
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImparting functional variety to cellulose ethers via olefin cross-metathesis†
Yifan
Dong
ab and
Kevin J.
Edgar
*ac
aDepartment of Sustainable Biomaterials, Virginia Tech, 230 Cheatham Hall, Blacksburg, VA 24061, USA. E-mail: kjedgar@vt.edu
bDepartment of Chemistry, Virginia Tech, 2018 Hahn Hall South, MC 0212, Blacksburg, VA 24061, USA
cMacromolecules and Interfaces Institute, Virginia Tech, Blacksburg, VA 24061, USA
First published on 9th April 2015
Abstract
Olefin cross-metathesis is a valuable new approach for imparting functional variety to cellulose ethers. Starting from commercially available ethyl cellulose, terminally unsaturated alkyl groups were appended as metathesis handles by reaction with allyl chloride, 5-bromo-1-pentene, 7-bromo-1-heptene and 11-bromo-1-undecene, employing sodium hydride catalyst. These olefin-terminal ethyl cellulose derivatives were then subjected to olefin cross-metathesis with a variety of electron-poor olefin substrates, including acrylic acid and acrylate esters under optimized conditions (5–10 mol% Hoveyda-Grubbs’ 2nd generation catalyst, 37 °C, 2 h). The effects of varying the length of the ω-unsaturated alkyl handle, and of the solvent systems used were evaluated. Ethyl cellulose containing a pent-4-enyl substituent performed best in cross metathesis reactions and a hept-6-enyl substituent gave similarly good results. Ethyl cellulose with allyl substituents gave low to moderate metathesis conversion (<50%), possibly due to steric effects and the proximity of the ether oxygen to the terminal olefin. Interestingly, longer tethers (undec-10-enyl) gave high conversions (up to 90%) but relatively slow reactions (ca. 12 h needed for high conversion). While limited in this study by the relatively low DS (OH) of the starting commercial ethyl cellulose materials, this methodology has strong promise for introduction of diverse functionality to cellulose ethers in chemospecific and mild fashion, enabling amorphous solid dispersion and other applications.
Introduction
Modifications of naturally occurring cellulose to prepare cellulose ethers are of substantial scientific and commercial value, not only imparting solubility (frequently in water) and processability, but also providing new properties that have greatly extended the range of applications of cellulose and its derivatives into coatings, personal care, cleaning products, foods, and pharmaceuticals,1–6 to name just a few. The historical development of cellulose ether synthetic methods has provided useful derivatives including methyl cellulose (MC), ethyl cellulose (EC), carboxymethyl cellulose (CMC), hydroxypropyl cellulose (HPC) and hydroxypropylmethyl cellulose (hypromellose or HPMC). Even though these cellulose ethers have great functional and commercial utility, and are produced on billions of kilograms per annum scale, the range of available cellulose ether derivatives is still surprisingly limited if we consider the high functionality of hydroxyl groups per anhydroglucose unit (AGU), and the increasing demands for and upon renewable biopolymers. To a significant extent this narrow scope has to do with the requirements of traditional synthesis methods, in which alkali cellulose is reacted in aqueous media with electrophiles like alkyl halides and epoxides. This methodology is limiting with respect to the solubility and reactivity of the electrophiles that may be used.Cellulose ether esters have also been of considerable recent interest, including in particular cellulose ether esters that possess pendent carboxylic acid functionality.7–9 This pendent carboxyl functionality can be of value for imparting pH responsiveness, promoting specific interactions with small molecules, and for other purposes.10 Synthesis of these carboxy-containing cellulose ether esters can be complicated, since they typically possess both hydroxyl and carboxyl groups, and thus can be prone to crosslinking via ester formation during synthesis and/or use. Thus carboxymethyl cellulose acetate butyrate is a very promising polymer for coatings11 and drug delivery applications,12–14 but since it is synthesized by esterification of carboxymethylcellulose under acidic and anhydrous conditions,15 it may be expected to be prone to crosslinking. Acylation of cellulose ethers under mildly basic catalysis can be more successful, especially when the acylating reagent is a cyclic anhydride; hydroxypropyl methyl cellulose acetate succinate (HPMCAS) is a commercially important cellulose ether ester that can be made in this way.8
Cellulose ether esters have been of particular interest of late for their ability to enhance aqueous solubility of hydrophobic drugs, by incorporation of the drug into an amorphous solid dispersion (ASD);16–20 that is, to form a miscible blend of polymer matrix and hydrophobic drug, thereby preventing drug crystallization and removing from the dissolution process the need to surmount the energy barrier of the drug heat of fusion. These ASDs thereby provide a supersaturated solution of the hydrophobic drug, increasing drug permeation and hence bioavailability in oral drug delivery.21 Cellulose derivatives have proven to be highly valuable for use in ASDs due to their generally low toxicities, inability to permeate through the gastrointestinal (GI) epithelium, and high glass transition temperatures (keeping the formulation in the glassy state prevents drug mobility which may lead to drug crystallization). Carboxylated cellulose derivatives like HPMCAS can be especially useful for ASDs since they are pH responsive and can participate through the carboxyl group in specific interactions with drug functional groups.22–26 HPMCAS has hydrophobic moieties that interact with drug molecules in the ASD matrix and hydrophilic moieties (hydroxypropyl and succinate groups) that provide sufficient aqueous solubility such that HPMCAS can form stable drug-polymer colloids in the aqueous media of the GI tract.27 ASD is of growing importance to the pharmaceutical industry, but its ability to help patients by reducing dosage, cost, side effects, and pill size is restricted due to the fact that only polymers already used for other pharmaceutical purposes have been exploited to date (e.g., HPMCAS, HPMC, poly(ethylene glycol), and poly(vinyl pyrrolidinones)). There is a strong need for new polymers that are specifically designed for ASD, possessing the correct hydrophobic/hydrophilic balance, Tg, release triggers, and other important ASD properties.9,28 Therefore, strong fundamental structure–property studies of cellulose ether derivatives for ASD should be intriguing for polysaccharide chemists. However, traditional cellulose etherification typically requires harsh alkaline catalysts and aqueous-soluble reagents, so how can we practically vary the ether substituent across a wide structural range in order to investigate these structure–property relationships?
Recently, Meng and co-workers have demonstrated that olefin metathesis (OM) is a mild, efficient, and versatile method for synthesis of cellulose ester derivatives.29 Grubbs and his group pioneered OM as a powerful and convenient synthetic tool which is being extensively applied in both organic and polymer chemistry.30–32 Compared with all other transition-metal catalyzed carbon–carbon bond formation reactions, metathesis enjoys the advantages of using commercially available and easily handled ruthenium carbene-based catalysts, and has broad tolerance of functional groups.33–38 While there are many variants of OM, we have focused on olefin cross-metathesis (CM) due to its high selectivity, excellent efficiency, and ability to append different functional groups. Fig. 1 illustrates the structure of Hoveyda-Grubbs’ 2nd generation catalyst which shows high catalyst reactivity and selectivity for olefin CM.39–43
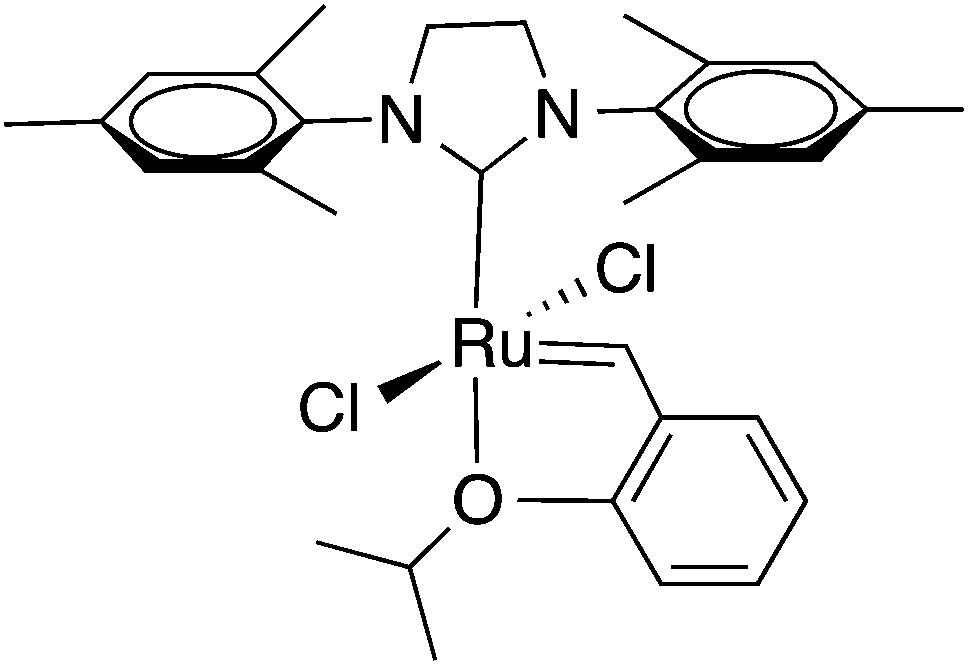 | ||
| Fig. 1 Hoveyda-Grubbs’ 2nd generation olefin metathesis catalyst. | ||
In using CM to modify polymers, the major issue is the selectivity for CM at the expense of self-metathesis (SM). SM leads to cross-linking, and cross-linking leads to poor processability, so is undesirable except in cases where loss of solubility, for example, is the goal. Fortunately Grubbs has categorized olefins39 according to their electron density and steric crowding into types that differ in CM reactivity, and has observed that by combining reactive and unreactive types, e.g. reactive Type I (electron rich and terminal olefins) with less reactive Type II (e.g., electron-deficient acrylates), highly selective CM can be achieved (Table 1), driven by the loss of volatile ethylene by-product. Recently, CM has also been explored as a method for synthesizing variously functionalized polysaccharide derivatives.29,44 Malzahn has reported an elegant preparation of hollow nanocapsules through interfacial olefin CM of hydrophilic acrylated dextran with a hydrophobic unsaturated organophosphate, employing a water-in-oil miniemulsion.45 Recently the Edgar group reported the first successful CM of cellulose ester derivatives that afforded soluble, discrete products.29 Successful CM of olefin terminated cellulose esters with different acrylates and hydrogenation of the product olefin to provide saturated derivatives afforded various modified cellulose esters with tailored functional groups.44
| Olefin type | Reactivity and selectivity | Examples |
|---|---|---|
| Type I | High reactivity, no selectivity | Terminal olefins, allyl silanes |
| Rapid homodimerization | ||
| Type II | Low reactivity and some selectivity | Acrylic acid and acrylates |
| Slow homodimerization | ||
| Type III | Low reactivity, high selectivity | Tertiary allylic alcohols |
| No homodimerization |
In this work we hypothesize that functionalized cellulose ethers may be excellent candidate ASD matrix polymers, and that olefin CM may be a novel, versatile, and efficient route for their synthesis. We put forward this hypothesis because of the far greater hydrolytic stability of ether linkages than ester linkages, which supports possible application in aqueous media, and because successful olefin CM would afford the opportunity to broadly vary the pendent functional groups, far beyond the variety available from conventional cellulose ether chemistry, thereby permitting detailed structure–property studies and tailoring of the polymer to high performance in ASD.4,46–48 At the same time these cellulose ether CM products, if properly designed, should be attractive for a wide variety of other applications due to retention of favorable cellulose ether properties like stability to ultraviolet radiation, high Tg, and low toxicity. In order to illuminate the key structure-reactivity-property-performance relationships, we explore synthesis and CM of a series of olefin-terminated ethyl cellulose derivatives with different side chain lengths (C3, C5, C7, and C11). We also probe the influence of the CM partner, beginning with acrylic acid and further extending to different acrylates like methyl acrylate (MA), hydroxyethyl acrylate (HEA), poly(ethylene glycol) methyl ether acrylate (PEGMEA) and poly(propylene glycol) acrylate (PPGA). The influences of catalyst load, reaction time and solvent system are also important and are fully explored.
