DOI:
10.1039/C4QI00129J
(Research Article)
Inorg. Chem. Front., 2015,
2, 278-289
1D chains, 2D networks and 3D interdigitated frameworks of isoorotic acid or 4,4′-bipyridyl and isoorotic acid: syntheses, structures, and sorption properties†
Received
3rd September 2014
, Accepted 14th January 2015
First published on 14th January 2015
Abstract
Five new coordination polymers, {[Cu(H2iso)2]·2H2O}n (1), {Mn(Hiso)(H2O)3}n (2), {Mn(Hiso)(H2O)2}n (3), {[Cu(Hiso)(4,4′-bipy)(H2O)]·2H2O}n (4) and {[Ni(Hiso)(4,4′-bipy)(H2O)}]·0.5(4,4′-bipy)}n (5) (H3iso = 2,4-dihydroxypyrimidine-5-carboxylic acid or isoorotic acid; 4,4′-bipy = 4,4′-bipyridyl), have been synthesized using hydrothermal as well as room temperature solvent diffusion techniques and structurally characterized. Compounds 1 and 2 are 1D coordination polymers which are further connected by O–H⋯N, O–H⋯O hydrogen bonding interactions forming 3D supramolecular frameworks. Compound 3 is a 2D coordination polymer where 1D chains of {Mn(Hiso)(H2O)2} are linked by an oxo (μ2-O) bridge. Compound 4 consists of isoorotic acid and 4,4′-bipy, a mixed linker system. It forms a 2D sheet like extended structure and these sheets stack on each other to give a 3D supramolecular porous structure. In case of 5, Ni(Hiso) 1D chains are bridged by 4,4′-bipy forming a 2D network and networks are further interdigitated through hydrogen bonding interactions resulting in a 3D supramolecular framework with a 1D channel where 4,4′-bipy molecules are accommodated as guests. Compound 5 undergoes a structural contraction after the removal of the guest 4,4′-bipy and metal bound water molecules and shows a non-porous behaviour toward N2 at 77 K. However, at 195 K, it shows a stepwise uptake for both CO2 and C2H2, suggesting the flexible nature and polar pore surface of the framework.
Introduction
Molecular self-assembly through crystal engineering has emerged as an attractive approach for the designing and fabrication of new functional materials.1 Organic molecular arrays with a desirable topology have been realized by H-bonds, π⋯π and C–H⋯π non-covalent interactions2 whereas extended architectures in metal–organic frameworks (MOFs) or porous coordination polymers (PCPs) are sustained by non-covalent as well as covalent interactions.1,3 However, designable and structurally controllable self-assembled solids consisting of covalent as well as noncovalent interactions are of great interest4 as such frameworks are more flexible and dynamic in nature compared to the frameworks which are sustained only by covalent bonds.
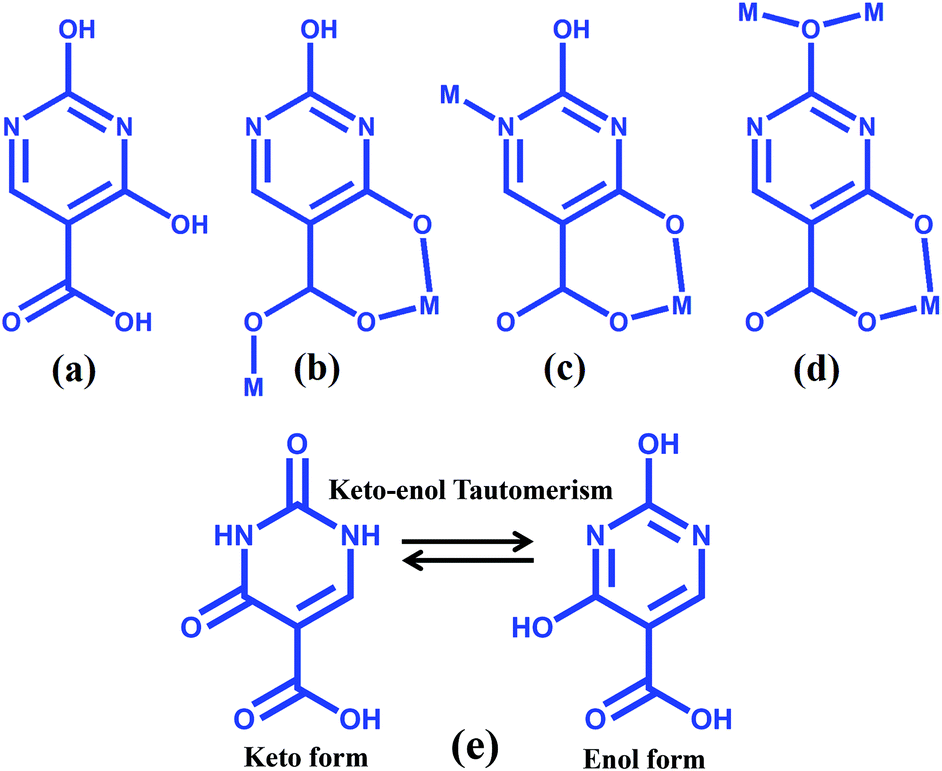
The manipulation of covalent and noncovalent interactions in MOFs may also provide structural flexibility and new functional molecular solids.4h In the realm of supramolecular chemistry, much attention has been paid for the synthesis of such kinds of materials due to their potential applications in molecular recognition,5 gas storage,6 separation,7 catalysis,8 and ion exchange.9 Novel structural motifs of MOFs, like honeycomb, grid, interdigitaion, different degrees of interpenetrations, three dimensional channels etc., have been synthesized by changing the binding modes, and the geometry of organic linkers and metal ions and they exhibit tunable properties.1–3,10 Using some specific organic linkers capable of exhibiting several H-bonding donor and acceptor sites, it has been possible to design supramolecular higher dimensional frameworks from both coordination bonds and the complementary H-bonds. 2,4-Dihydroxypyrimidine-5-carboxylic acid or isoorotic acid (H3iso) is one such linker and has hardly been exploited towards the synthesis of MOFs (Scheme 1).11 The ligand contains three pH dependent abstractable protons and two pyrimidine nitrogens, all together total five metal binding sites. For supramolecular interactions, isoorotic acid has three available H-bonding donor sites and two H-bonding acceptor sites for constructing a higher dimensional network. Depending upon the reaction conditions, e.g. pH, temperature, solvent etc., it is possible to exploit their different binding ability towards the synthesis of different architectures with different topologies and dimensionalities. Moreover, isoorotic acid is a substituted uracil, one of the four nucleobases present in the nucleic acid of RNA. Hence the assembly of such a versatile linker with suitable metal ions would result in bio-active frameworks. Furthermore, isoorotic acid is well known for its applications in medicine and on bonding with suitable metal ions it can be used as a therapeutic agent for cancer and has also showed antimicrobial activity.12 Moreover, isoorotic acid undergoes keto–enol tautomerism and we envisioned that organic linkers with such characteristics would facilitate toward the design and synthesis of tunable versatile supramolecular metal–organic structures by changing the reaction conditions with different metal ions.
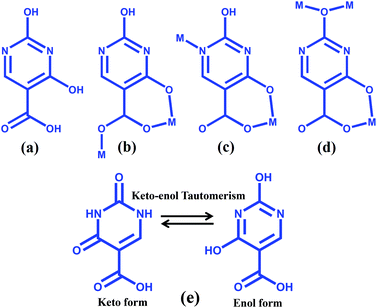 |
| | Scheme 1 (a)–(d) Schematic diagram of 2,4-dihydroxypyrimidine-5-carboxylic acid and its binding modes in the reported complexes; (e) Keto–enol tautomerism of H3iso. | |
Here, we present the synthesis and structural characterization of five new coordination polymers, {[Cu(H2iso)2]·2H2O}n (1), {Mn(Hiso)(H2O)3}n (2), {Mn(Hiso)(H2O)2}n (3), {[Cu(Hiso)(4,4′-bipy)(H2O)]·2H2O}n (4) and {[Ni(Hiso)(4,4′-bipy)(H2O)]·0.5(4,4′-bipy)}n (5) using the H3iso linker with different transition metal ions. 1 and 2 are 1D coordination polymers with different binding modes of H2/Hiso resulting in 2D and 3D supramolecular frameworks, respectively. 3 is a 2D coordination polymer and with the stacking of the 2D polymeric sheets it results in a porous framework with coordinatively unsaturated Mn(II) sites. On the other hand, compound 4 is a 2D supramolecular framework consisting of Hiso and 4,4′-bipy. In compound 5, rectangular sheets formed by the 1D Ni(Hiso) chains and 4,4′-bipy are interdigitated to form a 3D framework with the 1D rectangular channel filled by the guest 4,4′-bipy molecules. The desolvated framework shows a type-II N2 adsorption isotherm at 77 K but at 195 K it shows stepwise CO2 and C2H2 uptake. Moreover, at 293 K a two-step MeOH sorption with distinct hysteresis is observed.
Experimental section
Materials
All the reagents and solvents employed were commercially available and used as supplied without further purification. Ni(NO3)2·6H2O, Cu(NO3)2·2.5H2O, Mn(NO3)2·6H2O, 4,4′-bipyridyl and 2,4-dihydroxypyrimidine-5-carboxylic acid (H3iso) were obtained from Sigma Aldrich Chemical Co.
Physical measurements
Elemental analyses were carried out using a Perkin Elmer 2400 CHN analyzer. The IR spectra of all the compounds were recorded on a Bruker IFS 66v/S spectrophotometer using KBr pellets in the region 4000–400 cm−1. Thermogravimetric analyses (TGA) were carried out on METTLER TOLEDO TGA850 instrument in the temperature range of 30–600 °C under a nitrogen atmosphere (flow rate of 50 mL min−1) at a heating rate of 5 °C min−1. X-ray powder diffraction (PXRD) patterns of the compounds in different states were recorded on a Bruker D8 Discover instrument using Cu-Kα radiation.
Adsorption measurements
The adsorption isotherms of N2 (77 K) for compounds 3 and 5 and CO2 and C2H2 (195 K) for compound 5 were measured using QUANTACHROME QUADRASORB SI analyzer. The adsorbent samples (∼100–150 mg) were degassed at 200 and 180 °C for 3 and 5, respectively, under 0.1 Pa vacuum for about 12 h prior to the measurement of the isotherm. During the measurement helium gas at a certain pressure was introduced into the gas chamber and allowed to diffuse into the sample chamber by opening the valve. The change in pressure allowed an accurate determination of the volume of the total gas phase. The amount of gas adsorbed was calculated readily from the pressure difference (Pcal − Pe), where Pcal is the calculated pressure with no guest adsorption and Pe is the observed equilibrium pressure. The high pressure CO2 adsorption measurement for compound 5 at 298 K was carried out on a fully computer controlled volumetric BELSORP-HP, BEL JAPAN high pressure instrument. CO2 gas used for the measurement is of scientific/research grade with 99.999% purity. For the measurements, approximately 300 mg sample was taken in a stainless-steel sample holder and degassed at 180 °C for a period of 12 h under 0.1 Pa vacuum. The dead volume of the sample cell was measured with He gas of 99.999% purity. Non-ideal correction for CO2 and C2H2 gas was made by applying virial coefficients at 195 K. The P0 values of the CO2 and C2H2 measurements are 650 and 760 mmHg, respectively.
