
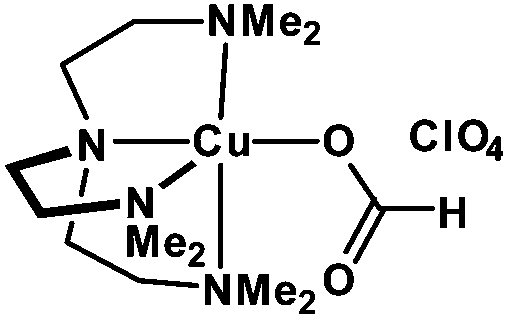
Discrete copper(II)-formate complexes as catalytic precursors for photo-induced reversible deactivation polymerization†
Vasiliki
Nikolaou‡
a,
Athina
Anastasaki‡
ab,
Francesca
Brandford-Adams
a,
Richard
Whitfield
a,
Glen R.
Jones
a,
Gabit
Nurumbetov
a and
David M.
Haddleton
*ab
aUniversity of Warwick, Chemistry Department, Library Road, CV4 7AL, Coventry, UK. E-mail: d.m.haddleton@warwick.ac.uk
bARC Centre of Excellence in Convergent Bio-Nano Science and Technology, Monash Institute of Pharmaceutical Sciences, Monash University (Parkville Campus), 399 Royal Parade, Parkville, Victoria 3152, Australia
First published on 20th October 2015
Abstract
Traditional copper-mediated reversible deactivation polymerization techniques (RDRP) employ various components mixed in situ (e.g. ligand, metal salt, additional deactivation species etc.) in order to achieve good control over the molecular weight distributions. In a previous communication we described a discrete copper(II)-formate/Me6-Tren complex to catalyse the polymerization of acrylates. Herein, we expand the scope of this complex by investigating the compatibility with various solvents, including acetonitrile (MeCN), dimethylformamide (DMF), methanol (MeOH), isopropanol (IPA), toluene, 2,2,2-trifluoroethanol (TFE) and water as well as mixtures thereof. A series of both hydrophilic and hydrophobic acrylic monomers are reported including n and tert butyl acrylate (n-BA and t-BA), poly ethylene glycol acrylate (PEGA), diethylene glycol ethyl ether acrylate (DEGEEA), lauryl acrylate (LA), octadecyl acrylate (ODA), hydroxyethyl acrylate (HEA), hydroxyl propyl acrylate (HPA) and solketal acrylate (SA). In most cases, narrow molecular weight distributions were attained (typically < 1.20), even when the polymerization was allowed to reach high conversions (>95%). High molecular weight polymers were targeted achieving poly(MA) with a final dispersity of 1.12 within 2 h (Mn ∼ 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) with additional NaBr being essential to obtain even higher molecular weight polymers. As Me6-Tren is relatively expensive to purchase commercially, an additional PMDETA complex was also synthesized, allowing for the polymerization of methacrylates (e.g. MMA) in addition to the polymerization of acrylates. Narrow molecular weight distributions, high monomer conversion and good spatiotemporal control could be achieved with this complex, demonstrating that it could be an efficient and less expensive alternative to obtain well-defined poly(acrylates) and poly(methacrylates).
000 g mol−1) with additional NaBr being essential to obtain even higher molecular weight polymers. As Me6-Tren is relatively expensive to purchase commercially, an additional PMDETA complex was also synthesized, allowing for the polymerization of methacrylates (e.g. MMA) in addition to the polymerization of acrylates. Narrow molecular weight distributions, high monomer conversion and good spatiotemporal control could be achieved with this complex, demonstrating that it could be an efficient and less expensive alternative to obtain well-defined poly(acrylates) and poly(methacrylates).