Results and discussion
We began our exploration of CM of cellulose ether derivatives using commercially available ethyl cellulose with DS(Et) of 2.58 (EC2.58), since the ethyl groups should provide needed hydrophobicity for attaining good miscibility with hydrophobic drugs, by analogy to our earlier design of cellulose esters for ASDs.10 The DS(Et) was measured as 2.58 by 1H NMR spectroscopy after perpropionylation (Fig. S2†).We investigated etherification of EC2.58 by use of sodium hydride base in THF solution, using alkyl halides as electrophiles. We used this method since its experimental simplicity on lab scale was conducive to the planned extensive structure–property studies, and since it would eliminate hydroxide/alkyl halide side reactions that would be expected had we used NaOH catalyst. Initial work was with the electrophiles allyl chloride, 5-bromo-1-pentene, 7-bromo-1-heptene and 11-bromo-1-undecene. We were gratified to learn that the reaction of available hydroxyl groups was nearly quantitative for C3, C5 and C7 with the ω-unsaturated alkyl halide used. Even with C11 the etherification was still efficient considering the steric bulk and hydrophobicity of the long chain alkyl bromide. As shown in Table 2, the DS values obtained by 1H NMR for these ω-unsaturated alkyl chains were 0.38, 0.38, 0.41 and 0.36, respectively, which correspond to efficiencies of 93%, 93%, 98% and 86%, respectively vs. the measured DS (OH) of 0.42 (Fig. S3–S5†).
| Starting material | Unsaturated alkyl halide | Producta | DSb (olefin) |
|---|---|---|---|
| a Reaction conditions: NaH (10 equiv./OH), unsaturated alkyl halide (10 equiv./OH), THF, R.T. for 1d and 50 °C for 3 d. b DS of terminal olefin determined by 1H NMR (see ESI S3–S5). | |||
| EC2.58 | Allyl chloride | EC2.58C3 | 0.38 |
| 5-Bromo-1-pentene | EC2.58C5 | 0.38 | |
| 7-Bromo-1-heptene | EC2.58C7 | 0.41 | |
| 11-Bromo-1-undecene | EC2.58C11 | 0.36 | |
With these cellulose ethers possessing metathesis “handles” in hand, we proceeded to study their potential as olefin CM substrates. In order to ensure against undesired SM and crosslinking, we made three experimental choices: (1) use of Grubbs type II, electron-poor, less reactive olefins as partners; (2) use of the partner in excess to limit self-reaction of the cellulose derivatives; and (3) use of the highly reactive and selective Hoveyda-Grubbs’ 2nd generation catalyst (Fig. 1). For initial studies we selected type II olefin partners acrylic acid (AA) and its ester methyl acrylate (MA), both electron-poor, relatively unreactive olefins. Initial CM reaction of EC2.58C3 employed 5 mol% Hoveyda-Grubbs’ 2nd generation catalyst in 0.1 mol L−1 solution of terminal olefin under nitrogen protection for 2 h. A minimal amount of radical scavenger 3,5-di-tert-4-butylhydroxytoluene (BHT) was added to each reaction to suppress any free-radical reactions that could result from the product α, β-unsaturation;29 hydrogen atoms in the γ position of these product α, β-unsaturated acids and esters are quite labile towards H-atom abstraction.49 The CM products were solids that were isolated by precipitation, in some cases following purification by dialysis and freeze-drying. Scheme 1 shows the preparation of olefin terminated cellulose ethers and the further CM reactions. Results of CM reactions with acrylic acid and different acrylate esters are summarized in Table 3. Each CM product is labeled with an additional number referring to different reaction conditions. All CM conversions were calculated based on 1H NMR of olefin peaks.
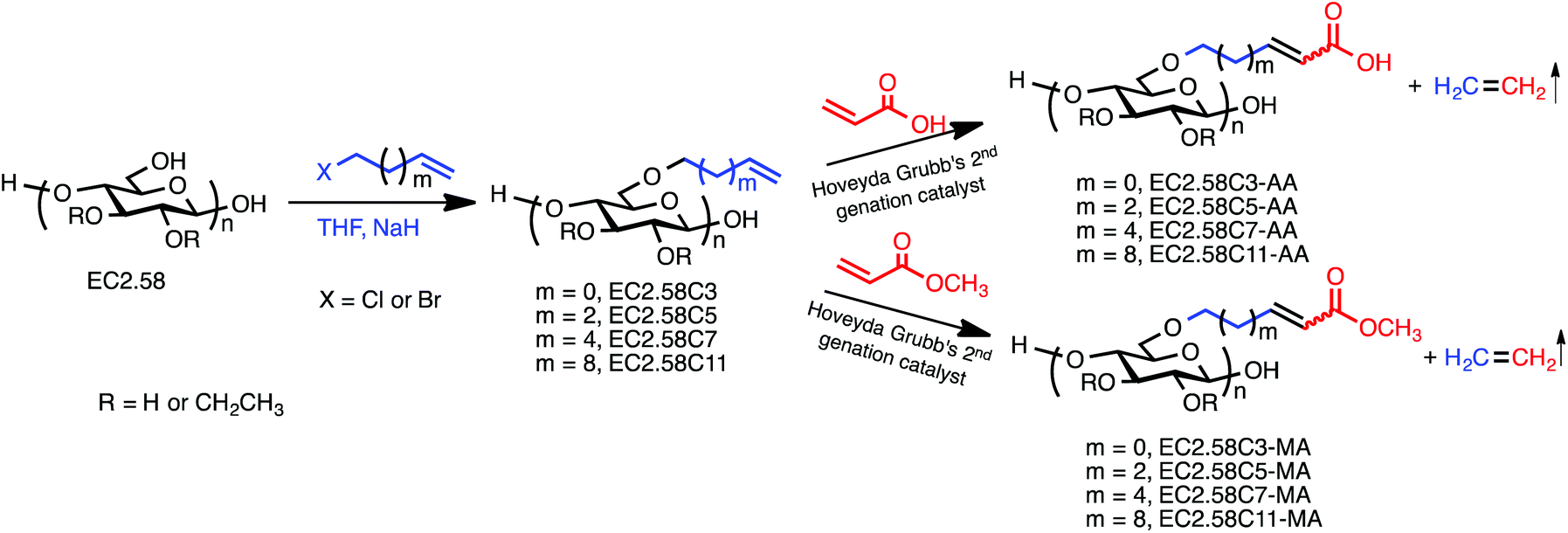 | ||
| Scheme 1 Two-step CM procedure using ethyl cellulose EC2.58. Note that structures are not meant to imply regioselectivity; particular positions of substitution in all schemes are only for convenience of depiction and clarity. | ||
| Cellulose ether (DSolefin) | CM partnera | Cat. load. (mol%) | Solvent | Time (h) | CM product | Conv. |
|---|---|---|---|---|---|---|
| a HEA: 2-hydroxyethyl acrylate, PEGMEA: poly(ethylene glycol) methyl ether acrylate; PPGA: poly(propylene glycol) acrylate; THF: tetrahydrofuran; DCM: dichloromethane; DMI: 1,3-dimethyl-2-imidazolidinone. b Reaction at 70 °C for 2 h. | ||||||
EC2.58C3 (DSC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C = 0.38) C = 0.38) |
AA | 5 | THF | 2 | EC2.58C3-AA-1 | 31% |
| 5 | DCM | 2 | EC2.58C3-AA-2 | 40% | ||
| 5 | AA | 2 | EC2.58C3-AA-3 | 23% | ||
| MA | 5 | THF | 2 | EC2.58C3-MA-1 | 47% | |
| 5 | DCM | 2 | EC2.58C3-MA-2 | 49% | ||
| 10 | DCM | 2 | EC2.58C3-MA-3 | 70% | ||
| EC2.58C5 (DSCC = 0.38) |
AA | 5 | THF | 2 | EC2.58C5-AA-1 | 60% |
| 5 | DCM | 2 | EC2.58C5-AA-2 | 45% | ||
| 5 | AA | 2 | EC2.58C5-AA-3 | 72% | ||
| 10 | THF | 2 | EC2.58C5-AA-4 | 87% | ||
| 10 | DCM | 2 | EC2.58C5-AA-5 | 100% | ||
| 10 | AA | 2 | EC2.58C5-AA-6 | 100% | ||
| MA | 5 | THF | 2 | EC2.58C5-MA-1 | 35% | |
| 5 | DMI | 2 | EC2.58C5-MA-2 | 38%b | ||
| 5 | DCM | 2 | EC2.58C5-MA-3 | 71% | ||
| 10 | THF | 2 | EC2.58C5-MA-4 | 51% | ||
| 10 | DCM | 2 | EC2.58C5-MA-5 | 100% | ||
| HEA | 10 | DCM | 2 | EC2.58C5-HEA | 100% | |
| PEGMEA | 10 | DCM | 2 | EC2.58C5-PEGMEA-1 | 85% | |
| 10 | DCM | 12 | EC2.58C5-PEGMEA-2 | 100% | ||
| PPGA | 10 | DCM | 2 | EC2.58C5-PPGA-1 | <20% | |
| 10 | DCM | 12 | EC2.58C5-PPGA-2 | <25% | ||
| 10 | THF | 12 | EC2.58C5-PPGA-3 | 42% | ||
| EC2.58C7 (DSCC = 0.41) |
AA | 10 | DCM | 2 | EC2.58C7-AA | >95% |
| MA | 10 | DCM | 2 | EC2.58C7-MA | >90% | |
| EC2.58C11 (DSCC = 0.36) |
AA | 5 | DCM | 2 | EC2.58C11-AA-1 | 44% |
| 5 | AA | 2 | EC2.58C11-AA-2 | 38% | ||
| 10 | AA | 2 | EC2.58C11-AA-3 | 59% | ||
| 5 | DCM | 12 | EC2.58C11-AA-4 | 83% | ||
| 10 | DCM | 12 | EC2.58C11-AA-5 | 100% | ||
| MA | 5 | DCM | 2 | EC2.58C11-MA-1 | 33% | |
| 5 | THF | 2 | EC2.58C11-MA-2 | 29% | ||
| 5 | DMI | 2 | EC2.58C11-MA-3 | 46%b | ||
| 5 | DCM | 12 | EC2.58C11-MA-4 | 95% | ||
| 10 | DCM | 12 | EC2.58C11-MA-5 | 100% | ||
Our initial CM reactions of EC2.58C3 with both acrylic acid and methyl acrylate in different solvent systems using 5 mol% Hoveyda-Grubbs’ 2nd generation catalyst gave low to moderate conversion (<50%). Even though by increasing the catalyst load to 10 mol%, reaction with methyl acrylate in dichloromethane gave improved (70%) conversion, the overall CM reaction was still not as efficient as those previously reported for long-chain ω-unsaturated cellulose esters. We thus propose that the partial conversion is due to the short olefin chain length of EC2.58C3 and the resulting steric effect from the cellulose backbone. The electron withdrawing effect from the ether oxygen also may reduce the electron density of the allyl group and thus the reactivity of terminal olefin. Therefore, we explored the CM of EC2.58C5, EC2.58C7, and EC2.58C11, with longer olefin terminated side chains (C5, C7 and C11 respectively).