The adsorption of different solvents like water (H2O) and ethanol (C2H5OH) at 298 K and methanol (CH3OH) at 293 K was measured in the vapour state by using a BELSORP-aqua-3 analyzer. In all the measurements, compounds 3 and 5 (∼100–150 mg) were placed in the sample tube which were prepared by heating at 200 and 180 °C, respectively, for about 12 h under vacuum prior to the measurement of the isotherms. Dead volume was measured with helium gas. The adsorbate vapour was placed into the sample tube, then the change of pressure was monitored and the degree of adsorption was determined by the decrease in pressure at the equilibrium state. All operations were computer controlled and automatic.
Synthesis of {[Cu(H2iso)2]·2H2O}n (1).
Cu(NO3)2·2.5H2O (0.116 g, 0.5 mmol), H3iso (0.078 g, 0.5 mmol) and KOH (0.056 g, 1 mmol) were mixed in 6 mL of water and placed in a 20 mL Teflon-lined vessel. This vessel was placed in a stainless steel autoclave, sealed and heated to 120 °C under autogenous pressure for 24 h and cooled to room temperature under an open atmosphere. Green coloured square shaped crystals were isolated by decanting the supernatant liquid and washed thoroughly with water and air dried before the single crystal X-ray diffraction measurement. The phase purity of the bulk compound prepared was checked by matching the PXRD pattern of the bulk compound with that of the simulated PXRD (Fig. S1†). Yield: 80% relative to Cu(II). Anal. Calcd for C10H8CuN4O10, C, 29.45; H, 1.97; N, 13.74. Found: C, 29.41; H, 1.92; N, 13.78. FT-IR (KBr cm−1): 3550 br; 2945 w; 2785 w; 1640 s, 1528 w, 1313 w, 1205 s (Fig. S2†).
Synthesis of {Mn(Hiso)(H2O)3}n (2).
A 12.5 mL aqueous solution of H3iso (0.078 g, 0.5 mmol) was mixed with a methanol solution of 4,4′-bipy (0.078 g, 0.5 mmol in 12.5 mL solution) and KOH (0.056 g, 1 mmol) and then stirred for 30 min. Mn(NO3)2·6H2O (0.143 g, 0.5 mmol) was dissolved in water (25 mL) and then a ligand solution (2 mL) was layered above the metal solution (2 mL) and after 5–6 days colourless cubic shaped crystals were obtained. The bulk compound was prepared and phase purity was checked by matching the PXRD pattern of the bulk compound with that of the simulated PXRD (Fig. S3†). Yield: 60% relative to Mn(II). Anal. Calcd for C5H8MnN2O7, C, 22.82; H, 3.05; N, 10.64. Found: C, 22.78; H, 3.00; N, 10.60. FT-IR (KBr cm−1): 3580 br; 2790 w; 1635 s, 1524 w, 1312 w, 1207 s (Fig. S4†).
Synthesis of {Mn(Hiso)(H2O)2}n (3).
The synthetic procedure for compound 3 is similar as that used for compound 1 except that Cu(NO3)2·2.5H2O was replaced by Mn(NO3)·6H2O (0.143 g, 0.5 mmol). Colourless needle shaped crystals were filtered, washed with deionized water and air dried. The bulk compound was prepared and phase purity was checked by matching the PXRD pattern of the bulk compound with that of the simulated PXRD (Fig. S5†). Yield: 75% relative to Mn(II). Anal. calcd for C5H6MnN2O6: C, 24.48; H, 2.44; N, 11.42. Found: C, 24.70; H, 2.56; N, 11.30. FT-IR (KBr cm−1): 3477 br; 2937 w; 2796 w; 1643 s, 1525 w, 1311 w, 1207 s (Fig. S6†).
{[Cu(Hiso)(4,4′-bipy)(H2O)]·2H2O}n (4).
A mixture of H3iso (0.078 g, 0.5 mmol), Cu(NO3)2·2.5H2O (0.116 g, 0.5 mmol), KOH (0.056 g, 1 mmol) and 8 mL water was stirred in a 23 mL Teflon container for 2 h. After the addition of 4,4′-bipy (0.078 g, 0.5 mmol) into the above mentioned reaction mixture, the Teflon container was kept in a steel autoclave, heated to 160 °C for 24 h. Green coloured single crystals were obtained, then separated and washed with water. The crystals were found to be unstable at room temperature, therefore single crystals of very good quality were covered with paraffin oil for the single crystal X-ray diffraction study. Pure phase bulk compound preparation is hardly achievable probably due to the instability of the framework at room temperature (Fig. S7†). Yield: 65% relative to Cu(II). Anal. Calcd for C10H11Cu4O7N3, C, 34.43; H, 3.17; N, 12.04. Found: C, 34.79; H, 3.21; N, 12.19. FT-IR (KBr cm−1): 3590 br; 2935 w; 2794 w; 1641 s, 1525 w, 1310 w, 1209 s (Fig. S8†).
{[Ni(Hiso)(4,4′-bipy)(H2O)]·0.5(4,4′-bipy)}n (5).
Compound 5 is synthesized using a similar methodology as that of compound 4 except that Cu(NO3)2·2.5H2O was replaced by Ni(NO3)2·6H2O (0.145 g, 0.5 mmol). Blue coloured crystals were isolated, washed thoroughly with water and EtOH and dried at room temperature. Yield: 78% relative to Ni(II). Anal. Calcd for C5H2NiN4O4, C, 50.85; H, 2.46; N, 15.11. Found: C, 51.64; H, 3.43; N, 15.06. FT-IR (KBr cm−1): 3324 br; 2796 w; 1625 s, 1527 w; 1315 s (Fig. S9†).
Single crystal X-ray diffraction
Suitable single crystals of compounds 1–5 were mounted on a thin glass fiber with commercially available super glue. X-ray single crystal structural data were collected on a Bruker Smart-CCD diffractometer equipped with a normal focus, 2.4 kW sealed tube X-ray source with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) operating at 50 kV and 30 mA. The programme SAINT13 was used for the integration of diffraction profiles and absorption correction was made with the SADABS14 programme. All the structures were solved by SIR 9215 and refined by the full matrix least squares method using SHELXL-97.16 All hydrogen atoms were fixed by HFIX and placed in ideal positions. The potential solvent accessible area or void space was calculated using the PLATON17 multipurpose crystallographic software. Selected bond distances, angles and hydrogen bonds are shown in Tables S1–S8.† The coordinates, anisotropic displacement parameters, and torsion angles for all compounds are submitted as ESI† in CIF format. All crystallographic and structure refinement data of the compounds are summarized in Table 1. All calculations were carried out using PLATON,17 and WinGX system, Ver 1.70.01.18 The CIF files have been submitted to CCDC. The CCDC numbers are 1022424–1022428 for compounds 1–5, respectively.
