DOI:
10.1039/C5PY00157A
(Paper)
Polym. Chem., 2015,
6, 3054-3062
Industrially-relevant polymerization-induced self-assembly formulations in non-polar solvents: RAFT dispersion polymerization of benzyl methacrylate†
Received
2nd February 2015
, Accepted 2nd March 2015
First published on 4th March 2015
Abstract
Industrially-sourced mineral oil and a poly(α-olefin) are used as solvents for the reversible addition–fragmentation chain transfer (RAFT) dispersion polymerization of benzyl methacrylate (BzMA) using a poly(lauryl methacrylate) macromolecular chain transfer agent (PLMA macro-CTA) at 90 °C. The insolubility of the growing PBzMA chains under such conditions leads to polymerization-induced self-assembly (PISA), whereby poly(lauryl methacrylate)-poly(benzyl methacrylate) (PLMA-PBzMA) diblock copolymer spheres, worms or vesicles are produced directly as concentrated dispersions. The particular diblock copolymer composition required to access each individual morphology depends on the nature of the oil. Moreover, the solvent type also affects important properties of the physical free-standing gels that are formed by the PLMA-PBzMA worm dispersions, including the storage modulus (G′), critical gelation temperature (CGT) and critical gelation concentration (CGC). Spherical PLMA-PBzMA diblock copolymer nanoparticles can be prepared at up to 50% w/w solids and an efficient ‘one-pot’ protocol involving solution polymerization of LMA followed immediately by dispersion polymerization of BzMA has been developed. The latter formulation enables high BzMA conversions to be achieved, with spherical nanoparticles being produced at 30% w/w solids.
Introduction
Traditionally, block copolymer self-assembly in solution to form various types of nanoparticles is conducted at high dilution (<1% w/w) and often involves post-polymerization processing via solvent1 or pH switching,2 or thin film rehydration.3 Over the last two decades or so, controlled radical techniques such as reversible addition–fragmentation chain transfer (RAFT) polymerization4–6 have enabled the convenient synthesis of a wide range of functional diblock copolymers.7–20 Currently, there is considerable academic interest in performing polymerization-induced self-assembly (PISA) syntheses at relatively high solids via RAFT dispersion polymerization.21–24 The final diblock copolymer nanoparticle morphology typically depends on the relative volume fractions of the two blocks, as dictated by the so-called packing parameter,25–27 and also the copolymer concentration.28 Purely spherical, worm-like or vesicular morphologies have been reported for aqueous,28–37 alcoholic38–50 and non-polar51–56 formulations, with the construction of phase diagrams enabling the reproducible targeting of each of these morphologies. Moreover, there appears to be some scope for developing ‘one-pot’ syntheses,57–60 which should provide a highly convenient and potentially industrially-relevant protocol for generating organic nanoparticles.
A wide range of potential applications have been explored for selected RAFT PISA syntheses, including coatings,61 drug delivery,15,47,62 sterilizable gels,63 contact lenses,64 and novel Pickering emulsifiers.65 In principle, block copolymer nanoparticles comprising an oil-soluble stabilizer such as poly(lauryl methacrylate) (PLMA) have various potential applications, including drag reduction,66 oil absorbency agents,67,68 and viscosity modifiers for engine oils.69–71 Of particular relevance to the present work, Zheng et al.72 reported that spherical block copolymer nanoparticles dispersed in non-polar solvents significantly reduced the friction coefficient of lubricant base oils in the boundary lubrication regime. In this case, copper-catalyzed atom transfer radical polymerization (ATRP) was utilized to synthesize all-acrylic block copolymer spheres in 2-butanone, with their subsequent redispersion in oil involving various post-polymerization modification and purification steps.73
Herein we revisit a RAFT-mediated dispersion polymerization formulation originally developed for the synthesis of poly(lauryl methacrylate)-poly(benzyl methacrylate) (PLMA-PBzMA) nanoparticles in n-alkanes and extend this formulation to include both mineral oil and a poly(α-olefin) (PAO) oil, see Fig. 1. Phase diagrams have been constructed for PISA syntheses conducted in both these industrially-sourced oils, and subtle differences are observed relative to pure n-alkanes, particularly with respect to the physical properties of PLMA-PBzMA worm gels. Moreover, a ‘one-pot’ synthesis protocol has been examined for the synthesis of spherical PLMA-PBzMA nanoparticles.
 |
| | Fig. 1 Synthesis of a poly(lauryl methacrylate) macro-CTA via RAFT solution polymerization in toluene at 70 °C, followed by RAFT dispersion polymerization of benzyl methacrylate (BzMA) in mineral oil or a poly(α-olefin) (PAO) at 90 °C. | |
Experimental
Materials
Monomers were purchased from Sigma-Aldrich (UK) and passed through basic alumina prior to use. tert-Butyl peroxy-2-ethylhexanoate (T21s) initiator was purchased from AkzoNobel (The Netherlands). CDCl3, cumyl dithiobenzoate (CDB) and all other reagents were purchased from Sigma-Aldrich (UK) and were used as received, unless otherwise noted. THF, n-heptane and toluene were purchased from Fisher Scientific (UK), CD2Cl2 was purchased from Goss Scientific (UK). Industrial-grade mineral (viscosity at 20 °C = 2.5 mPa s) and poly(α-olefin) (PAO; viscosity at 20 °C = 3.0 mPa s) oils were provided by The Lubrizol Corporation Ltd.
Synthesis of poly(lauryl methacrylate) macro-chain transfer agent
The synthesis of poly(lauryl methacrylate) (PLMA) macro-CTAs has been previously reported.54 A typical synthesis of a PLMA47 macro-CTA was conducted as follows. A 250 mL round-bottomed flask was charged with lauryl methacrylate (LMA; 20.0 g; 78.6 mmol), cumyl dithiobenzoate (CDB; 0.43 g; 1.57 mmol; target degree of polymerization = 50), 2,2′-azobisisobutyronitrile (AIBN; 51.6 mg, 314 μmol; CDB/AIBN molar ratio = 5.0) and toluene (30.7 g). The sealed reaction vessel was purged with nitrogen and placed in a pre-heated oil bath at 70 °C for 11 h. The resulting PLMA (LMA conversion = 81%; Mn = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 g mol−1, Mw/Mn = 1.24) was purified by precipitation into excess methanol. The mean degree of polymerization (DP) of this macro-CTA was calculated to be 47 using 1H NMR spectroscopy by comparing the integrated signals corresponding to the CDB aromatic protons at 7.1–8.1 ppm with that assigned to the two oxymethylene protons of PLMA at 3.7–4.2 ppm. Thus the CTA efficiency of the CDB was estimated to be 86%.