Introduction
Reversible deactivation radical polymerization methods have significantly evolved over the last few decades allowing access to narrow molecular weight distributions (MWDs), efficient regulation of the molecular weight and the monomer sequence and complex architectures.1,2 Transition metal methods in particular, including atom transfer radical polymerization (ATRP)3,4 and single electron transfer living radical polymerization (SET-LRP),5–7 have significantly contributed towards this direction exploiting the manipulation of the activation/deactivation equilibrium between active and dormant species.ATRP and SET-LRP utilize either CuI or Cu(0) to facilitate the activation of alkyl halide initiators in order to generate carbon-based radicals and allow for the propagation (activation and propagation have been reported to proceed via either an inner sphere or an outer sphere mechanism). Importantly, in both methods, the control over the polymerization is inferred by the accumulation of the higher oxidation species, CuII which has been reported to occur through either the persistent radical effect (PRE)8,9 or via disproportionation.10–12 CuII will thus act as deactivating species by adjusting the polymerization equilibrium towards the dormant species and limiting the termination events. The role of the deactivator is of upmost importance for copper-mediated polymerizations and external amounts of CuII are often added when high end group fidelity is required in both ATRP13,14 and SET-LRP protocols.15–17 Therefore, it is necessary to optimize the CuII concentration in order to achieve narrow MWDs and high end group fidelity as well as acceptable polymerization rates. In some cases, NaBr is also added to invoke further control over the molecular weight distributions and thus further adding on the complexity of the system.18,19
The nature of the ligand facilitates both the solubility of the transition metal salt and adjusts the redox potential and halogenophilicity of the metal.20–22 Even minor changes in the ligand concentration can lead to significant loss of end group fidelity and termination events and thus careful optimization of the reaction conditions is required.23–25
Thus, various components (e.g. type of activator, ligand and deactivator concentration etc.) have to be optimized for a successful copper-mediated radical polymerization which is not only time consuming but also challenging.26,27 CuIBr has to be stored under oxygen free conditions to avoid air oxidation and has to be purified prior to use as it often contains high amounts of CuII contamination despite its often stated high purity (a typical commercial bottle of CuI, even at >99% purity is usually green, arising from the CuII impurities). Cu(0) also needs to be purified prior to use with either hydrazine28 or acid29–31 while in many cases there has been a general confusion of whether Cu(0) powder or Cu(0) wire should be preferred.32 N-containing aliphatic amine ligands such as Me6-Tren are also susceptible to oxidation and degradation33 and have to be stored under dry, dark and inert conditions, although degradation will still occur in many cases which will require further purification methods prior to use (e.g. redistilling the ligand). Thus a discrete CuII complex offers an attractive and stable solution.
Different “variations of ATRP” have been employed in order to cope with different issues giving rise to a range of acronyms including activators regenerated by electron transfer (ARGET)34 and initiators for continuous activator regeneration (ICAR).35 However, the presence of reducing agents or free radical initiators further adds to the complexity of the system while the relative concentrations of CuII and ligand also requires careful optimization. It is noted that previous studies in ATRP has led to slow polymerization rates whilst the monomer conversion, in most cases, did not reach quantitative levels.36 Recently, attention has been drawn towards controlling the activation-deactivation step via external regulation.37 Photochemical stimuli have been used to mediate the controlled polymerization of various monomers, although these often require photo initiators, an excess of ligand or exotic catalysts.38–52
In a recent communication, we reported a discrete copper(II)-formate/Me6-Tren complex as an effective catalyst for the polymerization of acrylates.53 Good spatiotemporal control and narrow molecular weight distributions were demonstrated with MA. However, different monomers were not tested and only relatively low molecular weights were targeted (up to Mn ∼ 16000 g mol−1). In addition, DMSO was the only solvent employed, perhaps limiting the applicability of this complex. More importantly, the Me6-Tren which is incorporated in the coordination sphere of the complex is relatively expensive to purchase, and as such it would be of interest to replace it with a less expensive ligand, without compromising the features of the initial complex (e.g. narrow molecular weight distributions, near-quantitative conversions, good spatiotemporal control etc.).
In this present work, we report an expansion of the scope of the [Cu(Me6-Tren)(O2CH)](ClO4) complex. Different solvents were investigated including MeCN, DMF, MeOH, IPA, toluene, TFE, water and mixtures thereof. This range of solvents, allowed access to the polymerization of a large diversity of acrylates including functional and hydrophilic acrylates (HEA, HPA, PEGA and SA, hydrophobic acrylates (n-BA, t-BA, LA, ODA) and thermoresponsive acrylates (e.g. DEGEEA). Narrow MWDs even at quantitative or near-quantitative conversions have been obtained in all cases highlighting the versatility of the approach. Importantly, higher molecular weight polymers could also be attained (Mn ∼ 120000 g mol−1) within 2 h of reaction time. Finally, a second complex was synthesized, where PMDETA (1 ml ∼ £0.28) was used to replace Me6-Tren (1 ml ∼ £115) in the copper(II) complex as an inexpensive alternative.
Results and discussion
In order to assess the compatibility of the [Cu(Me6-Tren)(O2CH)](ClO4) complex with various solvents, methyl acrylate was used as the monomer (MA) utilizing previously optimized conditions ([I]:[M]:[complex] = [1]:[50]:[0.08]) (Scheme 1, Fig. S1,† source of UV light: OmniCure S2000, spot UV curing lamp system, 200 W).53 MeCN and DMF were initially tested giving rise to well-defined polymers with good agreement between theoretical and experimental values and narrow molecular weight distributions (Đ ∼ 1.09 in both solvents) (Fig. S2 and S3†). However, the polymerization rates were slower than in DMSO (∼4 h to reach >95% conversion as opposed to 2 h in DMSO) (Table 1). Alcohols were also found to be compatible with this complex, with very high conversions achieved within 6 h in MeOH and IPA solution, without compromising the control over the molecular weight distributions (Đ ∼ 1.09 and Đ ∼ 1.20 for MeOH and IPA respectively) (Table 1, Fig. S4 and S5†). TFE was also tested as fluorinated solvents have been shown that they can be utilized for the polymerization of a large diversity of monomers with both hydrophobic and hydrophilic moieties (Đ ∼ 1.08) while the end group fidelity was estimated to be ∼95% as calculated by 1H NMR (Table 1, Fig. S6†).31,54–56 In order to facilitate the polymerization of more hydrophobic monomers, toluene was investigated. Relatively fast polymerization rates were observed by NMR confirming >98% conversion within 4 h, albeit with a much higher dispersity attained (Đ ∼ 1.42). This is attributed to the limited solubility of [Cu(Me6-Tren)(O2CH)](ClO4) in toluene at ambient temperature, which is consistent with previously reported data (Table 1, Fig. S7†).43 However, addition of a small amount of MeOH ([MeOH]:[toluene] = [1]:[4]) resulted in a significant reduction in the dispersity, which resembled the dispersities obtained in MeOH (Đ ∼ 1.09), perhaps due to enhanced solubility of the copper complex (Table 1, Fig. S8†).