Table 1 Crystal structure refinement parameters of 1, 2, 3, 4 and 5
| Empirical formula |
C10H4CuN4O10 (1) |
C5H7MnN2O7 (2) |
C5H5MnN2O6 (3) |
C10H11N3CuO7 (4) |
C20H14N5NiO5 (5) |
|
R = ∑||Fo| − |Fc||/∑|Fo|.
R
w = [∑{w(Fo2 − Fc2)2}/∑{w(Fo2)2}]1/2.
|
|
M
r
|
403.72 |
263.07 |
244.05 |
348.77 |
462.04 |
| Crystal system |
Monoclinic |
Monoclinic |
Orthorhombic |
Monoclinic |
Monoclinic |
| Space group |
P21/n (no. 14) |
P21/n (no. 14) |
Pbca (no. 61) |
P21/n (no. 14) |
P
2/n (no. 13) |
|
a (Å) |
5.2386(10) |
5.2909(2) |
12.9385(3) |
14.154(3) |
10.3949(2) |
|
b (Å) |
11.3793(17) |
12.0901(5) |
6.7310(1) |
6.6209(13) |
11.3275(2) |
|
c (Å) |
10.8544(18) |
13.9601(5) |
17.0240(4) |
15.047(3) |
18.6129(3) |
|
α (°) |
90 |
90 |
90 |
90 |
90 |
|
β (°) |
92.222(11) |
90.199(2) |
90 |
117.39(3) |
102.99990(10) |
|
γ (°) |
90 |
90 |
90 |
90 |
90 |
|
V (Å3) |
646.56(19) |
892.99(6) |
1482.60(5) |
1252.0(6) |
2135.47(7) |
|
Z
|
2 |
4 |
8 |
4 |
4 |
|
T (K) |
293 |
293 |
293 |
293 |
293 |
|
D
c (g cm−3) |
2.074 |
1.957 |
2.187 |
1.850 |
1.437 |
|
μ (mm−1) |
1.763 |
1.500 |
1.789 |
1.785 |
0.949 |
|
F (000) |
402 |
532 |
976 |
708 |
944 |
|
θ
max (°) |
23.8 |
29.0 |
22.2 |
31.2 |
25.7 |
|
λ (Mo Kα)(Å) |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| Tot. data |
4707 |
5162 |
8983 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 533 533 |
16479 |
| Unique data, Rint |
964, 0.060 |
2106, 0.021 |
930, 0.041 |
3547, 0.069 |
4069, 0.036 |
| Data [I > 2σ(I)] |
749 |
1695 |
843 |
2946 |
3449 |
|
R
|
0.0610 |
0.0319 |
0.0260 |
0.0908 |
0.0389 |
|
R
wb |
0.1868 |
0.0826 |
0.1069 |
0.2509 |
0.0881 |
|
S
|
1.10 |
1.06 |
1.28 |
1.08 |
1.03 |
Results and discussion
Structural description of {[Cu(H2iso)2]·H2O}n (1)
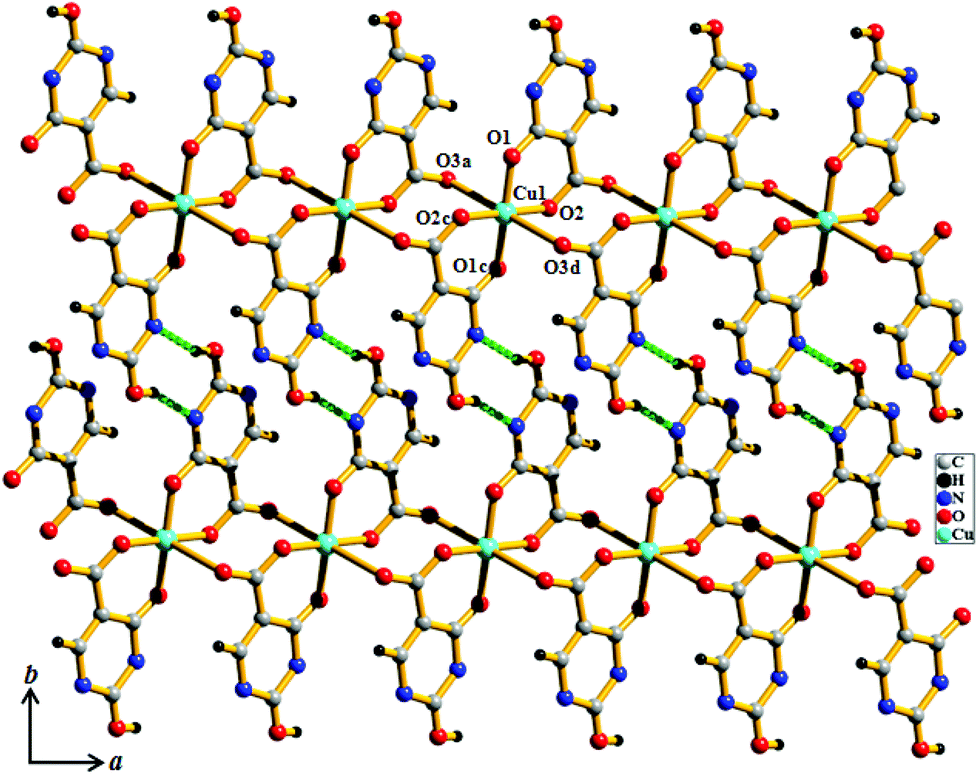
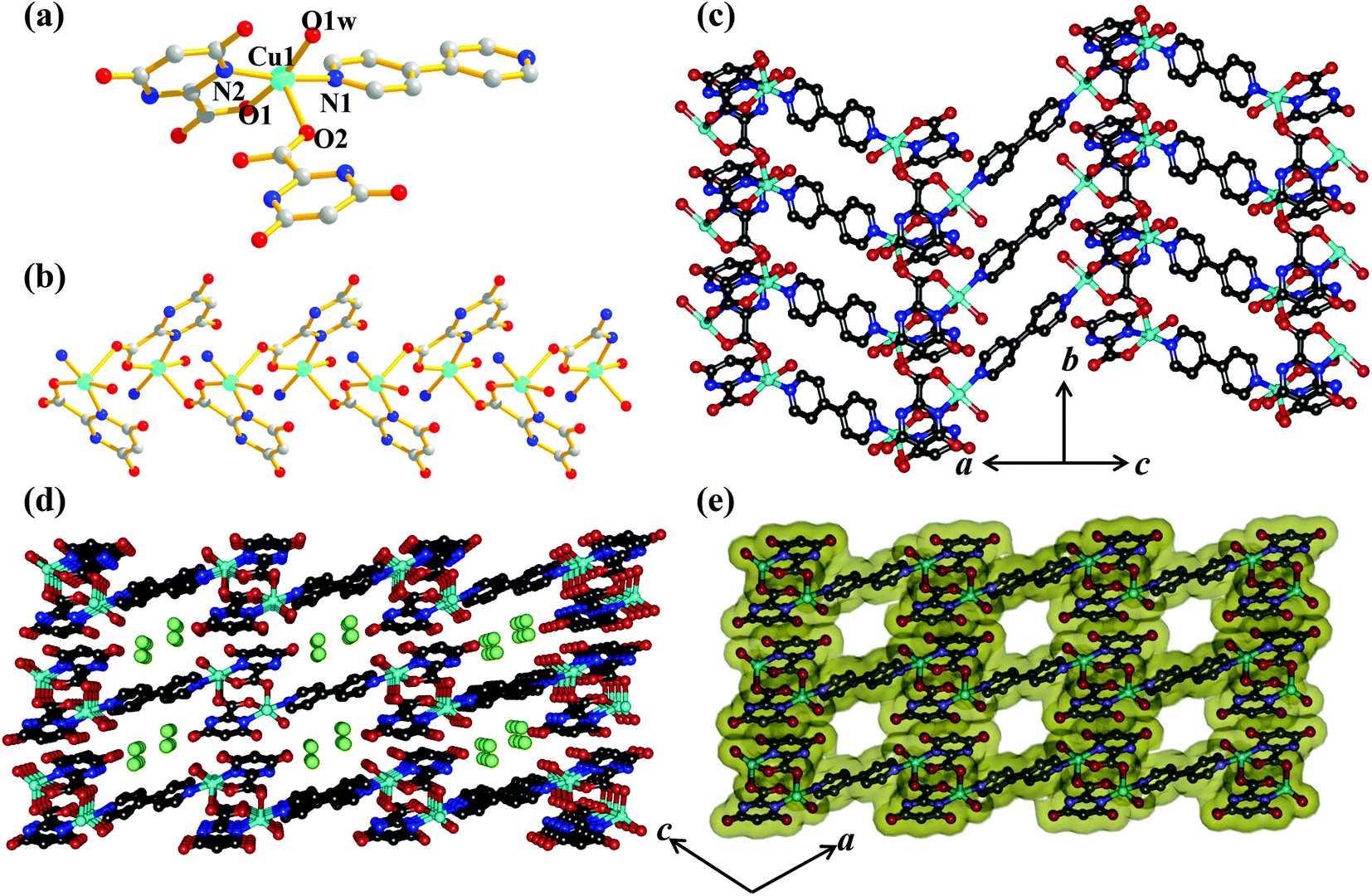
Compound 1 crystallizes in the P21/n space group. The structure determination of 1 reveals a 1D coordination chain of Cu(II) bridged through the H2iso ligand (Fig. 1), where each Cu(II) resides in a special position. Each Cu(II) is linked to two chelated symmetry related H2iso ligands through hydroxo oxygens (O1, O1c) and carboxylate oxygens (O2, O2c) in the equatorial plane. The trans axial positions are occupied by two other oxygen atoms (O3a, O3d) from the two different H2iso ligands, resulting in a 4 + 2 coordination environment around each Cu(II). Cu(II)–O bond distances are in the range of 1.904(5)–2.705(5) Å. Each H2iso linker acts as a monoanionic bridging tridentate ligand and chelates to one Cu(II) and connects another Cu(II) center through syn–anti carboxylate bridging resulting in a 1D coordination chain. The 1D chains are connected via O–H⋯N hydrogen bonding, through the pendent hydroxyl and pyrimidine nitrogen atoms, forming a 2D supramolecular sheet along the ab plane (Fig. 1 and 2). The 2D sheets are stacked along the crystallographic c-axis, that result in small channels occupied by the guest water molecules.
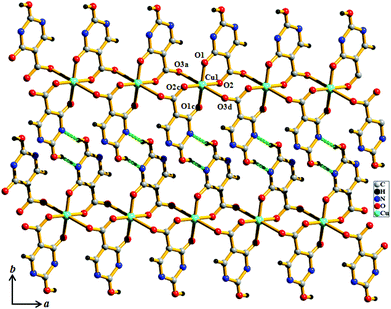 |
| | Fig. 1 View of the coordination environment of Cu(II) and H-bonding between the 1D chains along the ab plane in compound 1. | |
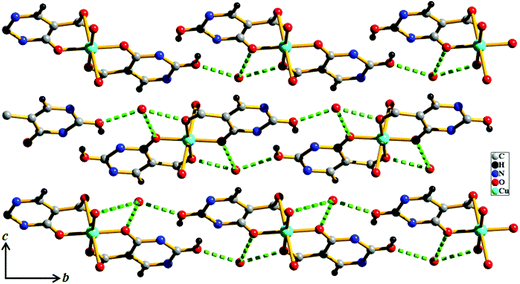 |
| | Fig. 2 View of H-bonding between guest water molecules and the Hiso linker along the a direction in compound 1. | |
Structural description of {Mn(Hiso)(H2O)3}n (2)
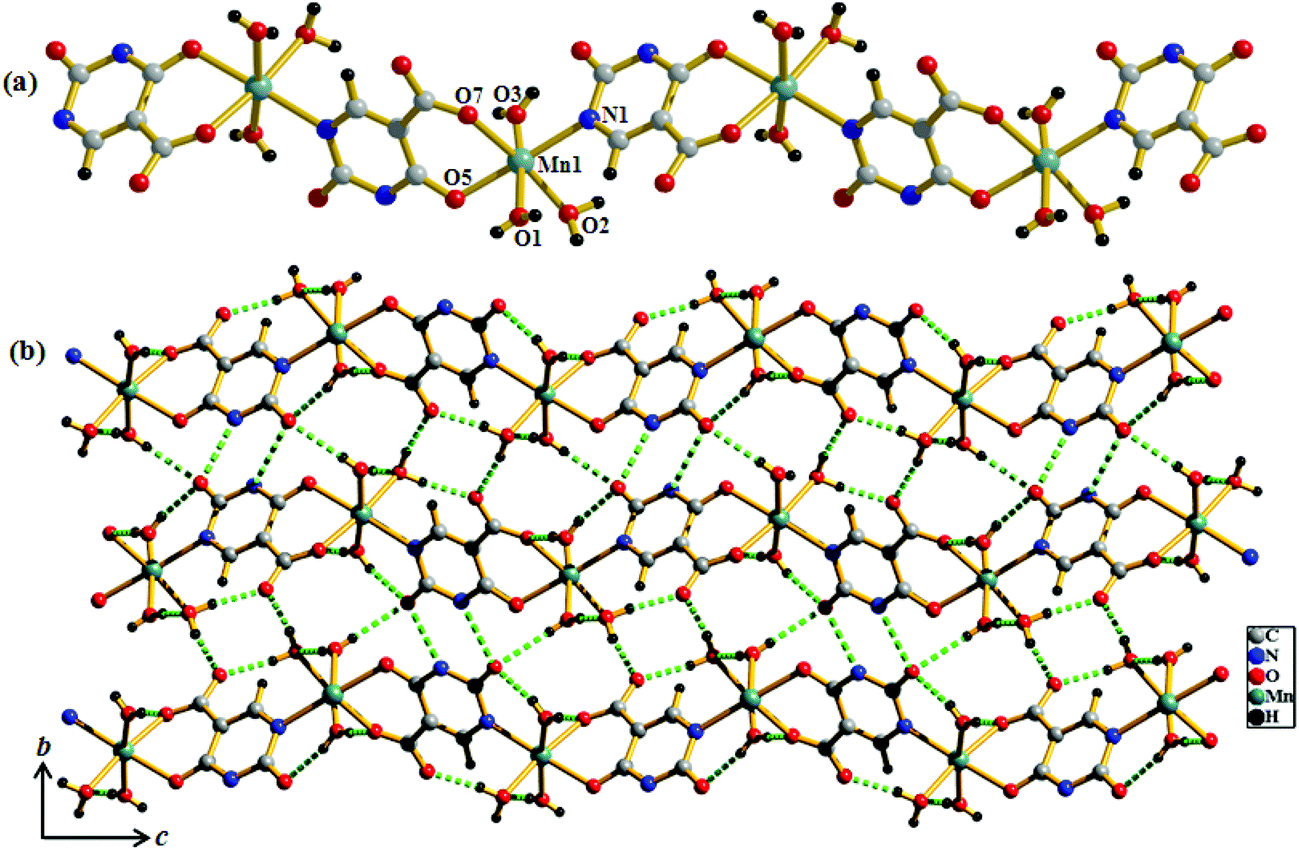
Compound 2 crystallizes in the monoclinic system with the space group P21/n. The structure determination reveals that a 1D coordination chain of Mn(II) is formed by bridging the Hiso ligand (Fig. 3a). Each Hiso ligand chelates to one Mn(II) by the hydroxyl oxygen (O5) and one carboxylate oxygen (O7) and connects to another Mn(II) through one pyrimidine nitrogen (N1), therefore acting as a bridging tridentate dianionic ligand. One water molecule O2, and two other oxygen atoms (O5, O7) and one nitrogen (N1) atom from the two Hiso ligands forming the equatorial plane around Mn(II) and the axial positions are occupied by two other water molecules (O1 and O3). The degree of distortion from the ideal octahedral geometry around each Mn(II) is reflected in the cisoid (83.23(7)–96.85(8)°) and transoid angles (170.51(9)–178.42(9)°). The Mn(II)–O and Mn(II)–N1 bond distances are in the range of 2.135 (2)–2.207(2) Å and 2.248(2) Å, respectively. The Mn(II)⋯Mn(II) separation along the coordination chain is 7.727 Å. There are several intra- and inter-chain H-bonding interactions among the coordinated water molecules through which 2D supramolecular sheets are formed. These 2D layers are stacked successively along the c-direction and grow the 3D supramolecular framework (Fig. 3b).
