence spectroscopy. The superoxide radical scavenging assay results showed that all the complexes possess significant activity. In addition the in vitro cytotoxicity of complexes 1–5 suggested that complexes 3 and 1 have better cytotoxic activity compared to the other complexes. The cytotoxic activity towards normal cells shows a high IC50 value which reveals that the complexes are cytotoxic only towards cancerous cells and it did not affect normal cells. Further docked models also confirmed the binding affinity of the complexes with DNA. There is a correlation between the DNA binding and cytotoxicity of the complexes proving that complexes 3 and 1 are better candidates as anticancer drugs. But studies like DNA cleavage and docking models proved that complex 5 is a better candidate. As a conclusion further mechanistic studies have to be dealt with in detail to understand the controversy prevailing regarding the DNA interactions with the complexes.
Acknowledgements
K. J. thanks DST for a doctoral fellowship under the DST-INSPIRE programme. R. K. gratefully acknowledges DST for financial support. We acknowledge the Sophisticated Analytical Instrumentation facility (SAIF), Indian Institute of Technology – Madras and Indian Institute of Technology – Bombay for NMR and EPR analyses respectively. We thank Dr T. Parimelazhagan, Bharathiar University for his help in carrying out the antioxidant study.
References
- I. Kostova, Recent Pat. Anti-Cancer Drug Discovery, 2006, 1, 1–22 CrossRef CAS.
- A. Kamal, R. Ramu, V. Tekumalla, G. B. R. Khanna, M. S. Barkume, A. S. Juvekar and S. M. Zingde, Bioorg. Med. Chem., 2007, 15, 6868–6875 CrossRef CAS PubMed.
- D. Dreher and A. F. Junod, Eur. J. Cancer, 1996, 32, 30–38 CrossRef.
- W. J. Chen, P. Guo, J. Song, W. Cao and J. Bian, Bioorg. Med. Chem. Lett., 2006, 16, 3582–3585 CrossRef CAS PubMed.
- B. P. Esposito and R. Najjar, Coord. Chem. Rev., 2002, 232, 137–149 CrossRef CAS.
- T. M. Sielecki, J. F. Boylan, P. A. Benfield and G. L. Trainor, J. Med. Chem., 1999, 43, 1–18 CrossRef.
- Z. Zang, L. Jin, X. Qian, M. Wei, Y. Wang, J. Wang, Y. Yang, Q. Xu, Y. Xu and F. Liu, ChemBioChem, 2007, 8, 113–121 CrossRef PubMed.
- P. K. M. Siu, D. L. Ma and C. M. Che, Chem. Commun., 2005, 1025–1027 Search PubMed.
- J. Chern, Y. Leu, S. Wang, R. Jou, S. Hsu, Y. Liaw and H. Lin, J. Med. Chem., 1997, 40, 2276–2286 CrossRef CAS PubMed.
- Z. Brzozowski, F. Saczewski and M. Gdaniec, Eur. J. Med. Chem., 2002, 37, 285–293 CrossRef CAS.
- A. V. Dolzhenko, B. J. Tan, A. V. Dolzhenko, G. N. C. Chiu and W. K. Chui, J. Fluorine Chem., 2008, 129, 429–434 CrossRef CAS PubMed.
- G. Murtaza, A. Badshah, M. Said, H. Khan, A. Khan, S. Khan, S. Siddiq, M. I. Choudhary, J. Boudreau and F. G. Fontaine, Dalton Trans., 2011, 40, 9202–9211 RSC.
- P. J. Bailey and S. Pace, Coord. Chem. Rev., 2001, 214, 91–141 CrossRef CAS.
- K. Jeyalakshmi, N. Selvakumaran, N. S. P. Bhuvanesh, A. Sreekanth and R. Karvembu, RSC Adv., 2014, 4, 17179–17195 RSC.
-
H. B. Kraatz and N. M. Nolte, Concepts and Models in Bioinorganic Chemistry, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed.
-
S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, CA, 1994 Search PubMed.
- Z. H. Harris and J. D. Gitlin, Am. J. Clin. Nutr., 1996, 63, 836S–841S CAS.
- P. U. Maheswari, S. Roy, H. D. Dulk, S. Barends, G. V. Wezel, B. Kozlevcar, P. Gamez and J. Reedijk, J. Am. Chem. Soc., 2006, 128, 710–711 CrossRef CAS PubMed.
- J. D. Ranford, P. J. Sadler and D. A. Tocher, J. Chem. Soc., Dalton Trans., 1993, 3393–3399 RSC.
- C. H. Ng, K. C. Kong, S. T. Von, P. Balraj, P. Jensen, E. Thirthagiri, H. Hamada and M. Chikira, Dalton Trans., 2008, 447–454 RSC.