600 g mol−1, Mw/Mn = 1.24) was purified by precipitation into excess methanol. The mean degree of polymerization (DP) of this macro-CTA was calculated to be 47 using 1H NMR spectroscopy by comparing the integrated signals corresponding to the CDB aromatic protons at 7.1–8.1 ppm with that assigned to the two oxymethylene protons of PLMA at 3.7–4.2 ppm. Thus the CTA efficiency of the CDB was estimated to be 86%.
Synthesis of poly(lauryl methacrylate)-poly(benzyl methacrylate) diblock copolymer nanoparticles
A typical RAFT dispersion polymerization synthesis of PLMA18-PBzMA45 diblock copolymer nanoparticles at 25% w/w solids was carried out as follows. Benzyl methacrylate (BzMA; 0.415 g; 2.36 mmol), T21s initiator (2.26 mg; 10.5 μmol; dissolved at 10.0% v/v in mineral oil) and PLMA18 macro-CTA (0.27 g; 52.3 μmol; macro-CTA/initiator molar ratio = 5.0; target degree of polymerization of PBzMA = 45) were dissolved in mineral oil (2.06 g). The reaction mixture was sealed in a 10 mL round-bottomed flask and purged with nitrogen gas for 30 min. The deoxygenated solution was then placed in a pre-heated oil bath at 90 °C for 5 h (final BzMA conversion = 99%; Mn = 9700 g mol−1, Mw/Mn = 1.24).
‘One-pot’ synthesis of poly(lauryl methacrylate)-poly(benzyl methacrylate) diblock copolymer spheres
A typical ‘one-pot’ synthesis of PLMA50-PBzMA100 diblock copolymer spheres was conducted as follows. Lauryl methacrylate (LMA; 0.700 g; 2.75 mmol), cumyl dithiobenzoate (CDB; 15.0 mg; 55.0 μmol; target degree of polymerization = 50; dissolved at 10.0% w/w in mineral oil) and T21s initiator (2.14 mg; 9.90 μmol; dissolved at 10% v/v in mineral oil) were dissolved in mineral oil (0.150 g). The reaction mixture was sealed in a 25 mL round-bottomed flask and purged with nitrogen gas for 30 min. The deoxygenated solution was then placed in a pre-heated oil bath at 90 °C for 5 h (final LMA conversion = 95%; Mn = 12500 g mol−1; Mw/Mn = 1.18). Benzyl methacrylate (BzMA; 0.970 g; 5.50 mmol; target degree of polymerization = 100) and T21s initiator (2.14 mg; 9.90 μmol; dissolved at 10% v/v in mineral oil) were dissolved in mineral oil (3.65 g) and purged with nitrogen gas for 30 min before being added to the original reaction vessel at high (>95%) LMA conversion (final BzMA conversion = 98%; Mn = 24500 g mol−1; Mw/Mn = 1.15).
Gel permeation chromatography
Molecular weight distributions were assessed by gel permeation chromatography (GPC) using THF eluent. The THF GPC system was equipped with two 5 μm (30 cm) Mixed C columns; a WellChrom K-2301 refractive index detector operating at 950 ± 30 nm. The mobile phase contained 2.0% v/v triethylamine and 0.05% w/v butylhydroxytoluene (BHT) with a toluene flow rate marker and the flow rate was fixed at 1.0 mL min−1. A series of ten near-monodisperse poly(methyl methacrylate) standards (Mp values ranging from 1280 to 330000 g mol−1) were used for calibration.
1H NMR spectroscopy
1H NMR spectra were recorded in either CD2Cl2 or CDCl3 using a Bruker AV1-400 or AV1-250 MHz spectrometer. Typically 64 scans were averaged per spectrum.
Dynamic light scattering
Dynamic light scattering (DLS) studies were performed using a Zetasizer Nano-ZS instrument (Malvern Instruments, UK) at a fixed scattering angle of 173°. Copolymer dispersions were diluted in n-heptane (0.10% w/w) prior to light scattering studies at 25 °C. Temperature-dependent DLS studies were performed using the same Zetasizer Nano-ZS instrument, which was equipped with a Peltier cell. Copolymer dispersions were diluted in n-heptane and heated from 20 to 90 °C at 5 °C intervals allowing 5 min for thermal equilibration between measurements. In both sets of experiments, the intensity-average diameter and polydispersity of the diblock copolymer particles were calculated by cumulants analysis of the experimental correlation function using Dispersion Technology Software version 6.20. Data were averaged over thirteen runs each of thirty seconds duration.
Transmission electron microscopy
Transmission electron microscopy (TEM) studies were conducted using a Philips CM 100 instrument operating at 100 kV and equipped with a Gatan 1 k CCD camera. Diluted block copolymer solutions (0.10% w/w) were placed on carbon-coated copper grids and exposed to ruthenium(VIII) oxide vapor for 7 minutes at 20 °C prior to analysis.74 This heavy metal compound acted as a positive stain for the core-forming PBzMA block to improve contrast. The ruthenium(VIII) oxide was prepared as follows: ruthenium(IV) oxide (0.30 g) was added to water (50 g) to form a black slurry; addition of sodium periodate (2.0 g) with stirring produced a yellow solution of ruthenium(VIII) oxide within 1 min.75
Rheology measurements
An AR-G2 rheometer equipped with a variable temperature Peltier plate and a 40 mm 2° aluminum cone was used for all experiments. The loss and storage moduli were measured as a function of temperature at a heating rate of 1.0 °C per minute, a fixed strain of 1.0% and an angular frequency of 10 rad s−1 so as to assess the gel strength and critical gelation temperature (CGT). During temperature sweeps, the temperature was varied at 5 °C intervals, with an equilibration time of five minutes being allowed prior to each measurement. In all cases the gap between the cone and plate was 58 μm.