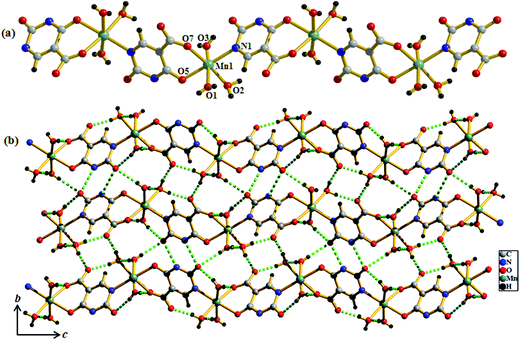 |
| | Fig. 3 (a) View of the coordination environment of Mn(II) in compound 2; (b) view of H-bonding interactions between the 1D chains on the bc plane in compound 2. | |
Structural description of {Mn(Hiso)(H2O)2}n (3)
Compound 3 crystallizes in the orthorhombic crystal system with the space group Pbca. The structure determination reveals a 2D coordination network of Mn(II) bridged by Hiso ligand (Fig. 4a). The binding mode of the Hiso ligand is different from 2 and each Hiso ligand chelates to one Mn(II) and binds the other two Mn(II) centers through μ-oxo bridging, thus acting as a tridentate dianionic linker. Two chelated oxygen atoms (O5 and O3) from one Hiso, one hydroxyl oxygen (O6) from another Hiso and one coordinated water molecule (O2) form an equatorial plane around each Mn(II). The axial sites are occupied by the coordinated water molecule (O1) and the hydroxyl oxygen (O6) from another Hiso ligand. Each Mn(II) is in a slightly distorted octahedral environment, reflected in the cisoid (79.81–104.74°) and the transoid angles (164.98(13)–175.70(12)°). The Mn1–O6–Mn1 angle is about 100.19(11)°. Mn(II)–O bond distances are in the range of 2.117(4)–2.337(4) Å. Each 2D sinusoidal network undergoes several H-bonding interactions among the coordinated water molecules (O1, O2), the free carboxylate oxygen atom (O4) (O1⋯O2, 2.884 Å, O1⋯O4, 2.738 Å, and O2⋯O4, 2.683 Å) and the hydroxyl oxygen (O5) atom (O1⋯O5, 2.7 Å) and finally forms a 3D supramolecular framework (Fig. 4b). These directional H-bonding interactions organize the 3D framework in such a way that upon the removal of the coordinated water molecules the 1D channel along the crystallographic b-axis with open Mn(II) sites is realized (Fig. 5). The dimension of the 1D channel is about 3.6 × 4.1 Å2.19 In the 2D network, the Mn(II)⋯Mn(II) separation along the hydroxyl bridge is 2.906 Å, the nearest distance along the Hiso ligand is 7.002 Å and in between the 2D network the nearest separation is about 7.014 Å.
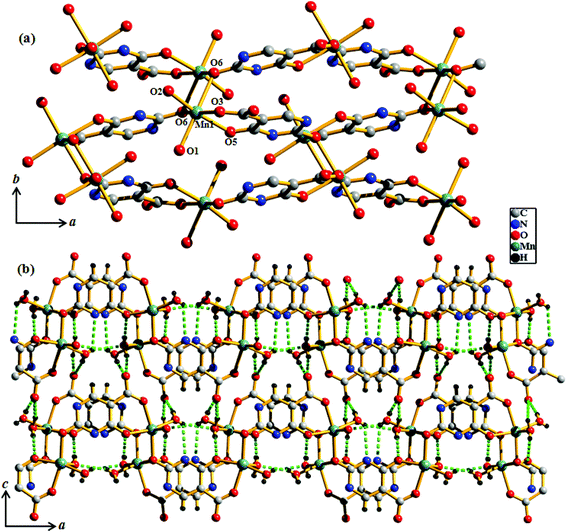 |
| | Fig. 4 (a) View of the coordination environment of Mn(II) in compound 3; (b) view of H-bonding between the 1D chains on the ac plane in compound 3. | |
 |
| | Fig. 5 View of the 1D channel along the b-axis after the removal of coordinated water molecules in compound 3. Cyan: Mn(II), grey: C, pale blue: N, red: O and white: H. | |
Structural description of {[Cu(Hiso)(4,4′-bipy)(H2O)]·2H2O}n (4)
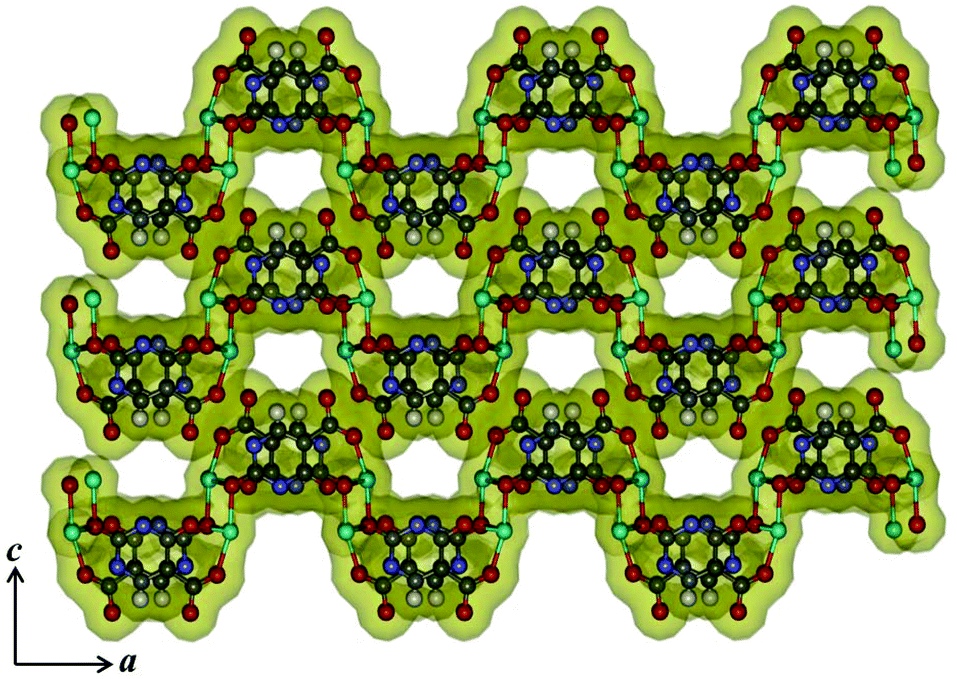
Unlike compounds 1–3, compound 4 consists of the mixed linker system where Hiso and 4,4′-bipy were assembled with Cu(II) to get a 2D supramolecular structure. Single crystal X-ray diffraction analysis suggests that each Cu(II) is five-coordinated with two oxygen (O1, O2) atoms from two Hiso, one oxygen from the water molecule (O1W) and two nitrogen atoms from one 4,4′-bipy and one Hiso (Fig. 6a). The Cu(II)–O and Cu(II)–N bond distances are in the range of 1.955(4)–2.287(4) and 1.997–1.989 Å, respectively. The carboxylate group of Hiso bridges the two Cu(II) centers along the b-axis to form a 1D chain like structure as shown in Fig. 6b. These 1D chains are further connected by 4,4′-bipy along the c-axis to form a 2D undulated sheet like extended structure (Fig. 6c). The unusual coordination behavior of Hiso actually restricts the structure to two dimensions. The view along the b-axis shows that the 2D sheet has a stair like shape due to the syn–anti bridging mode of Hiso (Fig. 6d). These 2D sheets stack along the (10–1) direction to form a 1D distorted square shaped (dimension ∼3.1 × 3.3 Å2) channel along the b-axis (Fig. 6d and e). These channels are filled with guest water molecules which are hydrogen bonded. Here, the pendent hydroxyl oxygen atom (O4) of Hiso forms a strong hydrogen bond with the hydrogen atoms of guest water (O3W and O4W) molecules and thus through the guest water molecules the 2D sheets are further tethered (Fig. S10†). The complete removal of the guest solvent molecules creates a void space of 23% of the total cell volume.