- A. Barve, A. Kumbhar, M. Bhat, B. Joshi, R. Butcher, U. Sonawane and R. Joshi, Inorg. Chem., 2009, 48, 9120–9132 CrossRef CAS PubMed.
- C. Fernandes, G. L. Parrilha, J. A. Lessa, L. J. M. Santiago, M. M. Kanashiro, F. S. Boniolo, A. J. Bortoluzzi, N. V. Vugman, M. H. Herbstand and A. Horn Jr., Inorg. Chim. Acta, 2006, 359, 3167–3176 CrossRef CAS PubMed.
- B. C. Bales, T. Kodama, Y. N. Weledji, M. Pitie, B. Meunier and M. M. Greenberg, Nucleic Acids Res., 2005, 33, 5371–5379 CrossRef CAS PubMed.
- L. Turecky, P. Kalina and E. Uhlikova, Klin. Wochenschr., 1984, 62, 187–189 CrossRef CAS.
- D. Yoshida, Y. Ikeda and S. Takanawa, J. Neurooncol., 1993, 16, 109–115 CrossRef CAS.
- D. H. Petering, Met. Ions Biol. Syst., 1980, 11, 197–229 CAS.
- F. A. French and E. J. Blanz Jr., Cancer Res., 1965, 25, 1454–1458 CAS.
- F. A. French and E. J. Blanz Jr., Cancer Res., 1966, 26, 1638–1640 CAS.
-
P. M. May and D. R. Williams, in Metal Ions in Biological Systems, ed. H. Sigel, Marcel Dekker, New York, 1981, vol. 12 Search PubMed.
-
P. M. May and D. R. Williams, in Metal Ions in Biological Systems, ed. H. Sigel, Marcel Dekker, New York, 1981, vol. 13 Search PubMed.
- T. Miura, A. Hori-i, H. Mototani and H. Takeuchi, Biochemistry, 1999, 38, 11560–11569 CrossRef CAS PubMed.
- G. Binzet, N. Kulcu, U. Florke and H. Arslan, J. Coord. Chem., 2009, 62, 3454–3462 CrossRef CAS PubMed.
- D. A. Powell, P. D. Ramsden and R. A. Batey, J. Org. Chem., 2003, 68, 2300–2309 CrossRef CAS PubMed.
- S. Cunha, M. B. Costa, H. B. Napolitano, C. Lauriucci and I. Vecanto, Tetrahedron, 2001, 57, 1671–1675 CrossRef CAS.
- J. G. Tojal, A. G. Orad, J. L. Serra, J. L. Pizarro, L. Lezama, M. I. Arriortua and T. Rojo, J. Inorg. Biochem., 1996, 75, 45–54 CrossRef.
- J. Binoy, C. James, I. H. Joe and V. S. Jayakumar, J. Mol. Struct., 2006, 784, 32–46 CrossRef CAS PubMed.
- R. Loganathan, S. Ramakrishnan, E. Suresh, A. Riyasdeen, M. A. Akbarsha and M. Palaniandavar, Inorg. Chem., 2012, 51, 5512–5532 CrossRef CAS PubMed.
- P. Murrayrust, H. B. Burgi and J. D. Dunitz, J. Am. Chem. Soc., 1975, 97, 921–922 CrossRef CAS.
- S. Keinan and D. Avnir, J. Chem. Soc., Dalton Trans., 2001, 941–947 RSC.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3051–3058 CrossRef CAS.
- N. Chitrapriya, T. Sathiyakamatchi, M. Zeller, H. Lee and K. Natarajan, Spectrochim. Acta, Part A, 2011, 81, 128–134 CrossRef CAS PubMed.
- J. R. Lakowicz and G. Webber, Biochemistry, 1973, 12, 4161–4170 CrossRef CAS.
- B. C. Baguley and M. Lebret, Biochemistry, 1984, 23, 937–943 CrossRef CAS.
- E. Nyarko, N. Hanada, A. Habib and M. Tabata, Inorg. Chim. Acta, 2004, 357, 739–745 CrossRef CAS PubMed.
- J. B. Lepecq and C. Paoletti, J. Mol. Biol., 1967, 27, 87–106 CrossRef CAS.
- K. S. Ghosh, B. K. Sahoo, D. Jana and S. Dasgupta, J. Inorg. Biochem., 2008, 102, 1711–1718 CrossRef CAS PubMed.
- S. Ramakrishnan, D. Shakthipriya, E. Suresh, V. S. Periasamy, M. A. Akbarsha and M. Palaniandavar, Inorg. Chem., 2011, 50, 6458–6471 CrossRef CAS PubMed.