Results and discussion
Synthesis of PLMA macro-CTAs
Low-polydispersity PLMA macro-CTAs with mean DPs of 16, 18, or 47 (see Table S1 in ESI†) were synthesized via RAFT solution polymerization in toluene at 70 °C using cumyl dithiobenzoate (CDB) as a CTA. In all macro-CTA syntheses, the polymerization was quenched at 71–81% conversion in order to avoid monomer-starved conditions and therefore ensure retention of the RAFT end-groups.76,77 This is usually considered to be desirable for high blocking efficiencies and hence well-defined PLMA-PBzMA diblock copolymers. As with previous studies reporting well-controlled RAFT syntheses conducted in non-polar solvents,54,55 each PLMA macro-CTA had a polydispersity (Mw/Mn) of less than 1.25. A representative kinetic study of the RAFT solution polymerization of LMA to prepare a PLMA18 macro-CTA indicated a linear evolution of molecular weight with conversion (see Fig. S1 in ESI†). After an initial induction period of ∼100 min, this reaction obeyed first-order kinetics and was quenched after 11 h (71% conversion).
PLMA-PBzMA block copolymer syntheses and phase diagrams
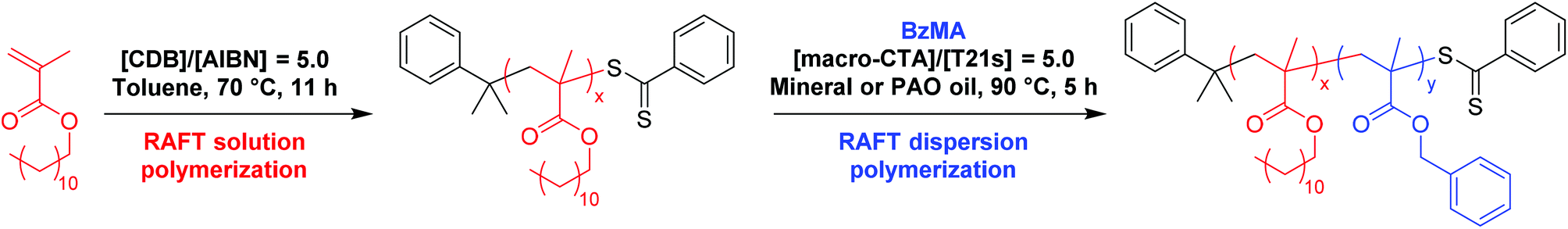
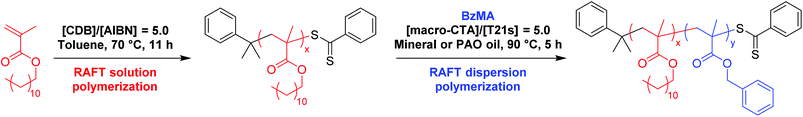
One important trend in the commercial engine oil sector is a general shift from mineral oil towards wholly synthetic oils such as poly(α-olefins). Preliminary experiments confirmed that both these industrially-sourced solvents were good solvents for PLMA and bad solvents for PBzMA. Hence phase diagrams for PLMA-PBzMA block copolymer nanoparticles prepared in these two oils were constructed in order to assess the effect of the nature of the solvent on the positions of the phase boundaries. Firstly, BzMA monomer was polymerized using a low-polydispersity PLMA18 macro-CTA via RAFT dispersion polymerization in mineral oil to produce a series of well-defined PLMA-PBzMA diblock copolymer nanoparticles at various copolymer concentrations. More than 95% BzMA conversion was achieved in all dispersion polymerizations within 5 h at 90 °C, as judged by 1H NMR spectroscopy. Pure spherical, worm-like, or vesicular morphologies (as judged by TEM) can be accessed when chain-extending the PLMA18 macro-CTA (see Fig. 2a, b and c, respectively). We have previously reported phase diagrams for PLMA-PBzMA diblock copolymer formulations in n-heptane54 and n-dodecane.55 Pure spherical and vesicular phases were obtained at copolymer concentrations ranging from 12.5 to 25% w/w, whereas solely worm-like micelles could only be obtained at copolymer concentrations at or above 18% w/w. At first sight, the phase diagram reported in the present work (Fig. 2) is similar to that reported for n-heptane, but the precise block copolymer compositions required to access each individual morphology are subtly different. This shift in phase boundaries is best highlighted when comparing the pure worm phase in each oil. In n-heptane, the worm phase for PLMA17-PBzMAx diblock copolymers corresponds to x ≈ 50–70, whereas worms are obtained at x ≈ 37–47 for a PLMA18-PBzMAx formulation in mineral oil (see Fig. 2), which is close to that observed for n-dodecane (x = 36–44).55
 |
| | Fig. 2 Phase diagram constructed for PLMA18-PBzMAx diblock copolymer nanoparticles prepared by RAFT dispersion polymerization of BzMA in mineral oil using T21s initiator at 90 °C ([PLMA]/[T21s] = 5.0). The post mortem diblock copolymer morphologies were assigned by TEM studies. TEM images (a), (b) and (c) represent examples of purely vesicular, worm-like or spherical morphologies, respectively. | |
Evaluation of the effect of solvent on PLMA-PBzMA worm gels
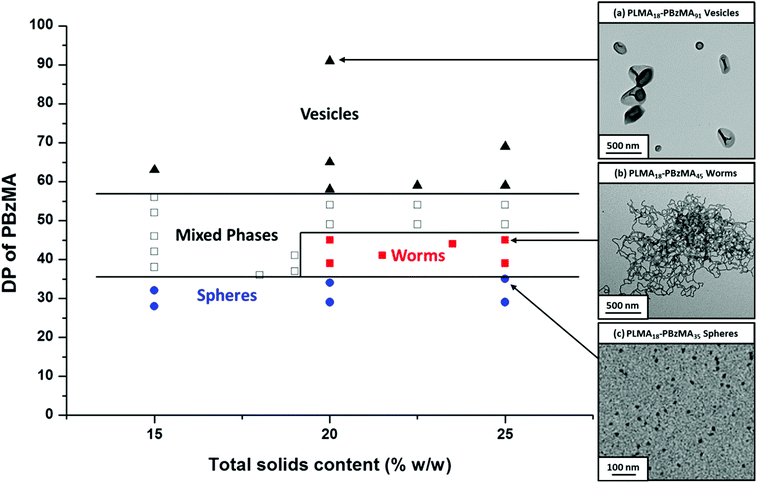