EC2.58C5 with its pent-4-enyl substituents reacts more efficiently in CM reactions with methyl acrylate than does the corresponding allyl derivative at 5 mol% catalyst (conversion in DCM >70%), and we were pleasantly surprised to observe that increasing the catalyst load from 5 mol% to 10 mol% afforded 100% of CM conversion with methyl acrylate within 2 h. Fig. 2 shows the FT-IR spectra of EC2.58C5 and the 100% CM product EC2.58C5-MA-5, where the ester CO stretch at 1726 cm−1 is quite distinct, confirming successful introduction of ester groups after CM.
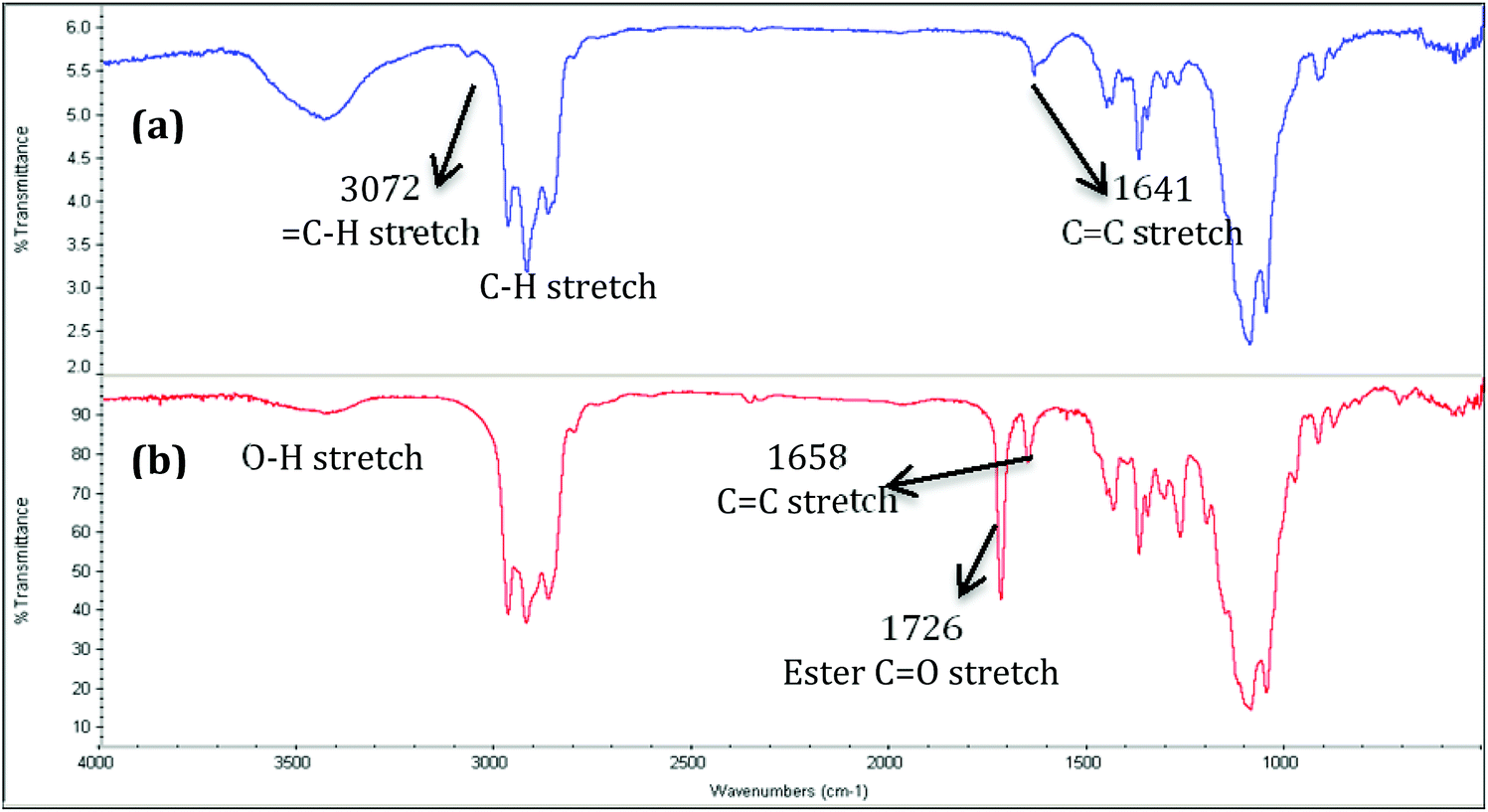 | ||
| Fig. 2 FT-IR spectra of EC2.58C5 (a) and EC2.58C5-MA-5 (b). | ||
1H NMR can often be diagnostic for reaction progression (Fig. 3, S2–S8†), since the terminal olefin protons resonate not only cleanly downfield of the backbone region, at 4.98 and 5.82 ppm in this case, but also are quite sharp since the olefin is distant from the cellulose main chain and therefore has rather free rotation compared to backbone protons and those more proximate to the main cellulose chain. Furthermore, the CM products now have conjugated olefins, so the olefin proton resonances appear even further downfield, in this case at 5.75 and 6.92 ppm. It is evident by comparing the 1H NMR spectrum of product EC2.58C5-MA-5 in Fig. 3 with that of the starting EC2.58C5 that the starting olefin was completely consumed, as shown by the complete disappearance of resonances at 4.98 and 5.82 ppm, to be replaced by the anticipated α, β-unsaturated ester CM product, with olefin proton resonances at 5.75 and 6.98 ppm, demonstrating essentially perfect CM conversion to the conjugated ester. The CM product olefin is predominantly in the E configuration, in accordance with results from previous CM reactions of cellulose esters.29
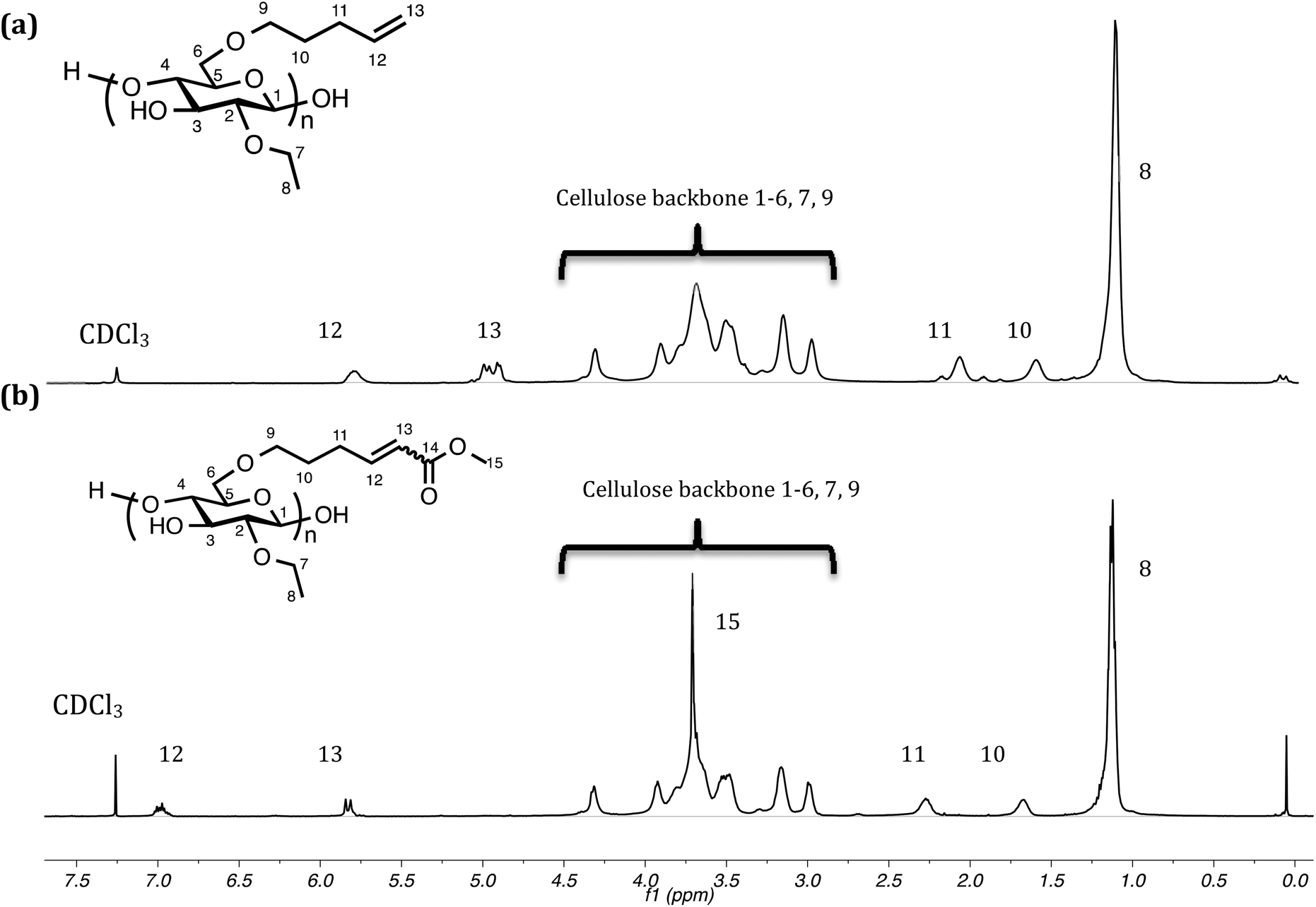 | ||
| Fig. 3 1H NMR spectra of EC2.58C5 (a) and EC2.58C5-MA-5 (b). | ||
13C NMR is also diagnostic for conversion to EC2.58C5-MA-5 (Fig. 4) since EC2.58C5 possesses no carbonyl group and, after CM, a distinct ester carbonyl resonance at 167.2 ppm appeared. At the same time, the two olefin resonances shifted downfield from 114.1 and 139.0 ppm to 121.2 and 150.2 ppm due to conjugation with the ester group. Moreover, the methoxy group from methyl acrylate also gave a sharp resonance at 51.7 ppm, strongly supporting successful CM. 1H–13C HSQC was also performed to help confirm the Z/E configuration. Fig. 5 showed clear correlation of the carbon signal at 150.2 ppm with proton signals at 6.98 and 6.28 ppm, and correlation of the carbon signal at 121.2 ppm with proton signals at 5.82 and 5.74 ppm, which confirmed the assignment of proton peaks of the E/Z isomers. The carbon β to the carbonyl group was shifted 11.2 ppm downfield after CM (from 139.0 ppm) due to conjugation with the ester group, and was correlated to proton peaks at 6.98 (E configuration) and 6.28 ppm (Z configuration). The α carbon was shifted 7.1 ppm downfield (from 114.1 ppm) and correlated with proton peaks at 5.82 (E configuration) and 5.74 ppm (Z configuration). The 1H–13C HSQC spectra also helped identify the cellulose backbone peaks; the small peak at 72.5 ppm was found to be a methylene, assigned as the backbone C6, and the backbone C2 peak at 77.4 ppm, overlapping with the CDCl3 solvent peak (77.2 ppm), was also revealed by the HSQC spectra.
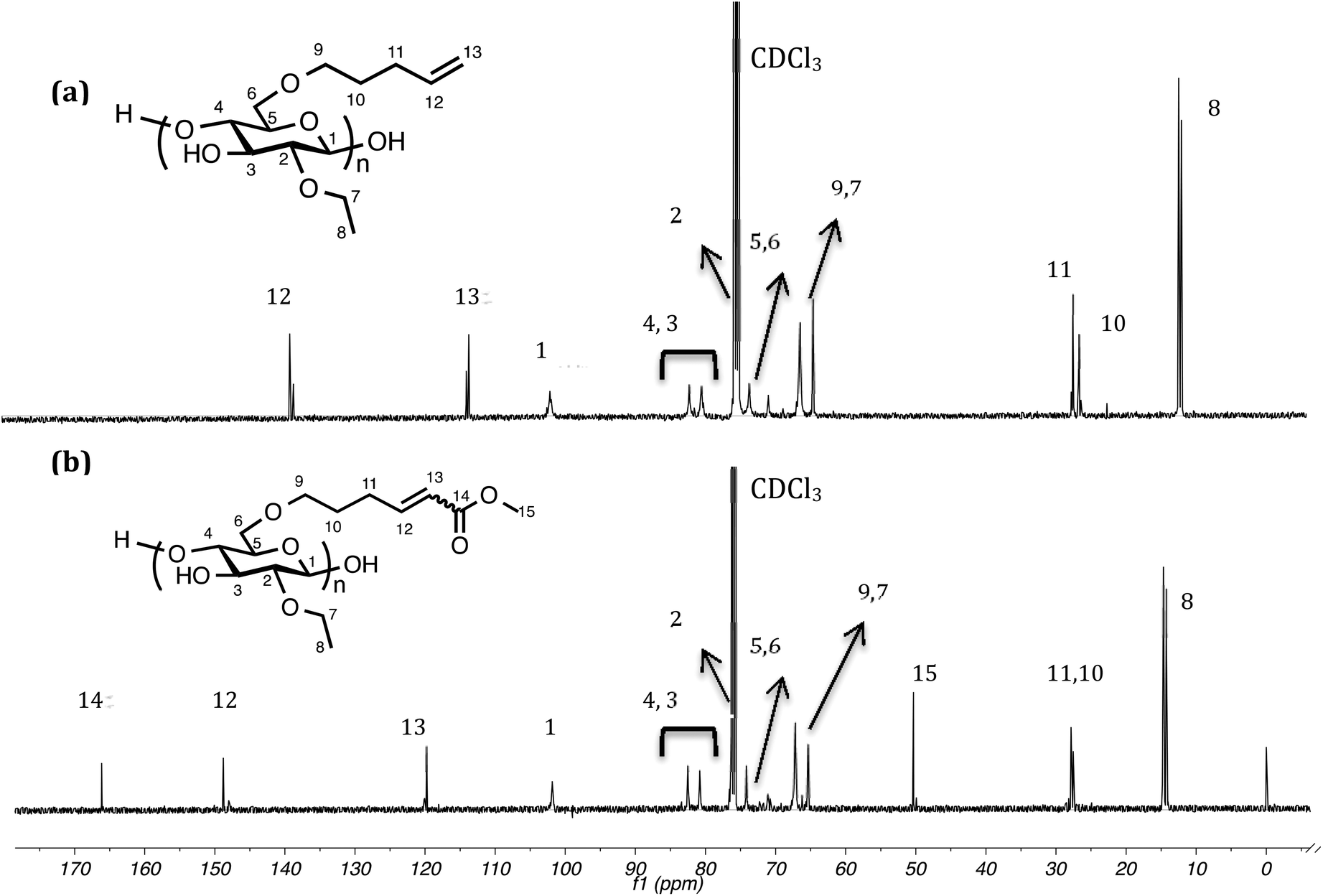 | ||
| Fig. 4 13C NMR spectra of EC2.58C5 (a) and EC2.58C5-MA-5 (b). | ||
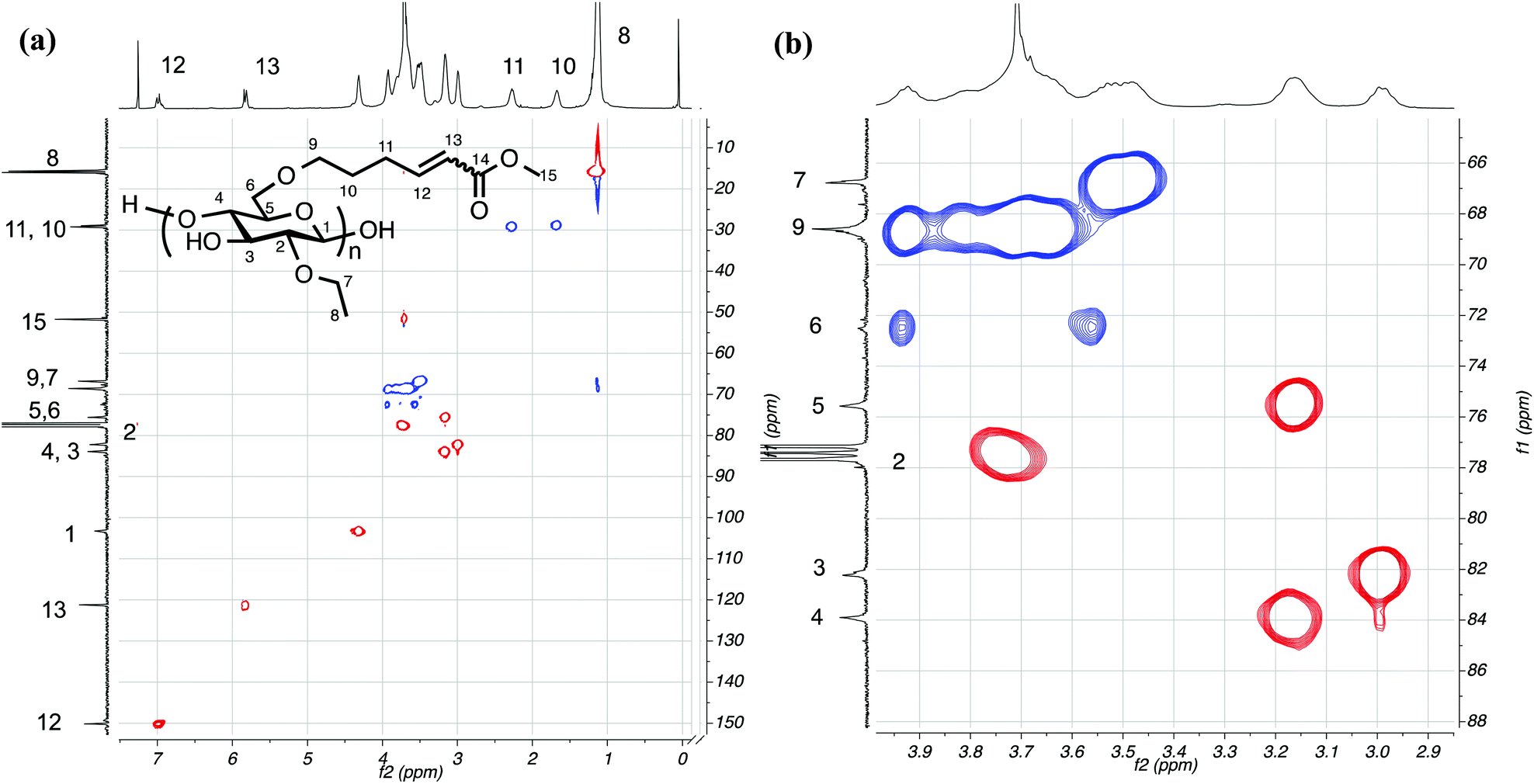 | ||
| Fig. 5 1H-13C HSQC spectrum of EC2.58C5-MA-5 (a) and enlarged cellulose backbone region (b). | ||