- X. S. Xiao, S. Antony, Y. Pommier and M. Cushman, J. Med. Chem., 2005, 48, 3231–3238 CrossRef CAS PubMed.
- X. S. Xiao and M. Cushman, J. Am. Chem. Soc., 2005, 127, 9960–9961 CrossRef CAS PubMed.
- D. S. Raja, G. Paramaguru, N. S. P. Bhuvanesh, J. H. Reibenspies, R. Renganathan and K. Natarajan, Dalton Trans., 2011, 40, 4548–4559 RSC.
- D. S. Raja, N. S. P. Bhuvanesh and K. Natarajan, Eur. J. Med. Chem., 2011, 46, 4584–4594 CrossRef CAS PubMed.
-
J. R. Lakowicz, Fluorescence Quenching: Theory and Applications. Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, New York, 1999, pp. 53–127 Search PubMed.
- X. Z. Feng, Z. Yang, L. J. Wang and C. Bai, Talanta, 1998, 47, 1223–1229 CrossRef CAS.
- V. Rajendiran, R. Karthik, M. Palaniandavar, H. S. Evans, V. S. Periasamy, M. A. Akbarsha, B. S. Srinag and H. Krishnamurthy, Inorg. Chem., 2007, 46, 8208–8221 CrossRef CAS PubMed.
- M. Bhattacharyya, U. Chaudhuri and R. K. Poddar, Biochem. Biophys. Res. Commun., 1990, 167, 1146–1153 CrossRef CAS.
- J. N. Miller, Proc. Anal. Div. Chem. Soc., 1979, 16, 199–208 RSC.
- C. X. Wang, F. F. Yan, Y. X. Zhang and L. Ye, J. Photochem. Photobiol., A, 2007, 192, 23–28 CrossRef CAS PubMed.
- F. Zhang, Q. Y. Lin, S. K. Li, Y. L. Zhao, P. P. Wang and M. M. Chen, Spectrochim. Acta, Part A, 2012, 98, 436–443 CrossRef CAS PubMed.
- W. C. Albert, W. M. Gregory and G. S. Allan, Anal. Biochem., 1993, 213, 407–413 CrossRef PubMed.
- R. Rajan, R. Rajaram, B. U. Nair, T. Ramasami and S. K. Mandal, J. Chem. Soc., Dalton Trans., 1996, 2019–2021 RSC.
- U. Weser, L. M. Schubotz and E. Lengfelder, J. Mol. Catal., 1981, 13, 249–261 CrossRef CAS.
- S. B. Etcheverry, E. G. Ferrer, L. Naso, J. Rivadeneira, V. Salinas and P. A. M. Williams, J. Biol. Inorg. Chem., 2008, 13, 435–447 CrossRef CAS PubMed.
- I. Schepetkin, A. Potapov, A. Khlebnikov, E. Korotkova, G. A. Lukina, L. Malovichko, M. Kirpotina and T. Quinn, J. Biol. Inorg. Chem., 2006, 11, 499–513 CrossRef CAS PubMed.
- N. A. Roberts and P. A. Robinson, Br. J. Rheumatol., 1985, 24, 128–136 CrossRef CAS PubMed.
- E. G. Ferrer, N. Baeza, L. G. Naso, E. E. Castellano, O. E. Piro and P. A. M. Williams, J. Trace Elem. Med. Biol., 2010, 24, 20–26 Search PubMed.
- M. Blagosklonny and W. S. EIdiery, Int. J. Cancer, 1996, 67, 386–392 CrossRef CAS.
-
APEX2, Program for Data Collection on Area Detectors, BRUKER AXS Inc., 5465 East Cheryl Parkway, Madison, WI 53711-5373, USA Search PubMed.
-
SADABS and G. M. Sheldrick, Program
for Absorption Correction of Area Detector Frames, BRUKER AXS Inc., 5465 East Cheryl Parkway, Madison, WI 53711-5373, USA Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3057 CrossRef CAS.
-
http://autodock.scripps.edu/resources/references
.
- C. Beauchamp and I. Fridovich, Anal. Biochem., 1971, 44, 276–287 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Electronic absorption spectra of complexes 2, 4 and 5 upon addition of DNA (Fig. S1), emission spectra of DNA-EB in the presence of complexes 2, 4 and 5 (Fig. S2), fluorescence quenching curves of BSA with complexes 1, 2 and 4 (Fig. S3), synchronous spectra (Δλ = 15 and 60 nm) of complexes 1, 2 and 4 with BSA (Fig. S4 and S5) and cytotoxicity of the complexes against normal cells (Fig. S6). CCDC 1023785–1023791. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00234b |
|
| This journal is © the Partner Organisations 2015 |