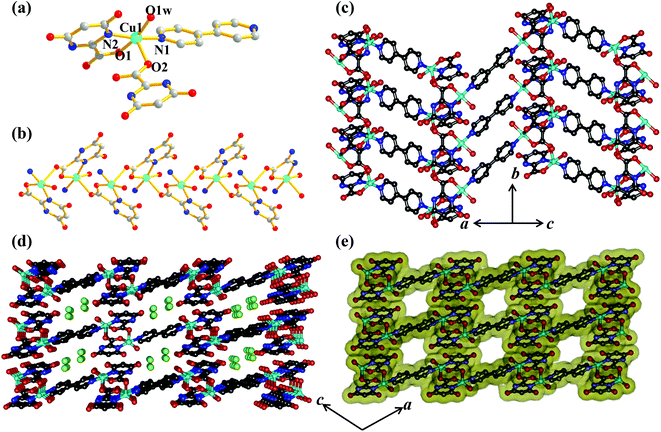 |
| | Fig. 6 (a) View of the coordination environment of Cu(II) in compound 4; (b) 1D chain of [Cu(Hiso)]n formed by a syn–anti carboxylate bridging; (c) view of the 2D sheet of {[Cu(Hiso)(4,4′-bipy)(H2O)]}n; (d) stacking of the 2D sheet along the b-axis showing the stairs like shape of a 2D layer; (e) surface added view along the b-axis showing 1D supramolecular channels occupied by guest water molecules. Cyan: Cu(II), grey: C, red: O blue: N and green: guest water molecules. | |
Structural description of {[Ni(Hiso)(4,4′-bipy)(H2O)]·0.5(4,4′-bipy)}n (5)
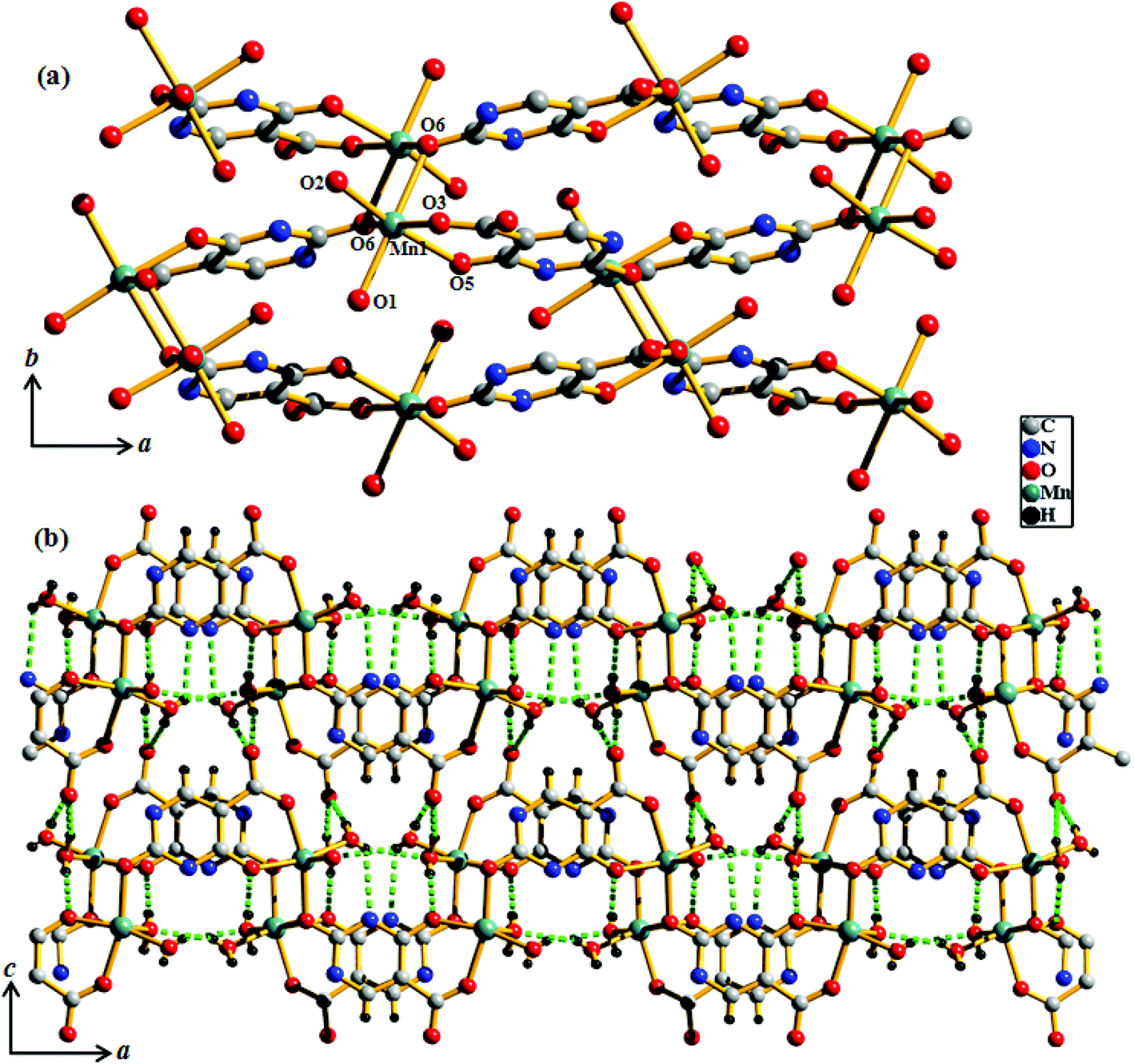
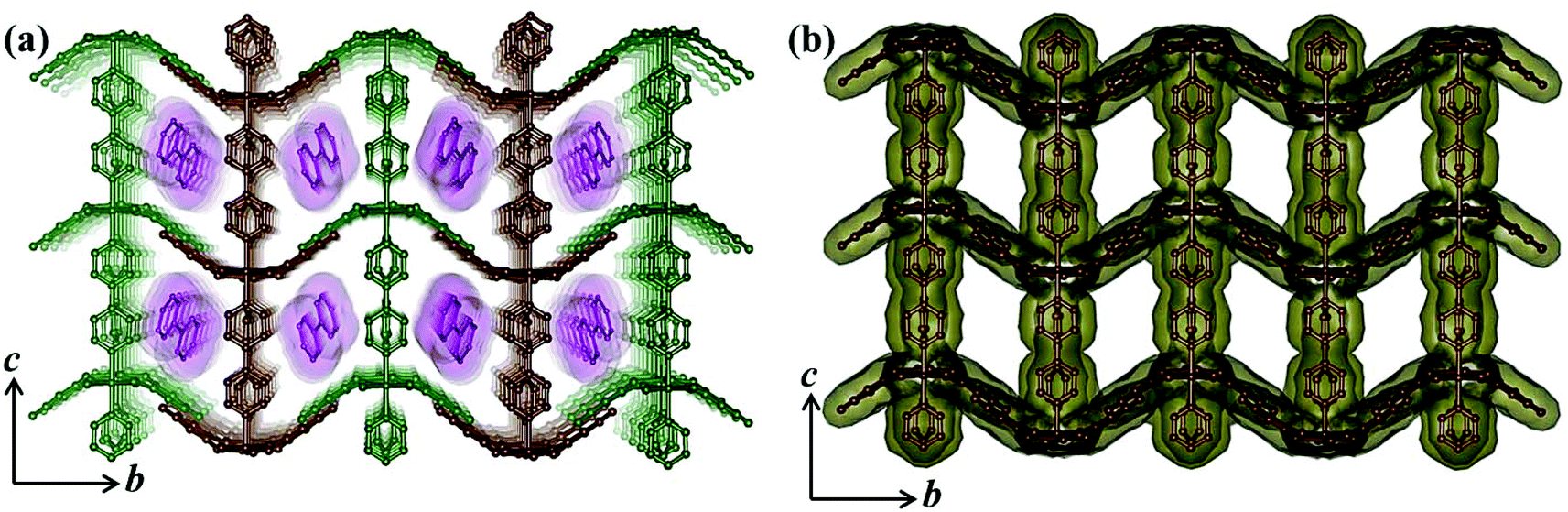
Compound 5 crystallizes in the monoclinic crystal system with the P2/n space group. The structure determination by the single crystal X-ray diffraction experiment reveals a 3D supramolecular interdigitated coordination framework of Ni(II) composed of Hiso and 4,4′-bipy linkers. In the asymmetric unit, there are two crystallographically independent Ni(II) centers residing in the special position, one and half 4,4′-bipy, one Hiso and one water molecule (Fig. 7). Each H3iso ligand chelates to a Ni2 through a carboxylate oxygen (O3) and a hydroxyl oxygen (O4) and connects to Ni1 through another carboxylate oxygen (O2). Thus tridentate Hiso forms 1D {Ni(Hiso)} chains along the crystallographic a-axis, which are connected by the 4,4′-bipy pillar forming a 2D rectangular grid like coordination network in the ab plane (Fig. S11†). Two water molecules (O1, O1_a) and two carboxylate oxygen atoms (O2, O2_a) (a = −1 + x, y, z) symbolize that this oxygen molecule (O2_a) is symmetrically equivalent to O2 and forms an equatorial plane around each Ni1 and the axial positions are occupied by two nitrogen atoms (N1, N2) from the two 4,4′-bipy pillars. In the case of Ni2, four oxygen atoms (O3, O4, O3_a, O4_a) from two chelated Hiso form the equatorial plane and the axial positions are occupied by the two nitrogen atoms (N3, N4) from the two 4,4′-bipy. The Ni1–O bond distances are in the range of 2.063(3)–2.083(4) Å which are slightly higher than the Ni2–O bond distances (2.014(3)–2.025(4) Å), whereas the Ni–N bond distances are in the range of 2.093(7)–2.158(7) Å. It is interesting to note that the orientation of the 4,4′-bipy pillars connected to Ni1 and Ni2 is perpendicular and parallel to the ab plane, respectively. One rectangular 2D grid is connected by H-bonding interactions between the pendent OH– of Hiso and the coordinated water molecule O1 (O5⋯O1 = 2.723 Å) is attached to Ni1 atoms and pyrimidine N-atoms (O5⋯N5 = 2.826 Å) from another 2D grid to architect a 3D interdigitated supramolecular framework (Fig. 8). The resulting supramolecular framework contains a 1D rhombic channel along the crystallographic a-axis which is occupied by guest 4,4′-bipy molecules and there is no effective channel along the b and c-axes. The dimension of the channel is about 5.6 × 7.7 Å2, which provides a void space of ∼38.4% to the total crystal volume.17 The Ni⋯Ni separation in the 2D grid is 5.214 and 11.328 Å along the Hiso and 4,4′-bipy, respectively.
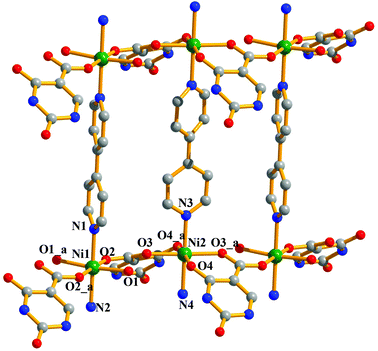 |
| | Fig. 7 View of the coordination environment of Ni(II) in the 2D network of compound 5. Green: Ni, grey: C, red: O, blue: N. | |
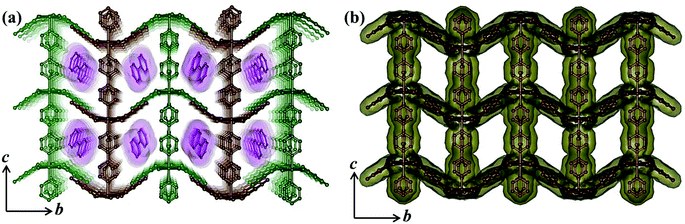 |
| | Fig. 8 (a) View of the 3D interdigitated framework of 5 showing 1D channels occupied by the guest 4,4′-bipy molecules; (b) view of the 1D channels along the a-axis after the removal of guest molecules. | |
Framework stability (TGA and PXRD analysis)
The TGA curve of compound 1 (Fig. S12†) shows a 1.4% weight loss in the temperature range of 30–80 °C corresponding to the loss of one water molecule (calc. 1.7% weight) and it is thermally stable up to 215 °C. Compound 2 shows a 2.98% weight loss in the temperature range of 30–100 °C corresponding to three water molecules (calc. 3.1% weight) and starts degrading at 110 °C into an unidentified product (Fig. S12†). The TGA curve for 3 shows that the compound is stable up to 195 °C without any weight loss and then ∼13.3% of weight loss is observed in the temperature range of ∼195–260 °C which corresponds to the loss of two coordinated water molecules (calc. 14.7% weight) (Fig. S12†). The dehydrated framework is stable up to ∼310 °C and at higher temperatures the release of the coordinated water molecules is attributed to the strong binding with Mn(II) as well as the H-bonding interaction with the O– atoms of Hiso. Compound 4 shows 4.7% weight loss in the temperature range of 30–90 °C corresponding to three water molecules (calc. 14.7% weight) followed by slow degradation leading to thermal stability up to 180 °C (Fig. S12†). In the case of compound 5, the loss of the guest 4,4′-bipy and coordinated water molecules occurs in the temperature range of ∼145–245 °C (obs. 19.55% and calc. 20.57%) and then the guest removed framework is stable up to ∼360 °C and upon further heating the framework decomposes (Fig. S12†).