Knowing that CM of the pent-4-enyl ether with methyl acrylate could be efficient in DCM, we explored the flexibility with regard to solvent (i.e. tetrahydrofuran (THF) and 1,3-dimethyl-2-imidazolidinone (DMI)). We found that DCM gave the highest conversion among all these solvents, even vs. increased temperature in DMI (38% CM conversion when using 5 mol% catalyst in DMI at 70 °C for 2 h, vs. 71% in DCM in 2 h under same catalyst load at 37 °C). We further explored CM of EC2.58C5 using acrylic acid as the partner, since this would afford ω-carboxy substituents that are known to be favorable structural elements for ASD.9,50 At 5 mol% catalyst load, reaction in DCM or THF gave moderate conversion (45% and 60% separately), but upon increasing to 10 mol% catalyst CM of EC2.58C5 with acrylic acid in DCM was completely successful, affording 100% conversion to the desired CM product EC2.58C5-AA-5. On the other hand, we found that acrylic acid could be used as both solvent and reagent, thereby promoting high conversion. Under these somewhat acidic conditions, reaction of EC2.58C5 in acrylic acid afforded relatively high conversion (72%) with 5 mol% catalyst, and gratifyingly 100% conversion to the desired, α, β-unsaturated acid CM product EC2.58C5-AA-6 with 10 mol% catalyst within 2 h. Therefore the optimized CM conditions for successful CM of EC2.58C5 to afford desired product EC2.58C5-AA and EC2.58C5-MA are 10 mol% catalyst, DCM solvent for methyl acrylate, and either DCM or acrylic acid for CM with acrylic acid. Successful CM of EC2.58C5 with both acrylic acid and methyl acrylate led us to extend the range of CM partners. We further explored the CM of EC2.58C5 with a variety of hydrophilically substituted acrylates, since hydrophilicity is important for drug release and polymer solubility: 2-hydroxyethyl acrylate (HEA), poly(ethylene glycol) methyl ether acrylate (PEGMEA) (average Mn = 480 Da) and poly(propylene glycol) acrylate (PPGA) (average Mn = 475 Da). 2-Hydroxyethyl acrylate also reacts efficiently with EC2.58C5 under conditions optimized for the other acrylates (10 mol% catalyst in DCM, 2 h). We were also delighted by the high CM conversion with PEGMEA (85%), given concerns about potential steric interactions between the polymers, the relatively short tether between cellulose and olefin, and the high solution viscosity due to the polymeric metathesis partner. With extended reaction time (12 h), CM with the high molecular weight PEGMEA also reached 100%. Reaction with PPGA gave relatively low CM conversion even with 12 h reaction time (<25%), perhaps due to the more viscous nature of the PPGA and the poor homogeneity of the reaction system. For better solubility of the polymer CM partner, the CM reaction with PPGA was also performed in THF under the same conditions, and slightly increased but still moderate conversion (42%) was observed. Overall the pent-4-enyl substituted EC2.58C5 undergoes rapid, high conversion at mild temperatures to discrete CM products with various acrylates (AA, MA, HEA and PEGMEA), with no evidence of crosslinking (initial CM products were very solvent-soluble, see Table 4).
| Sample | CH2Cl2 | CHCl3 | THF | Acetone | DMSO | DMF |
|---|---|---|---|---|---|---|
| a (+) soluble; (−) insoluble; (P) partially soluble. b Solubility determined by visual examination using ∼10 mg dried sample in 2–3 mL of solvent after vortex mixing for 5 min, followed by gentle rolling overnight. | ||||||
| EC2.58 | P | + | + | P | P | P |
| EC2.58C5 | + | + | + | + | + | + |
| EC2.58C5-AA-5 | P | P | + | + | + | + |
| EC2.58C5-MA-5 | + | + | + | P | − | + |
| EC2.58C5-HEA | + | + | + | + | + | + |
| EC2.58C5-PEGMEA-2 | + | + | + | + | + | + |
| EC2.58C11 | + | + | + | P | + | + |
| EC2.58C11-AA-5 | + | + | + | + | + | + |
| EC2.58C11-MA-5 | + | + | + | P | + | + |