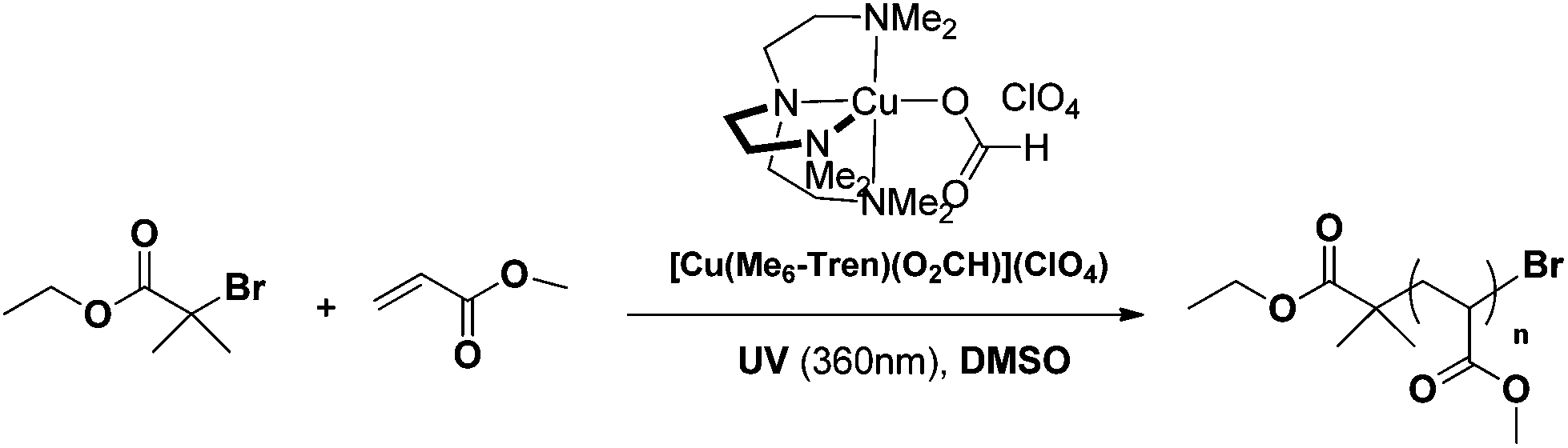 | ||
| Scheme 1 Photo-induced polymerization of MA, utilizing [Cu(Me6-Tren)(O2CH)](ClO4) as the precursor catalyst. | ||
| Solvent | Monomer | t (h) | Conv. (%) | M n,th (g mol−1) | M n,SEC (g mol−1) | Đ |
|---|---|---|---|---|---|---|
| DMSO53 | MA | 2 | 97 | 4400 | 5100 | 1.07 |
| MeCN | 4 | 96 | 4300 | 5600 | 1.09 | |
| DMF | 4 | 95 | 4300 | 4600 | 1.09 | |
| MeOH | 6 | 96 | 4300 | 4200 | 1.09 | |
| IPA | DP = 50 | 6 | 92 | 4100 | 3200 | 1.20 |
| TFE | 2 | 96 | 4300 | 5500 | 1.08 | |
| Toluene | 4 | 98 | 4400 | 3300 | 1.42 | |
| Tol:MeOH |
6 | 97 | 4400 | 4300 | 1.09 |
Finally, water was also tested as the solvent, but as MA is not soluble in aqueous media, PEGA was selected as an alternative monomer. Under these conditions, the aqueous polymerization of PEGA presented near quantitative conversion in 2 h (∼98% by NMR), although broad molecular weight distributions were attained (Đ ∼ 1.47) suggesting limited control over the polymerization (Table 2, Fig. S9†). In order to facilitate a direct comparison, the polymerization of PEGA in DMSO was also conducted. Although the polymerization rates were comparable, achieving also near quantitative conversion within 2 h, the dispersity was significantly improved (Đ ∼ 1.13) suggesting that the solvent (water) was responsible for the loss of control (Table 2, Fig. S10†). We postulate that this is due to the inefficient reduction of CuII in aqueous media under these conditions. However, for applications where water is desirable, mixtures with any of the aforementioned solvents could be alternatively employed.
