
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymerization of 5-alkyl δ-lactones catalyzed by diphenyl phosphate and their sequential organocatalytic polymerization with monosubstituted epoxides†
Junpeng
Zhao
a and
Nikos
Hadjichristidis
*b
aFaculty of Materials Science and Engineering, South China University of Technology, Guangzhou, People's Republic of China 510640
bPolymer Synthesis Laboratory, KAUST Catalysis Center, Physical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia. E-mail: nikolaos.hadjichristidis@kaust.edu.sa
First published on 4th February 2015
Abstract
Organocatalytic ring-opening polymerization (ROP) reactions of three renewable 5-alkyl δ-lactones, namely δ-hexalactone (HL), δ-nonalactone (NL) and δ-decalactone (DL), using diphenyl phosphate (DPP) were investigated. Room temperature, together with a relatively high monomer concentration (≥3 M), was demonstrated to be suitable for achieving a living ROP behavior, a high conversion of the lactone, a controlled molecular weight and a low dispersity of the polyester. HL, containing a 5-methyl substituent, showed a much higher reactivity (polymerization rate) and a slightly higher equilibrium conversion than the compounds with longer alkyl substituents (NL and DL). The effectiveness of DPP-catalyzed ROP of 5-alkyl δ-lactones facilitated the one-pot performance following the t-BuP4-promoted ROP of monosubstituted epoxides. It has been shown in an earlier study that substituted polyethers acted as “slow initiators” for non-substituted lactones. However, efficient initiations were observed in the present study as substituted lactones were polymerized from the substituted polyethers. Therefore, this reinforces the previously developed “catalyst switch” strategy, making it a more versatile tool for the synthesis of well-defined polyether–polyester block copolymers from a large variety of epoxide and lactone monomers.
Introduction
Polyether–polyester type block copolymers have shown great potential and promising prospects in the fabrication of micro- or nanostructured biomaterials for e.g. drug delivery and controlled release, gene therapy, tissue engineering, etc., owing to the combination, complementation and interplay of the respective physicochemical properties such as biodegradability, biocompatibility, solubility/miscibility, rigidity/flexibility, and crystallinity derived from each of the block components.1–5 Ring-opening polymerization (ROP) of epoxides6,7 and cyclic esters (lactones or lactides)8–11 is a commonly used method to synthesize polyethers and polyesters, respectively, with controlled molecular weights, low dispersities and tailored macromolecular structures. One-pot sequential ROP of the corresponding epoxide and cyclic ester is clearly the most ideal route for the synthesis of the polyether–polyester block copolymer, and it seems straightforward and facile at first glance as the polyether and polyester chains both grow maintaining an alkoxide/hydroxyl end group. However, it remains a major challenge as the monomers are suited to different initiating/catalytic systems, i.e. the active species that work for epoxides (or cyclic esters) can either be inactive or lead to uncontrolled polymerization for the other monomer, especially in the case of conventional metal-based initiating/catalytic systems. Therefore, the preparation of polyether–polyester block copolymers is generally tedious as multiple steps of synthesis, isolation and purification are required.12The recent development of organocatalytic polymerization methodologies13–16 has offered new opportunities for the fulfillment of such synthetic tasks. Similar to their metallic counterparts, organic catalysts also need to be appropriately chosen for each specific monomer type to achieve the best compromise between the polymerization rate and control. For example, a strong phosphazene base (t-BuP4) is well suited for the ROP of epoxides (either ethylene oxide or the monosubstituted ones),17–25 while strong organic acids (such as trifluoromethanesulfonic acid, sulfonimide derivatives and phosphoric acids) appear more appropriate for the ROP of cyclic esters.26–34 Although neither can act as an ideal single catalyst for both types of monomer,35–37 a combination (sequential performance) of t-BuP4-promoted ROP of epoxides and acid-catalyzed ROP of lactones, i.e. the “catalyst switch” strategy, has been successfully used to achieve one-pot preparations of well-defined polyether–polyester block copolymers.35,38
Previously it has been found that when a monosubstituted epoxide (e.g. 1,2-butylene oxide) and a non-substituted lactone (e.g. ε-caprolactone and δ-valerolactone) were sequentially polymerized, the polyether chain end (a secondary alcohol) acted as a “slow initiator” for the lactone as the polyester chain grew through a more nucleophilic primary-alcohol end group.35 Hence we are interested to examine the sequential ROP of a monosubstituted epoxide and a ω-substituted lactone, which both polymerize with the chain end maintained as a secondary alcohol. Two monosubstituted epoxides and three commercially available 5-alkyl δ-lactones, as listed in Fig. 1, are involved in the present study. The use of such bio-sourced lactone monomers,39,40 associated with organocatalytic polymerization methods, adds significantly to the interest in the corresponding polyester-based materials due to their renewability and sustainability.41–46 The effectiveness of the organic acid catalyst (diphenyl phosphate) for the ROP of the substituted lactones was first investigated, aiming to achieve conditions resulting in high monomer conversions and controlled molecular weights. Then the one-pot sequential ROP of the two types of monomer was performed using the suitable conditions for each via the “catalyst switch” strategy.
 | ||
| Fig. 1 Structures of the monosubstituted epoxides and 5-alkyl δ-lactones used in this study. | ||
Experimental section
Chemicals
All chemicals were purchased from Aldrich. N-Ethyldiisopropylamine (EDIPA; 99%), acetic acid (AcOH; 99%) and phosphazene base (t-BuP4; 0.8 M in n-hexane) were used as received. Toluene (HPLC grade) was dried and distilled successively over calcium hydride and n-butyllithium. 1,2-Butylene oxide (BO; 99%) and 2-ethylhexyl glycidyl ether (EHGE; 98%) were dried and distilled successively over calcium hydride and sodium hydride. 3-Phenyl-1-propanol (PPA; 98%), δ-hexalactone (HL; 98%), δ-nonalactone (NL; 98%) and δ-decalactone (DL; 98%) were dried over calcium hydride and distilled twice under vacuum. Diphenyl phosphate (DPP; 99%) was first dissolved in toluene (HPLC grade), followed by slow cryo-evaporation of toluene on the vacuum line, and then dissolved in purified toluene to prepare a 2.0 M solution.Instrumentation