To further explore the influence of olefin tether length, we explored CM of EC2.58C7 (hept-6-enyl ether) with acrylic acid and methyl acrylate, with interesting initial results. Firstly, it was evident that the reactivity of EC2.58C7 is slightly lower than that of EC2.58C5. Using the earlier identified optimized conditions, CM with either acrylic acid and methyl acrylate afforded nearly quantitative conversion (>95% and >90%), but product 1H NMR spectra still reveal small peaks from unreacted terminal olefins (δ 4.96 and 5.80, respectively, see Fig. S8). It is likely that 100% conversion of EC2.58C7 would be achievable, e.g. with longer reaction time. The apparently slightly lower CM reactivity of the hept-6-enyl substituted ether may be due to enhanced self-association driven by side chain hydrophobic interactions.51,52 The CM reactivity of EC2.58C11 was even more intriguing. Using 5 mol% catalyst 37 °C, CM conversions with either acrylic acid or methyl acrylate were no better than moderate (<50%). We first considered that the low CM conversions may be due to the fact that THF or DCM are weaker solvents for EC2.58C11 than for the shorter chain ethers, leading to polymer self-association. Hence we looked at solvent variation, employing both more (DMI) and less polar (toluene) solvents at increased temperature (70 °C) but under otherwise similar conditions (2 h reaction, 5 mol% catalyst); however with these solvents of varying polarity, CM conversion remained below 50%. Even with increased catalyst (10 mol%, 2 h) the conversion was still less than 60%. We next explored whether we might simply be experiencing a kinetics problem. We found that after 12 h reaction in DCM (5% catalyst, 37 °C), CM conversion with acrylic acid rose to 83%, and with methyl acrylate reached near complete conversion (95%). Upon increasing to 10 mol% catalyst in DCM for 12 h, CM reactions with both acrylic acid and methyl acrylate reached 100% conversion. Therefore, CM of EC2.58C11 does proceed, but more slowly than with other tether lengths. It is possible that EC2.58C11 may tend to aggregate and self-assemble through hydrophobic interaction of the side chains, and therefore requires extended reaction time.
An overall comparison of the observed reactivity of the ω-unsaturated cellulose ethers prepared reveals that the pent-4-enyl substituted ether (EC2.58C5) reacts most quickly and completely in CM with acrylic acid and various acrylate esters including MA, HEA and PEGMEA. The hept-6-enyl ether EC2.58C7 has similar but slightly lower reactivity than the pent-4-enyl ether, while the allyl and undec-10-enyl ethers show lower CM conversions under the same conditions. We propose that the pent-4-enyl has side chain length adequate to minimize steric concerns, while not so long as to negatively impact solubility, catalyst interactions, or cause excessive self-association.
We used SEC to monitor the change of polymer molecular weight during CM process, and the results are summarized in Table 5. Only products with 100% CM conversion were characterized for comparison. For both C5 and C11 chain length, the molecular weight and degree of polymerization (DP) of starting materials EC2.58 decreased during the four-day etherification due to alkaline peeling from the end of cellulose chain. However, the DP of EC2.58C5 CM products did not greatly decrease during the 2 h CM reactions at 37 °C in DCM or even acetic acid solvents. Dispersity (Đ) was used to evaluate the molecular weight distribution and from the results we can see that with the same starting material EC2.58C5, the CM products EC2.58C5-AA-5, EC2.58C5-HEA and EC2.58C5-PEGMEA-2 have almost the same dispersity (Đ), demonstrating the mild nature of the CM conditions and their minimal impact on molecular weight during the 2 h reaction. CM of EC2.58C11 also gave products with relatively unchanged molecular weight and dispersity, providing further support for the conclusion that successful intermolecular CM had occurred to the exclusion of SM; SM of the cellulose ω-olefins would result in rapid cross-linking and sharply increasing molecular weight.