| Monomer | Solvent | DP | t (h) | Conv. (%) | M n,th (g mol−1) | M n,SEC (g mol−1) | Đ |
|---|---|---|---|---|---|---|---|
| a 1 equiv. of NaBr in respect to the complex. | |||||||
| n-BA | DMSO | 50 | 4 | 98 | 6500 | 9200 | 1.18 |
| DMF | 4 | 95 | 6300 | 6600 | 1.07 | ||
| t-BA | DMSO | 2 | 93 | 6100 | 7400 | 6.40 | |
| DMF | 4 | 99 | 6600 | 8300 | 1.09 | ||
| PEGA | DMSO | 20 | 2 | 98 | 9600 | 10400 |
1.13 |
| H2O | 2 | 98 | 9600 | 10000 |
1.47 | ||
| DEGEEA | DMSO | 6 | 99 | 3000 | 3000 | 1.15 | |
| HEA | DMSO | 6 | 95 | 2400 | 2800 | 1.18 | |
| HPA | DMSO | 6 | 99 | 2800 | 3000 | 1.32 | |
| ODA | Tol:IPA |
50 | 12 | 90 | 14800 |
9900 | 1.13 |
| LA | Tol:MeOH |
12 | 98 | 11900 |
9100 | 1.10 | |
| SA | DMSO | 2 | 95 | 9000 | 8000 | 1.09 | |
| MMA | DMSO | 10 | 27 | 1400 | 3400 | 2.10 | |
| MMAa | DMSO | 20 | 0 | — | — | — | |
| Sty | Tol:MeOH |
6 | 10 | 540 | — | — | |
In order to further expand the scope of this system, n-BA was polymerized in DMSO under the following reaction conditions: [I]:[n-BA]:[complex] = [1]:[50]:[0.08]). Very high conversion (>98%) could be achieved in 4 h with SEC revealing a symmetrical, monomodal molecular weight distribution with low dispersity (Đ ∼ 1.18) (Table 2, Fig. S11†). This slightly higher dispersity (in comparison with MA) was attributed to the heterogeneity of the system as poly(butyl acrylate) phase separates under these conditions (solvent/monomer).30,57,58 As it was shown earlier, DMF was also found to be compatible with this polymerization protocol and as such, it was chosen as an alternative solvent since it would maintain full solubility of the monomer and polymer throughout the polymerization. Interestingly, the polymerization under homogeneous conditions proceeded in a more controlled manner and an observed reduction in the dispersity was evident (Đ ∼ 1.07) (Table 2, Fig. S12†). Under identical conditions, the polymerization of t-BA was also attempted in DMSO. However, a gel-like polymer was reproducibly observed within 2 h with broad molecular weight distributions attained by SEC (Đ > 6) (Table 2, Fig. S13†). Conversely, when DMF was chosen as the solvent the control over the molecular weight distribution was restored and low dispersity was attained (Đ ∼ 1.09) suggesting that maintaining the solubility of all components is crucial to mediate this polymerization (Table 2, Fig. S14†).
Hydrophilic monomers were also polymerized in DMSO, including HEA and HPA with good agreement between theoretical and experimental molecular weights and relatively narrow molecular weight distributions (Table 2, Fig. S15 and S16†). Monomers synthesized in our laboratory were also subjected to these polymerization conditions, e.g. SA, demonstrating fast polymerization rates (95% in 2 h) and low dispersity (Đ ∼ 1.09) (Table 2, Fig. S17†). Pleasingly, DEGEEA also afforded a controlled polymerization yielding well-defined materials with narrow molecular weight distributions (Đ ∼ 1.15) (Table 2, Fig. S18†). The controlled photo-induced polymerization of increasingly hydrophobic acrylates was also demonstrated. As LA is insoluble in DMSO and other polar solvents, a mixture of toluene/methanol was selected to facilitate monomer solubility, polymer and catalyst. Under these conditions, well-controlled polymers could be obtained, albeit slower polymerization rates were observed (∼12 h). Nevertheless, low dispersities were attained even when the reaction was pushed to reach quantitative or near-quantitative levels (Đ ∼ 1.10) (Table 2, Fig. S19†). Similarly, ODA was also polymerized by simply tuning the solvent system from toluene/methanol to toluene/IPA in order to facilitate the solubility of this longer alkyl chain acrylate. Again 1H NMR confirmed high conversions (∼90%) and SEC revealed symmetrical traces without any obvious low or high molecular weight shoulders (Table 2, Fig. S20†). Although different families of monomers were also tested, including MMA and styrene, broad molecular weight distributions and poor conversion was attained respectively (Table 2, Fig. S21†), while the inclusion of additional NaBr gave rise to 0% conversion, even when the reaction was left to proceed overnight.