Size exclusion chromatography (SEC) was conducted in chloroform (CHCl3) at 30 °C using two 7.8 mm × 300 mm (5 μm) Styragel columns (Styragel HR 2 and Styragel HR 4) at a flow rate of 1.0 mL min−1. Calibration was done with a series of polystyrene (PS) standards to obtain the apparent number-average molecular weights (Mn,SEC) of the (co)polymers and their dispersities (Mw/Mn). Nuclear magnetic resonance (NMR) measurements were carried out at room temperature using a Bruker AVANCE III 600 spectrometer operating at 600 MHz; CDCl3 (Aldrich) was used as the solvent. 1H NMR spectra were used to calculate the molecular weights (Mn,NMR) of the isolated (co)polymers using the integrals of the characteristic signals from the end groups and main bodies of the (co)polymers. Differential scanning calorimetry (DSC) measurements were performed with a Mettler Toledo DSC1/TC100 system in a nitrogen atmosphere. The samples were first heated from room temperature (RT) to 100 °C in order to erase the thermal history, then cooled to −150 °C and finally heated again to 100 °C at a heating/cooling rate of 10 °C min−1. The second heating curve was used to acquire the glass transition temperature (Tg), which was determined as the temperature corresponding to the middle (half-height) of the glass transition.DPP-catalyzed ROP of 5-alkyl δ-lactones
A typical procedure for PDL41 and PDL42 (Table 1): 0.057 mL of PPA (0.42 mmol), 0.21 mL of DPP solution (0.42 mmol of DPP) and 1.6 mL of clean toluene were put into a reaction flask. Then 3.0 mL of DL (16.8 mmol) was added to start the polymerization. Aliquots were withdrawn (0.05 mL each) in an argon flow at different time intervals. Each aliquot was injected into a mixture of 1.0 mL of CDCl3 and two drops of EDIPA. This solution was used for the 1H NMR measurement to determine the conversion of DL. 0.1 mL of this CDCl3 solution was diluted with 1.0 mL of CHCl3 for SEC analysis. After 72 h, the conversion of DL reached 78%. The polymer is termed PDL41 (Table 1). Mn,theor = 5.3 kg mol−1. Mn,SEC = 9.1 kg mol−1, Mw/Mn = 1.20.| Polymer | Temp.a (°C) | [M]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (mol L−1) a (mol L−1) |
[M]0/[OH]0a | Timea (h) | Conv.a (%) |
M
n,theorb (kg mol−1) |
M
n,NMRc (kg mol−1) |
M
n,SECd (kg mol−1) |
M
w/Mnd |
|---|---|---|---|---|---|---|---|---|---|
| a Temperature, monomer concentration, feed ratio of monomer to the alcoholic initiator (hydroxyl group), time and monomer conversion for the ROP of the epoxide or lactone in the case of the polyether or polyester homopolymer, and for the ROP of the lactone (the second monomer) in the case of the polyether–polyester diblock copolymer. The ratio of the catalyst to the alcoholic initiator was maintained at 0.1 for the ROP of the epoxides and 1.0 for the ROP of the lactones ([t-BuP4]0/[OH]0 = 0.1 and [DPP]0/[OH]0 = 1). b Theoretical number-average molecular weight calculated from the feed and monomer conversion. c Number-average molecular weight calculated from 1H NMR spectrum of the isolated polymer using the integrals of the characteristic signals from the end group and polymer main body. d Number-average molecular weight and dispersity obtained from SEC analysis (CHCl3, 30 °C, PS standards) of the non-precipitated polymer. e PDL42, PNL12 and PHL12 are from chain extension experiments (second monomer feed) of PDL41, PNL11 and PHL11, respectively. The reaction time here represents the time used for the chain extension. | |||||||||
| PDL1 | RT | 1.5 | 50 | 48 | 6 | — | — | — | — |
| PDL2 | RT | 3.0 | 50 | 48 | 68 | 5.8 | 6.1 | 8.3 | 1.12 |
| PDL3 | RT | 3.5 | 50 | 114 | 80 | 6.8 | 7.4 | 10.9 | 1.16 |
| PDL41 | RT | 3.5 | 40 | 72 | 78 | 5.3 | — | 9.1 | 1.20 |
| PDL42e | RT | 4.5 | 107 | 168 | 83 | 15.1 | 16.2 | 26.4 | 1.09 |
| PDL5 | 40 | 3.5 | 40 | 72 | 68 | 4.6 | — | 8.5 | 1.36 |
| PBO1 | 40 | Bulk | 41 | 24 | >99 | 3.0 | 3.1 | 4.6 | 1.04 |
| PBO2 | 40 | Bulk | 41 | 24 | >99 | 3.0 | 2.9 | 4.5 | 1.05 |
| PBO2PDL | RT | 3.5 | 40 | 72 | 77 | 8.2 | 8.3 | 11.7 | 1.09 |
| PNL11 | RT | 3.5 | 40 | 72 | 78 | 4.9 | — | 9.7 | 1.18 |
| PNL12e | RT | 4.5 | 80 | 72 | 80 | 10.0 | 11.3 | 18.4 | 1.10 |
| PEHGE1 | 40 | Bulk | 27 | 24 | >99 | 5.0 | 5.4 | 5.1 | 1.07 |
| PEHGE2 | 40 | Bulk | 27 | 24 | >99 | 5.0 | 5.4 | 5.3 | 1.05 |
| PEHGE2PNL | RT | 3.7 | 53 | 120 | 80 | 11.6 | 12.3 | 15.0 | 1.09 |
| PHL11 | RT | 3.5 | 40 | 24 | 85 | 3.9 | — | 8.8 | 1.17 |
| PHL12e | RT | 5.7 | 107 | 24 | 89 | 10.9 | 11.4 | 23.6 | 1.09 |
| PBO3 | 40 | Bulk | 55 | 24 | >99 | 4.0 | 4.2 | 6.7 | 1.04 |
| PBO3PHL | RT | 3.7 | 71 | 31 | 87 | 11.1 | 10.9 | 18.6 | 1.09 |
| PEHGE3 | 40 | Bulk | 27 | 24 | >99 | 5.0 | 5.5 | 5.1 | 1.08 |
| PEHGE3PHL | RT | 3.5 | 69 | 31 | 88 | 11.9 | 12.2 | 16.3 | 1.16 |
As the second monomer feed, 5.0 mL of DL (28.0 mmol) was added to the PDL41 solution. Aliquots were withdrawn from time to time for 1H NMR and SEC analysis. After another 168 h, the polymerization was quenched by the addition of 1 mL of EDIPA. Then the solution was diluted with 10 mL of toluene and poured into cold (−20 °C) methanol to precipitate the polymer. The viscous liquid polymer (PDL42, Table 1) was then collected and dried substantially under vacuum. Conv.(DL) = 83%. Theoretical number-average molecular weight (Mn,theor) = 15.1 kg mol−1. Mn,SEC = 26.4 kg mol−1. Mw/Mn = 1.09. 1H NMR (600 MHz, CDCl3): δ/ppm = 7.20–7.15 (aromatic protons on the PPA end group), 5.01–4.71 (–OCOCH2CH2CH2CH(CH2CH2CH2CH2CH3)–), 4.12–4.07 (PhCH2CH2CH2OCO–PDL), 3.62–3.55 (PDL–OCOCH2CH2CH2CH(CH2CH2CH2CH2CH3)OH), 2.71–2.65 (PhCH2CH2CH2OCO–PDL), 2.43–2.13 (–OCOCH2CH2CH2CH(CH2CH2CH2CH2CH3)–), 1.99–1.92 (PhCH2CH2CH2OCO–PDL), 1.83–1.37 (–OCOCH2CH2CH2CH(CH2CH2CH2CH2CH3)–), 1.36–1.13 (–OCOCH2CH2CH2CH(CH2CH2CH2CH2CH3)–), 1.00–0.74 (–OCOCH2CH2CH2CH(CH2CH2CH2CH2CH3)–); Mn,NMR = 16.2 kg mol−1.
Sequential ROP of a monosubstituted epoxide and a 5-alkyl δ-lactone
A typical procedure for PBO2 and PBO2PDL (Table 1): 0.17 mL of PPA (1.3 mmol) and 4.5 mL of BO (51.7 mmol) were put into a reaction flask and cooled at 0 °C. Then 0.16 mL of t-BuP4 solution (0.13 mmol of t-BuP4) was added to start the polymerization. The flask was then sealed using a stopcock and the temperature was slowly elevated to 40 °C. After heating and stirring for 24 h, 0.05 mL of the reaction mixture was withdrawn in an argon flow and injected into a mixture of 1 mL of CDCl3 and a few drops of AcOH for 1H NMR analysis. 0.1 mL of this solution was diluted with 2 mL of CHCl3 for SEC measurement. Conv.(BO) > 99%. Mn,theor = 3.0 kg mol−1. Mn,SEC = 4.5 kg mol−1. Mw/Mn = 1.05.Subsequently, 0.71 mL of DPP solution (1.42 mmol of DPP) was added in an argon flow to the living PBO2 solution (1.3 mmol of PBO–OH + PBO–O−, 0.13 mmol of PBO–O−), causing the brownish yellow living PBO to turn white and opaque, indicating the neutralization of the alkoxide PBO chain ends. 15 min later, the flask was cooled to RT and 9.0 mL of DL (50.4 mmol) was added in an argon flow, which turned the reaction mixture into a transparent and slightly yellowish solution. Aliquots were withdrawn at different time intervals for 1H NMR and SEC analysis. After the withdrawal of the last aliquot at 72 h, the polymerization was quenched by the addition of 1 mL of EDIPA. Then the solution was poured into cold (−20 °C) methanol and kept at −20 °C overnight to precipitate the PBO-b-PDL diblock copolymer. The white viscous liquid was then collected and dried substantially under vacuum. Conv.(DL) = 77%. Mn,theor(PDL) = 5.2 kg mol−1. Mn,theor(PBO-b-PDL) = 8.2 kg mol−1. Mn,SEC = 11.7 kg mol−1. Mw/Mn = 1.09. 1H NMR (600 MHz, CDCl3): δ/ppm = 7.20–7.15 (aromatic protons on the PPA end group), 5.01–4.71 (–OCOCH2CH2CH2CH(CH2CH2CH2CH2CH3)–), 3.75–3.15 (–CH2CH(CH2CH3)O–), 2.70–2.66 (PhCH2CH2CH2O–PBO–), 2.43–2.16 (–OCOCH2CH2CH2CH(CH2CH2CH2CH2CH3)–), 1.92–1.85 (PhCH2CH2CH2O–PBO–), 1.82–1.37 (–CH2CH(CH2CH3)O–) and (–OCOCH2CH2CH2CH(CH2CH2CH2CH2CH3)–), 1.36–1.13 (–OCOCH2CH2CH2CH(CH2CH2CH2CH2CH3)–), 1.05–0.74 (–CH2CH(CH2CH3)O–) and (–OCOCH2CH2CH2CH(CH2CH2CH2CH2CH3)–); Mn,NMR(PBO) = 2.9 kg mol−1, Mn,NMR(PDL) = 5.4 kg mol−1, Mn,NMR(PBO-b-PDL) = 8.3 kg mol−1.