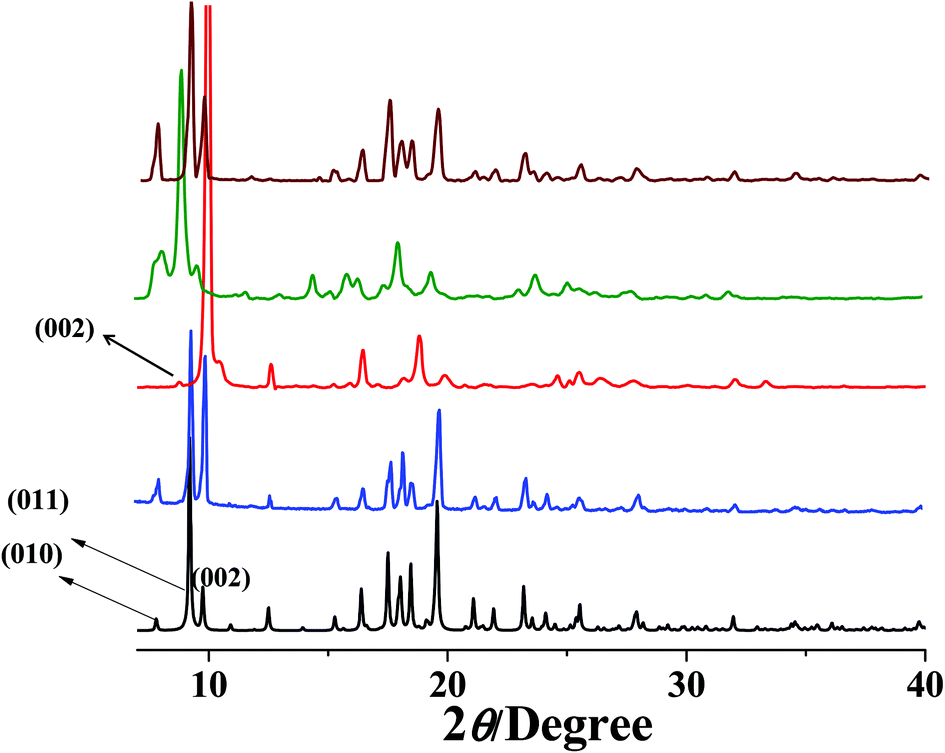
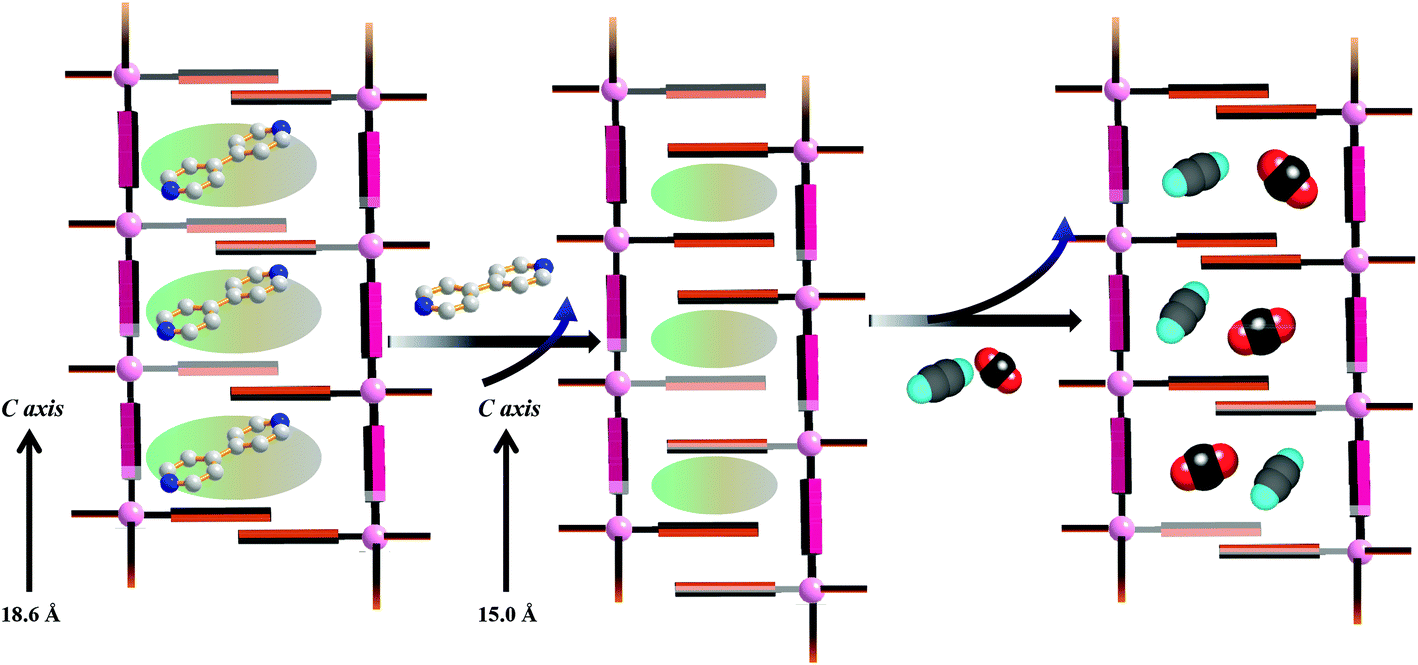
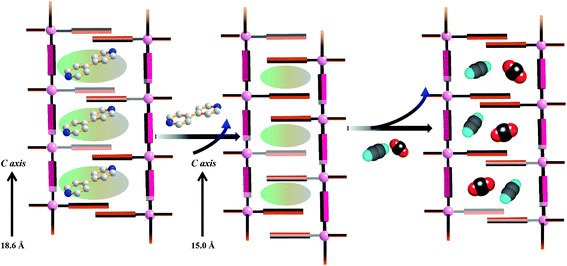
PXRD measurements were carried out to understand the phase purity and stability of all the compounds. The PXRD patterns of all the as-synthesized compounds 1–5 are completely matching with the simulated pattern indicating the phase purity of the samples (Fig. S1, S3, S5, S7† and Fig. 9). The desolvated compound 3 (3′) shows changes in the diffraction patterns compared to that of the as-synthesized one (Fig. S5†). The peak corresponding to the (002) reflection shifts to a lower angle in the desolvated phase and after rehydration it shows a similar PXRD pattern as that of the as-synthesized one indicating structural reversibility. Similarly, a change in the peak position of the (011) reflection and a disappearance of the peak corresponding to the (010) reflection can be observed in the desolvated framework of 5 (i.e., 5′) (Fig. 9). To get a better insight into the desolvated state we further indexed the powder pattern using the TREOR 90 program20 and it suggests distinct changes in the cell parameters. It shows an orthorhombic crystal system and the cell parameters are a = 10.8106 Å, b = 10.8106 Å, c = 15.0286 Å, V = 1756.373 Å3 (see ESI†). As can be seen from the indexing of PXRD data the c-axis shortens upon desolvation, hence we expect an overall structural contraction on desolvation. It is assumed that the H-bonded interdigitated 2D networks can easily slide on one another along the c-axis upon the removal of guest molecules. This type of structural change is characteristic of an interdigitated structure.2d On rehydration it does not come back to the as-synthesized structure and even exposure to methanol vapour shows different PXRD patterns. These guest dependent different structural changes clearly point to the framework flexibility on different guest loadings. A possible rearrangement path is shown in Scheme 2 where the 2D layers shift with respect to each other.
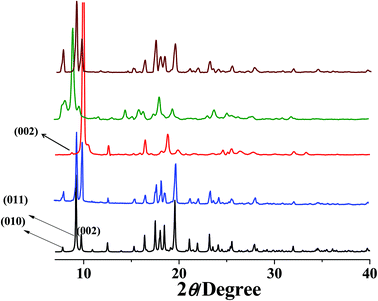 |
| | Fig. 9 PXRD patterns of compound 5 in different states (a) simulated from X-ray single crystal data; (b) as-synthesized; (c) dehydrated at 150 °C; (d) rehydrated by exposing the water vapour for 72 h; (e) exposed to the MeOH vapour for 72 h. | |
 |
| | Scheme 2 Possible guest responsive structural rearrangement in compound 5. | |
Adsorption properties
Sorption experiments with different adsorbates were carried out to confirm the permanent porosity in the desolvated state of compound 3 (i.e., 3′). At 77 K, 3′ exhibits a type-II adsorption profile with N2 (kinetic diameter = 3.64 Å),21 suggesting only surface adsorption (Fig. S13†). Probably the pore size of desolvated 3′ is not sufficient enough for the diffusion of N2. We have also measured the solvent vapour adsorption properties of compound 3′. At 298 K the desolvated framework shows an almost linear uptake of water vapour and at a relative vapour pressure of P/P0 ∼ 0.98, the uptake amount is ∼162 mL g−1 (Fig. S14†). This final uptake amount corresponds to 1.5 water molecules per formula. Interestingly we observed a large hysteresis in the desorption path and till a very low pressure there is almost no desorption of water molecules. This observation suggests a very strong interaction of water molecules with the unsaturated Mn(II) sites in the pore surface. But an increase in the kinetic diameter of the adsorbate molecules totally restricts the diffusion into the pores as can be realized from the adsorption measurement of methanol vapour (kinetic diameter ∼4 Å). Almost no uptake was observed for methanol vapour at 293 K.