We were also interested to explore the potential of this compound to obtain higher molecular weight polymers. For this reason, a range of polymerizations were conducted targeting various degrees of polymerization (DP = 400–3200). For DP = 400 ([I]:[MA]:[complex] = [1]:[400]:[0.08]), high conversion (∼95% by 1H NMR) and narrow molecular weight distributions (Đ ∼ 1.10) were attained in 2 h with good correlation between the theoretical and experimental molecular weights (Table 3, Fig. 1). Similar results were obtained when DP = 800 was subsequently targeted (Đ ∼ 1.10, 95% conversion in 2 h) and a final Mn = 67000 g mol−1 was evident as shown by SEC (Table 3, Fig. 1). However, when identical conditions were applied for the synthesis of even higher MW polymers (DP = 1600) the polymerization rate decreased reaching a final conversion of 86% after 14 h (Table 3, Fig. S22†). In order to circumvent this, the complex concentration was increased to 0.16 equiv. with respect to initiator ([I]:[MA]:[complex] = [1]:[1600]:[0.16]), which lead to a large increase on the rate yielding well-controlled polymers with Mn ∼ 120000 g mol−1 and Đ ∼ 1.12 (Table 3, Fig. 1). Interestingly the same conditions led to uncontrolled polymers when a polymerization degree of 3200 was targeted (bimodal peak). A similar scenario was evident (bimodal peaks) even when higher concentrations of copper complexes were utilized (0.32 equiv. with respect to initiator) suggesting that the limitations of the system had been reached. Although when a small amount of NaBr was externally added at the beginning of the polymerization (1 equiv. with respect to the complex) higher molecular weight polymers could be attained, a low molecular weight shoulder was still visible by SEC, showing unavoidable premature termination under the conditions employed (Mn ∼ 160000 g mol−1, 85% conversion) (Table 3, Fig. S23–S25†).
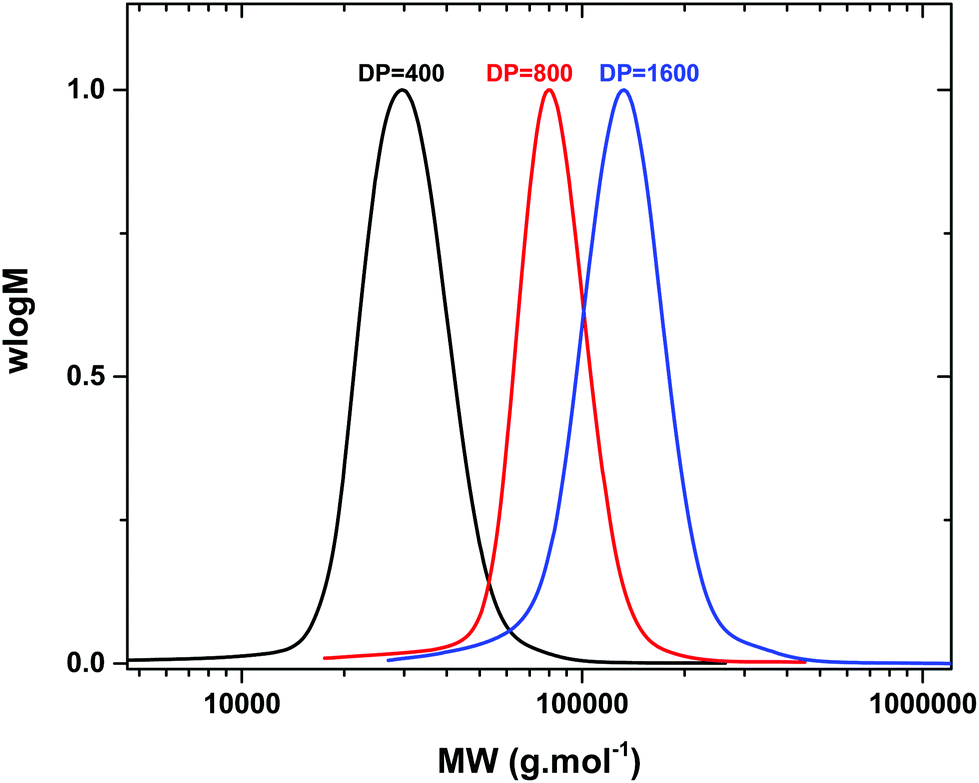 | ||
| Fig. 1 High molecular weight poly(MA) synthesized via photo-induced polymerization utilizing [Cu(Me6-Tren)(O2CH)](ClO4) as the precursor catalyst. | ||
| Complex | DP | t (h) | Conv. (%) | M n,SEC (g mol−1) | Đ | |
|---|---|---|---|---|---|---|
| a 0.16 equiv. b 0.32 equiv. of the complex. c 1 equiv. NaBr was added in the solution. | ||||||
| Methyl acrylate |
|
400 | 2 | 95 | 28800 |
1.10 |
| 800 | 2 | 95 | 67000 |
1.10 | ||
| 1600 | 6 | 90 | 94000 |
1.10 | ||
| 1600a | 2 | 95 | 120000 |
1.12 | ||
| 3200a | 12 | 94 | 110000 |
1.35 | ||
| 3200b | 4 | 95 | 110000 |
1.18 | ||
| 3200c | 6 | 85 | 160000 |
1.13 | ||
|
200 | 10 | 99 | 20000 |
1.13 | |
| 800 | 10 | 88 | 50000 |
1.38 | ||
| 800a | 10 | 95 | 60000 |
1.20 | ||
| 1600a | 10 | 70 | 69000 |
1.60 | ||
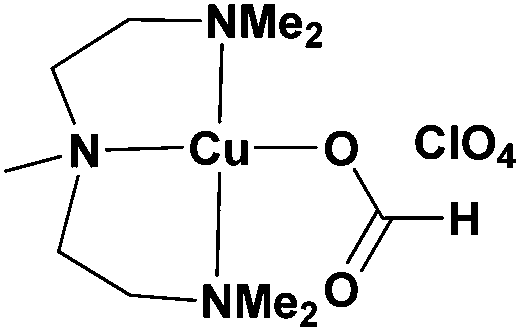