| Sample | M n (kDa) | DP | PDI | T g (°C) |
|---|---|---|---|---|
| a By SEC in THF solvent. b Transition very broad, see ESI (Fig. S9). c EC2.58C11-AA-5 was measured by a TA Q100 instrument and EC2.58C11-MA-5 was measured using a TA Q200 instrument. | ||||
| EC2.58 | 56.0a | 240 | 1.51 | 127 |
| EC2.58C5 | 27.9 | 107 | 1.45 | 80b |
| EC2.58C5-AA-5 | 23.5a | 85 | 1.35 | 96 |
| EC2.58C5-MA-5 | 21.4 | 76 | 2.24 | 81 |
| EC2.58C5-HEA | 23.0a | 78 | 1.33 | 88 |
| EC2.58C5-PEGMEA-2 | 42.0a | 102 | 1.23 | 82 |
| EC2.58C11 | 29.0a | 97 | 1.30 | 117 |
| EC2.58C11-AA-5 | 40.0a | 127 | 1.15 | 96c |
| EC2.58C11-MA-5 | 34.6 | 107 | 1.81 | 120c |
Glass transition temperature (Tg) is an important parameter for polymer materials. For amorphous solid dispersion applications, a Tg that is ≥50 °C higher than any likely ambient temperature is used as a rule of thumb to predict the ability of the polymer to keep the formulation in the glassy state, preventing drug mobility and therefore recrystallization.53 We used DSC to determine polymer Tg values. Commercial cellulose ethers are complex mixtures of monosaccharides and monosaccharide sequences, making DSC glass transitions quite broad (sometimes ranging across ca. 40 °C) with low resolution; therefore modulated DSC was applied where necessary to improve resolution and clarity. The starting material EC2.58 showed a broad transition at 127 °C, which is comparable to the previous reported value for ethyl cellulose.54 After etherification the EC2.58C5 showed an even broader DSC transition (analysis by MDSC also gave no clear transition) (Fig. S9†). However, by applying MDSC, all EC2.58C5 CM products showed lower Tg compared to the starting material EC2.58, which is as expected since the C5 side chains should increase the free volume of the polymer and thus enhance the main chain mobility, resulting in a lower Tg (Table 5 and Fig. S9†). The better resolution of CM products than the olefin terminated cellulose ethers may be due to the dispersive interaction between the terminal polar functional groups.55–57 On the other hand, the EC2.58C11 CM products showed higher Tg than EC2.58C5 CM products, which we suppose is due to the hydrophobic interaction of hydrocarbon chain and the interaction between the acid/ester terminal groups promoting the C11 side chain stacking, which stiffen the backbone, decrease free volume, and eventually contribute to higher measured Tg when compared to EC2.58C5 CM products.57,58 Overall the Tg values of many CM products are ≥50 °C higher than any likely ambient temperature (maximum ca. 40–50 °C), indicating these cellulose ethers are suitable for ASD application with regard to maintaining the amorphous state and preventing drug recrystallization.
An issue of potential concern is the stability of these pendant α, β-unsaturated carboxylic acid and ester products, given the previously mentioned lability of the γ-hydrogen atoms to radical abstraction. The previous report by Meng revealed that free radical oligomerization can lead to crosslinking and loss of solubility in analogous cellulose ester CM products.29 With regard to the current cellulose ether CM products, adding BHT as radical scavenger for the CM reaction prevented crosslinking and maintained product solubility during CM reaction and purification (either precipitation or dialysis). Additional BHT was also added to the products for storage, which helped keep these cellulose ether CM products soluble for extended periods. Ultimately, the key to permanently eliminating this instability will be to eliminate the α, β-unsaturation, either by means of hydrogenation44 or by further double bond elaboration, e.g. by Michael addition.59,60
Experimental
Materials
Ethyl cellulose N50 (EC2.58, Mw = 56 KDa, DP = 240 as determined by size exclusion chromatography) was from Ashland Specialty Chemical Company. DS(Et) was determined by perpropionylation (Fig. S2†). Allyl chloride, 5-bromo-pent-1-ene, 11-bromo-undec-1-ene, anhydrous tetrahydrofuran (THF), acrylic acid, methyl acrylate, 2-hydroxyl ethyl acrylate, poly(ethylene glycol) methyl ether acrylate, poly(propylene glycol) methyl ether acrylate, 3,5-di-tert-butylhydroxytoluene (BHT), and Hoveyda-Grubb's 2nd generation catalyst were purchased from Sigma-Aldrich. 1-Bromo-7-heptene was purchased from Oakwood Products. Monopotassium phosphate (KH2PO4), dimethyl sulfoxide (DMSO), dichloromethane, ethanol and isopropanol were purchased from Fisher Scientific. Sodium hydride (NaH) and potassium phosphate dibasic (K2HPO4) were from Acros Organics. Ethyl vinyl ether was purchased from Fluka Analytical. All reagents were used as received without further purification. Dialysis tubing (MWCO 3500 Da) was purchased from Fisher Scientific.Measurements
1H NMR spectra were acquired on a Bruker Avance 500 spectrometer operating at 500 MHz. Samples were analyzed as solutions in CDCl3 or DMSO-d6 (ca. 10 mg mL−1) at 25 °C in standard 5 mm o.d. tubes. Three drops of trifluoroacetic acid were added to shift the water peak in DMSO-d6 downfield from the spectral region of interest. 13C NMR spectra were acquired on Bruker 500 MHz spectrometer with a minimum of 5000 scans in DMSO-d6 (ca. 40 mg mL−1). FT-IR spectra were obtained on a Nicolet 8700 instrument using KBr powder as matrix. Tg values were obtained by differential scanning calorimetry (DSC) on a TA instruments Q100 apparatus using heat/cool/heat mode. Dry powders (ca. 5 mg) were loaded in Tzero aluminum pans; each sample was equilibrated at −50 or −20 °C and then heated to 150 °C at 20 °C min−1. The sample was then quenched at 100 °C min−1 to −50 °C. During the second heating cycle the sample was heated to 190 °C at 20 °C min−1. For transitions that were not sufficiently resolved, modulated DSC (MDSC) was performed: sample first equilibrated at −25 °C and then gradually heated up to 190 °C with the underlying ramp heating rate as 3 °C min−1 and the oscillation amplitude ±0.5 °C with oscillation period 60 s. Size exclusion chromatography (SEC) in DMAc/LiCl was performed at 1 mL min−1 flow rate at 40 °C on a Agilent 1260 Infinity Multi-Detector SEC using 3 PLgel 10 μm mixed-B columns connected in series with multiple detectors: a Wyatt Dawn Helios multi-angle laser light scattering (MALS) detector, a viscometer detector and a Wayatt Optilab Rex refractive index detector. Monodisperse polystyrene standard (Mw ∼ 21k, PDI ∼ 1.02) was used for calibration. SEC in THF was carried out at 1 mL min−1 at 30 °C on two Agilent PLgel 10 μm mixed-B columns connected in series with a Wyatt Dawn Helios light scattering detector and a Wayatt Optilab Rex refractive index detector. For SEC in THF no calibration standards were used and dn/dc values were obtained by assuming 100% mass elution from the columns.Abbreviation
We employ abbreviations for the products obtained; for example EC2.58C3 refers to the product of reaction of EC2.58 with allyl chloride, that is the “C3” electrophile. EC2.58C5, EC2.58C7 and EC2.58C11 are named using a similar code and have C5, C7 and C11 ω-unsaturated alkyl substituents, respectively. The CM products are named using a term for the CM partner after the cellulose ether term; for example EC2.58C5-AA refers to the CM product of EC2.58C5 with acrylic acid (AA). Each sample was labeled with an additional number referring to the sample prepared under specific reaction conditions.Solubility tests
Dried sample (∼10 mg) was added to a 10 mL glass vial, then 2–3 mL of solvent was added. The mixture was subjected to vortex mixing for 5 min (moderate heating was applied) and placed on a roller overnight; the solubility was determined by visual examination.Preparation of allyl ethyl cellulose (EC2.58C3)
Ethyl cellulose EC2.58 (1.0 g, 4.30 mmol AGU, 1.85 mmol –OH) was dissolved in 10 mL anhydrous THF at room temperature with mechanical stirring. NaH (95%, 0.47 g, 18.5 mmol, 10 equiv.) was added. The solution was cooled to 0 °C, then allyl chloride (1.42 g, 18.5 mmol, 10 equiv.) was added dropwise with vigorous stirring. The solution was stirred at room temperature for 1 day and at 50 °C for three days. Solidification occurred during this process, but after the temperature reached 50 °C, the solid gradually disappeared. The reaction was quenched by isopropanol to destroy residual NaH, and then the mixture was poured into 500 mL pH 7.4 buffer (7.14 g K2HPO4 and 3.54 g KH2PO4 in 1000 mL deionized water). The resulting precipitate was collected by vacuum filtration, washed with water and ethanol, and dried overnight under vacuum at 40 °C. 1H NMR (500 MHz, CDCl3): 1.14 (br s, OCH2CH3), 2.85–5.58 (m, cellulose backbone, OCH2CH3 and OCH2CHCH2), 4.95–5.35 (dq, OCH2CHCH2), 5.80–6.05 (m, OCH2CHCH2). 13C NMR (500 MHz, CDCl3): 16.0 (OCH2CH3), 66.8 (OCH2CH3), 68.6 (OCH2CHCH2), 72.4 (C6), 74.3 (C5), 75.6 (C2), 82.3 (C3), 83.9 (C4), 103.3 (C1), 115.6 (OCH2CHCH2), 136.7 (OCH2CHCH2). DS by 1H NMR: DS (Et) 2.58, DS (allyl) 0.38; yield: 93%.
Similar procedures were followed for the preparation of EC2.58C5, EC2.58C7 and EC2.58C11 using 5-bromo-1-pentene, 7-bromo-1-heptene and 11-bromo-1-undecene, respectively, and NaH catalyst. The last two polymers were purified as follows: EC2.58C7 was centrifuged after precipitation into buffer, the upper liquid was then discarded and the polymer was collected and washed sufficiently by hexane to remove the alkyl halide reagent before vacuum drying. EC2.58C11 was dialyzed against ethanol for three days with ethanol replaced five times and in water for another two days before freeze-drying.
EC2.58C5
1H NMR (500 MHz, CDCl3): 1.14 (br s, OCH2CH3), 1.63 (s, OCH2CH2CH2CHCH2), 2.08 (s, OCH2CH2CH2CHCH2), 2.85–4.50 (cellulose backbone, OCH2CH3 and OCH2CH2CH2CHCH2), 4.80–5.12 (m, OCH2CH2CH2CHCH2), 5.81 (m, OCH2CH2CH2CHCH2). 13C NMR (500 MHz, CDCl3): 15.6 (OCH2CH3), 29.5 (OCH2CH2CH2CHCH2), 30.3 (OCH2CH2CH2CHCH2), 66.4 (OCH2CH3), 68.2 (OCH2CH2CH2CHCH2), 72.6 (C6), 75.5 (C5), 77.5 (C2), 81.8 (C3), 83.6 (C4), 102.8 (C1), 114.1(OCH2CH2CH2CHCH2), 139.0 (OCH2CH2CH2CHCH2). DS by 1H NMR: DS (Et) 2.58, DS (C5) 0.38; yield: 89%.