Using an industrially-sourced PAO as the continuous phase for the synthesis of PLMA-PBzMA diblock copolymer nano-objects enables the role of the solvent in such PISA syntheses to be examined. Accordingly, a PLMA16 macro-CTA was chain-extended with varying amounts of BzMA at various solids contents in order to construct a phase diagram (Fig. 3). Although this exhibits obvious similarities to the phase diagram obtained for PLMA18-PBzMAx in mineral oil (Fig. 2), nevertheless some differences are observed. In particular, for PLMA16-PBzMAx syntheses conducted at ≥20% w/w solids, the PAO worm phase is observed for x ≈ 28–35. In contrast, the worm phase obtained for PLMA18-PBzMAx when using mineral oil occurs for x ≈ 37–47. Even allowing for the small differences between the mean DPs of the PLMA stabilizer blocks, it is clear that a somewhat longer PBzMA block is required to access the worm phase in mineral oil. This suggests that PAO is a slightly poorer solvent for the growing PBzMA chains than the mineral oil. In order to further illustrate this point, PLMA18-PBzMA45 nano-objects were prepared at 20% w/w solids in both PAO and mineral oil (see Fig. S2 in ESI†). Inspecting Fig. 2, PLMA18-PBzMA45 forms a pure worm phase in the latter solvent. However, precisely the same diblock copolymer composition forms a mixed phase of spheres, worms and vesicles in PAO (Fig. S2†). In view of these subtle solvent effects, the physical properties of free-standing worm gels prepared in n-dodecane,55 mineral oil and PAO oil were compared via rheology (Table 1).
 |
| | Fig. 3 Phase diagram constructed for PLMA16-PBzMAx diblock copolymer nanoparticles prepared by RAFT dispersion polymerization of BzMA in poly(α-olefin) oil using T21s initiator at 90 °C ([PLMA]/[T21s] = 5.0). The post mortem diblock copolymer morphologies were assigned by TEM studies. TEM images (a), (b) and (c) represent examples of pure vesicular, worm-like or spherical morphologies, respectively. | |
Table 1 Summary of physical properties of 20% w/w PLMA-PBzMA worm gels prepared in various non-polar solvents: initial storage modulus (G′) at 20 °C, critical gelation temperature (CGT) and critical gelation concentration (CGC). Measurements were obtained at a heating rate of 1.0 °C min−1, a fixed strain of 1.0% and an angular frequency of 10 rad s−1. The temperature was varied at 5 °C intervals, with a 5 min equilibration time at each temperature
| Solvent |
Block composition |
Initial G′ at 20 °C/Pa |
CGT/°C |
CGC/% w/w |
|
n-Dodecane55 |
PLMA18-PBzMA37 |
2300 |
47 |
∼11 |
| Mineral oil |
PLMA18-PBzMA45 |
21000 |
49 |
9 |
| PAO oil |
PLMA16-PBzMA32 |
41000 |
44 |
9 |
PLMA-PBzMA worm gels in the mineral and PAO oils provided G′ values that are approximately an order of magnitude greater than in n-dodecane. This suggests that the worms are either significantly longer and/or that there are stronger inter-worm interactions in these gels. The G′ for the 20% w/w PLMA16-PBzMA32 worm gel in PAO oil is significantly greater than that of the PLMA18-PBzMA45 worm gel in mineral oil. The critical gelation temperature (CGT) of worm gels is defined as the temperature at which the dispersion no longer forms a gel (i.e. when G′′ > G′).78 In our previous study,55 we found that a worm-to-sphere transition was responsible for the degelation of such worm gels above the CGT. The same morphological transition is believed to be responsible for the degelation that occurs on heating in the present study. This hypothesis is supported by TEM and DLS studies (see Fig. S3 in ESI†). Thus the sphere-equivalent particle diameter of 163 nm (polydispersity = 0.39) obtained for the initial PLMA18-PBzMA40 worms formed in mineral oil is reduced to 28 nm (polydispersity = 0.10) for spheres on heating to 90 °C. For PLMA-PBzMA worm gels prepared in the three oils, the CGT ranged from 44 to 49 °C. Interestingly, the PLMA16-PBzMA32 worm gel produced in PAO oil, which provided the highest G′, possessed the lowest CGT. The specific DP of the PBzMA core-forming block may influence this parameter, since higher CGTs are observed for longer core-forming PBzMA blocks for the three worm gels characterized in this study. The dispersion is a free-flowing fluid below the critical gelation concentration (CGC), which is slightly lower for the industrially-sourced oils than for n-dodecane. However, all of the non-polar worm gels investigated in the present work exhibit much higher CGCs than previously reported aqueous worm gels.78 This might perhaps reflect the lack of inter-worm hydrogen bonding in the non-aqueous formulations.