As Me6-Tren is a relatively expensive ligand, an additional strategy was also employed to provide a less expensive alternative, PMDETA replaced Me6-Tren as the ligand. The [Cu(Me5-Dien)(O2CH)](ClO4) complex was synthesized according to a literature procedure59 and subsequently used for the polymerization of MA. Although slower polymerization rates were obtained (95% in 5 h), the control over the molecular weight distribution was not compromised with SEC showing low dispersities (Đ ∼ 1.18) (Table 4, Fig. S26†). In order to probe the versatility of the complex, EGA was also polymerized in DMF, yielding high conversions and low dispersities and thus demonstrating the potential of this complex to be used as an inexpensive and efficient alternative (Table 4, Fig. S27†). Higher molecular weight polymers (DP = 800) could also be attained, although Đ < 1.20 could be achieved only with the use of additional NaBr (Table 3, Fig. S28–S31†). The polymerization of MMA was also attempted, showing higher conversion and better control over the molecular weight distribution in comparison with the Me6-Tren complex, although the dispersities were still broad for a controlled polymerization (Đ ∼ 1.50) (Table 4, Fig. S32†). However, we were able to circumvent this by adding 1 equiv. of NaBr with respect to the complex, giving rise to quite well-defined poly(MMA) with Đ ∼ 1.25 (Table 4, Fig. S33†).
In order to further explore the potential of this complex, the possibility of “on/off” temporal control was also investigated throughout the polymerization. Intermittent “light/dark” cycles for alternating periods were conducted, where the polymerization mixture was initially exposed under UV irradiation for 45 min, achieving 36% of conversion. A “dark” period of another 45 min was followed in which no polymerization was observed by either 1H NMR or SEC. Upon re-exposure of the mixture to UV irradiation switched the polymerization back “on” and approximately 58% conversion was attained in an additional 1 h (Fig. 2 and S34†). These cycles were repeated several times (with no or minimal polymerization (1–2%) observed during the dark periods), demonstrating not only the necessity of photo exposure at an appropriate wavelength for the polymerization to commence but also the potential of utilizing spatiotemporal control for future applications. Although no mechanistic insight is presented in this contribution, the reader is referred to our previously reported studies with respect to the mechanism in the photo-induced polymerization in the presence of either CuBr2 or discrete copper complexes.45,53
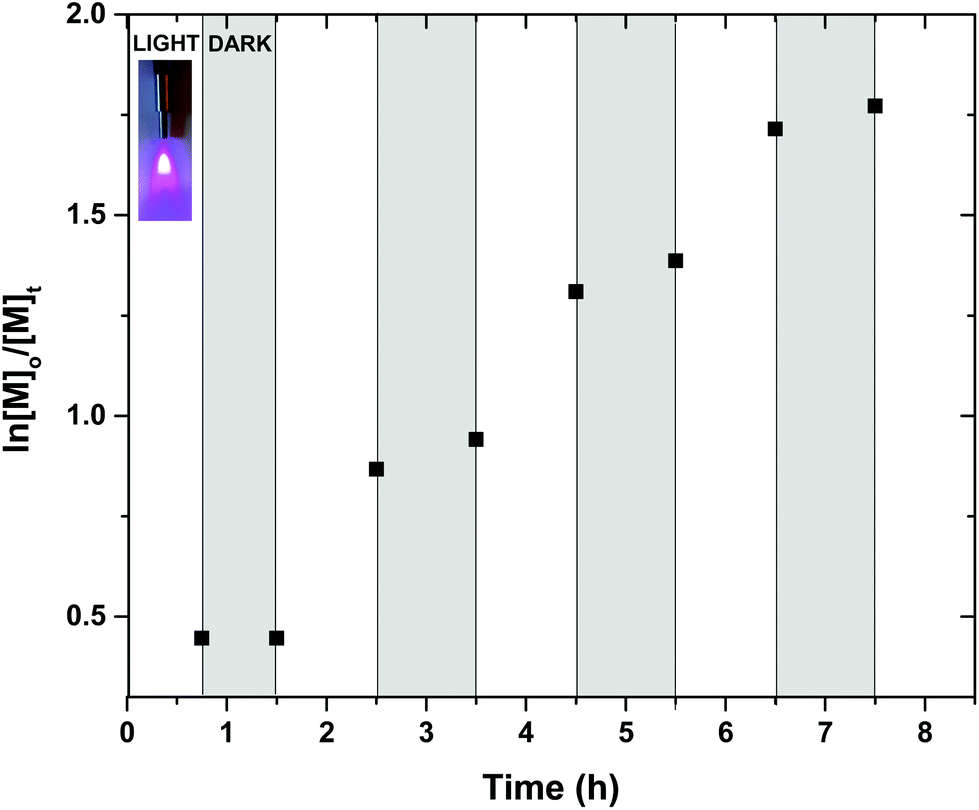 | ||
| Fig. 2 Evidence of temporal control via concecutive light and dark exposure. Initial conditions: [MA]:[I]:[[Cu(Me5-Dien)(O2CH)](ClO4)] = [50]:[1]:[0.08] in DMSO 50% v/v. | ||
Conclusions
In summary, two discrete copper complexes, [Cu(Me6-Tren)(O2CH)](ClO4) and [Cu(Me5-Dien)(O2CH)](ClO4) have been used for the controlled polymerization of acrylates and methacrylates is reported. In the case of [Cu(Me6-Tren)(O2CH)](ClO4), different solvents were investigated such as MeCN, DMF, MeOH, IPA, toluene, TFE, water and solvent mixtures. Functional and hydrophilic acrylates (e.g. HEA, HPA, PEGA and SA), hydrophobic acrylates (e.g. n-BA, t-BA, LA and ODA) and thermoresponsive