EC2.58C7
1H NMR (500 MHz, CDCl3): 1.15 (m, OCH2CH3), 1.25–1.37 (m, OCH2CH2CH2CH2CH2CHCH2), 1.54 (m, OCH2CH2CH2CH2CH2CHCH2), 2.05 (q, OCH2CH2CH2CH2CH2CHCH2), 2.83–4.50 (m, cellulose backbone, OCH2CH2 and OCH2CH2CH2CH2CH2CHCH2), 4.85–5.05 (dd, OCH2CH2CH2CH2CH2CHCH2), 5.79 (m, OCH2CH2CH2CH2CH2CHCH2). DS by 1H NMR: DS (Et) 2.58, DS (C7) 0.41; yield: 86%.
EC2.58C11
1H NMR (500 MHz, CDCl3): 1.15 (m, OCH2CH3), 1.25–1.37 (m, OCH2CH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 1.80–1.92 (m, OCH2CH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 2.04 (q, OCH2CH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 2.85–4.50 (m, cellulose backbone, OCH2CH2 and OCH2CH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 4.88–5.04 (dd, OCH2CH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 5.82 (m, OCH2CH2CH2CH2CH2CH2CH2CH2CH2CHCH2). DS by 1H NMR: DS (Et) 2.58, DS (C11) 0.36; yield: 74%.
General procedure for olefin metathesis of pent-4-enyl ethylcellulose with acrylic acid in DCM or THF
To a flask charged with EC2.58C5 (100 mg, 0.38 mmol AGU, 0.14 mmol terminal olefin), 5 mg BHT and 3 mL DCM or THF were added. The mixture was stirred under N2 protection at 37 °C for DCM system (or THF) until all reagents were completely dissolved. Then acrylic acid (0.42 g, 5.8 mmol, 40 equiv.) was added, followed by Hoveyda-Grubbs’ 2nd generation catalyst (4.4 mg, 5 mol% based on olefin, dissolved in DCM or THF). After stirring at 40 °C for 2 h, the reaction was stopped by adding 2 drops of ethyl vinyl ether. For DCM as solvent, after quenching the reaction with ethyl vinyl ether, the reaction solution was dialyzed in a 600 mL beaker against EtOH overnight, then the medium was changed to water and dialyzed for another day, with both EtOH and water changed twice during dialysis. When THF was the solvent, the product was precipitated in water and sufficiently washed by water before vacuum drying at 40 °C.General procedure for olefin metathesis of pent-4-enyl ethylcellulose with acrylic acid as solvent and reagent
To a flask charged with EC2.58C5 (100 mg, 0.38 mmol AGU, 0.14 mmol terminal olefin), 5 mg BHT and 3 mL acrylic acid were added. The mixture was stirred under N2 protection at 40 °C until all reagents were completely dissolved. Then Hoveyda-Grubbs’ 2nd generation catalyst (4.4 mg, 5 mol%, dissolved in 3 mL acrylic acid) was added. After stirring at 40 °C for 2 h, the reaction was stopped by adding 2 drops of ethyl vinyl ether. The solution was added to water to precipitate the product. The product was collected by vacuum filtration, then was sufficiently washed with water before vacuum drying at 40 °C.General procedure for olefin metathesis of pent-4-enyl ethylcellulose EC2.58C5 with methyl acrylate in DCM
To a flask charged with EC2.58C5 (100 mg, 0.38 mmol AGU, 0.14 mmol terminal olefin), 5 mg BHT and 3 mL DCM were added, and the mixture was stirred under N2 protection at 37 °C until all reagents were completely dissolved. Then methyl acrylate (0.50 g, 5.8 mmol, 40 equiv.) was added followed by Hoveyda-Grubbs’ 2nd generation catalyst (4.4 mg, 5 mol%, dissolved in DCM). After 2 h reaction, two drops of ethyl vinyl ether was added to stop CM. The product was dialyzed in a 600 mL beaker against EtOH overnight, then against water for one more day, with both EtOH and water changed twice every 24 h. The CM product precipitated during dialysis, then was collected by filtration and vacuum dried overnight at 40 °C.Similar procedures were performed for CM of EC2.58C3, EC2.58C7 and EC2.58C11 in other solvent systems. Each sample is labeled with an additional number referring to specific reaction conditions. Analysis of CM conversion is based on the ratio of olefin peak integrations in the 1H NMR spectra, and one example calculation of EC2.58C5-MA-3 with partial conversion is shown in ESI S6.†
EC2.58C3-AA-2
1H NMR (500 MHz, CDCl3): 1.15z (br s, OCH2CH3), 2.85–5.58 (m, cellulose backbone, OCH2CH3 and OCH2CHCH2), 4.95–5.35 (dq, unreacted OCH2CHCH2), 5.80–6.05 (m, unreacted OCH2CHCH2), 6.05–6.18 (m, OCH2CHCHCOOH), 6.96 (m, OCH2CHCHCOOH).
EC2.58C5-AA-5
1H NMR (500 MHz, CDCl3): 1.14 (br s, OCH2CH3), 1.67 (s, OCH2CH2CH2CHCHCOOH), 2.28 (s, OCH2CH2CH2CHCHCOOH), 2.85–4.50 (cellulose backbone, OCH2CH3 and OCH2CH2CH2CHCHCOOH), 5.75 (m, OCH2CH2CH2CHCHCOOH, Z configuration), 5.83 (m, OCH2CH2CH2CHCHCOOH, E configuration), 6.29 (m, OCH2CH2CH2CHCHCOOH, Z configuration), 6.99 (m, OCH2CH2CH2CHCHCOOH, E configuration). 13C NMR (500 MHz, CDCl3): 16.0 (OCH2CH3), 28.9 (OCH2CH2CH2CHCHCOOH), 29.2 (OCH2CH2CH2CHCHCOOH), 66.8 (OCH2CH3), 68.6 (OCH2CH2CH2CHCHCOOH), 72.5 (C6), 75.6 (C5), 77.5 (C2), 82.2 (C3), 83.9 (C4), 103.2 (C1), 121.2 (OCH2CH2CH2CHCHCOOH), 150.2 (OCH2CH2CH2CHCHCOOH), 167.6 (OCH2CH2CH2CHCHCOOH). CM conversion by 1H NMR: 100%, E/Z ratio 11.1; yield: 92%.
EC2.58C5-MA-5
1H NMR (500 MHz, CDCl3): 1.12 (br s, OCH2CH3), 1.68 (s, OCH2CH2CH2CHCHCOOCH3), 2.27 (s, OCH2CH2CH2CHCHCOOCH3), 2.85–4.50 (cellulose backbone, OCH2CH3 and OCH2CH2CH2CHCHCOOCH3), 5.73 (d, OCH2CH2CH2CHCHCOOCH3, Z configuration), 5.82 (d, OCH2CH2CH2CHCHCOOCH3, E configuration), 6.27 (m, OCH2CH2CH2CHCHCOOCH3, Z configuration), 6.97 (m, OCH2CH2CH2CHCHCOOCH3, E configuration). CM conversion by 1H NMR: 100%, E/Z ratio 12.5; yield: 90%.
EC2.58C5-HEA
1H NMR (500 MHz, CDCl3): 1.13 (br s, OCH2CH3), 1.67 (s, OCH2CH2CH2CHCHCOOCH2CH2OH), 2.12–2.38 (m, OCH2CH2CH2CHCHCOOCH2CH2OH), 2.85–4.50 (cellulose backbone, OCH2CH3 and OCH2CH2CH2CHCHCOOCH2CH2OH), 5.78 (d, OCH2CH2CH2CHCHCOOCH2CH2OH, Z configuration), 5.95 (d, OCH2CH2CH2CHCHCOOCH2CH2OH, E configuration), 6.34 (m, OCH2CH2CH2CHCHCOOCH2CH2OH, Z configuration), 6.97 (m, OCH2CH2CH2CHCHCOOCH2CH2OH, E configuration). CM conversion by 1H NMR: 100%, E/Z ratio 11.8; yield: 95%.
Conclusions
Cellulose ω-unsaturated alkyl ethers were synthesized and olefin cross-metathesis of this series of cellulose ω-unsaturated alkyl ether derivatives was successful under very mild conditions, with high efficiency and selectivity in many cases. Conversions were modest for the sterically crowded and rather electron poor C3 (allyl) ethers, whereas they were high under the right conditions for longer chain unsaturated ethers (C5, C7, and C11). Free-radical initiated crosslinking of α, β-unsaturated carboxylic acid and ester products could be suppressed by using a minimal amount of a radical scavenger such as BHT. Each type II olefin examined could afford high CM conversion under the appropriate, mild reaction conditions, which were readily determined. The molecular weight, solubility and glass transition temperatures of the CM products were studied and were also related to structural features in predictable fashion.This first successful olefin CM of cellulose ether derivatives opens broad and novel pathways, enabling us to increase the diversity of accessible cellulose ether derivatives, thus providing ready access for example to a varied group of carboxy-terminal substituents for use in ASD and other forms of drug delivery.21 Certain limitations revealed themselves in this initial study as well. The relatively low DS (OH) available in commercial EC samples limits the amount of CM “handle” group that can be incorporated, thereby limiting the DS of other functional groups that can be appended by CM. This could for example cripple ASD structure–property studies, where a higher DS of carboxyl functionality has been found to be desirable in cellulose ester derivatives. Furthermore, the residual unsaturation and resulting reactivity towards H-atom abstraction of these derivatives can be managed in the short term by adding radical scavengers, but in the longer term may prove problematic. Our future studies will seek to address these issues, for example through hydrogenation and/or Michael addition, and will seek to illuminate in detail the structure–property-performance relationships of these new cellulose ether derivative families, especially with regard to amorphous solid dispersion.