Synthesis of PLMA-PBzMA diblock copolymer spheres at high solids
In many RAFT dispersion polymerization formulations, it has been reported that only spherical nanoparticles are obtained when chain-extending a sufficiently long macro-CTA.42,54,55 This is presumably because inter-sphere fusion must first occur if a so-called ‘higher order’ non-spherical morphology (such as worms) is to be generated. Thus using a longer steric stabilizer block confers more effective steric stabilization, which inevitably leads to a higher proportion of elastic inter-particle collisions. Using a PLMA47 macro-CTA is sufficient to ensure an exclusively spherical morphology for all PLMA47-PBzMAx diblock copolymers prepared by RAFT dispersion polymerization in mineral oil, regardless of the targeted degree of polymerization (x). For example, well-defined spherical nanoparticles are obtained for both PLMA47-PBzMA99 and PLMA47-PBzMA495 diblock copolymer nanoparticles prepared at 20% w/w solids. This copolymer concentration allows access to pure worm-like and vesicular morphologies when using shorter PLMA macro-CTAs.54,55 PLMA47-PBzMA495 diblock copolymer spheres are significantly larger than PLMA47-PBzMA99, as confirmed by TEM (Fig. 4). Given the diblock copolymer asymmetry, the former particles most likely represent a kinetically-trapped morphology.35,79 Interestingly, such spherical nanoparticles can be synthesized at copolymer concentrations up to 50% w/w, with the smaller PLMA47-PBzMA99 spheres producing a viscous free-flowing dispersion. The synthesis of relatively small spherical nanoparticles at high solids bodes well for the industrial relevance of such PISA formulations. In contrast, the larger PLMA47-PBzMA495 spheres lead to stirring problems at concentrations as low as 30% w/w solids, with a gel-like paste being obtained (see Fig. 4b, inset digital images).
 |
| | Fig. 4 (a) PLMA47-PBzMA99 and (b) PLMA47-PBzMA495 diblock copolymer spheres prepared via RAFT dispersion polymerization of benzyl methacrylate at 90 °C using PISA formulations conducted at 30, 40 or 50% w/w solids. Inset digital photos depict physical appearance of each dispersion. | |
‘One-pot’ synthesis of PLMA-PBzMA spheres at high solids
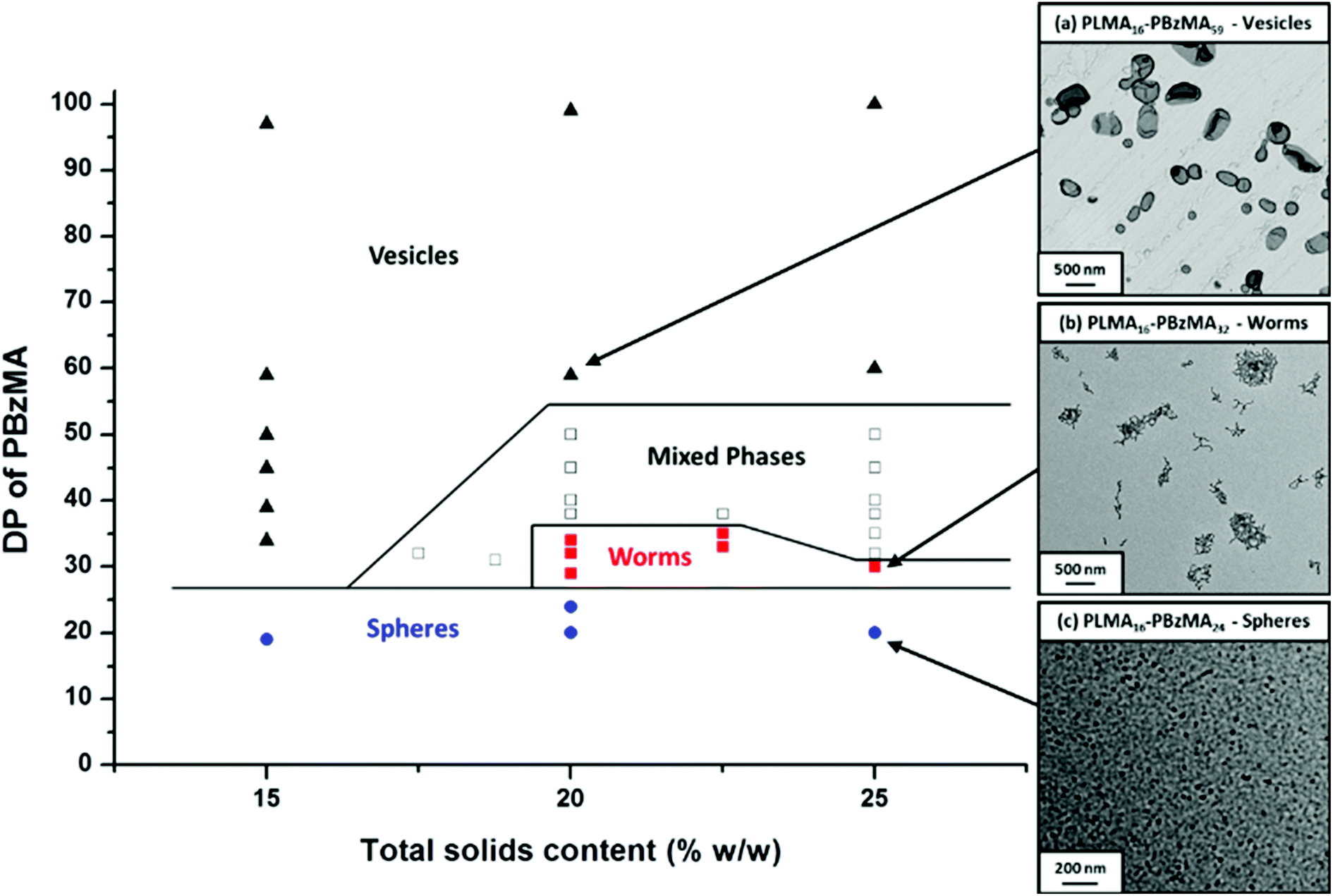
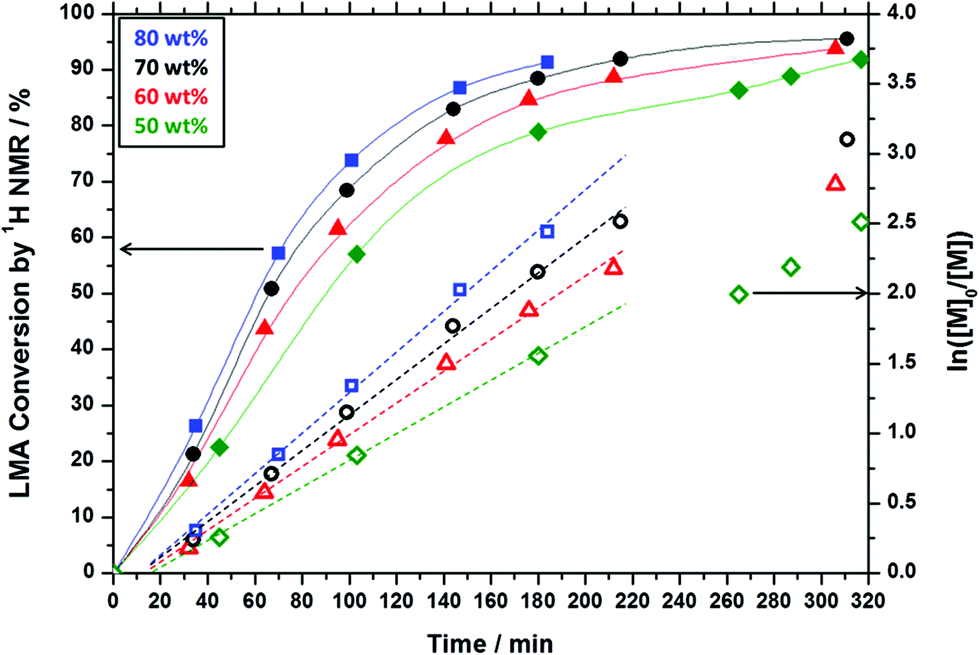
To further examine the robust nature (and hence potential industrial relevance) of this particular PISA formulation, a series of ‘one-pot’ polymerizations were conducted in mineral oil. Firstly, a kinetic study of the RAFT solution polymerization of LMA in mineral oil was conducted (Fig. 5). Efficient polymerizations could be conducted at up to 70% w/w solids when targeting a PLMA50 macro-CTA: a linear semi-logarithmic plot indicated first-order kinetics up to 90% LMA conversion. At 80% w/w solids, the solution viscosity became too high for efficient stirring above 90% LMA conversion, but similar first-order polymerization kinetics were observed up to this point. As expected, polymerizations conducted at higher LMA concentrations exhibited faster rates of polymerization, with conversions reaching 90% within 180 min at 80% w/w, 220 min at 70% w/w, 300 min at 60% w/w, and >320 min at 50% w/w. The homopolymerization of LMA conducted at 70% w/w reached high (>95%) conversion within 320 min. Moreover, this reaction solution remained sufficiently fluid for efficient stirring. Hence these conditions were selected for the first stage of a ‘one-pot’ synthesis of PLMA50-PBzMA100 diblock copolymer nanoparticles. The subsequent RAFT dispersion polymerization of BzMA was conducted at 30% w/w solids in order to enable the reaction mixture to be efficiently stirred throughout the polymerization. We had previously observed that similar PLMA47-PBzMA99 block copolymer spheres prepared via a traditional two-step synthesis exhibited relatively high viscosities above 30% w/w solids, see Fig. 4. Kinetic data were obtained for both the