acrylates (e.g. DEGEEA) were successfully polymerized yielding narrow molecular weight distributions even at very high conversions and high molecular weight polymers in a matter of 2 h (Mn ∼ 120000 g mol−1, Đ ∼ 1.12). [Cu(Me5-Dien)(O2CH)](ClO4) was also synthesized and employed for the controlled polymerization of acrylates as an inexpensive alternative yielding well-defined polymers with narrow molecular weight distributions. Importantly, the incorporation of PMDETA in the complex afforded the polymerization of methacrylates in addition to the polymerization of acrylates allowing access to a wider range of materials. Spatiotemporal control was also demonstrated highlighting the potential of simple and cost effective complexes to be used for copper mediated processes in numerous applications, including synthesis of these materials in an industrial level.
Acknowledgements
We appreciate financial support from the University of Warwick (A.A., G.N, F.B.A.), Syngenta (R.W.) and The Lubrizol Corporation (V.N., G.R.J.). D.M.H. is a Royal Society/Wolfson Fellow.References
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed.
- A. Anastasaki, V. Nikolaou, G. S. Pappas, Q. Zhang, C. Wan, P. Wilson, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Sci., 2014, 5, 3536–3859 RSC.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2015 DOI:10.1021/acs.chemrev.5b00191.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS PubMed.
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed.
- H. Fischer, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1885–1901 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6880–6895 CrossRef CAS.
- B. M. Rosen, X. Jiang, C. J. Wilson, N. H. Nguyen, M. J. Monteiro and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5606–5628 CrossRef CAS.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed.
- H. Zhang, B. Klumperman, W. Ming, H. Fischer and R. van der Linde, Macromolecules, 2001, 34, 6169–6173 CrossRef CAS.
- K. Matyjaszewski, M. Wei, J. Xia and S. G. Gaynor, Macromol. Chem. Phys., 1998, 199, 2289–2292 CrossRef CAS.
- A. Anastasaki, C. Waldron, P. Wilson, C. Boyer, P. B. Zetterlund, M. R. Whittaker and D. Haddleton, ACS Macro Lett., 2013, 2, 896–900 CrossRef CAS.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed.
- F. Nyström, A. H. Soeriyadi, C. Boyer, P. B. Zetterlund and M. R. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5313–5321 CrossRef.
- V. Nikolaou, A. Anastasaki, F. Alsubaie, A. Simula, D. Fox and D. Haddleton, Polym. Chem., 2015, 6, 3581–3585 RSC.
- A. Simakova, S. E. Averick, D. Konkolewicz and K. Matyjaszewski, Macromolecules, 2012, 45, 6371–6379 CrossRef CAS.
- W. Tang and K. Matyjaszewski, Macromolecules, 2006, 39, 4953–4959 CrossRef CAS.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS PubMed.
- A. Simula, V. Nikolaou, F. Alsubaie, A. Anastasaki and D. M. Haddleton, Polym. Chem., 2015, 6, 5940–5950 RSC.
- A. Anastasaki, C. Waldron, P. Wilson, R. McHale and D. M. Haddleton, Polym. Chem., 2013, 4, 2672–2675 RSC.