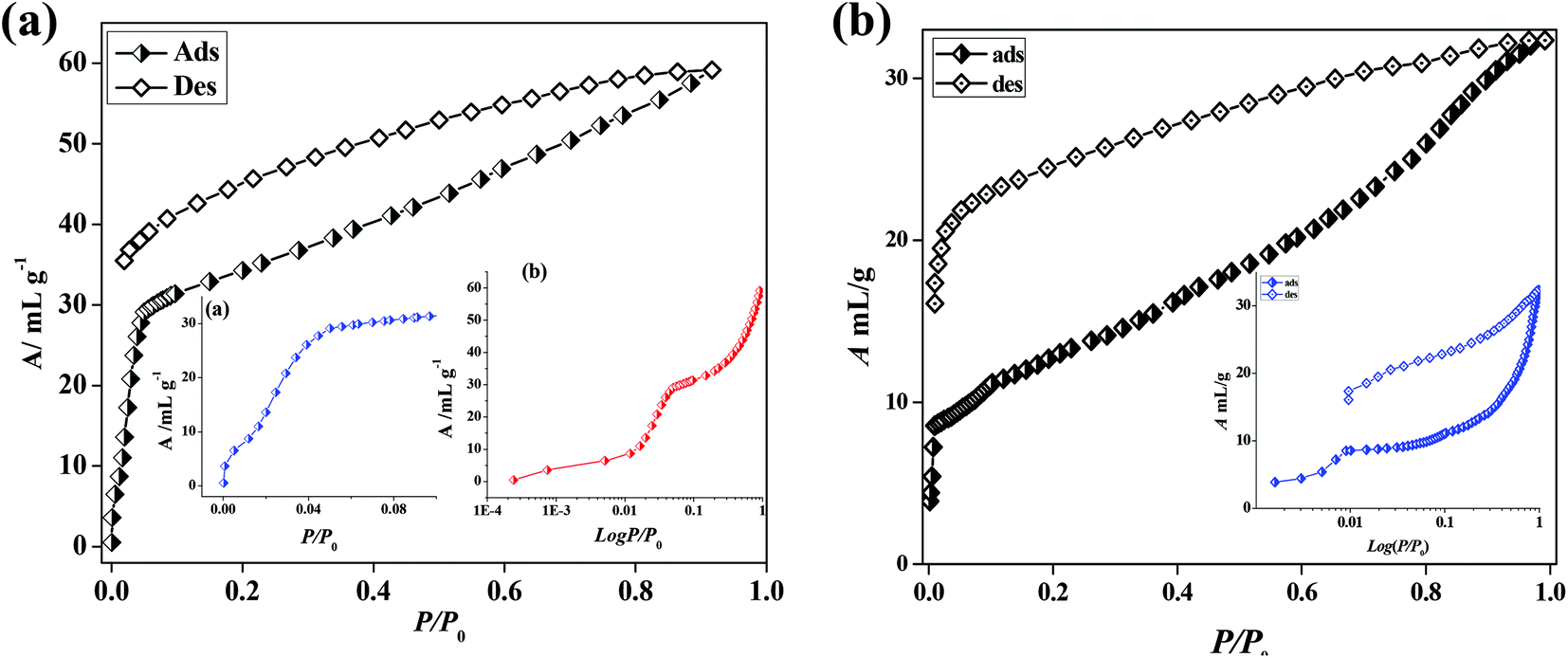
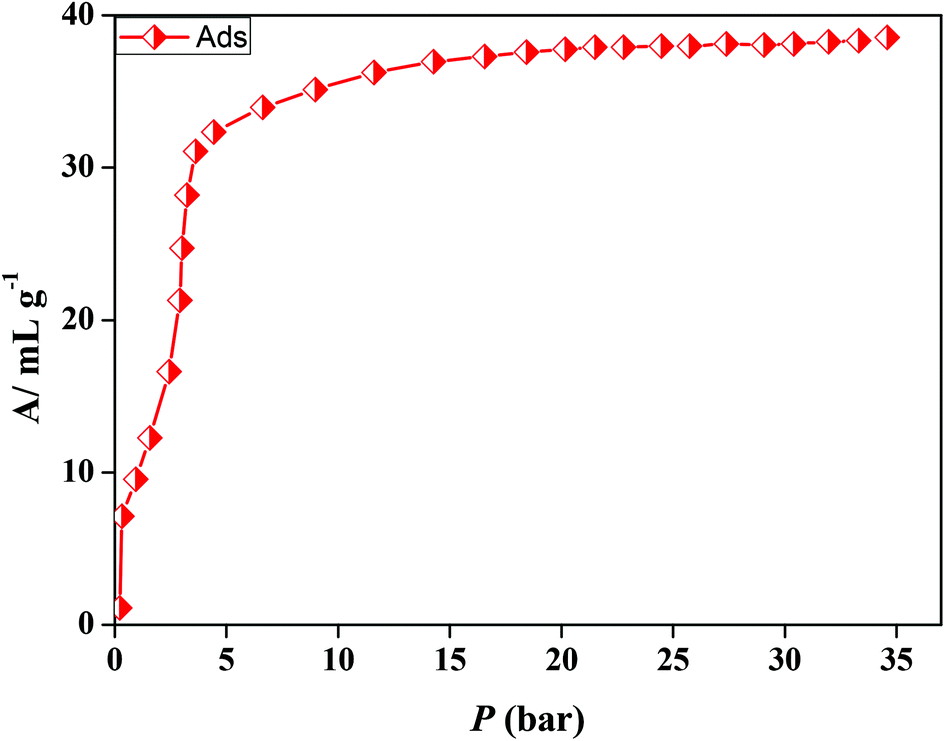
In the case of 5′, similarly, the N2 (77 K) adsorption profile suggests a type-II profile indicating only surface adsorption (Fig. S15†). Here also, the interdigitation of 2D networks causes an overall structural contraction (also evident from PXRD) and probably the pore size is not sufficient for N2 diffusion. Interestingly, we observed a stepwise uptake of CO2 at 195 K and the desorption path did not follow the adsorption path leading to a distinct hysteresis (Fig. 10a). At a relative pressure of P/P0 ∼ 0.01 a sharp uptake was observed indicating a structural transformation. The final uptake amount at P/P0 ∼ 0.9 pressure is ∼60 mL g−1 (∼11.8 wt%). The isosteric heat of adsorption calculated using the DR (Dubinin–Radushkevich)22 equation at 195 K was found to be ∼27.3 kJ mol−1. The high pressure CO2 adsorption at 293 K shows an uptake of 46 mL g−1 at 35 bar (Fig. 11). The adsorption profile is not a typical type-I rather a stepwise uptake is observed. The initial uptake of CO2 is slow and after 2 bar there is a distinct step which can be due to structural expansion. We have further measured C2H2 at 195 K that revealed a similar kind of stepwise uptake profile with a large hysteresis compared to CO2 (Fig. 10b). The step is observed exactly at P/P0 ∼ 0.01 followed by a gradual uptake up to P/P0 ∼ 0.9 with a total storage capacity of ∼32 mL g−1 (∼3.7%). The broad hysteretic nature in both cases signifies a strong interaction of the C2H2 and CO2 molecules with the pore surface of the framework. The pore surface is decorated with unsaturated Ni(II) sites and other polar sites such as uncoordinated pyrimidine and free carboxylate oxygen atoms which can interact well with CO2 molecules possessing a high quadrupole moment and C2H2 molecules with a π-electron cloud in addition to acidic acetylenic hydrogens. The step observed at a very low pressure is characteristic of a flexible framework.23 It is worth mentioning that on the removal of solvent molecules the c axis shortens from 18.6 to 15.0 Å which suggests a contraction in the pore size along with the overall structural contraction. However, such contracted interdigitated structures may exhibit a guest dependent dynamic structural transformation based on the sliding of the 2D networks through specific host–guest interactions. Such a stepwise uptake of CO2 and C2H2 clearly indicates that the diffusion of gas molecules open up the pores, possibly by the sliding of the 2D interdigitated networks (Scheme 2). The contracted pore in 5′ opens up for the CO2 and C2H2 molecules through strong interactions with polar pore surfaces and this further emphasizes the structural transformation at a particular pressure.
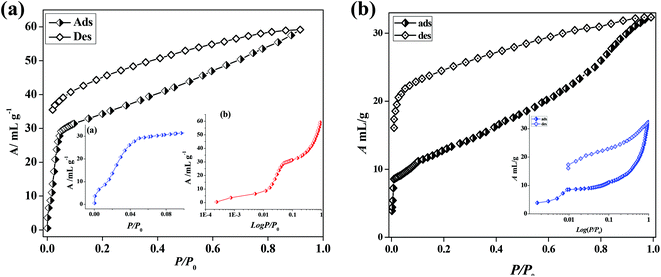 |
| | Fig. 10 (a) CO2 gas adsorption profiles of compound 5′ at 195 K, inset: (a) showing low pressure range uptake; (b) logarithmic scale plot; (b) C2H2 gas adsorption profile of compound 5′ at 195 K and inset: logarithmic scale plot. | |
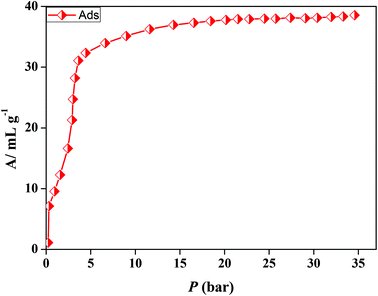 |
| | Fig. 11 CO2 (293 K) gas adsorption profile of compound 5′ at high pressure. | |
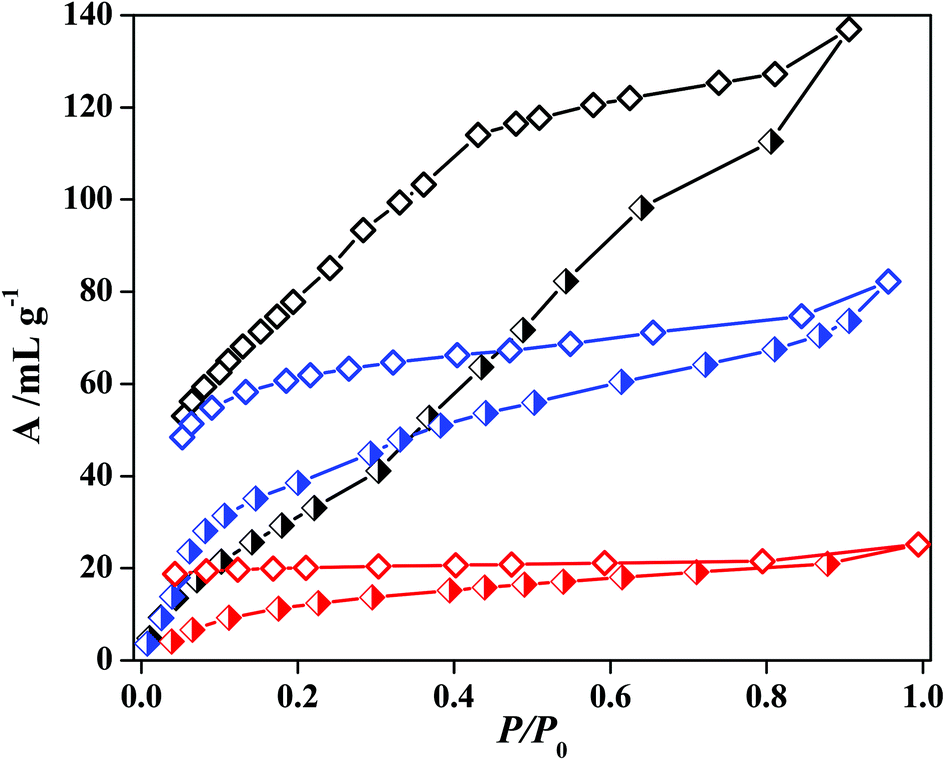
The water vapour adsorption isotherm of 5′ at 298 K shows a stepwise uptake which further establishes the flexible nature of the framework (Fig. 12). Up to a relative pressure of P/P0 ∼ 0.2, the uptake amount rises slowly and after P/P0 ∼ 0.3 there is a further increase which reaches to an amount of 130 mL g−1 at P/P0 ∼ 0.95. At a very low pressure all the adsorbed water molecules are not released and also the hysteresis indicates a strong interaction of water molecules with the unsaturated metal sites on the pore surface. For methanol a small step at a low pressure is observed which again supports structural flexibility. This fact can be easily correlated to the difference in the PXRD pattern of the solvent exposed samples with the as-synthesized ones. The final uptake amount is 82 mL g−1 at P/P0 ∼ 0.95. The fact is that with an increasing kinetic diameter of the adsorbate the uptake amount decreases and the trend is maintained in the case of ethanol also. The uptake of ethanol vapour at 298 K is about 20 mL g−1.