RAFT solution polymerization of LMA at 70% w/w solids and the RAFT dispersion polymerization of BzMA at 30% w/w solids in mineral oil (Fig. 6). A degassed solution of BzMA in mineral oil containing additional T21s initiator was added to the reaction mixture once the LMA conversion had reached 95%. Initially, a relatively slow rate of BzMA polymerization was observed for the first 40 min until a critical degree of polymerization of the growing PBzMA chains is reached, at which point micellar nucleation occurred (see stage (b) in Fig. 6). Thereafter, the polymerization proceeds much faster, because the unreacted BzMA migrates into the PBzMA micelle cores, thus producing a relatively high local monomer concentration as suggested by Blanazs et al.33 Thereafter, first-order kinetics was observed from 30 to 90% conversions (see Fig. 6). The final diblock copolymer spheres were relatively monodisperse with an intensity-average diameter of 39 nm, as judged by DLS (see Fig. S4 in ESI†). Molecular weight distributions for each of the data points shown in Fig. 6 were assessed using GPC (Fig. 7a). A linear evolution in number-average molecular weight (expressed in poly(methyl methacrylate) equivalents) was observed for both solution and dispersion polymerizations (Fig. 7b), with relatively narrow molecular weight distributions (Mw/Mn < 1.20) being achieved throughout both polymerizations. Thus excellent RAFT control can be achieved in such ‘one-pot’ syntheses.
 |
| | Fig. 5 Kinetic data obtained for the RAFT solution polymerization of a PLMA50 macro-CTA in mineral oil at 90 °C conducted at 50 (green diamonds), 60 (red triangles), 70 (black circles) and 80 (blue squares) % w/w solids using CDB as a RAFT chain transfer agent and T21s initiator. | |
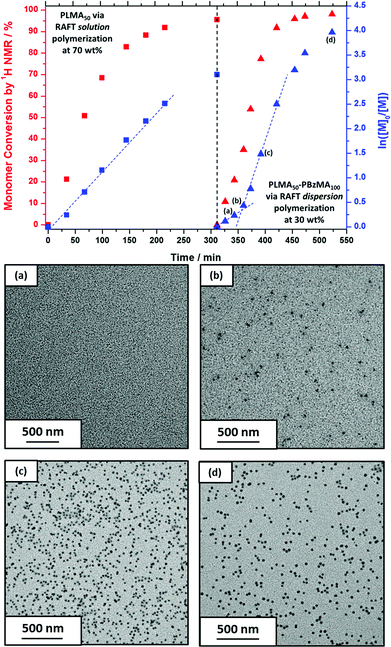 |
| | Fig. 6 Conversion vs. time curves obtained for the ‘one-pot’ synthesis of PLMA50-PBzMA100 diblock copolymer spheres in mineral oil: RAFT solution polymerization of a PLMA50 macro-CTA at 70% w/w solids (squares) followed by the RAFT dispersion polymerization of BzMA at 30% w/w solids (triangles). BzMA was added after 310 min. TEM images (a), (b), (c), and (d) represent various points in the kinetic data and indicate the onset of micellization at (b). | |
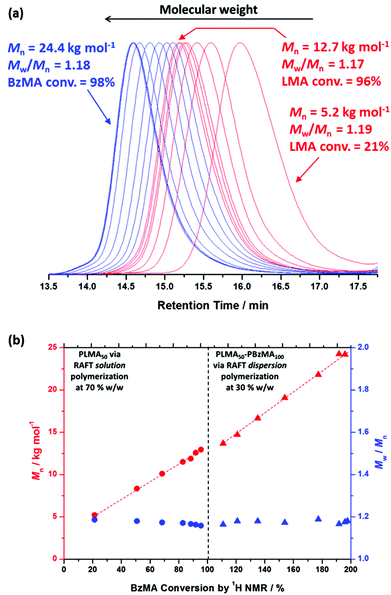 |
| | Fig. 7 (a) THF gel permeation chromatograms (vs. poly(methyl methacrylate) standards) obtained for the ‘one-pot’ synthesis of PLMA50-PBzMA100 diblock copolymer spheres in mineral oil: solution polymerization of LMA (red traces) and dispersion polymerization of BzMA (blue traces). (b) Number-average molecular weight vs. monomer conversion plots for the RAFT solution polymerization of LMA at 70% w/w solids (circles) and the subsequent RAFT dispersion polymerization of BzMA at 30% w/w solids in mineral oil (triangles). | |
Conclusions
In summary, well-defined PLMA-PBzMA block copolymer nano-objects can be reproducibly prepared in the form of spheres, worms and vesicles via polymerization-induced self-assembly in industrially-sourced mineral oil at 90 °C, provided that the mean degree of polymerization of the PLMA stabilizer block is sufficiently low (e.g., DP = 18). The phase diagram constructed for PLMA18-PBzMAx diblock copolymers in mineral oil is very similar to that previously reported for n-dodecane.55 However, subtle variation in the precise location of phase boundaries is observed for PLMA16-PBzMAx diblock copolymers in a second industrially-relevant solvent, poly(α-olefin) (PAO) oil. The nature of the oil also affected the physical properties of the worm gels, with a worm gel in PAO oil exhibiting a higher storage modulus (G′) than those in mineral oil and n-dodecane. The critical gelation temperature (CGT) for each worm gel was found to be 44–49 °C, with the strongest worm gel (in PAO oil) having the lowest CGT. The critical gelation concentration (CGC) was lower for PLMA-PBzMA worm gels synthesized