- N. H. Nguyen, M. E. Levere and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 35–46 CrossRef CAS.
- N. H. Nguyen, X. Jiang, S. Fleischmann, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5629–5638 CrossRef CAS.
- A. Anastasaki, A. J. Haddleton, Q. Zhang, A. Simula, M. Droesbeke, P. Wilson and D. M. Haddleton, Macromol. Rapid Commun., 2014, 35, 965–970 CrossRef CAS PubMed.
- A. Simula, V. Nikolaou, A. Anastasaki, F. Alsubaie, G. Nurumbetov, P. Wilson, K. Kempe and D. M. Haddleton, Polym. Chem., 2015, 6, 2226–2233 RSC.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5109–5119 CrossRef CAS.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4241–4252 CAS.
- A. Anastasaki, C. Waldron, V. Nikolaou, P. Wilson, R. McHale, T. Smith and D. M. Haddleton, Polym. Chem., 2013, 4, 4113–4119 RSC.
- S. R. Samanta, A. Anastasaki, C. Waldron, D. M. Haddleton and V. Percec, Polym. Chem., 2013, 4, 5555–5562 RSC.
- N. H. Nguyen, B. M. Rosen, G. Lligadas and V. Percec, Macromolecules, 2009, 42, 2379–2386 CrossRef CAS.
- A. L. Aleksandrov, Bull. Acad. Sci. USSR. Div. Chem. Sci. (Engl. Transl)., 1980, 29, 1740–1744 CrossRef.
- K. Min, H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2005, 127, 3825–3830 CrossRef CAS PubMed.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS PubMed.
- S. Dadashi-Silab, M. Atilla Tasdelen and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2878–2888 CrossRef CAS.
- F. A. Leibfarth, K. M. Mattson, B. P. Fors, H. A. Collins and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 199–210 CrossRef CAS PubMed.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- N. J. Treat, B. P. Fors, J. W. Kramer, M. Christianson, C.-Y. Chiu, J. R. d. Alaniz and C. J. Hawker, ACS Macro Lett., 2014, 3, 580–584 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 2271–2275 CrossRef CAS.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox, V. Percec, P. Wilson and D. M. Haddleton, J. Am. Chem. Soc., 2013, 136, 1141–1149 CrossRef PubMed.
- A. Anastasaki, V. Nikolaou, A. Simula, J. Godfrey, M. Li, G. Nurumbetov, P. Wilson and D. M. Haddleton, Macromolecules, 2014, 47, 3852–3859 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, N. W. McCaul, A. Simula, J. Godfrey, C. Waldron, P. Wilson, K. Kempe and D. M. Haddleton, Macromolecules, 2015, 48, 1404–1411 CrossRef CAS.
- E. Frick, A. Anastasaki, D. M. Haddleton and C. Barner-Kowollik, J. Am. Chem. Soc., 2015, 137, 6889–6896 CrossRef CAS PubMed.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- J. Xu, A. Atme, A. F. Marques Martins, K. Jung and C. Boyer, Polym. Chem., 2014, 5, 3321–3325 RSC.
- Y.-M. Chuang, A. Ethirajan and T. Junkers, ACS Macro Lett., 2014, 3, 732–737 CrossRef CAS.
- J. Vandenbergh, G. Reekmans, P. Adriaensens and T. Junkers, Chem. Sci., 2015, 6, 5753–5761 RSC.
- J. A. Burns, C. Houben, A. Anastasaki, C. Waldron, A. A. Lapkin and D. M. Haddleton, Polym. Chem., 2013, 4, 4809–4813 RSC.
- S. Shanmugam, J. Xu and C. Boyer, Chem. Sci., 2015, 6, 1341–1349 RSC.
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, F. Brandford-Adams, G. Nurumbetov, Q. Zhang, G. J. Clarkson, D. J. Fox, P. Wilson, K. Kempe and D. M. Haddleton, Chem. Commun., 2015, 51, 5626–5629 RSC.
- S. R. Samanta, M. E. Levere and V. Percec, Polym. Chem., 2013, 4, 3212–3224 RSC.
- S. R. Samanta, H.-J. Sun, A. Anastasaki, D. M. Haddleton and V. Percec, Polym. Chem., 2014, 5, 89–95 RSC.
- S. R. Samanta, A. Anastasaki, C. Waldron, D. M. Haddleton and V. Percec, Polym. Chem., 2013, 4, 5563–5569 RSC.
- C. Boyer, A. Atme, C. Waldron, A. Anastasaki, P. Wilson, P. B. Zetterlund, D. Haddleton and M. R. Whittaker, Polym. Chem., 2013, 4, 106–112 RSC.
- C. Waldron, A. Anastasaki, R. McHale, P. Wilson, Z. Li, T. Smith and D. M. Haddleton, Polym. Chem., 2014, 5, 892–898 RSC.
- M. J. Scott, C. A. Goddard and R. H. Holm, Inorg. Chem., 1996, 35, 2558–2567 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py01578b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |