
A two-dimensional flexible porous coordination polymer based on Co(II) and terpyridyl phosphine oxide†
Juan
Feng
,
Hongmiao
Li
,
Qiuli
Yang
,
Shi-Chao
Wei
,
Jianyong
Zhang
* and
Cheng-Yong
Su

School of Chemistry and Chemical Engineering, Sun Yat-Sen University, Guangzhou, 510275, China. E-mail: zhjyong@mail.sysu.edu.cn; Tel: +86 20 8411 0539
First published on 4th February 2015
Abstract
Two porous coordination polymers, Co-Py2PO and Co-Py2PO′, have been obtained via reactions of in situ oxidized terpyridyl phosphines and cobalt(II) nitrate. Single-crystal structure analysis reveals that Co-Py2PO′ has a three-dimensional (3D) structure with (8·102)(83) net topology, while the structural analysis of Co-Py2PO reveals a 2D polycatenated framework consisting of two sets of parallel 2D networks of (63) net topology. For Co-Py2PO′, guest CHCl3 molecules are locked in the channels along the b axis by P(O)Ph2 phenyl rings and coordinated nitrate ions, and cannot be released out of the host framework. In contrast, the as-synthesized 2D framework Co-Py2PO readily loses its solvated guest molecules to give an activated amorphous material for sorption. Activated Co-Py2PO has a flexible structure and shows dynamic adsorption behaviours towards various guests (N2 and benzene). The flexibility may arise from the combination of layer–layer motion and the local flexibility of the P–C bond of –P(O)Ph2 groups.
Introduction
Porous coordination polymers (PCPs) or metal–organic frameworks (MOFs) composed of metal ions and organic ligands have been developed as an important class of porous materials. Among them, dynamic/flexible MOFs and/or soft porous materials have received much attention due to their flexible and responsive properties.1 Based on the materials’ ‘softness’ and stimuli responsiveness, unique functional properties have been obtained for this family of materials. Possible applications of flexible metal–organic frameworks include separation, catalysis, sensing, and biomedicine.1 Coordination polymers made up of two-dimensional (2D) structures are a group of interesting flexible porous materials.2 The subnets of interdigitated and stacked 2D frameworks are connected to each other only by weak forces, and thus can drift, relocate, or shift. Therefore 2D frameworks have a more marked flexibility than 3D structures.3 Because the subnetwork movements can be easily induced by guest inclusion, 2D frameworks show the breathing phenomenon and responsiveness to guest adsorption.4 They can respond to external stimuli by changing their porosity and show flexibility-associated properties such as gas adsorption,5 stepwise gas adsorption,6 selective adsorption/separation,7 and ion exchange.8 These dynamic responses are closely related to the structural changes, including sliding of 2D grids,9 structural expansion/shrinkage along the stacking direction,10 and bond formation/cleavage.11In the design and synthesis of flexible 2D frameworks, N-donor or O-donor-based ligands have been used widely. Although phosphines and phosphine oxides are important ligands in catalysis and coordination chemistry and have recently been used to construct porous coordination polymers,12,13 phosphine-based ligands have rarely been exploited in flexible 2D frameworks.14 Phosphine-based 2D frameworks may be particularly interesting because the rotation of –PPh2 phenyl rings is an important, specific source of flexibility. Herein we report the syntheses and structures of one 2D and one 3D porous coordination polymers based on Co(II) and terpyridyl phosphine oxides. Their sorption properties were investigated and interestingly the 2D framework shows remarkable flexibility behaviour.
Results and discussion
Synthesis and crystal structures