in the industrially-sourced oils compared to those previously studied in n-dodecane. Purely spherical nanoparticles are obtained when using a PLMA stabilizer with a relatively high DP. In this case, PISA syntheses can be conducted at up to 50% w/w solids when targeting PLMA47-PBzMA100, with efficient stirring being maintained throughout the polymerization. A convenient ‘one-pot’ synthesis protocol has also been explored. Thus the PLMA macro-CTA is first synthesized at 70% w/w solids in mineral oil via RAFT solution polymerization, with subsequent chain extension via RAFT dispersion polymerization of BzMA being conducted at 30% w/w solids. The final PLMA50-PBzMA100 spherical nanoparticles were obtained at 98% conversion with a relatively narrow size distribution, as judged by TEM and DLS studies. Moreover, relatively high blocking efficiencies and low final copolymer polydispersities (Mw/Mn < 1.20) were indicated by GPC studies.
Acknowledgements
The EPSRC (EP/J007846/1) is thanked for providing postdoctoral support for LAF. SPA also acknowledges an ERC Advanced Investigator grant (PISA 320372).
Notes and references
- L. F. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- V. Bütün, S. P. Armes and N. C. Billingham, Polymer, 2001, 42, 5993–6008 CrossRef.
- J. R. Howse, R. A. L. Jones, G. Battaglia, R. E. Ducker, G. J. Leggett and A. J. Ryan, Nat. Mater., 2009, 8, 507–511 CrossRef CAS PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248–2256 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562–11563 CrossRef CAS PubMed.
- C. Barner-Kowollik, T. P. Davis, J. P. A. Heuts, M. H. Stenzel, P. Vana and M. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 365–375 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- M. Mertoglu, S. Garnier, A. Laschewsky, K. Skrabania and J. Storsberg, Polymer, 2005, 46, 7726–7740 CrossRef CAS.
- D. Quémener, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Chem. Commun., 2006, 5051–5053 RSC.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715–5723 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Q. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- R. K. O'Reilly, Polym. Int., 2010, 59, 568–573 Search PubMed.
- C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551–595 CrossRef CAS.
- A. Lu, T. P. Smart, T. H. Epps, D. A. Longbottom and R. K. O'Reilly, Macromolecules, 2011, 44, 7233–7241 CrossRef CAS PubMed.
- Y. Kang, A. Pitto-Barry, H. Willcock, W. D. Quan, N. Kirby, A. M. Sanchez and R. K. O'Reilly, Polym. Chem., 2015, 6, 106–117 RSC.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- M. J. Monteiro and M. F. Cunningham, Macromolecules, 2012, 45, 4939–4957 CrossRef CAS.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2013, 4, 873–881 RSC.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans., 1976, 72, 1525–1568 RSC.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- Z. An, Q. Shi, W. Tang, C.-K. Tsung, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2007, 129, 14493–14499 CrossRef CAS PubMed.
- J. Rieger, C. Grazon, B. Charleux, D. Alaimo and C. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2373–2390 CrossRef CAS.
- S. Boisse, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M.-H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950–1952 RSC.
- Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- G. Liu, Q. Qiu, W. Shen and Z. An, Macromolecules, 2011, 44, 5237–5245 CrossRef CAS.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS PubMed.
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. M. Haddleton and B. S. Sumerlin, Chem. Sci., 2015, 6, 1230–1236 RSC.
- W.-M. Wan and C.-Y. Pan, Polym. Chem., 2010, 1, 1475–1484 RSC.
- W.-M. Wan, X.-L. Sun and C.-Y. Pan, Macromol. Rapid Commun., 2010, 31, 399–404 CrossRef CAS PubMed.
- C.-Q. Huang and C.-Y. Pan, Polymer, 2010, 51, 5115–5121 CrossRef CAS.
- W. Cai, W. Wan, C. Hong, C. Huang and C. Pan, Soft Matter, 2010, 6, 5554–5561 RSC.