Acknowledgements
We thank Ashland Specialty Chemical Company for their kind donation of the ethyl cellulose used in this work. We thank the National Science Foundation for funding this work through award DMR-1308276. We thank the Institute of Critical Technologies and Applied Science at Virginia Tech for facility support, and the Department of Chemistry and the Macromolecules and Interfaces Institute for educational support. We also thank Shreya Roy Choudhury, Mark Flynn and Scott Radzinski of Virginia Tech for performing SEC analyses.Notes and references
- K. J. Edgar, C. M. Buchanan, J. S. Debenham, P. A. Rundquist, B. D. Seiler, M. C. Shelton and D. Tindall, Prog. Polym. Sci., 2001, 26, 1605–1688 CrossRef CAS.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- K. J. Edgar, Cellulose, 2007, 14, 49–64 CrossRef CAS.
- S. Kamel, N. Ali, K. Jahangir, S. M. Shah and A. A. El-Gendy, eXPRESS Polym. Lett., 2008, 2, 758–778 CrossRef CAS.
- C. Chang and L. Zhang, Carbohydr. Polym., 2011, 84, 40–53 CrossRef CAS PubMed.
- K. Devarayan, H.-Y. Kim and B.-S. Kim, Carbohydr. Polym., 2014, 117, 408–413 CrossRef PubMed.
- Y. Bao, J. Ma and N. Li, Carbohydr. Polym., 2011, 84, 76–82 CrossRef CAS PubMed.
- W. K. Miller, D. K. Lyon, D. T. Friesen, W. B. Caldwell, D. T. Vodak and D. E. Dobry, Hydroxypropyl methyl cellulose acetate succinate with enhanced acetate and succinate substitution, U.S. Patent, US20130102691, 2011 Search PubMed.
- L. Yin and M. A. Hillmyer, Mol. Pharm., 2013, 11, 175–185 CrossRef PubMed.
- H. Liu, G. A. Ilevbare, B. P. Cherniawski, E. T. Ritchie, L. S. Taylor and K. J. Edgar, Carbohydr. Polym., 2014, 100, 116–125 CrossRef CAS PubMed.
- J. Amim, Jr., D. S. Petri, F. B. Maia and P. Miranda, Cellulose, 2009, 16, 773–782 CrossRef.
- J. D. Posey-Dowty, T. L. Watterson, A. K. Wilson, K. J. Edgar, M. C. Shelton and L. R. Lingerfelt, Cellulose, 2007, 14, 73–83 CrossRef CAS PubMed.
- M. C. Shelton, J. D. Posey-Dowty, L. R. Lingerfelt, S. K. Kirk, S. Klein and K. J. Edgar, in Polysaccharide Materials: Performance by Design, ed. K. J. Edgar, T. Heinze and T. Liebert, American Chemical Society, Washington, D.C., 2009, pp. 93–113 Search PubMed.
- J. M. Pereira, R. Mejia-Ariza, G. A. Ilevbare, H. E. McGettigan, N. Sriranganathan, L. S. Taylor, R. M. Davis and K. J. Edgar, Mol. Pharmaceutics, 2013, 10, 4640–4653 CrossRef CAS PubMed.
- J. M. Allen, A. K. Wilson, P. L. Lucas and L. G. Curtis, Slurrying water-wet carboxyalkyl cellulose in alkanoic acid, treating with alkanoic acid anhydride in presence of strong acid catalyst, heating, adding water, acid, acid salt to neutralize catalyst, U.S. Patent, 5792856, 1998 Search PubMed.
- C. Leuner and J. Dressman, Eur. J. Pharm. Biopharm., 2000, 50, 47–60 CrossRef CAS.
- T. Vasconcelos, B. Sarmento and P. Costa, Drug Discovery Today, 2007, 12, 1068–1075 CrossRef CAS PubMed.
- P. J. Marsac, T. Li and L. S. Taylor, Pharm. Res., 2009, 26, 139–151 CrossRef CAS PubMed.
- F. Qian, J. Huang and M. A. Hussain, J. Pharm. Sci., 2010, 99, 2941–2947 CAS.
- J. Posey-Dowty, K. Seo, K. Walker and A. Wilson, Surf. Coat. Int. PT B-C, 2002, 85, 203–208 CrossRef CAS.
- G. A. Ilevbare, H. Liu, K. J. Edgar and L. S. Taylor, Mol. Pharmaceutics, 2013, 10, 2381–2393 CrossRef CAS PubMed.
- E. Fukui, N. Miyamura and M. Kobayashi, J. Controlled Release, 2001, 70, 97–107 CrossRef CAS.
- F. Tanno, Y. Nishiyama, H. Kokubo and S. Obara, Drug Dev. Ind. Pharm., 2004, 30, 9–17 CrossRef CAS PubMed.
- W. Curatolo, J. A. Nightingale and S. M. Herbig, Pharm. Res., 2009, 26, 1419–1431 CrossRef CAS PubMed.
- F. Tajarobi, A. Larsson, H. Matic and S. Abrahmsén-Alami, Eur. J. Pharm. Biopharm., 2011, 78, 125–133 CrossRef CAS PubMed.
- S. Sotthivirat, C. McKelvey, J. Moser, B. Rege, W. Xu and D. Zhang, Int. J. Pharm., 2013, 452, 73–81 CrossRef CAS PubMed.
- D. T. Friesen, R. Shanker, M. Crew, D. T. Smithey, W. Curatolo and J. Nightingale, Mol. Pharmaceutics, 2008, 5, 1003–1019 CrossRef CAS PubMed.
- G. A. Ilevbare, H. Liu, K. J. Edgar and L. S. Taylor, Cryst. Growth Des., 2012, 12, 3133–3143 CAS.
- X. T. Meng, J. B. Matson and K. J. Edgar, Biomacromolecules, 2014, 15, 177–187 CrossRef CAS PubMed.
- R. H. Grubbs, Tetrahedron, 2004, 60, 7117–7140 CrossRef CAS PubMed.
- A. D. Piscopio and J. E. Robinson, Curr. Opin. Chem. Biol., 2004, 8, 245–254 CrossRef CAS PubMed.
- J. B. Binder and R. T. Raines, Curr. Opin. Chem. Biol., 2008, 12, 767–773 CrossRef CAS PubMed.
- J. Huang, H.-J. Schanz, E. D. Stevens and S. P. Nolan, Organometallics, 1999, 18, 5375–5380 CrossRef CAS.
- J. S. Kingsbury, J. P. A. Harrity, P. J. Bonitatebus and A. H. Hoveyda, J. Am. Chem. Soc., 1999, 121, 791–799 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS.
- S. Gessler, S. Randl and S. Blechert, Tetrahedron Lett., 2000, 41, 9973–9976 CrossRef CAS.
- P. S. Swaroop, S. Tripathy, G. Jachak and D. S. Reddy, Tetrahedron Lett., 2014, 55, 4777–4779 CrossRef CAS PubMed.
- H. Mutlu, L. M. de Espinosa and M. A. Meier, Chem. Soc. Rev., 2011, 40, 1404–1445 RSC.
- A. K. Chatterjee, T. L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS PubMed.
- S. J. Connon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900–1923 CrossRef CAS PubMed.
- H. Bilel, N. Hamdi, F. Zagrouba, C. Fischmeister and C. Bruneau, Catal. Sci. Technol., 2014, 4, 2064–2071 CAS.
- O. Kreye, D. Kugele, L. Faust and M. A. Meier, Macromol. Rapid Commun., 2014, 35, 317–322 CrossRef CAS PubMed.
- M. Winkler and M. A. R. Meier, Green Chem., 2014, 16, 3335–3340 RSC.
- X. T. Meng, J. B. Matson and K. J. Edgar, Polym. Chem., 2014, 5, 7021–7033 RSC.
- K. Malzahn, F. Marsico, K. Koynov, K. Landfester, C. K. Weiss and F. R. Wurm, ACS Macro Lett., 2014, 3, 40–43 CrossRef CAS.
- C. Ferrero, D. Massuelle and E. Doelker, J. Controlled Release, 2010, 141, 223–233 CrossRef CAS PubMed.
- C. L. Li, L. G. Martini, J. L. Ford and M. Roberts, J. Pharm. Pharmacol., 2005, 57, 533–546 CrossRef CAS PubMed.
- T. Ozeki, H. Yuasa and H. Okada, AAPS PharmSciTech, 2005, 6, 231–236 CrossRef PubMed.
- H. Yin, L. Xu and N. A. Porter, Chem. Rev., 2011, 111, 5944–5972 CrossRef CAS PubMed.
- B. Li, S. Konecke, K. Harich, L. Wegiel, L. S. Taylor and K. J. Edgar, Carbohydr. Polym., 2013, 92, 2033–2040 CrossRef CAS PubMed.
- L. Landoll, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 443–455 CrossRef CAS PubMed.
- M. A. Winnik and A. Yekta, Curr. Opin. Colloid Interface Sci., 1997, 2, 424–436 CrossRef CAS.
- J. A. Baird and L. S. Taylor, Adv. Drug Delivery Rev., 2012, 64, 396–421 CrossRef CAS PubMed.
- C. Entwistle and R. Rowe, J. Pharm. Pharmacol., 1979, 31, 269–272 CrossRef CAS PubMed.
- Y. H. Kim and R. Beckerbauer, Macromolecules, 1994, 27, 1968–1971 CrossRef CAS.
- C.-F. Shu and C.-M. Leu, Macromolecules, 1999, 32, 100–105 CrossRef CAS.
- D. W. Lim, S. H. Choi and T. G. Park, Macromol. Rapid Commun., 2000, 21, 464–471 CrossRef CAS.
- V. A. Kusuma, B. D. Freeman, M. A. Borns and D. S. Kalika, J. Member Sci., 2009, 327, 195–207 CrossRef CAS PubMed.
- A. H. Soeriyadi, G.-Z. Li, S. Slavin, M. W. Jones, C. M. Amos, C. R. Becer, M. R. Whittaker, D. M. Haddleton, C. Boyer and T. P. Davis, Polym. Chem., 2011, 2, 815–822 RSC.
- G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196–1204 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR, 1H NMR spectra and DSC curves. See DOI: 10.1039/c5py00369e |
| This journal is © The Royal Society of Chemistry 2015 |