A reaction of Py2P with Co(NO3)2·6H2O in MeCN–CHCl3 at 80 °C for two days led to the formation of (Py2PO)4Co4(NO3)8·xMeCN·xCHCl3·xH2O (denoted Co-Py2PO). When Py2P′ was used as the starting ligand, (Py2PO′)Co(NO3)2·CHCl3 (denoted Co-Py2PO′) was obtained. During the reactions, Py2P and Py2P′ were oxidized in situ to form Py2PO and Py2PO′, respectively. Py2PO and Py2PO′ are considered as 3-connecting rigid terpyridyl phosphine oxide ligands for Co(II) (and other harder 3d ions) with two outer pyridyl N donors and one P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O donor, leaving the N donor of the central pyridine ring uncoordinated15 (Scheme 1). In comparison to this, the spacer between terpyridyl and –P(O)Ph2 groups is one phenyl ring longer for Py2P′ than for Py2P. The oxidation of Py2P and Py2P′ was evidenced using FT-IR and mass spectra. FT-IR spectra showed characteristic bands of –PO at 1167 and 1162 cm−1 for Co-Py2PO and Co-Py2PO′, respectively (Fig. S1 and S2†). Electrospray ionization time-of-flight (ESI TOF) mass spectroscopy showed only the signals of oxidized ligands (no signals of the ligands) after digesting the Co(II) complexes with HCl, suggesting complete oxidation. In addition, both the FT-IR spectra showed the strong characteristic band of nitrate anions at around 1384 cm−1. Interestingly, Co-Py2PO′ could be prepared directly using the oxidized ligand Py2PO′, while Co-Py2PO could not be obtained using Py2PO under similar conditions (see ESI and Fig. S3–S6 for characterization data of Py2PO and Py2PO′†). A similar phenomenon was observed in another phosphine oxide framework.16
O O donor, leaving the N donor of the central pyridine ring uncoordinated15 (Scheme 1). In comparison to this, the spacer between terpyridyl and –P(O)Ph2 groups is one phenyl ring longer for Py2P′ than for Py2P. The oxidation of Py2P and Py2P′ was evidenced using FT-IR and mass spectra. FT-IR spectra showed characteristic bands of –PO at 1167 and 1162 cm−1 for Co-Py2PO and Co-Py2PO′, respectively (Fig. S1 and S2†). Electrospray ionization time-of-flight (ESI TOF) mass spectroscopy showed only the signals of oxidized ligands (no signals of the ligands) after digesting the Co(II) complexes with HCl, suggesting complete oxidation. In addition, both the FT-IR spectra showed the strong characteristic band of nitrate anions at around 1384 cm−1. Interestingly, Co-Py2PO′ could be prepared directly using the oxidized ligand Py2PO′, while Co-Py2PO could not be obtained using Py2PO under similar conditions (see ESI and Fig. S3–S6 for characterization data of Py2PO and Py2PO′†). A similar phenomenon was observed in another phosphine oxide framework.16
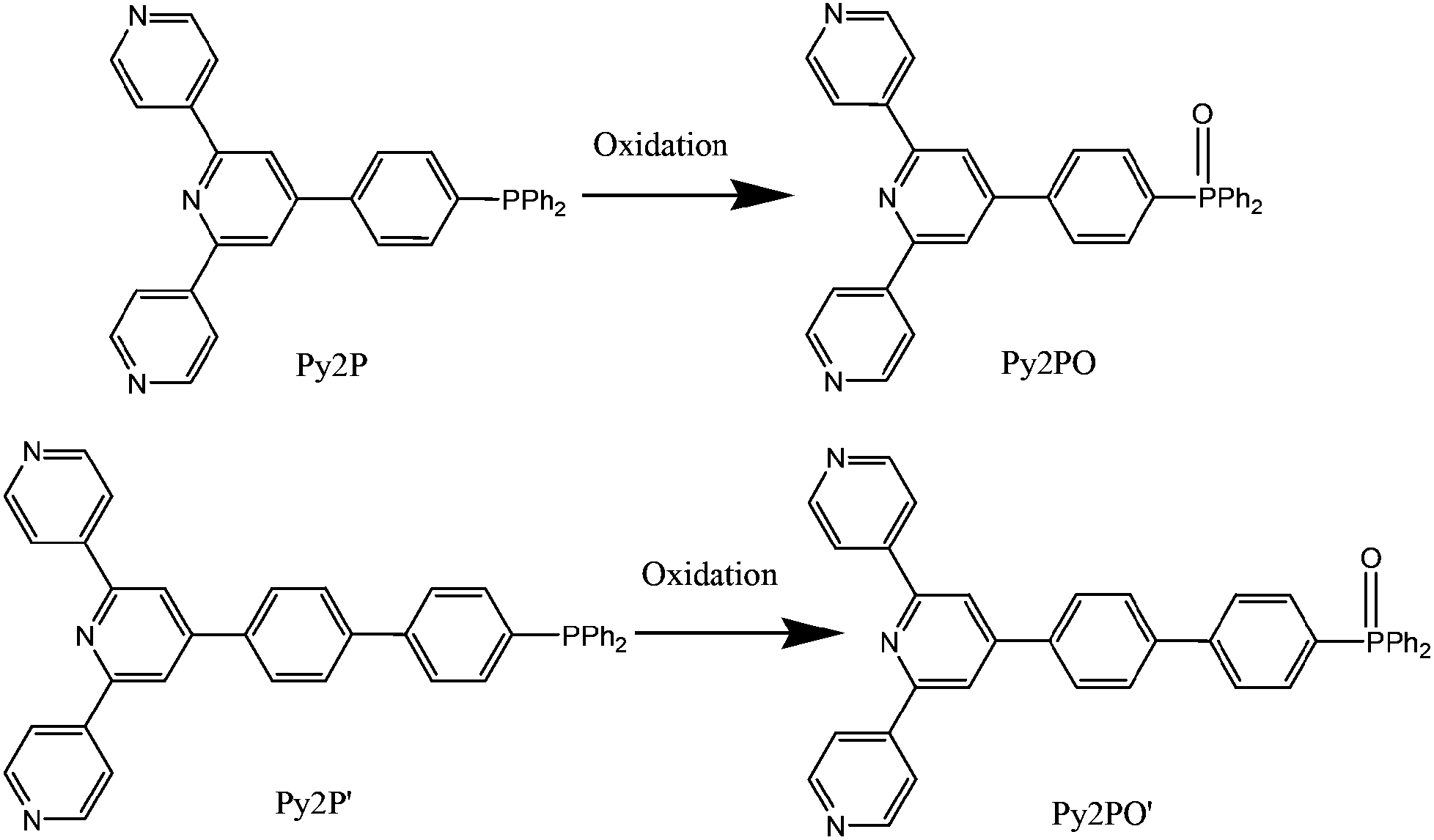 | ||
| Scheme 1 Oxidation of Py2P and Py2PO to form Py2P′ and Py2PO′, respectively. | ||
The structure of Co-Py2PO was unambiguously identified by single-crystal X-ray analysis. Co-Py2PO crystallizes in the triclinic crystal system within space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit contains two oxidized phosphine ligands Py2PO, two Co2+ and four NO3− coordinated anions. Two independent Co2+ ions in the asymmetric unit have similar coordination environments, adopting a CoO4N2 distorted octahedral geometry. Each Co2+ ion is six-coordinated by one PO oxygen atom and two pyridyl nitrogen atoms from three different Py2PO ligands and three oxygen atoms from two nitrate ions (Fig. 1a). For the nitrate ions, one nitrate ion connects with Co2+ through a monodentate O atom in a η1-mode and the other nitrate ion binds through two oxygen atoms in a η2-mode. Each Py2PO ligand binds to three Co2+ ions with one PO O and two outer pyridyl N donors, leaving the central pyridyl N donor uncoordinated. Overall, the Co2+ ions as three-connected nodes are bridged by 3-connected Py2PO ligands to form a 2D honeycomb (hcb) structure of (63) net topology (Fig. 1b). The Co⋯Co distances separated by Py2PO are different depending on different donors, and are 14.07 Å (separated by PO O and pyridyl N donors) and 12.93 Å (separated by two pyridyl N donors). Every two undulating 2D layers are interlaced in a parallel fashion by stacking with offset of their average planes to result in 2D→2D 2-fold parallel polycatenation (Fig. 1c–e). The resulting flat catenated 2D layers show a thickness of ca. 15.04 Å. The 2D catenated layers stack in an ABCABC packing mode. Solvated molecules (MeCN, CHCl3 and H2O) are located between adjacent 2D catenated layers. Upon removal of the solvated molecules, there is 25.7% void space to the total crystal volume (total potential solvent area volume 967.9 Å3 per unit cell volume 3765.9 Å3) calculated using PLATON.17 Thus Co-Py2PO represents a 2D framework with interlayer cavity rather than intralayer cavity.18
. The asymmetric unit contains two oxidized phosphine ligands Py2PO, two Co2+ and four NO3− coordinated anions. Two independent Co2+ ions in the asymmetric unit have similar coordination environments, adopting a CoO4N2 distorted octahedral geometry. Each Co2+ ion is six-coordinated by one PO oxygen atom and two pyridyl nitrogen atoms from three different Py2PO ligands and three oxygen atoms from two nitrate ions (Fig. 1a). For the nitrate ions, one nitrate ion connects with Co2+ through a monodentate O atom in a η1-mode and the other nitrate ion binds through two oxygen atoms in a η2-mode. Each Py2PO ligand binds to three Co2+ ions with one PO O and two outer pyridyl N donors, leaving the central pyridyl N donor uncoordinated. Overall, the Co2+ ions as three-connected nodes are bridged by 3-connected Py2PO ligands to form a 2D honeycomb (hcb) structure of (63) net topology (Fig. 1b). The Co⋯Co distances separated by Py2PO are different depending on different donors, and are 14.07 Å (separated by PO O and pyridyl N donors) and 12.93 Å (separated by two pyridyl N donors). Every two undulating 2D layers are interlaced in a parallel fashion by stacking with offset of their average planes to result in 2D→2D 2-fold parallel polycatenation (Fig. 1c–e). The resulting flat catenated 2D layers show a thickness of ca. 15.04 Å. The 2D catenated layers stack in an ABCABC packing mode. Solvated molecules (MeCN, CHCl3 and H2O) are located between adjacent 2D catenated layers. Upon removal of the solvated molecules, there is 25.7% void space to the total crystal volume (total potential solvent area volume 967.9 Å3 per unit cell volume 3765.9 Å3) calculated using PLATON.17 Thus Co-Py2PO represents a 2D framework with interlayer cavity rather than intralayer cavity.18
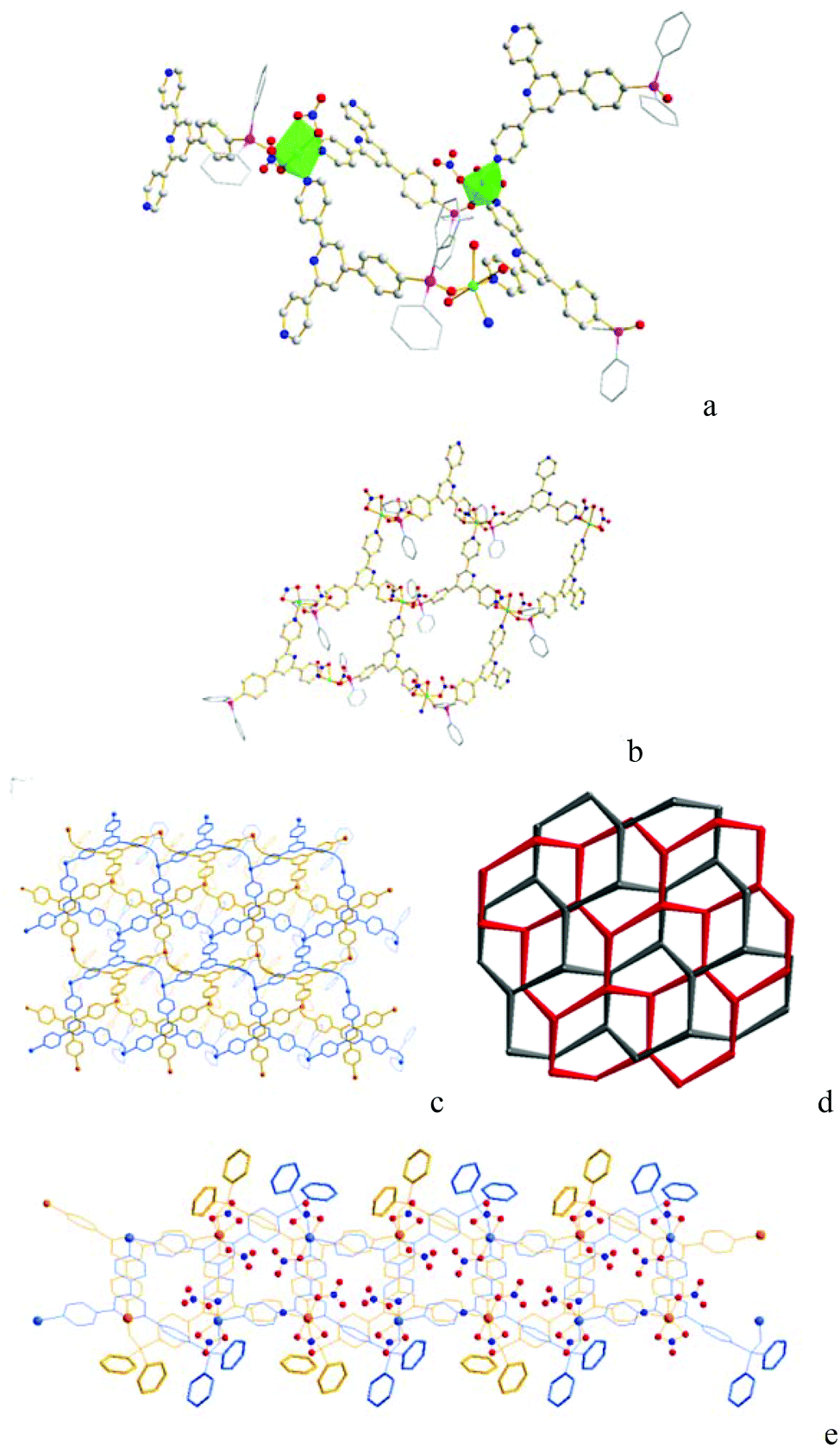 | ||
| Fig. 1 X-ray structure of Co-Py2PO showing (a) the distorted octahedral coordination geometry around Co2+ ions highlighting two independent Co2+ ions in the asymmetric unit, (b) part of the 2D layer viewed along the ac plane of the 2D honeycomb (63) network with the solvated molecules omitted for clarity, (c) part of the 2D catenated layers viewed along the ac plane with nitrate ions and solvated molecules omitted for clarity, (d) topological diagram of 2D catenated layers, (e) part of the 2D catenated layers viewed along the ac plane, highlighting the –P(O)Ph2 phenyl groups on the surface. | ||
Single crystal X-ray diffraction analysis shows that Co-Py2PO′ crystallizes in the orthorhombic group Pna21. The asymmetric unit contains one oxidized phosphine ligand Py2PO′, one Co2+ and two NO3− coordinated anions. Each Py2PO′ ligand binds to three Co2+ ions with one PO O and two outer pyridyl N donors like Py2PO, while each Co2+ is coordinated with three ligands to form a 3D network. The Co2+ ion is six-coordinated with a distorted octahedral CoO4N2 coordination geometry (Fig. 2a) like that in Co-Py2PO. Every Co2+ ion coordinates with one PO O and two pyridyl N donors from three different ligands and three O donors from a chelating nitrate and a monodentate nitrate ion. Net topological analysis reveals a 3D 2-nodal 3-connected net with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry if both Co2+ and Py2PO′ are simplified as 3-connecting nodes. The point (Schläfli) symbol is (8·102)(83) calculated using OLEX19 (Fig. 2c). Its vertex symbol is (82·106·102)(8·8·8). 1D channels are formed along the b axis (Fig. 2b). –P(O)Ph2 phenyl rings and coordinated nitrate ions are pointed to the channel center. CHCl3 guest molecules are accommodated inside the channels (Fig. 2d), which accounts for 16.2% solvent-accessible voids (total potential solvent area volume is 631.3 Å3 per unit cell volume 3907.9 Å3) calculated using PLATON.17
:1 stoichiometry if both Co2+ and Py2PO′ are simplified as 3-connecting nodes. The point (Schläfli) symbol is (8·102)(83) calculated using OLEX19 (Fig. 2c). Its vertex symbol is (82·106·102)(8·8·8). 1D channels are formed along the b axis (Fig. 2b). –P(O)Ph2 phenyl rings and coordinated nitrate ions are pointed to the channel center. CHCl3 guest molecules are accommodated inside the channels (Fig. 2d), which accounts for 16.2% solvent-accessible voids (total potential solvent area volume is 631.3 Å3 per unit cell volume 3907.9 Å3) calculated using PLATON.17
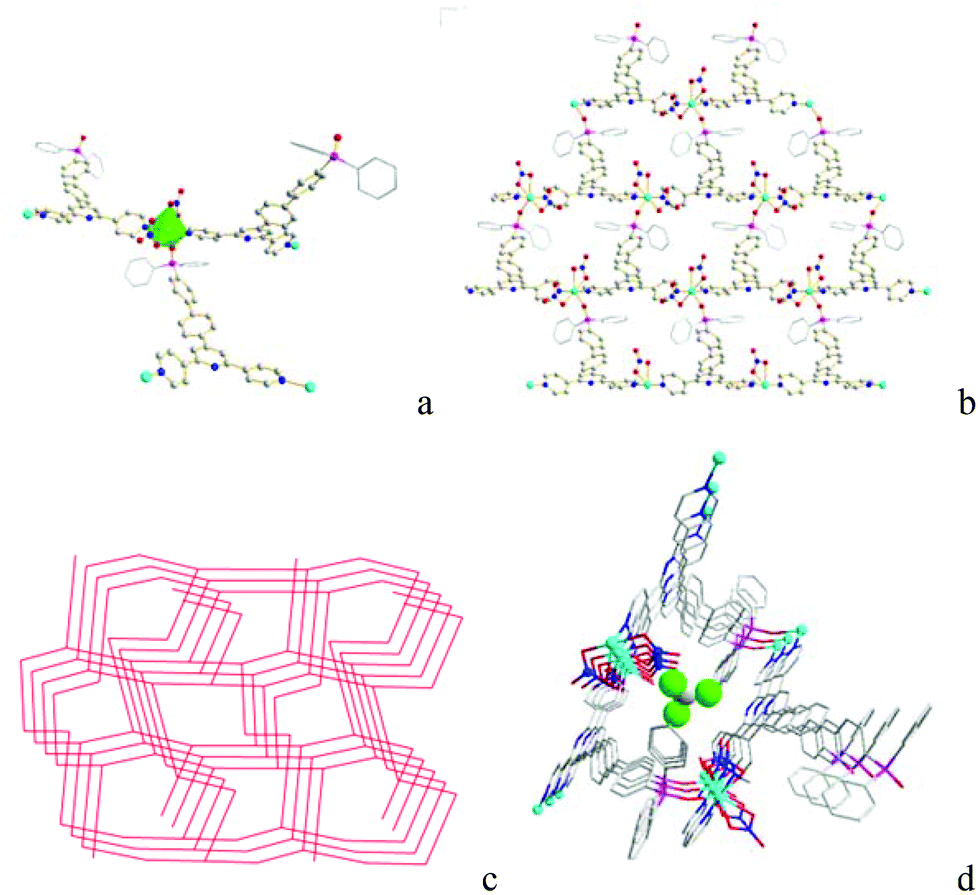 | ||
| Fig. 2 X-ray structure of Co-Py2PO′ showing (a) the distorted octahedral coordination geometry around Co2+ ions, (b) tubular channels along the b axis, (c) (8·102)(83) topological diagram, and (d) one tubular channel along the b axis to host CHCl3 guest molecules. | ||
Framework stability
Thermogravimetric analysis (TGA) showed that Co-Py2PO released the guest solvated molecules from room temperature to ca. 200 °C (Fig. 3). The weight loss of 19.4% corresponds to the loss of four CHCl3, two MeCN and ten H2O solvated molecules per Co4(Py2PO)4 unit (calcd: 21.0%). The framework of Co-Py2PO was stable up to 280 °C. In contrast, unexpectedly no loss of solvated CHCl3 molecules was observed for Co-Py2PO′ up to ca. 250 °C and then the framework of Co-Py2PO′ began to collapse upon further heating (Fig. S7†). Different thermostability of Co-Py2PO and Co-Py2PO′ may be explained by their structures. For Co-Py2PO the solvated molecules are located between 2D catenated layers and easily escape from the cavity. However, for Co-Py2PO′ the solvated CHCl3 molecules are firmly locked in the channels along the b axis by P(O)Ph2 phenyl rings and coordinated nitrate ions.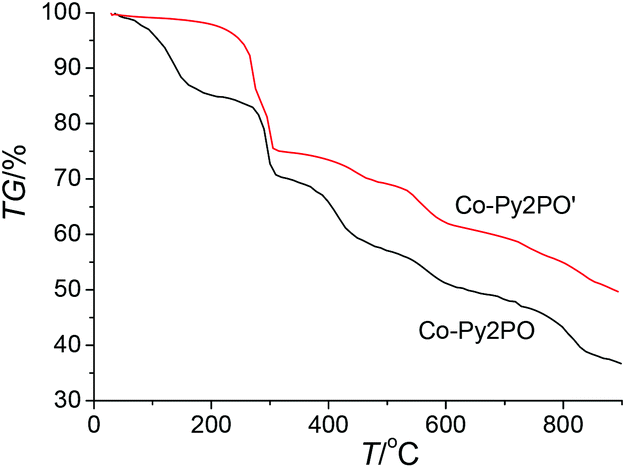 | ||
| Fig. 3 TG curves of Co-Py2PO and Co-Py2PO′. | ||
X-ray power diffraction (XRPD) was performed to study the framework stability of Co-Py2PO (Fig. 4) and Co-Py2PO′ (Fig. 5). For Co-Py2PO′, the pattern for the as-synthesized bulk material closely matches the simulated one from the single-crystal analysis, indicating stable framework and good phase purity at room temperature. In comparison, the pattern of Co-Py2PO at room temperature was found to deviate from the simulated to some degree, probably due to its 2D nature and easy loss of solvent molecules. In addition, XRPD for the activated Co-Py2PO (activated at 50 °C under vacuum for 16 h) reveals a broad and featureless pattern. This suggests that the long-range order in the as-synthesized material was lost and a crystalline-to-amorphous phase transformation occurred. The amorphous material could not be reversed to crystalline even after the activated Co-Py2PO was immersed in the mother liquor where Co-Py2PO crystallized for one week. Considering the structural characteristics of the present framework, we speculate that the activated material may undergo an interlayer shift/distortion of the 2D catenated layers after the guest molecules are removed.
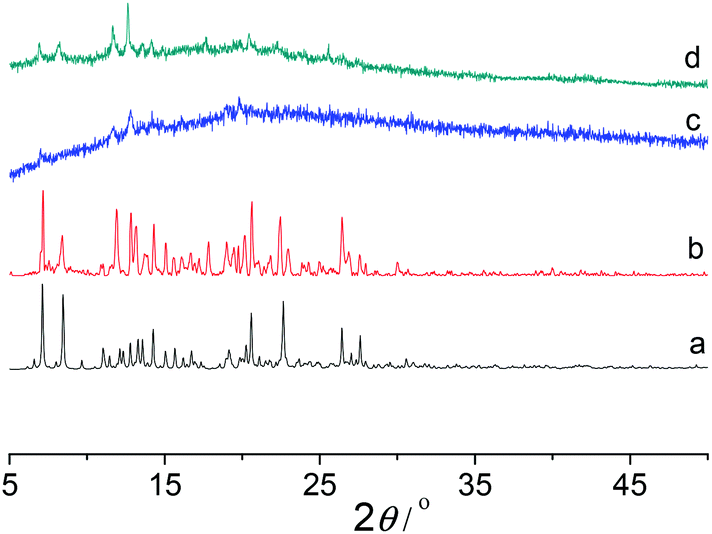 | ||
| Fig. 4 XRPD patterns of Co-Py2PO, (a) simulated from CIF, (b) as-synthesized, (c) activated at 50 °C under vacuum for 16 h, and (d) immersing the activated sample in mother liquor for one week. | ||
 | ||
| Fig. 5 XRPD patterns of Co-Py2PO′, (a) simulated from CIF and (b) as-synthesized. | ||
Sorption study
The above results reveal that Co-Py2PO readily loses guest molecules between its 2D networked layers to get activated Co-Py2PO, but Co-Py2PO′ firmly holds guest molecules in its pore channels. To study the porosity of Co-Py2PO, N2 sorption analysis at 77 K was performed for activated Co-Py2PO (activated at 50 °C under vacuum for 16 h) (Fig. 6). Interestingly, the isotherm shows a gradual increase in gas uptake and exhibits a large desorption hysteresis loop. The uptake reaches 114 cm3 g−1 without saturation at 1 bar. Moreover, the desorption does not trace the adsorption one. It loses 55 cm3 N2 gradually up to P/P0 = 0.016 and rapidly loses 59 cm3 N2 below P/P0 = 0.016. Hysteresis in the sorption profiles suggests the presence of a high diffusion barrier for N2 in Co-Py2PO. Such a remarkable breathing hysteric behaviour is rarely observed for non-polar N2.20 In comparison, CO2 adsorption of Co-Py2PO at 195 K revealed a type I profile indicative of its microporous nature. The adsorption capacity at 1 bar is 123 cm3 g−1. The Langmuir and Brunauer–Emmett–Teller surface areas calculated from the CO2 profile are 689 and 479 m2 g−1, respectively. The pore volume was calculated to be 0.157 cm3 g−1 and the pore width is centred around 7.8 Å according to the Horvath–Kawazoe method. Selective sorption of CO2 over N2 indicates that the interlayer micropore surface is overall polar due to coordinated nitrate ions.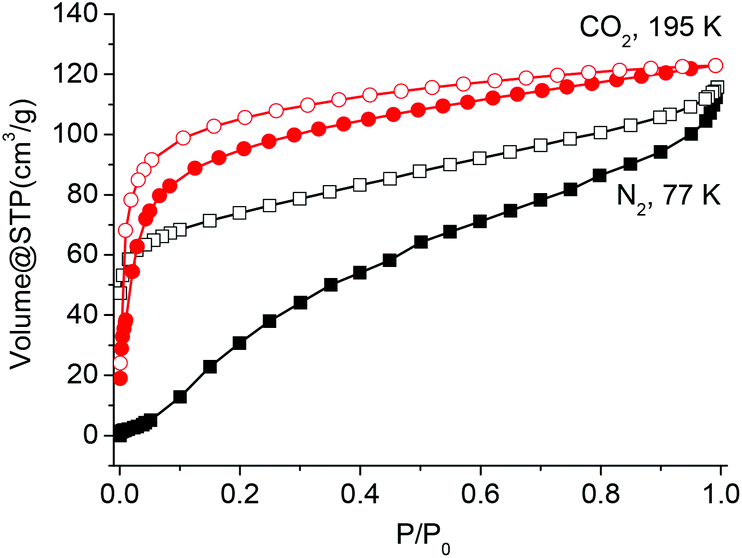 | ||
| Fig. 6 N2 and CO2 adsorption (solid symbols) and desorption (open symbols) isotherms of Co-Py2PO. | ||
Co-Py2PO was further subjected to vapour adsorption (Fig. 7). The sorption isotherms of methanol and acetonitrile vapours measured at 298 K exhibit type I profiles with hysteresis, showing that polar molecules may readily diffuse into the interlayer cavity. The MeCN isotherm displays a more marked hysteresis than the MeOH one. The overall uptake capacities are 17.3 wt% and 18.3 wt% for MeOH and MeCN, respectively.
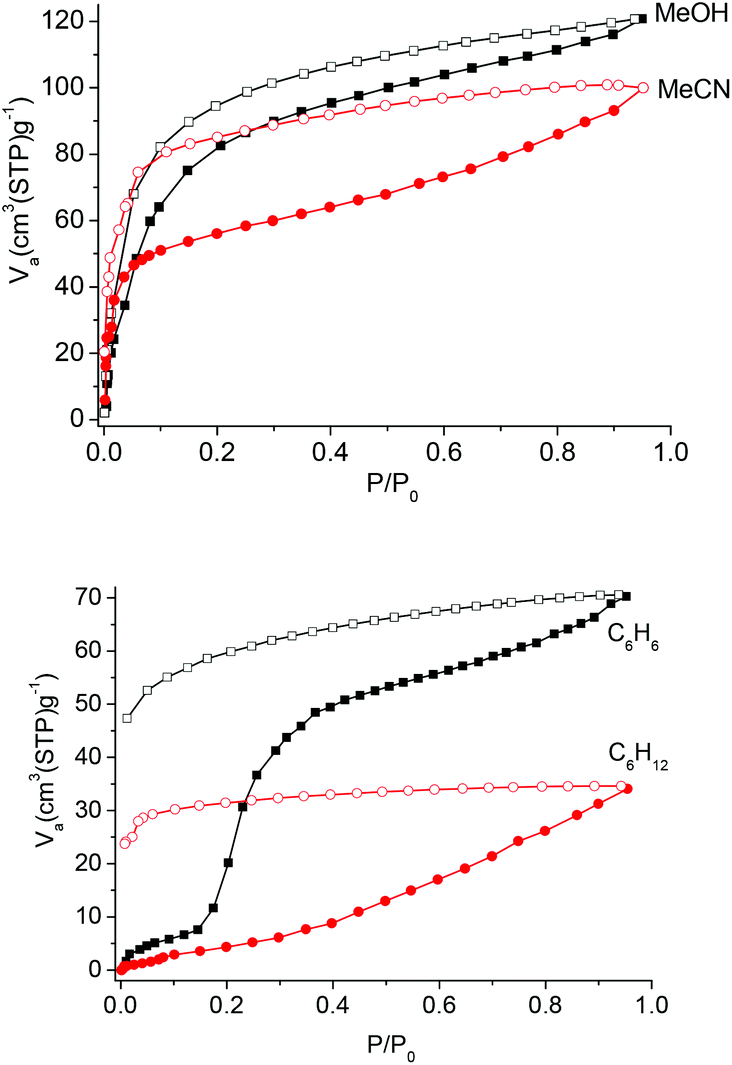 | ||
| Fig. 7 Methanol, acetonitrile, benzene and cyclohexane adsorption (solid symbols) and desorption (open symbols) isotherms of Co-Py2PO at 298 K. | ||
The sorption studies of benzene and cyclohexane were also carried out. At the beginning range of the sorption isotherm, both the amounts of benzene and cyclohexane adsorbed are negligible. This suggests that it is difficult for both the molecules to diffuse into the interlayer cavity. For benzene, a sudden increase in uptake appears when the relative pressure P/P0 is 0.15. The amount adsorbed is 24.5 wt% when P/P0 reaches 1, which is equivalent to the adsorption of 9.0 benzene molecules per Co4(Py2PO)4 unit. In contrast to the stepwise benzene adsorption, the amount adsorbed for cyclohexane increased gradually in the tested pressure range and a final uptake is 12.8 wt% (4.1 molecules per Co4(Py2PO)4 unit) at P/P0 = 1. The different sorption behaviours for benzene and cyclohexane are probably due to different interactions between the adsorbent and the adsorbates. For Co-Py2PO, π–π interactions between guest benzene molecules and the –P(O)Ph2 phenyl rings located on the surface of 2D catenated layers provide the favourable driving force for the adsorption of guest benzene molecules. As P/P0 increases and approaches the gate point (P/P0 = 0.15), the π–π interactions trigger the dynamic movement of 2D catenated layers to lead to a sudden increase in benzene uptake. In addition, the dynamic motion is probably related to the rotation of –P(O)Ph2 phenyl rings via the P–C bond. In comparison, it is difficult to form such interactions between cyclohexane and the 2D catenated layers, so a different isotherm with a gradually increasing amount was obtained. Thus the flexibility herein arises from the combination of layer–layer motion and local flexibility of P–C bonds of –P(O)Ph2 groups. Additionally, the large hysteresis loop for both benzene and cyclohexane indicates that the 2D catenated layers restrict the release of guest molecules at high pressure. Such a 2D structure may be developed for the selective sorption of benzene over cyclohexane, which is important in industrial separation.21
Conclusions
In summary, two porous coordination polymers, Co-Py2PO′ and Co-Py2PO, have been synthesized based on Co(II) and terpyridyl phosphine oxides. The terpyridyl phosphine oxides were generated by in situ oxidation of terpyridyl phosphines, Py2P and Py2P′. The terpyridyl phosphine oxides coordinated with Co(II) to yield the porous materials. Both the porous coordination polymers are based on 3-connected Co(II) ions and 3-connected terpyridyl phosphines. Co-Py2PO′ has a 3D structure with (8·102)(83) net topology, while Co-Py2PO has a 2D polycatenated framework consisting of two sets of parallel 2D networks of (63) net topology. Co-Py2PO′ is not able to release its guest CHCl3 molecules out of the host framework because they are locked in the channels along the b axis by –P(O)Ph2 phenyl rings and coordinated nitrate ions. In contrast, as-synthesized crystalline Co-Py2PO readily loses its guest solvated molecules to obtain an amorphous activated material for sorption. Activated Co-Py2PO has a flexible structure and shows gated adsorption behaviours towards various guests (N2 and benzene). The unprecedented sorption behaviours are related to layer–layer motion and the rotational motion of –P(O)Ph2 phenyl groups with respect to the P–C bond.Experimental
Chemicals and solvents were obtained from commercial sources and used as received unless otherwise stated. Py2P22 and Py2P′23 were synthesized according to the reported methods. NMR spectra were obtained using a Bruker Avance III spectrometer. Mass spectra were recorded using a Bruker maXis 4G ESI-Q-TOF mass spectrometer. X-ray powder diffraction data were collected using a Bruker D8 ADVANCE diffractometer at 40 kV and 40 mA with a Cu-target tube and a graphite monochromator (λ = 1.5418 Å). Thermo analyses were performed under an N2 atmosphere at a heating rate of 10 °C min−1 with a NETZSCH TG 209F3 system. Adsorption measurements were performed using a Quantachrome Autosorb-iQ2 or Autosorb-iQ analyzer. Before sorption measurements, the as-synthesized sample of Co-Py2PO was degassed at 50 °C for 16 h under high vacuum.Synthesis of Co-Py2PO
A solution of Co(NO3)2·6H2O (2.9 mg, 0.01 mmol) in MeCN (2 mL) was added to a solution of Py2P (5.0 mg, 0.01 mmol) in CHCl3 (2 mL) while stirring. The reaction mixture was stirred for 20 min at room temperature to yield a pink suspension. Then the suspension was heated in a closed vial at 80 °C. Red block crystals were obtained after five days (3.5 mg, 40%). Microanal. found (calcd) for C140H126N22P4Co4O38Cl12 ((Py2PO)4Co4(NO3)8·4CHCl3·2MeCN·10H2O): C, 47.70 (47.97); H, 3.79 (3.63); N, 8.32 (8.80)%. IR (KBr, cm−1): 3402br, 1603m, 1534w, 1436w, 1384s, 1166.6w, 1120w, 1066w, 1022w, 880w, 827m, 725w, 696w, 655s, 547m.Synthesis of Co-Py2PO′
Method 1: A solution of Co(NO3)2·6H2O (2.9 mg, 0.01 mmol) in MeCN (1 mL) was added to a solution of Py2P′ (4.3 mg, 0.075 mmol) in CHCl3 (1 mL) while stirring. The reaction mixture was stirred for 20 min at room temperature to yield a pink suspension. Then the suspension was heated in a closed vial at 80 °C. Red block crystals were obtained after five days (3.5 mg, 53% based on ligand). Method 2: A solution of Co(NO3)2·6H2O (2.9 mg, 0.01 mmol) in MeCN (1 mL) was added to a solution of Py2PO′ (4.3 mg, 0.075 mmol) in CHCl3 (1 mL) while stirring. The reaction mixture was stirred for 20 min at room temperature to yield a pink suspension. Then the suspension was heated in a closed vial at 80 °C. Red block crystals were obtained after two days (2.5 mg, 38% based on ligand). Microanal. found (calcd) for C40H29N5PCoO7Cl3 ((Py2PO′)Co(NO3)2·CHCl3): C 53.76 (54.17); H, 3.47 (3.30); N, 7.97 (7.90)%. IR (KBr pellets, cm−1): 3389br, 2916br, 1797w, 1603s, 1545w, 1459s, 1384s, 1307m, 1162m, 1120m, 1067w, 1023w, 874w, 822m, 750w, 555m, 529w.X-ray structure determination
X-ray crystallographic data of Co-Py2PO and Co-Py2PO′ were collected at 150(2) K using an Oxford Gemini S Ultra diffractometer equipped with a graphite monochromated Enhance Cu X-ray source (λ = 1.54178 Å). The structure was solved by direct methods following difference Fourier syntheses, and refined by the full-matrix least-squares method against F02 using SHELXTL software. All non-hydrogen atoms were refined with anisotropic thermal parameters while hydrogen atoms were introduced into the final refinement model in calculated positions with isotropic thermal parameters. For Co-Py2PO, one CHCl3 and one H2O solvated molecules display disorder, which are refined with fractional occupancy and modelled, and the hydrogen atoms of solvated water molecules are not added. Selected bond lengths and bond angles are given in Tables S1 and S2†.Crystallographic data for Co-Py2PO: C143H111N24P4Co4O31Cl9Mw = 3340.21, triclinic, P, a = 14.1110(5), b = 15.3337(5), c = 18.7087(6) Å, α = 78.679(3), β = 84.134(2), γ = 71.745(3)°, V = 3765.9(2) Å3, Z = 1, D = 1.548 g cm−3, μ = 5.942 mm−1, λ = 1.54178 Å, T = 150(2) K, F(000) = 1706, 24009 reflections were collected (12690 were unique) for 2.41 < θ < 66.03, R(int) = 0.0644, R = 0.0974, wR2 = 0.2077 [I > 2σ(I)], R = 0.0700, wR2 = 0.1851 (all data) for 993 parameters with 3 restraints, GOF = 1.025. CCDC 1023746.
Crystallographic data for Co-Py2PO′: C40H29N5PCoO7Cl3Mw = 887.93, orthorhombic, Pna21, a = 23.8620(6), b = 8.5930(1), c = 19.0584(5) Å, α = β = γ = 90°, V = 3907.85 (15) Å3, Z = 4, D = 1.509 g cm−3, μ = 6.204 mm−1, λ = 1.54178 Å, T = 150(2) K, F(000) = 1812, 6609 reflections were collected (4484 were unique) for 3.70 < θ < 62.36, R(int) = 0.0513, R = 0.0611, wR2 = 0.1472 [I > 2σ(I)], R = 0.0812, wR2 = 0.1588 (all data) for 514 parameters with 1 restraint, GOF = 1.027. CCDC 1023747.
Acknowledgements
This work was funded by the 973 Program (2012CB821701), the NSFC (21273007, 21350110212 and 91222201), the Program for New Century Excellent Talents in University (NCET-13-0615) and the Fundamental Research Funds for the Central Universities (14lgpy05).Notes and references
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed; L. J. May and G. K. H. Shimizu, Z. Kristallogr., 2005, 220, 364–372 CrossRef.
- S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109–119 RSC; S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed; H. Kajiro, A. Kondo, K. Kaneko and H. Kanoh, Int. J. Mol. Sci., 2010, 11, 3803–3845 CrossRef PubMed; L. Carlucci, G. Ciani, D. M. Proserpio, T. G. Mitina and V. A. Blatov, Chem. Rev., 2014, 114, 7557–7580 CrossRef PubMed.
- A. Kondo, H. Kajiro, H. Noguchi, L. Carlucci, D. M. Proserpio, G. Ciani, K. Kato, M. Takata, H. Seki, M. Sakamoto, Y. Hattori, F. Okino, K. Maeda, T. Ohba, K. Kaneko and H. Kanoh, J. Am. Chem. Soc., 2011, 133, 10512–10522 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- S. Onishi, T. Ohmori, T. Ohkubo, H. Noguchi, L. Di, Y. Hanzawa, H. Kanoh and K. Kaneko, Appl. Surf. Sci., 2002, 196, 81–88 CrossRef CAS; Y. Cheng, A. Kondo, H. Noguchi, H. Kajiro, K. Urita, T. Ohba, K. Kaneko and H. Kanoh, Langmuir, 2009, 25, 4510–4513 CrossRef PubMed; Y. Cheng, H. Kajiro, H. Noguchi, A. Kondo, T. Ohba, Y. Hattori, K. Kaneko and H. Kanoh, Langmuir, 2011, 27, 6905–6909 CrossRef PubMed; P. Kanoo, G. Mostafa, R. Matsuda, S. Kitagawa and T. K. Maji, Chem. Commun., 2011, 47, 8106–8108 RSC; S. S. Nagarkar, R. Das, P. Poddar and S. K. Ghosh, Inorg. Chem., 2012, 51, 8317–8321 CrossRef PubMed; B. Liu, L. Hou, Y. Y. Wang, H. Miao, L. Bao and Q. Z. Shi, Dalton Trans., 2012, 41, 3209–3213 RSC; J. Yang, Q. Yu, Q. Zhao, J. Liang, J. Dong and J. Li, Microporous Mesoporous Mater., 2012, 161, 154–159 CrossRef PubMed; A. Demessence and J. R. Long, Chem. – Eur. J., 2010, 16, 5902–5908 CrossRef PubMed; P. Kanoo, R. Sambhu and T. K. Maji, Inorg. Chem., 2011, 50, 400–402 CrossRef PubMed; A. Kondo, A. Chinen, H. Kajiro, T. Nakagawa, K. Kato, M. Takata, Y. Hattori, F. Okino, T. Ohba, K. Kaneko and H. Kanoh, Chem. – Eur. J., 2009, 15, 7549–7553 CrossRef PubMed; T. Fukushima, S. Horike, Y. Inubushi, K. Nakagawa, Y. Kubota, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2010, 49, 4820–4824 CrossRef PubMed.
- A. Kondo, H. Noguchi, L. Carlucci, D. M. Proserpio, G. Ciani, H. Kajiro, T. Ohba, H. Kanoh and K. Kaneko, J. Am. Chem. Soc., 2007, 129, 12362–12363 CrossRef CAS PubMed.
- Y. Inubushi, S. Horike, T. Fukushima, G. Akiyama, R. Matsuda and S. Kitagawa, Chem. Commun., 2010, 46, 9229–9231 RSC; M. Sadakiyo, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2011, 133, 11050–11053 CrossRef CAS PubMed; S. Horike, Y. Inubushi, T. Hori, T. Fukushima and S. Kitagawa, Chem. Sci., 2012, 3, 116–120 RSC; S.-i. Noro, Y. Hijikata, M. Inukai, T. Fukushima, S. Horike, M. Higuchi, S. Kitagawa, T. Akutagawa and T. Nakamura, Inorg. Chem., 2013, 52, 280–285 CrossRef PubMed; Y. Hijikata, S. Horike, M. Sugimoto, M. Inukai, T. Fukushima and S. Kitagawa, Inorg. Chem., 2013, 52, 3634–3642 CrossRef PubMed.