- E. R. Jones, M. Semsarilar, A. Blanazs and S. P. Armes, Macromolecules, 2012, 45, 5091–5098 CrossRef CAS.
- M. Semsarilar, E. R. Jones, A. Blanazs and S. P. Armes, Adv. Mater., 2012, 24, 3378–3382 CrossRef CAS PubMed.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Polym. Chem., 2014, 5, 3466–3475 RSC.
- C. Gonzato, M. Semsarilar, E. R. Jones, F. Li, G. J. P. Krooshof, P. Wyman, O. O. Mykhaylyk, R. Tuinier and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 11100–11106 CrossRef CAS PubMed.
- Y. Pei and A. B. Lowe, Polym. Chem., 2014, 5, 2342–2351 RSC.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- B. Karagoz, C. Boyer and T. P. Davis, Macromol. Rapid Commun., 2014, 35, 417–421 CrossRef CAS PubMed.
- W. Zhao, G. Gody, S. M. Dong, P. B. Zetterlund and S. Perrier, Polym. Chem., 2014, 5, 6990–7003 RSC.
- S. Dong, W. Zhao, F. P. Lucien, S. Perrier and P. B. Zetterlund, Polym. Chem., 2015 10.1039/C4PY01632G.
- L. Houillot, C. Bui, M. Save, B. Charleux, C. Farcet, C. Moire, J.-A. Raust and I. Rodriguez, Macromolecules, 2007, 40, 6500–6509 CrossRef CAS.
- L. Houillot, C. Bui, C. Farcet, C. Moire, J.-A. Raust, H. Pasch, M. Save and B. Charleux, ACS Appl. Mater. Interfaces, 2010, 2, 434–442 CAS.
- J. A. Raust, L. Houillot, M. Save, B. Charleux, C. Moire, C. Farcet and H. Pasch, Macromolecules, 2010, 43, 8755–8765 CrossRef CAS.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS PubMed.
- Y. Pei, L. Thurairajah, O. R. Sugita and A. B. Lowe, Macromolecules, 2015, 48, 236–244 CrossRef CAS.
- I. Chaduc, W. J. Zhang, J. Rieger, M. Lansalot, F. D'Agosto and B. Charleux, Macromol. Rapid Commun., 2011, 32, 1270–1276 CrossRef CAS PubMed.
- L. P. D. Ratcliffe, A. J. Ryan and S. P. Armes, Macromolecules, 2013, 46, 769–777 CrossRef CAS.
- W. Zhang, F. D'Agosto, P.-Y. Dugas, J. Rieger and B. Charleux, Polymer, 2013, 54, 2011–2019 CrossRef CAS.
- L. Guo, Y. Jiang, T. Qiu, Y. Meng and X. Li, Polymer, 2014, 55, 4601–4610 CrossRef CAS.
- H. De Brouwer, M. A. J. Schellekens, B. Klumperman, M. J. Monteiro and A. L. German, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3596–3603 CrossRef CAS.
- J. Liu, H. Duong, M. R. Whittaker, T. P. Davis and C. Boyer, Macromol. Rapid Commun., 2012, 33, 760–766 CrossRef CAS PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
-
C. W. Scales, E. R. George, C. D. Anderson, R. D. Gleim and B. M. Healy, WO, 2013085814-A2013085812, 2013 Search PubMed.
- K. L. Thompson, C. J. Mable, A. Cockram, N. J. Warren, V. J. Cunningham, E. R. Jones, R. Verber and S. P. Armes, Soft Matter, 2014, 10, 8615–8626 RSC.
- Y. X. Ma, X. L. Zheng, F. M. Shi, Y. Li and S. J. Sun, J. Appl. Polym. Sci., 2003, 88, 1622–1626 CrossRef CAS.
- J. Jang and B. S. Kim, J. Appl. Polym. Sci., 2000, 77, 903–913 CrossRef CAS.
- Y. Feng and C. F. Xiao, J. Appl. Polym. Sci., 2006, 101, 1248–1251 CrossRef CAS.
- Z. Janovic, K. Saric and K. Sertic-Bionda, Chem. Biochem. Eng. Q., 1998, 12, 19–24 CAS.
- A. Jukic, E. Vidovic and Z. Janovic, Chem. Technol. Fuels Oils, 2007, 43, 386–394 CrossRef CAS.
-
R. L. Stambaugh and B. G. Kinker, Viscosity Index Improvers and Thickeners, Springer-Verlag Berlin, Berlin, 2010 Search PubMed.
- R. Zheng, G. Liu, M. Devlin, K. Hux and T.-C. Jao, Tribol. Trans., 2010, 53, 97–107 CrossRef CAS.
- R. Zheng, G. Liu and T.-C. Jao, Polymer, 2007, 48, 7049–7057 CrossRef CAS.
- J. S. Trent, Macromolecules, 1984, 17, 2930–2931 CrossRef CAS.
- Our previously reported protocol for TEM staining using RuO4 (Chem. Sci., 4, 2081–2087 and J. Am. Chem. Soc., 2014, 136, 5790–5798) gave incorrect oxidation states. Those quoted in this manuscript are correct.
- P. Cacioli, D. G. Hawthorne, R. L. Laslett, E. Rizzardo and D. H. Solomon, J. Macromol. Sci., Chem., 1986, 23, 839–852 CrossRef.
- M. Rodlert, E. Harth, I. Rees and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4749–4763 CrossRef CAS.
- R. Verber, A. Blanazs and S. P. Armes, Soft Matter, 2012, 8, 9915–9922 RSC.
- J. van Stam, S. Creutz, F. C. De Schryver and R. Jérôme, Macromolecules, 2000, 33, 6388–6395 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table summarizing macro-CTAs; TEM evidence for different morphologies with the same diblock copolymer composition in each oil; DLS and TEM evidence for the worm-to-sphere transition of PLMA18-PBzMA40 worms upon heating to 90 °C; DLS size distribution of spheres synthesized via a ‘one-pot’ protocol. See DOI: 10.1039/c5py00157a |
|
| This journal is © The Royal Society of Chemistry 2015 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a