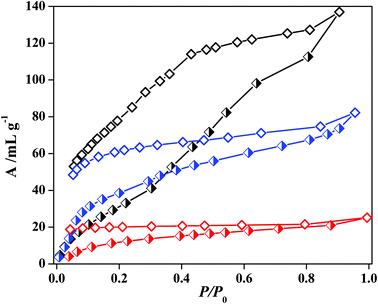 |
| | Fig. 12 Solvent vapour sorption isotherms for 5′; black curve represents H2O (298 K), blue colour curve represents MeOH (293 K) and red colour curve is for EtOH (298 K). | |
Conclusion
In conclusion, we have synthesized and structurally characterized five new supramolecular frameworks of H3iso with different transition metal ions (Mn(II), Ni(II) and Cu(II)). Following a bottom up approach, from a single H3iso linker based system to a mixed linker system of H3iso and bipy, higher dimensional porous structures have been synthesized. Such systematic tuning of dimensionality and also porosity is rare in coordination polymers. Compound 5 reveals a stepwise uptake of CO2 and C2H2 at 195 K which is attributed to structural expansion based on the strong interactions of gas molecules with the polar pore surface. Interesting structural dynamics were also observed for high pressure CO2 gas measurements and solvent vapour measurements. Such a flexible framework with a stepwise adsorption behaviour is of paramount importance for separation or recognition purposes. Apart from that, the H3iso linker is a biologically active organic precursor (H3iso) that with the assembly of different metal ions can open a new route towards the synthesis of bio-MOFs and also their applications.
Notes and references
-
(a) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef;
(b) P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38, 2638 CrossRef;
(c) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS PubMed;
(d) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS PubMed;
(e) C. Janiak, Dalton Trans., 2003, 2781 RSC;
(f) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed;
(g) S. Kitagawa, R. Kitaura and S. I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed;
(h) S. E. James, Chem. Soc. Rev., 2003, 32, 276 RSC;
(i) D. Maspoch, D. Ruiz-Molina and J. Veciana, J. Mater. Chem., 2004, 14, 2713 RSC;
(j) G. Férey, C. M. Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 217 CrossRef PubMed;
(k) G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC;
(l) J. J. Perry, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400 RSC.
-
(a) J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670 CrossRef CAS PubMed;
(b) X. Lin, J. Jia, X. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358 CrossRef CAS PubMed;
(c) S. Ma, D. Sun, J. M. Simmons, C. D. Collier, D. Yuan and H. C. Zhou, J. Am. Chem. Soc., 2007, 130, 1012 CrossRef PubMed;
(d) Y. Inubushi, S. Horike, T. Fukushima, G. Akiyama, R. Matsuda and S. Kitagawa, Chem. Commun., 2010, 46, 9229 RSC.
-
(a) D. Bradshaw, T. J. Prior, E. J. Cussen, J. B. Claridge and M. J. Rosseinsky, J. Am. Chem. Soc., 2004, 126, 6106 CrossRef CAS PubMed;
(b) L. Pan, K. M. Adams, H. E. Hernandez, X. Wang, C. Zheng, Y. Hattori and K. Kaneko, J. Am. Chem. Soc., 2003, 125, 3062 CrossRef CAS PubMed;
(c) T. K. Maji, R. Matsuda and S. Kitagawa, Nat. Mater., 2007, 6, 142 CrossRef CAS PubMed;
(d) D. N. Dybtsev, H. Chun, S. H. Yoon, D. Kim and K. Kim, J. Am. Chem. Soc., 2004, 126, 32 CrossRef CAS PubMed.
-
(a) S. Das and P. K. Bharadwaz, Inorg. Chem., 2006, 45, 5257 CrossRef CAS PubMed;
(b) P. Kanoo, R. Matsuda, R. Kitaura, S. Kitagawa and T. K. Maji, Inorg. Chem., 2012, 51, 9141 CrossRef CAS PubMed;
(c) C. Li, W. Qui, W. Shi, H. Song, G. Bai, H. He, J. Li and M. J. Zaworotko, CrystEngComm, 2012, 14, 1929 RSC;
(d) T. Friščič, Chem. Soc. Rev., 2012, 41, 3493 RSC;
(e) S. S. Chen, M. Chen, S. Takamizawa, P. Wang, G. C. Lv and W. Y. Sun, Chem. Commun., 2011, 47, 4902 RSC;
(f) B. K. Tripuramallu, P. Manna, S. N. Reddy and S. K. Das, Cryst. Growth Des., 2012, 12, 777 CrossRef CAS;
(g) V. Madhu and S. K. Das, Dalton Trans., 2011, 40, 12901 RSC;
(h) S. I. Noro, S. Kitagawa, T. Nakamura and T. Wada, Inorg. Chem., 2005, 11, 3960 CrossRef PubMed.
-
(a) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. V. Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed;
(b) P. L. Feng, K. Leong and M. Allendorf, Dalton Trans., 2011, 41, 8869 RSC;
(c) Y. Takashima, V. M. Martínez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nature, 2011, 2, 168 Search PubMed;
(d) B. Chen, L. Wang, F. Zapata, G. Qian and E. B. A. Lobkovsky, J. Am. Chem. Soc., 2008, 130, 6718 CrossRef CAS PubMed;
(e) R. Haldar, R. Matsuda, S. Kitagawa, S. J. George and T. K. Maji, Angew. Chem., Int. Ed., 2014, 53, 11772 CrossRef CAS PubMed.
-
(a) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC;
(b) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bye and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed;
(c) J. An, O. K. Farha, J. T. Hupp, E. Pohl, J. I. Yeh and N. L. Rosi, Nature, 2012, 3, 304 Search PubMed;
(d) S. Mohapatra, K. P. S. S. Hembram, U. Waghmore and T. K. Maji, Chem. Mater., 2009, 21, 5406 CrossRef CAS;
(e) R. Haldar and T. K. Maji, CrystEngComm, 2012, 14, 684 RSC;
(f) T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056 CrossRef CAS PubMed;
(g) C. M. Nagaraja, R. Haldar, T. K. Maji and C. N. R. Rao, Cryst. Growth Des., 2012, 12, 975 CrossRef CAS;
(h) R. Haldar, S. K. Reddy, M. V. Sursh, S. Mohapatra, S. Balasubramanian and T. K. Maji, Chem. – Eur. J., 2014, 20, 4347 CrossRef CAS PubMed;
(i) N. Sikdar, A. Hazra and T. K. Maji, Inorg. Chem., 2014, 53, 5993 CrossRef CAS PubMed;
(j) Q. Chen, Z. Chang, W. C. Song, H. Song, H. B. Song, T. L. Hu and X. H. Bu, Angew. Chem., Int. Ed., 2013, 52, 11550 CrossRef CAS PubMed;
(k) Y. Wu Li, J. R. Li, L. F. Wang, B. Y. Zhou, Q. Chen and X. H. Bu, J. Mater. Chem. A, 2013, 1, 495 RSC.
-
(a) M. C. Das, Q. Guo, Y. He, J. Kim, J. C. G. Zhou, K. Hong, X. Xiang, Z. Zhnag, K. M. Thomas, R. Krishna and B. Chen, J. Am. Chem. Soc., 2012, 134, 8703 CrossRef CAS PubMed;
(b) J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed;
(c) J. Liu, P. K. Thallapally and D. M. Strachan, Langmuir, 2012, 28, 11584 CrossRef CAS PubMed;
(d) Z. Chang, D. S. Zhang, Q. Chen and X. H. Bu, Phys. Chem. Chem. Phys., 2013, 15, 5430 RSC;
(e) S. M. Zhang, Z. Chang, T. L. Hu and X. H. Bu, Inorg. Chem., 2010, 49, 11581 CrossRef CAS PubMed.
-
(a) R. K. Das, A. Aijaz, M. K. Sharma, P. Lama and P. K. Bharadwaj, Chem. – Eur. J., 2012, 18, 6866 CrossRef CAS PubMed;
(b) S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607 CrossRef CAS PubMed;
(c) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC;
(d) J. Long, L. Wang, X. Gao, C. Bai, H. Jiang and Y. Li, Chem. Commun., 2012, 48, 12109 RSC;
(e) K. Jayramulu, K. K. R. Datta, M. V. Suresh, G. Kumari, R. Datta, C. Narayana, M. Eswaramoorthy and T. K. Maji, ChemPlusChem, 2012, 77, 743 CrossRef.
-
(a) D. N. Dybtsev, H. Chun, S. H. Yoon, D. Kim and K. Kim, J. Am. Chem. Soc., 2004, 126, 32 CrossRef CAS PubMed;
(b) S. Muthu, J. H. K. Yip and J. J. Vittal, Dalton Trans., 2002, 4561 RSC;
(c) B. C. Tzeng, T. H. Chiu, B. S. Chen and G. H. Lee, Chem. – Eur. J., 2008, 14, 5237 CrossRef CAS PubMed.
-
(a) V. N. Vukotic, K. J. Harris, K. Zhu, R. W. Schurko and S. J. Loeb, Nat. Chem., 2012, 4, 456 CrossRef CAS PubMed;
(b) Y. Inubushi, S. Horike, T. Fukushima, G. Akiyama, R. Matsuda and S. Kitagawa, Chem. Commun., 2010, 46, 9229 RSC;
(c) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS PubMed;
(d) P. Kanoo, G. Mostafa, R. Matsuda, S. Kitagawa and T. K. Maji, Chem. Commun., 2011, 47, 8106 RSC;
(e) K. L. Gurunatha and T. K. Maji, Inorg. Chem., 2009, 48, 10886 CrossRef CAS PubMed.
-
(a) C. Y. Sun and L. P. Jin, Polyhedron, 2004, 23, 2227 CrossRef CAS PubMed;
(b) C. Y. Sun and L. P. Jin, Polyhedron, 2004, 23, 2085 CrossRef CAS PubMed.
-
(a) O. Z. Yesilel, H. Erer and O. Buyukgungor, CrystEngComm, 2011, 13, 1339 RSC;
(b) H. Erer, O. Z. Yesilel, C. Darcan and O. Buyukgungor, Polyhedron, 2009, 28, 3087 CrossRef CAS PubMed;
(c) N. K. Davidenko and N. N. Zinich, Russ. J. Chem., 1979, 24, 891 Search PubMed.
-
SMART (V 5.628), SAINT (V 6.45a), XPREP, SHELXTL, Bruker AXS Inc., Madison, Wisconsin, USA, 2004 Search PubMed.
-
G. M. Sheldrick, Siemens Area Detector Absorption Correction Program, University of Götingen, Götingen, Germany, 1994 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Gualaradi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
-
G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Solution and Refinement, University of Götingen, Götingen, Germany, 1997 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- L. J. Farrugia, WinGX-A Windows Program for Crystal Structure Analysis, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- The sizes of the channels were calculated by considering the van der Waals radii of the atoms.
- P. E. Warner, L. Erikson and M. Westdahl, J. Appl. Crystallogr., 1985, 18, 367 CrossRef.
- D. E. Webster, R. S. Drago and M. C. Zerner, J. Am. Chem. Soc., 1998, 120, 5509 CrossRef.
- M. M. Dubinin, Chem. Rev., 1960, 60, 235 CrossRef CAS.
-
(a) P. Ju, L. Jiang and T. B. Lu, Chem. Commun., 2013, 49, 1820 RSC;
(b) K. S. Walton, A. R. Millward, D. Dubbeldum, H. Frost, J. J. Low, O. M. Yaghi and R. Q. Snurr, J. Am. Chem. Soc., 2008, 130, 406 CrossRef CAS PubMed;
(c) T. K. Maji, G. Mostafa, R. Matsuda and S. Kitagawa, J. Am. Chem. Soc., 2005, 127, 17152 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1022424–1022428. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00129j |
|
| This journal is © the Partner Organisations 2015 |