- Y. Wen, T. Sheng, S. Hu, X. Ma, C. Tan, Y. Wang, Z. Sun, Z. Xue and X. Wu, Chem. Commun., 2013, 49, 10644–10646 RSC.
- K. L. Gurunatha and T. K. Maji, Inorg. Chem., 2009, 48, 10886–10888 CrossRef CAS PubMed; P. Kanoo, R. Sambhu and T. K. Maji, Inorg. Chem., 2011, 50, 400–402 CrossRef PubMed.
- T. K. Maji, G. Mostafa, R. Matsuda and S. Kitagawa, J. Am. Chem. Soc., 2005, 127, 17152–17153 CrossRef CAS PubMed; R. Kitaura, K. Seki, G. Akiyama and S. Kitagawa, Angew. Chem., Int. Ed., 2003, 42, 428–431 CrossRef PubMed; A. Kondo, H. Noguchi, S. Ohnishi, H. Kajiro, A. Tohdoh, Y. Hattori, W.-C. Xu, H. Tanaka, H. Kanoh and K. Kaneko, Nano Lett., 2006, 6, 2581–2584 CrossRef PubMed; A. Kondo, T. Nakagawa, H. Kajiro, A. Chinen, Y. Hattori, F. Okino, T. Ohba, K. Kaneko and H. Kanoh, Inorg. Chem., 2010, 49, 9247–9252 CrossRef PubMed; D. Xiao, H. Chen, D. Sun, J. He, S. Yan, J. Yang, X. Wang, R. Yuan and E. Wang, CrystEngComm, 2012, 14, 2849–2858 RSC; A. Kondo, N. Kojima, H. Kajiro, H. Noguchi, Y. Hattori, F. Okino, K. Maeda, T. Ohba, K. Kaneko and H. Kanoh, J. Phys. Chem. C, 2012, 116, 4157–4162 Search PubMed.
- Y. Jiang, J. Huang, B. Kasumaj, G. Jeschke, M. Hunger, T. Mallat and A. Baiker, J. Am. Chem. Soc., 2009, 131, 2058–2059 CrossRef CAS PubMed.
- S. L. James, Chem. Soc. Rev., 2009, 38, 1744–1758 RSC; R. J. Puddephatt, Chem. Soc. Rev., 2008, 37, 2012–2027 RSC; P. W. Miller, M. Nieuwenhuyzen, J. P. H. Charmant and S. L. James, Inorg. Chem., 2008, 47, 8367–8379 CrossRef CAS PubMed; X. Xu, M. Nieuwenhuyzen and S. L. James, Angew. Chem., Int. Ed., 2002, 41, 764–767 CrossRef; J. Zhang, Q. Yang, Y. Zhu, H. Liu, Z. Chi and C.-Y. Su, Dalton Trans., 2014, 43, 15785–15790 RSC; X. Tan, L. Li, J. Zhang, X. Han, L. Jiang, F. Li and C.-Y. Su, Chem. Mater., 2012, 24, 480–485 CrossRef; J. Huang, X. Wang and J. Zhang, J. Inorg. Organomet. Polym. Mater., 2012, 22, 686–691 CrossRef; A. M. Bohnsack, I. A. Ibarra, V. I. Bakhmutov, V. M. Lynch and S. M. Humphrey, J. Am. Chem. Soc., 2013, 135, 16038–16041 CrossRef PubMed.
- A. M. Bohnsack, I. A. Ibarra, P. W. Hatfield, J. W. Yoon, Y. K. Hwang, J.-S. Chang and S. M. Humphrey, Chem. Commun., 2011, 47, 4899–4901 RSC; I. A. Ibarra, K. E. Tan, V. M. Lynch and S. M. Humphrey, Dalton Trans., 2012, 41, 3920–3923 RSC; I. A. Ibarra, J. W. Yoon, J.-S. Chang, S. K. Lee, V. M. Lynch and S. M. Humphrey, Inorg. Chem., 2012, 51, 12242–12247 CrossRef CAS PubMed; M. J. Plater, M. R. S. J. Foreman, E. Coronado, C. J. Gómez-García and A. M. Z. Slawin, J. Chem. Soc., Dalton Trans., 1999, 4209–4216 RSC; Q. Gao, M.-Y. Wu, L. Chen, F.-L. Jiang and M.-C. Hong, Inorg. Chem. Commun., 1999, 12, 1238–1241 CrossRef PubMed.
- X. Wang, J. Feng, J. Huang, J. Zhang, M. Pan and C.-Y. Su, CrystEngComm, 2010, 12, 725–729 RSC.
- E. C. Constable, G. Zhang, E. Coronado, C. E. Housecroft and M. Neuburger, CrystEngComm, 2010, 12, 2139–2145 RSC.
- S. M. Humphrey, S. E. Oungoulian, J. W. Yoon, Y. K. Hwang, E. R. Wisea and J.-S. Chang, Chem. Commun., 2008, 2891–2893 RSC.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. R. Murdock, N. W. McNutt, D. J. Keffer and D. M. Jenkins, J. Am. Chem. Soc., 2014, 136, 671–678 CrossRef CAS PubMed; S. Wang, Q. Yang, J. Zhang, X. Zhang, C. Zhao, L. Jiang and C.-Y. Su, Inorg. Chem., 2013, 52, 4198–4204 CrossRef PubMed; Q. Wang, J. Zhang, C.-F. Zhuang, Y. Tang and C.-Y. Su, Inorg. Chem., 2009, 48, 287–295 CrossRef PubMed.
- O. V. Dolomanov, A. J. Blake, N. R. Champness and M. Schröder, J. Appl. Crystallogr., 2003, 36, 1283–1284 CrossRef CAS.
- G. Mukherjee and K. Biradha, Chem. Commun., 2014, 50, 670–672 RSC.
- G. Ren, S. Liu, F. Ma, F. Wei, Q. Tang, Y. Yang, D. Liang, S. Li and Y. Chen, J. Mater. Chem., 2011, 21, 15909–15913 RSC; C.-X. Ren, L.-X. Cai, C. Chen, B. Tan, Y.-J. Zhang and J. Zhang, J. Mater. Chem. A, 2014, 2, 9015–9019 Search PubMed.
- X. Tan, X. Chen, J. Zhang and C.-Y. Su, Dalton Trans., 2012, 41, 3616–3619 RSC.
- H. Li, X. Tan and J. Zhang, Chin. J. Chem., 2015, 33, 141–146 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR spectra, characterization data of Py2PO and Py2PO′, selected bond lengths and bond angles. CCDC 1023746 and 1023747. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00138a |
| This journal is © the Partner Organisations 2015 |