Results and discussion
It has been demonstrated previously that DPP is an effective organic catalyst for the ROP of non-substituted lactones, giving rise to well-defined polyesters at nearly complete monomer conversions.33 Organocatalytic ROP of renewable 5-alkyl δ-lactones has also been studied,41,43,45 however, significantly less than the non-substituted ones. To assess the feasibility of their sequential ROP with monosubstituted epoxides using the base→acid “catalyst switch” strategy, the DPP-catalyzed ROP reactions of 5-alkyl δ-lactones (HL, NL and DL in Fig. 1) were first investigated (Scheme 1). Similar to the organic catalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), which has been successfully used to polymerize DL,41,45 phosphate types of organic catalysts are also considered to function by a monomer/chain-end dual activation mechanism.32,47–49 | ||
| Scheme 1 Schematic illustration of DPP-catalyzed ROP of 5-alkyl δ-lactones. | ||
The DPP-catalyzed ROP of DL was first performed in toluene at RT using different concentrations ([DL]0/[PPA]0/[DPP] = 50/1/1). At 1.5 M, a DL conversion of only 6% was reached after 48 h (PDL1 in Table 1). When the DL concentration was elevated to ≥3 mol L−1 (PDL2 and PDL3 in Table 1), reasonable conversions were reached (ca. 70% after 48 h). The polymers had relatively low dispersities, which slightly increased upon extending the reaction time (PDL3, from 1.12 to 1.16 as the reaction time was extended from 48 h to 114 h). These tests showed that a relatively high monomer concentration and a long polymerization time are essential for the DPP-catalyzed ROP of 5-alkyl δ-lactones, compared to the non-substituted ones,33,35 for achieving good monomer conversions due to their much lower reactivity.41,43,50–52
A detailed kinetic study was performed with a DL concentration of 3.5 mol L−1 ([DL]0/[PPA]0/[DPP] = 40/1/1). Fig. 2 shows the kinetic plots obtained under different experimental conditions. In each case, the kinetic plot appears to be linear during the early stages and slowly turns into a plateau after a certain conversion due to the monomer/polymer equilibrium.41 The polymerization at the early stages appears to be faster at 40 °C (PDL5 in Table 1) in terms of DL conversion, however, a higher equilibrium conversion of DL is eventually reached at RT (PDL41 in Table 1), indicating that the formation of PDL is more favored at lower temperatures in the present catalytic system.
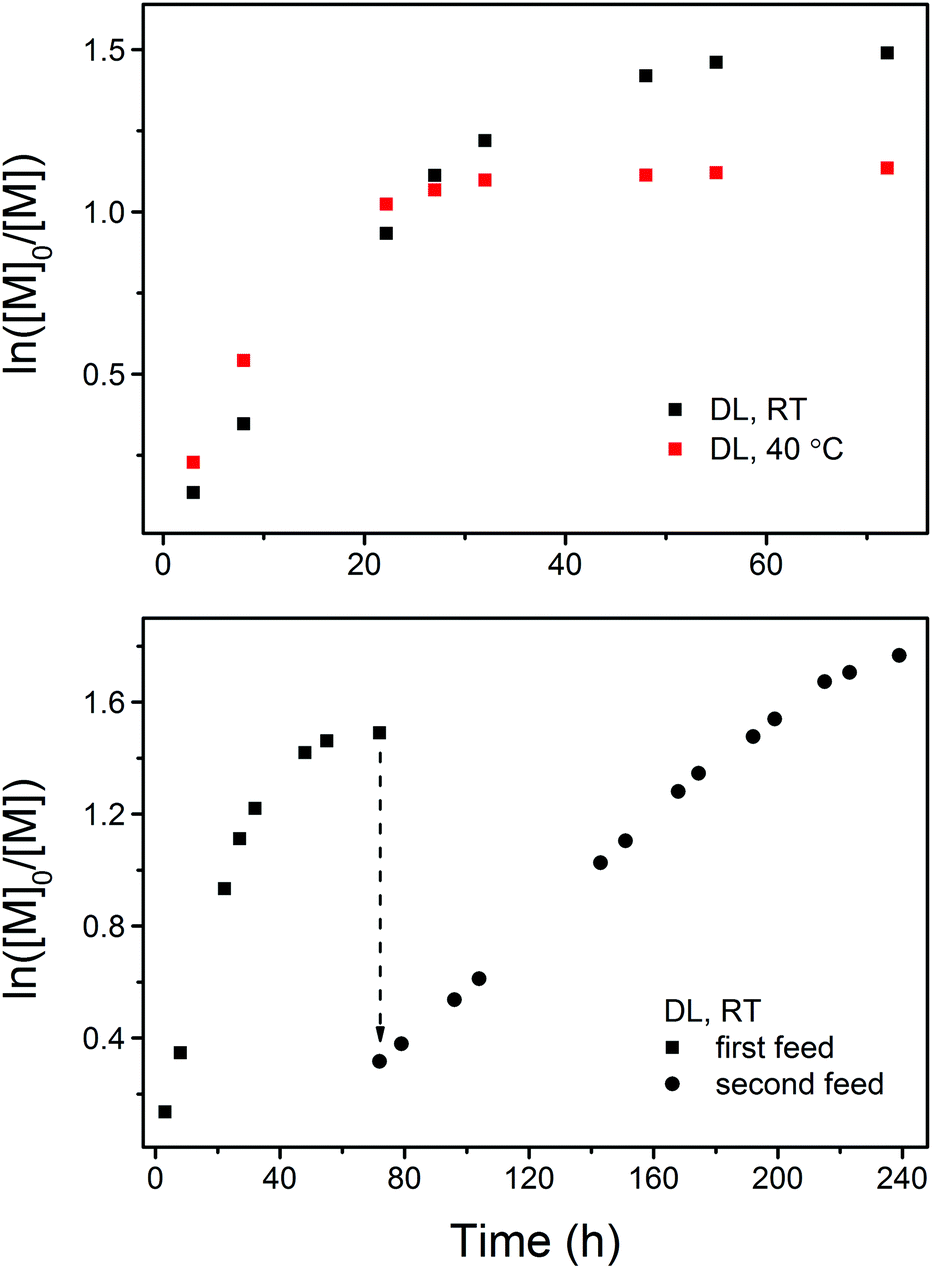 | ||
| Fig. 2 Kinetic plots of DPP-catalyzed ROP of DL at RT and 40 °C (upper figure, PDL41 and PDL5 in Table 1) and at RT with the first and second monomer feed (lower figure, PDL41 and PDL42 in Table 1). | ||
As shown by the SEC analysis of the withdrawn aliquots, the apparent molecular weight (Mn,SEC) of PDL increases linearly with DL conversion (Fig. 3). A steady increase in the PDL dispersity is also observed after the kinetic plot starts to approach the plateau, which is ascribed to the monomer–polymer (polymerization–depolymerization) equilibrium.41 Such an effect is more profound at an elevated temperature as PDL5 obtained at 40 °C has a significantly higher dispersity (Table 1 and Fig. 4).
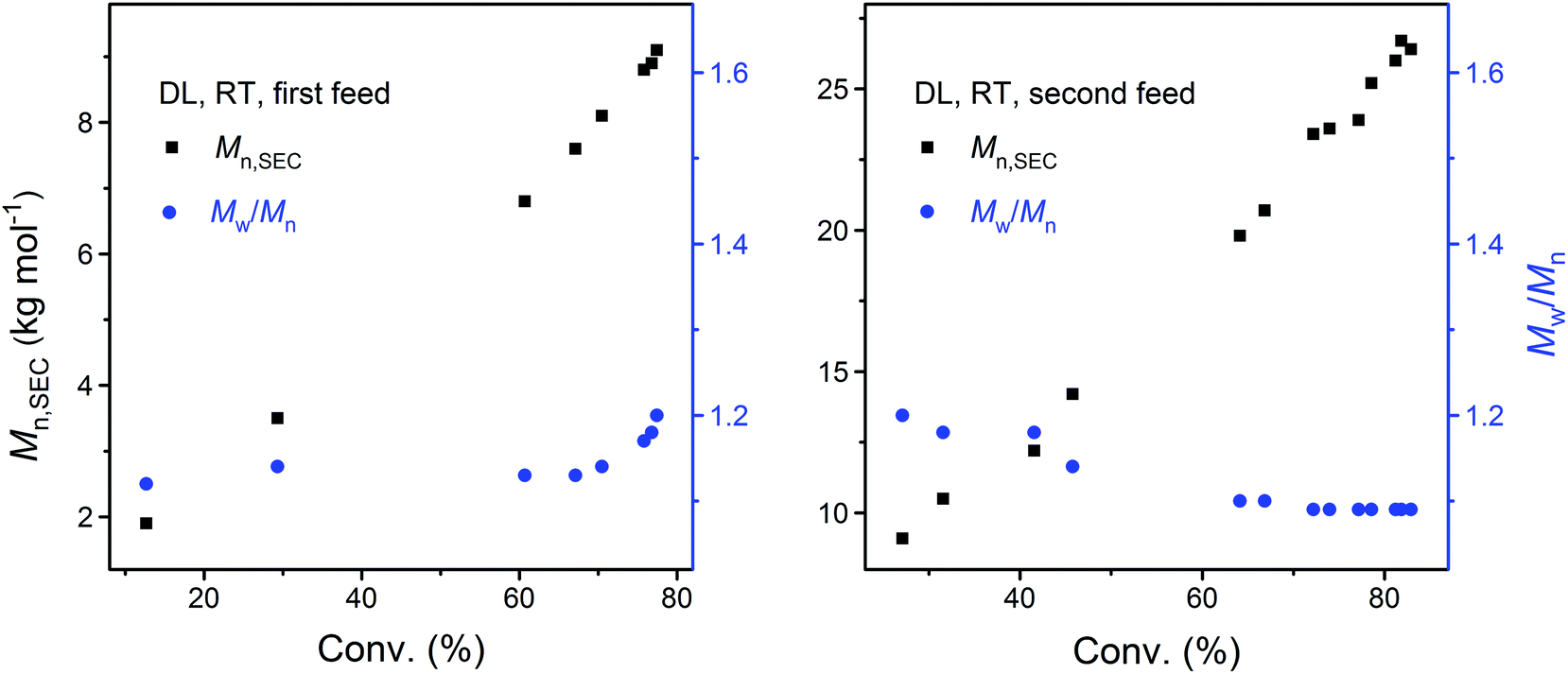 | ||
| Fig. 3 Dependence of apparent molecular weight (Mn,SEC) and dispersity (Mw/Mn) of PDL on monomer conversion during the DPP-catalyzed ROP of DL upon the first (left) and second (right) monomer feed (corresponding to PDL41 and PDL42 in Table 1, respectively). | ||
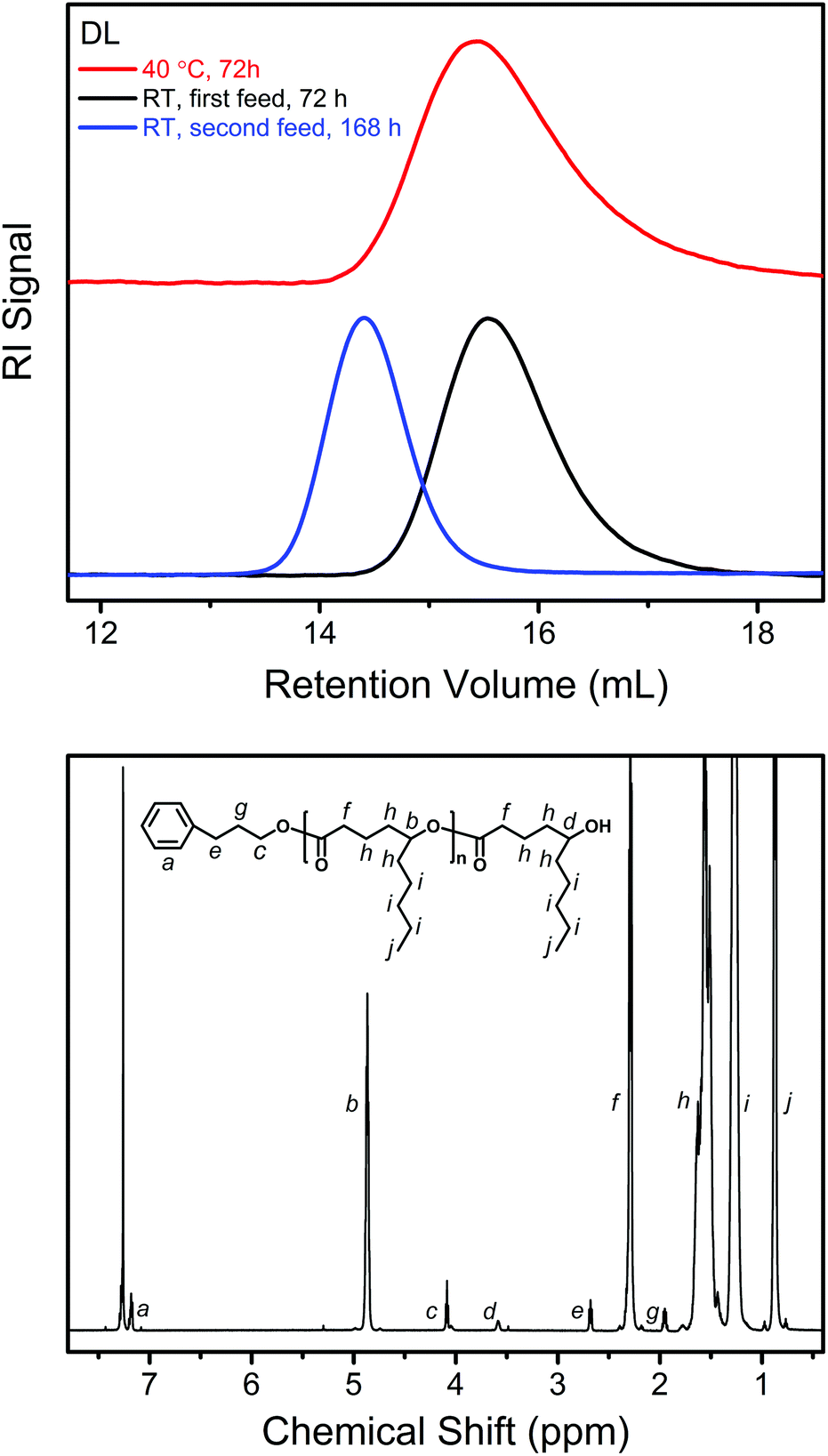 | ||
| Fig. 4 Upper: SEC traces of the products from the DPP-catalyzed ROP of DL under different conditions (PDL5, PDL41 and PDL42 in Table 1). Lower: 1H NMR spectrum obtained from a representative isolated PDL (PDL2 in Table 1). | ||
To confirm the living behavior of the ROP system, another batch of DL (second monomer feed) was added to PDL41 for a chain extension, upon which the polymerization was clearly “restarted” (Fig. 2 and 3, PDL42 in Table 1). Due to the lowered concentration of the catalyst (DPP) and hydroxy species, the polymerization proceeded slower this time (Fig. 2). However, linear Mn,SEC–conversion dependence was still present (Fig. 3). The equilibrium DL conversion appears to be higher (Fig. 2), presumably due to the higher monomer concentration (Table 1) which leads to more favored formation of the polymer in the polymerization–depolymerization equilibrium. The dispersity of PDL is maintained at a lower level this time (Table 1, Fig. 3 and 4), most probably because of the extended length of the polyester chain and the fact that the polymerization was quenched before the kinetic plot completely plateaued. Such results clearly verify the “livingness” of the DPP-catalyzed ROP of DL.
Fig. 4 also shows the 1H NMR spectrum of a representative PDL after isolation. All the characteristic signals from the PPA end group and PDL main body are clearly presented. The molecular weights (Mn,NMR) of the PDLs calculated from the signal integrals (e.g. c and f in Fig. 4) are slightly higher than the corresponding theoretical values (Mn,theor) calculated from the feed and monomer conversion (Table 1). This is because a bit of the low-molecular-weight part of the polymer is removed by the precipitation in methanol (the substituted polyesters have a better solubility in methanol than the non-substituted ones). However, the removal is less than 10 wt% in all cases, as indicated by the mass of the polymers finally collected.
Kinetic studies of DPP-catalyzed ROP of HL and NL under similar conditions (RT, [M]0 = 3.5 mol L−1, [M]0/[PPA]0/[DPP] = 40/1/1) were also conducted. For a better comparison, Fig. 5 presents the kinetic plots of all three 5-alkyl δ-lactones investigated (PDL41, PNL11 and PHL11 in Table 1). NL, having a 5-butyl substituent, shows a similar polymerization rate to that of DL (with a 5-pentyl substituent) and reaches nearly the same equilibrium conversion (78%, Table 1). On the other hand, the monomer carrying a 5-methyl substituent (HL) polymerizes much faster and reaches a higher equilibrium monomer conversion (85%, Table 1), indicating that higher reactivity of the lactone monomer leads to more favored formation of the polyester in the monomer–polymer equilibrium.50 This is consistent with the fact that the ROP of much more active non-substituted lactones (e.g. δ-valerolactone) can reach practically complete monomer conversion.33,35
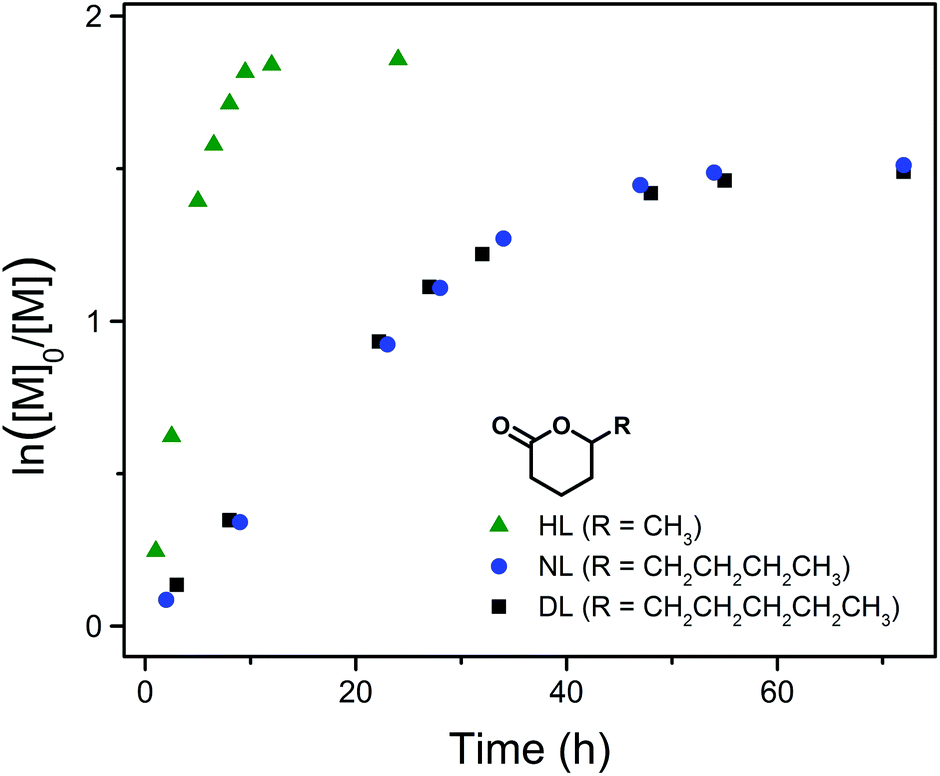 | ||
| Fig. 5 Kinetic plots of DPP-catalyzed ROP of HL, NL and DL at RT ([M]0 = 3.5 mol L−1, [M]0/[PPA]0/[DPP] = 40/1/1), corresponding to PHL11, PNL11 and PDL41 in Table 1, respectively. | ||
Linear Mn,SEC–conversion dependence, together with increased dispersities at high conversions, is also observed in the ROP of NL and HL (Fig. S1†). Chain extension experiments were also performed for these two monomers (Table 1, from PNL11 to PNL12 and from PHL11 to PHL12). These also showed the restart of the apparently ceased chain growth, further increased molecular weights and lowered dispersities (Table 1, Fig. S2 and S3†). The well-defined structures and controlled molecular weights of the obtained PNL and PHL were also confirmed by 1H NMR analysis (Fig. S2 and S3,†Table 1). These results have disclosed some common features shared by the ROPs of different 5-alkyl δ-lactones, regardless of the length of the alkyl substituent and the corresponding monomer reactivity.
After confirming the effectiveness of the DPP-catalyzed ROP of the 5-alkyl δ-lactones, the one-pot sequential polymerization reactions with monosubstituted epoxides were performed in a similar way to those previously developed.35,38 In order to ensure adequately high concentrations for the lactones to polymerize efficiently, the t-BuP4-promoted ROP reactions of the epoxides (BO and EHGE, Fig. 1) were performed in bulk. As previously demonstrated, the existence of phosphazenium diphenyl phosphate salt decelerates the ROP of lactones.35 Therefore, in the present study the ratio of t-BuP4 to the alcoholic initiator was reduced to 0.1 ([t-BuP4]0/[PPA]0 = 0.1, Scheme 2). Nevertheless, complete conversions of the epoxides and well-defined polyethers were still achieved in 24 h (PBO1/2/3 and PEHGE1/2/3 in Table 1, also see Experimental section), which are essential requirements for the successful performance of the “catalyst switch” strategy.35
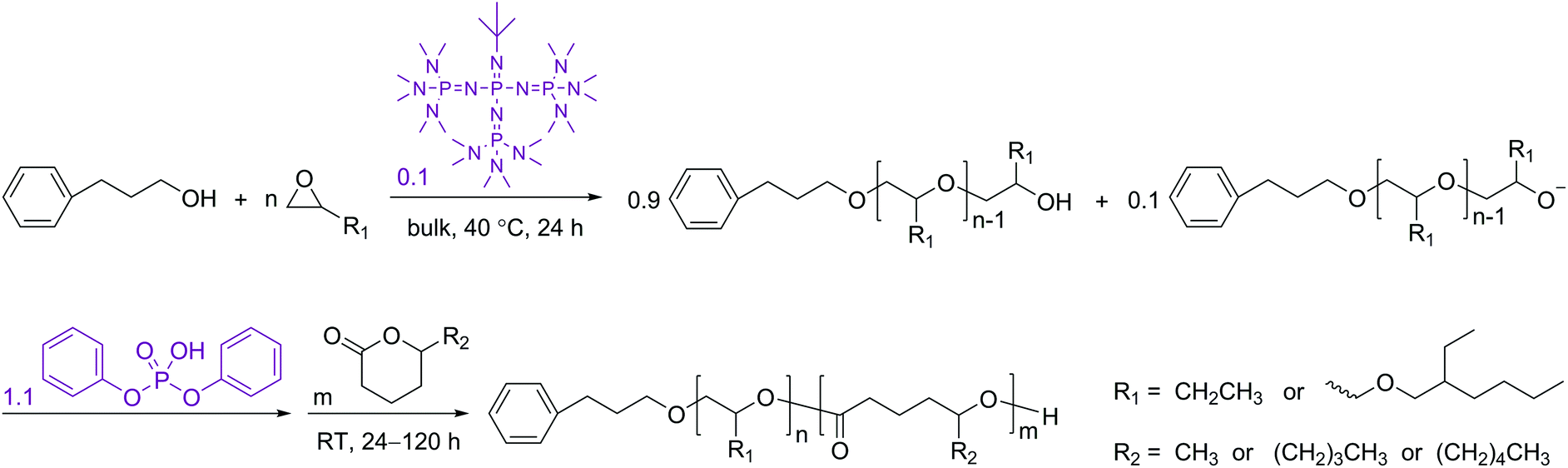 | ||
| Scheme 2 Reaction scheme for the synthesis of a polyether–polyester diblock copolymer via sequential organocatalytic ROP of a monosubstituted epoxide and a 5-alkyl δ-lactone using the base→acid “catalyst switch” strategy. | ||
After the completed ROP of BO (PBO2 in Table 1), excess DPP was added to neutralize the phosphazenium PBO-alkoxide and to catalyze the subsequent ROP of the second monomer (DL). The eventual ratio of DPP to the hydroxy species was maintained at 1.0 in all cases (Scheme 2). The complete dissolution of viscous PBO precursor in liquid DL took ca. 1 min, which did not show any influence on the formation of the diblock copolymer as the ROP of DL is a much slower process. Similar to the case of homopolymerization, the kinetic plot obtained from the ROP of DL appears to be linear during the early stages and bends down later on, indicating that the monomer–polymer equilibrium also exists when the ROP starts from a macroinitiator (Fig. 6, upper). Linear Mn,SEC–conversion dependence was observed and the dispersity of PBO-b-PDL was maintained at a low level (Fig. 6, lower) until the polymerization was quenched at a DL conversion of 77%.
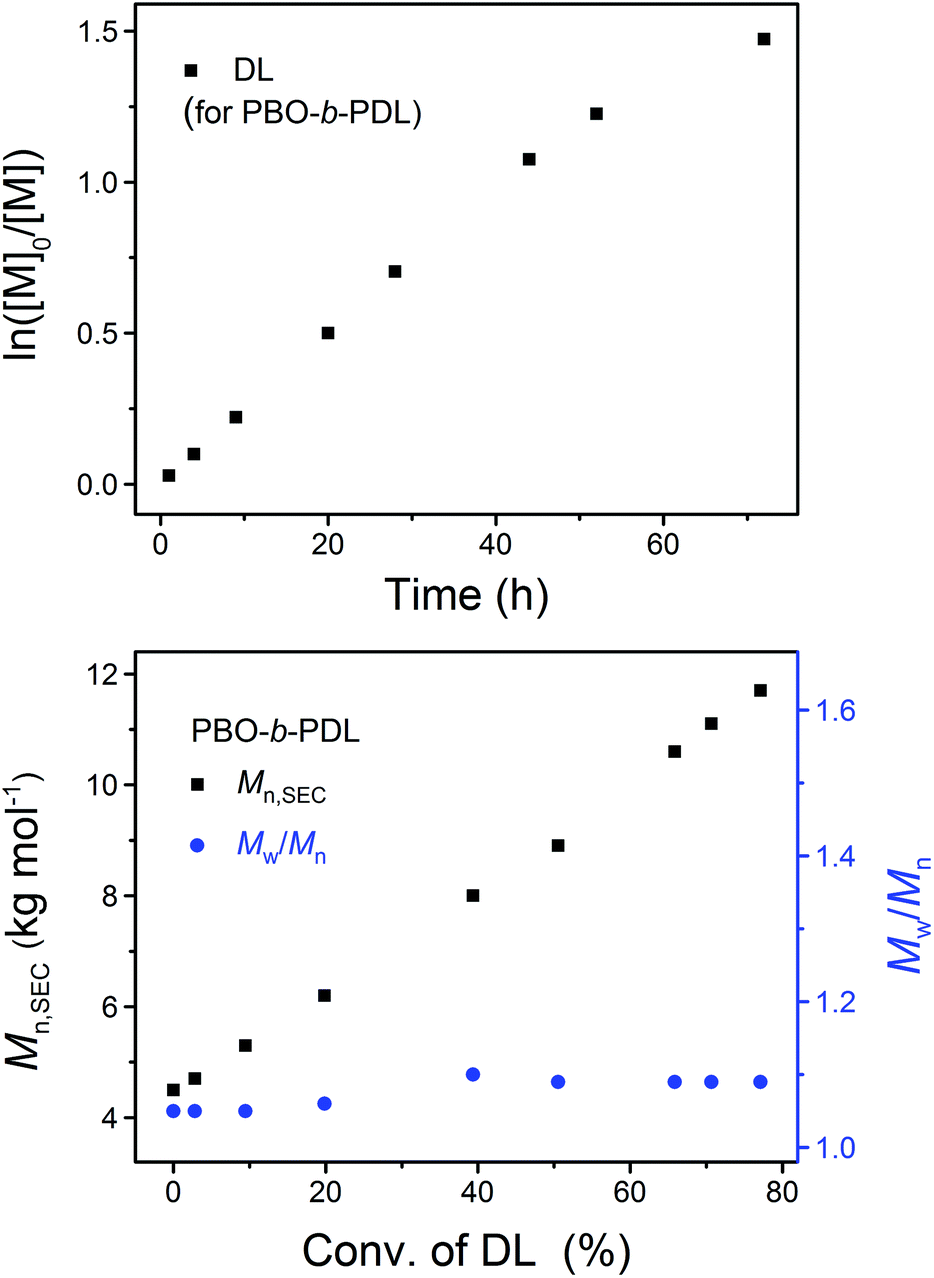 | ||
| Fig. 6 Upper: kinetic plot of DL. Lower: the dependence of the apparent molecular weight (Mn,SEC) and dispersity (Mw/Mn) of PBO-b-PDL on the DL conversion in its sequential ROP with BO using the “catalyst switch” strategy (PBO2PDL in Table 1). | ||
The linear kinetic plot implies that the slow initiation encountered in the sequential ROP of a monosubstituted epoxide and a non-substituted lactone may not be the case here.35Fig. 7 (upper) shows the evolution of the SEC traces during the transformation of PBO to PBO-b-PDL. It can be seen that the entire peak shifts slowly to the high-molecular-weight side while maintaining a narrow distribution as DL polymerizes, without the emergence of a bimodal distribution. The 1H NMR spectrum of the isolated PBO-b-PDL shows all the characteristic signals from the expected diblock copolymer structure (Fig. 7, lower) with the molecular weight of each block calculated to be close to the theoretical values (Table 1). These results point to the fact that the secondary-alcohol end group of PBO is an efficient initiator for DL, as the polymerization of DL also proceeds through a secondary-alcohol end group and in a sufficiently slow manner.
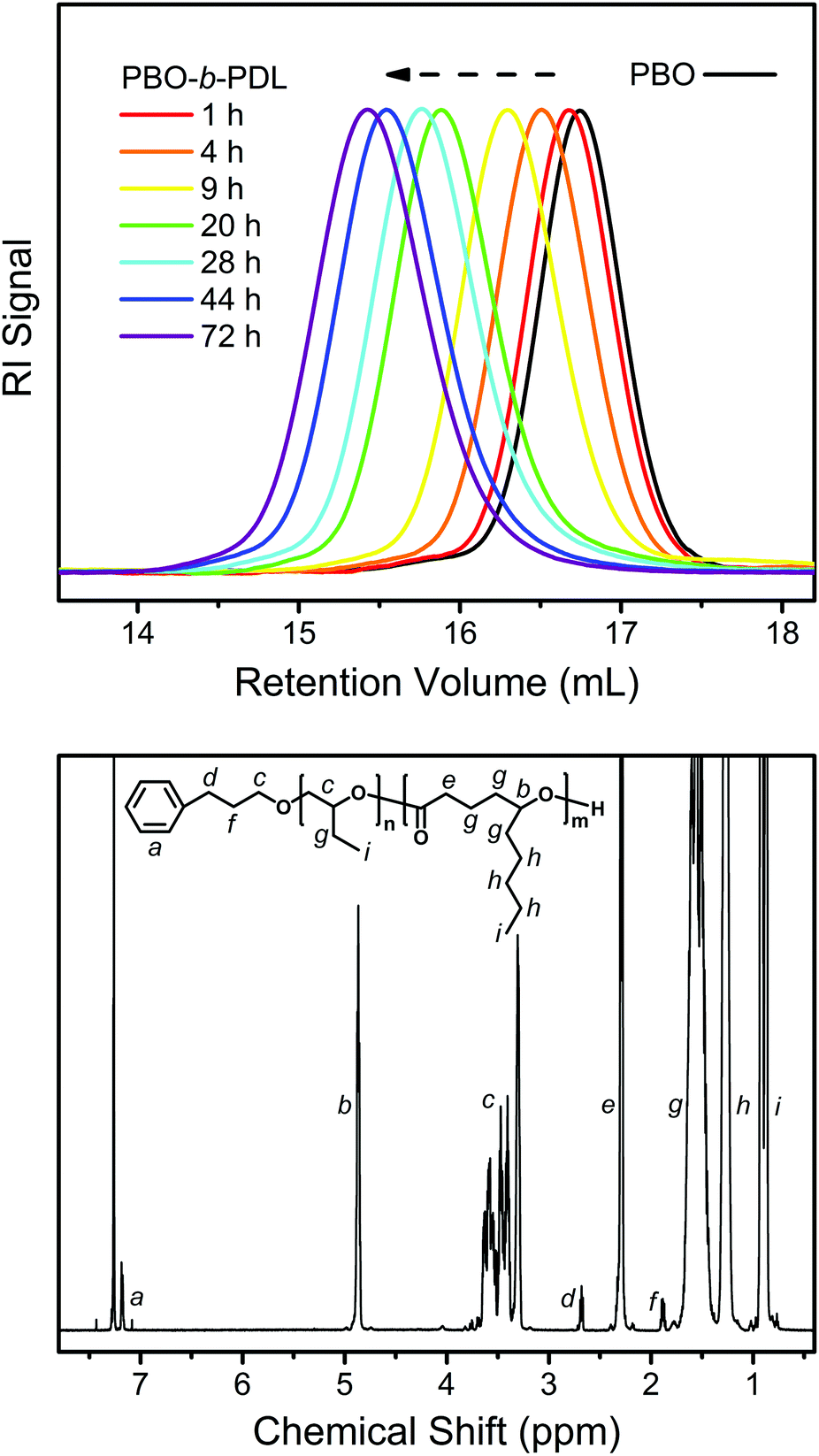 | ||
| Fig. 7 Upper: evolution of the SEC traces during the transformation of PBO to PBO-b-PDL (PBO2 and PBO2PDL in Table 1) using the “catalyst switch” strategy. Lower: 1H NMR spectrum obtained from the isolated PBO-b-PDL. | ||
The sequential ROP of EHGE and NL (Fig. 1) proceeded similarly (Fig. S4†), which implies that PEHGE, carrying bulky 2-ethylhexyl pendent groups, is also capable of acting as an efficient initiator for the lactone with a low reactivity. The isolated PEHEG-b-PNL (PEHGE2PNL in Table 1) has a low dispersity and a Mn,NMR value close to the theoretical value (Table 1 and Fig. S4†). For the ROP of the more active HL, the less bulky PBO still seems to be an efficient initiator (Fig. S5 and S6†). However, PEHGE shows some evidence of being a “slow initiator”, i.e. a shoulder towards the low-molecular-weight side in the SEC peak (Fig. 8, 4–11 h) and a distinctly non-linear kinetic plot during the early stages of the ROP of HL (Fig. S6†). Note that the shoulder towards the high-molecular-weight side in the SEC peaks is due to the existence of impurities (e.g. water) that function as difunctional initiators during the ROP of EHGE.53
 | ||
| Fig. 8 Evolution of the SEC traces during the transformation of PEHGE to PEHGE-b-PHL (PEHEG3 and PEHEG3PHL in Table 1) using the “catalyst switch” strategy. | ||
To better illustrate the initiation efficiency for the DPP-catalyzed ROP of the 5-alkyl δ-lactones, quantitative analysis was carried out utilizing the 1H NMR spectra from the aliquots withdrawn during the polymerization (Fig. 9). For the ROP of NL and HL initiated by PPA or PEHGE, the chemical shift of the ester group linking the initiator moiety and the polyester main body is distinct from that of the lactone monomer and polyester (Fig. 9, upper). Such a linkage is formed only in the initiation step, which enables us to use the signal integral ratio, Linkage/(Monomer + Polymer), to illustrate the evolution of the initiation using the combined integral of (Monomer + Polymer) as a constant (Fig. 9, lower).
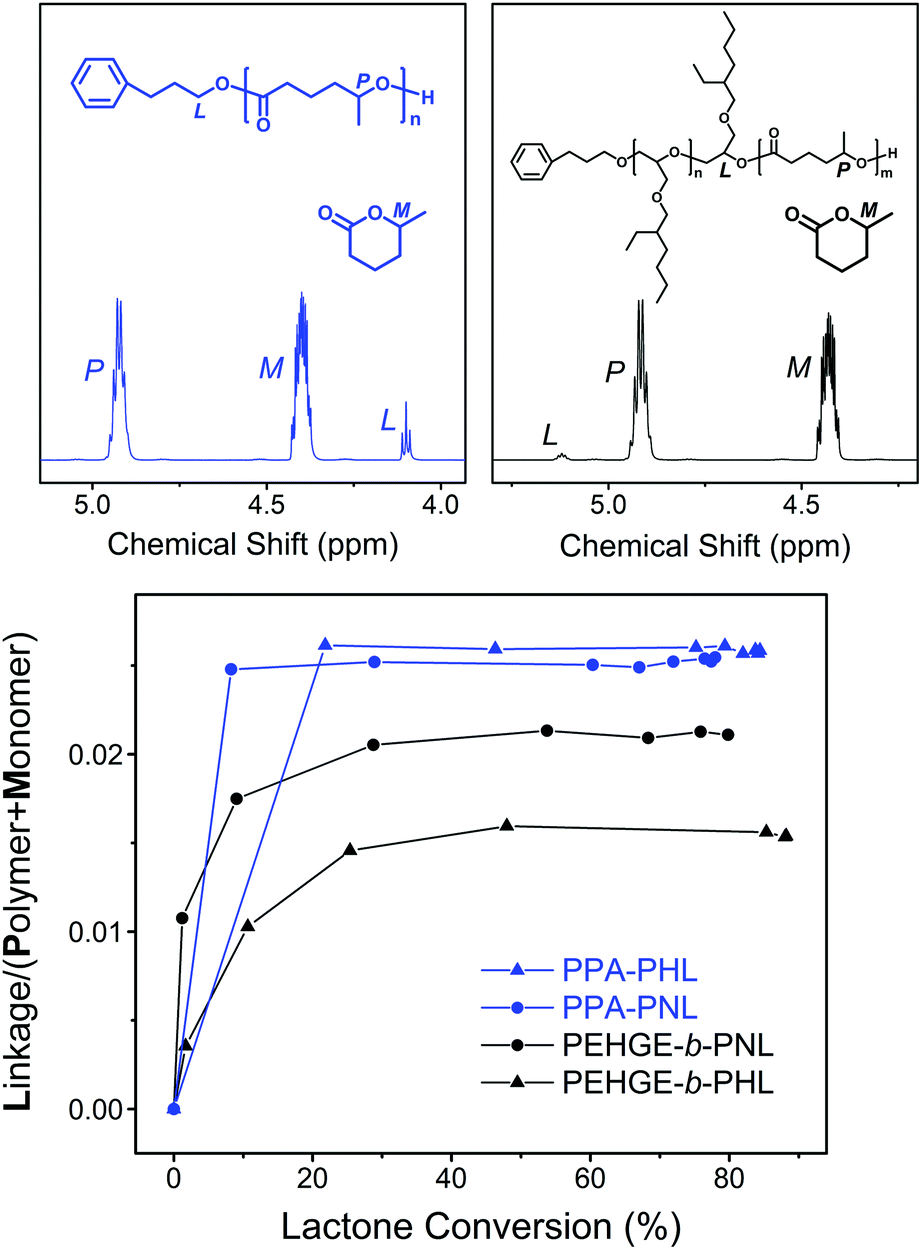 | ||
| Fig. 9 Illustration of the initiation efficiency for the DPP-catalyzed ROP of the 5-alkyl δ-lactones initiated by a small-molecule primary alcohol (PPA, blue) and a macromolecular secondary alcohol (PEHGE, black) using 1H NMR spectra from the aliquots withdrawn during the polymerization (upper) and the evolution of the signal integral ratios (lower). | ||
For the PPA-initiated ROP of NL/HL, the initiation step is completed before the withdrawal of the first aliquot (monomer conversion <20%). However, when the polymerization starts from PEHGE, a slower initiation mode is evidently presented for both NL and HL, and the initiation is completed at a monomer conversion of between 30% and 40%. Such a slow initiation is quite different from the case observed with PBO and a non-substituted lactone, in which the initiation step remains unfinished even after the full conversion of the monomer.35 In other words, for the ROP of a 5-alkyl δ-lactone the initiation from a macromolecular secondary alcohol is relatively slow compared to that from a small-molecule primary alcohol, but not slow enough to have a significant impact on the product as the isolated diblock copolymers still have well-defined structures, controlled molecular weights and dispersities (Fig. 7, S4, S5, S7† and Table 1). It has to be noted that such analysis cannot be performed for the polymerization from a PBO initiator, as the 1H NMR signal of the linking group overlaps with other signals (Fig. 7 and S5†).
The thermal properties (glass transition) of the isolated polyethers, polyesters and polyether–polyester diblock copolymers were analyzed by DSC (Fig. 10). PEHGE, carrying a larger alkyl pendent group, shows a lower Tg than PBO. The Tg of the substituted polyester also decreases with the increasing length of the alkyl pendent group. PEHGE-b-PNL and PBO-b-PDL each show only one glass transition with the Tg lying between those of the corresponding polyether and polyester homopolymers, indicating that the long alkyl pendent groups, especially those on the polyesters, result in a good miscibility of the polyether and polyester blocks. On the other hand, PEHGE-b-PHL and PBO-b-PHL each present two glass transitions with Tg values close to the values of the corresponding polyether or polyester homopolymer, respectively. It is thus evident that PHL, carrying a short alkyl pendent group, is immiscible with the polyethers, which is consistent with the fact that non-substituted polyethers and polyesters are usually immiscible and their block copolymers readily undergo nano- or microphase segregation in bulk or solution.4
 | ||
| Fig. 10 DSC traces (second heating curves) of the isolated polyethers (PEHGE1 and PBO1 in Table 1), polyesters (PHL12, PNL12 and PDL2 in Table 1) and polyether–polyester diblock copolymers (PEHGE3PHL, PBO3PHL, PEHGE2PNL and PBO2PDL in Table 1). | ||
Conclusions
DPP has been demonstrated to be an effective organic catalyst for the ROP of renewable 5-alkyl δ-lactones. In spite of the monomer–polymer equilibrium, reasonable monomer conversions and controlled molecular weights can still be reached provided suitable polymerization conditions are used, i.e. room temperature, sufficiently high monomer concentrations and relatively long reaction times. The sequential ROP of 5-alkyl δ-lactones and monosubstituted epoxides via the base→acid “catalyst switch” strategy is consequently feasible. Although slow initiation from the substituted polyethers is still manifested, the influence is practically insignificant due to the much slower polymerization of the 5-alkyl δ-lactones, and thus well-defined polyether–polyester block copolymers can still be achieved. This study has strengthened the “catalyst switch” strategy by involving epoxides and lactones with different substituents, and has further proven that it is an efficient and versatile synthetic method for polyether–polyester types of renewable and sustainable materials with variable thermal properties, miscibilities, phase segregation, morphological behavior, etc.Notes and references
- R. T. Liggins and H. M. Burt, Adv. Drug Delivery Rev., 2002, 54, 191–202 CrossRef CAS.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS PubMed.
- M. Sokolsky-Papkov, K. Agashi, A. Olaye, K. Shakesheff and A. J. Domb, Adv. Drug Delivery Rev., 2007, 59, 187–206 CrossRef CAS PubMed.
- R. V. Castillo and A. J. Müller, Prog. Polym. Sci., 2009, 34, 516–560 CrossRef CAS PubMed.
- X. Wei, C. Gong, M. Gou, S. Fu, Q. Guo, S. Shi, F. Luo, G. Guo, L. Qiu and Z. Qian, Int. J. Pharm., 2009, 381, 1–18 CrossRef CAS PubMed.
- H. Keul and M. Möller, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3209–3231 CrossRef CAS.
- A.-L. Brocas, C. Mantzaridis, D. Tunc and S. Carlotti, Prog. Polym. Sci., 2013, 38, 845–873 CrossRef CAS PubMed.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- O. Coulembier, P. Degee, J. L. Hedrick and P. Dubois, Prog. Polym. Sci., 2006, 31, 723–747 CrossRef CAS PubMed.
- A. P. Dove, Chem. Commun., 2008, 6446–6470 RSC.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- N. Hadjichristidis, S. Pispas and G. Floudas, Block Copolymers, John Wiley & Sons, Hoboken, 2003 Search PubMed.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- M. Fevre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142–2172 RSC.
- B. Esswein and M. Möller, Angew. Chem., Int. Ed., 1996, 35, 623–625 CrossRef CAS.
- B. Eßwein, N. M. Steidl and M. Möller, Macromol. Rapid Commun., 1996, 17, 143–148 CrossRef.
- H. Schlaad, H. Kukula, J. Rudloff and I. Below, Macromolecules, 2001, 34, 4302–4304 CrossRef CAS.
- H. Misaka, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 9099–9107 CrossRef CAS.
- J. Zhao and H. Schlaad, Macromolecules, 2011, 44, 5861–5864 CrossRef CAS.
- H. Misaka, E. Tamura, K. Makiguchi, K. Kamoshida, R. Sakai, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1941–1952 CrossRef CAS.
- T. Isono, K. Kamoshida, Y. Satoh, T. Takaoka, S. Sato, T. Satoh and T. Kakuchi, Macromolecules, 2013, 46, 3841–3849 CrossRef CAS.
- J. Zhao, H. Alamri and N. Hadjichristidis, Chem. Commun., 2013, 49, 7079–7081 RSC.
- J. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2014, 47, 1693–1698 CrossRef CAS.
- D. Bourissou, B. Martin-Vaca, A. Dumitrescu, M. Graullier and F. Lacombe, Macromolecules, 2005, 38, 9993–9998 CrossRef CAS.
- J. M. Ren, Q. Fu, A. Blencowe and G. G. Qiao, ACS Macro Lett., 2012, 1, 681–686 CrossRef CAS.
- R. Kakuchi, Y. Tsuji, K. Chiba, K. Fuchise, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2010, 43, 7090–7094 CrossRef CAS.
- K. Makiguchi, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3769–3777 CrossRef CAS.
- K. Makiguchi, S. Kikuchi, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2455–2463 CrossRef CAS.
- Y. Jin, Y. Ji, X. He, S. Kan, H. Xia, B. Liang, J. Chen, H. Wu, K. Guo and Z. Li, Polym. Chem., 2014, 5, 3098–3106 RSC.
- D. Delcroix, A. Couffin, N. Susperregui, C. Navarro, L. Maron, B. Martin-Vaca and D. Bourissou, Polym. Chem., 2011, 2, 2249–2256 RSC.
- K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999–2005 CrossRef CAS.
- J. Xu, J. Song, S. Pispas and G. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1185–1192 CrossRef CAS.
- J. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2014, 47, 3814–3822 CrossRef CAS.
- K. Brzezińska, R. Szymański, P. Kubisa and S. Penczek, Makromol. Chem., Rapid Commun., 1986, 7, 1–4 CrossRef.
- T. Biela and P. Kubisa, Makromol. Chem., 1991, 192, 473–489 CrossRef CAS.
- J. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 304–312 CrossRef CAS.
- T. B. Adams, D. B. Greer, J. Doull, I. C. Munro, P. Newberne, P. S. Portoghese, R. L. Smith, B. M. Wagner, C. S. Weil, L. A. Woods and R. A. Ford, Food Chem. Toxicol., 1998, 36, 249–278 CrossRef CAS.
- Y. Gounaris, Flavour Fragrance J., 2010, 25, 367–386 CrossRef CAS.
- M. T. Martello, A. Burns and M. Hillmyer, ACS Macro Lett., 2012, 1, 131–135 CrossRef CAS.
- J.-O. Lin, W. Chen, Z. Shen and J. Ling, Macromolecules, 2013, 46, 7769–7776 CrossRef CAS.
- P. Olsén, T. Borke, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2013, 14, 2883–2890 CrossRef PubMed.
- L. Jasinska-Walc, M. R. Hansen, D. Dudenko, A. Rozanski, M. Bouyahyi, M. Wagner, R. Graf and R. Duchateau, Polym. Chem., 2014, 5, 3306–3310 RSC.
- D. Tang, C. W. Macosko and M. A. Hillmyer, Polym. Chem., 2014, 5, 3231–3237 RSC.
- A. L. Holmberg, K.H. Reno, R. P. Wool and I. I. I. T. H. Epps, Soft Matter, 2014, 10, 7405–7424 RSC.
- K. Makiguchi, T. Yamanaka, T. Kakuchi, M. Terada and T. Satoh, Chem. Commun., 2014, 50, 2883–2885 RSC.
- K. Makiguchi, T. Saito, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2032–2039 CrossRef CAS.
- Y. Miao, Y. Phuphuak, C. Rousseau, T. Bousquet, A. Mortreux, S. Chirachanchai and P. Zinck, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2279–2287 CrossRef CAS.
- W. H. Carothers, G. L. Dorough and F. J. van Natta, J. Am. Chem. Soc., 1932, 54, 761–772 CrossRef CAS.
- H. Kikuchi, H. Uyama and S. Kobayashi, Polym. J., 2002, 34, 835–840 CrossRef CAS.
- J. van Buijtenen, B. A. C. van As, M. Verbruggen, L. Roumen, J. A. J. M. Vekemans, K. Pieterse, P. A. J. Hilbers, L. A. Hulshof, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2007, 129, 7393–7398 CrossRef CAS PubMed.
- J. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, Polym. Chem., 2014, 5, 3750–3753 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional SEC traces, 1H NMR spectra and kinetic plots. See DOI: 10.1039/c5py00019j |
| This journal is © The Royal Society of Chemistry 2015 |