
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design, synthesis and DNA interactions of a chimera between a platinum complex and an IHF mimicking peptide†
Harita
Rao
a,
Mariana S.
Damian
ab,
Alak
Alshiekh
b,
Sofi K. C.
Elmroth
b and
Ulf
Diederichsen
*a
aInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstrasse 2, 37077 Göttingen, Germany. E-mail: udieder@gwdg.de
bBiokemi, Kemicentrum, Lund Universitet, P. O. Box 124, 22100, Lund, Sweden
First published on 8th October 2015
Abstract
Conjugation of metal complexes with peptide scaffolds possessing high DNA binding affinity has shown to modulate their biological activities and to enhance their interaction with DNA. In this work, a platinum complex/peptide chimera was synthesized based on a model of the Integration Host Factor (IHF), an architectural protein possessing sequence specific DNA binding and bending abilities through its interaction with a minor groove. The model peptide consists of a cyclic unit resembling the minor grove binding subdomain of IHF, a positively charged lysine dendrimer for electrostatic interactions with the DNA phosphate backbone and a flexible glycine linker tethering the two units. A norvaline derived artificial amino acid was designed to contain a dimethylethylenediamine as a bidentate platinum chelating unit, and introduced into the IHF mimicking peptides. The interaction of the chimeric peptides with various DNA sequences was studied by utilizing the following experiments: thermal melting studies, agarose gel electrophoresis for plasmid DNA unwinding experiments, and native and denaturing gel electrophoresis to visualize non-covalent and covalent peptide–DNA adducts, respectively. By incorporation of the platinum metal center within the model peptide mimicking IHF we have attempted to improve its specificity and DNA targeting ability, particularly towards those sequences containing adjacent guanine residues.
Introduction
One of the very well established mechanisms by which cisplatin and related chemotherapeutics manifest their cytotoxic effects is by forming covalent crosslinks mostly with adjacent guanine residues of nuclear DNA.1–5 The resulting kinked structure of DNA is stabilized by the binding of the High Mobility Group (HMG) domain proteins which block the cellular repair mechanism.6–8 Subsequently, inhibition of essential cellular processes such as replication and transcription lead ultimately to induction of apoptosis.9–12 Apart from nuclear DNA, the platinum drugs can potentially target various other cellular components such as cytosolic proteins and can in parallel trigger biochemical pathways resulting in cytotoxicity.13–16 However, their side reactions with certain cytoskeletal nucleophiles such as thiols, phospholipids, microfilaments or RNA either deactivate the drugs or lead to several undesired secondary effects.17–20 These include toxic effects such as neurotoxicity, nephrotoxicity, ototoxicity, emetogenesis21–24 as well as onset of resistance in cancer cells.25,26Drug candidates with enhanced targeting of nuclear DNA in comparison with other cellular components can be considered a promising approach to foster the clinical applicability of platinum complexes. Conjugation of metal complexes with suitable carrier molecules having intrinsic binding affinity and recognition properties for nucleic acid sequences has proven to be an efficient delivery system for specifically targeting DNA.27–30 Thus, implementing peptide backbones as biological carriers offers a plethora of advantages due to their high bioavailability and cell penetrating, target directing and recognition properties.31–33 In our previous work, platinum complex/peptide chimera consisting of positive charged lysine residues showed enhanced binding to the polyanionic DNA.34 This approach to develop chimeric platinum complexes offers the possibility to have a unique DNA interaction mode that is distinct from the metal complex alone.35 We extended this work towards a more specific DNA targeting, by designing a platinum complex/peptide chimera using model peptides mimicking the IHF, a protein known for its ability to bind to the DNA minor groove with a high degree of sequence specificity.36
IHF is a chromatin architectural protein belonging to the family of bacterial type II DNA binding proteins.36,37 It plays a crucial role in processes demanding higher order structural organization of DNA such as replication, transcription and site specific recombination.38,39 The IHF-DNA cocrystal (Fig. 1A),40 consists of two intertwined subunits, the α- and the β-subunit, forming a compact body from which two long flexible ribbon like arms protrude and wrap around the DNA minor groove. Intercalation of a proline residue located at the tip of each arm between the base pairs, disrupts the stacking interactions and leads to the formation of two sharp kinks of >160° in the DNA structure. Combination of the IHF mimicking model peptide with a platinum chelating unit is expected to have a synergistic effect with respect to the interaction with DNA (Fig. 1B). Here we present a synthetic route to access the IHF mimicking platinum complex/peptide chimera for achieving enhanced DNA recognition properties, and performed preliminary DNA interaction studies.
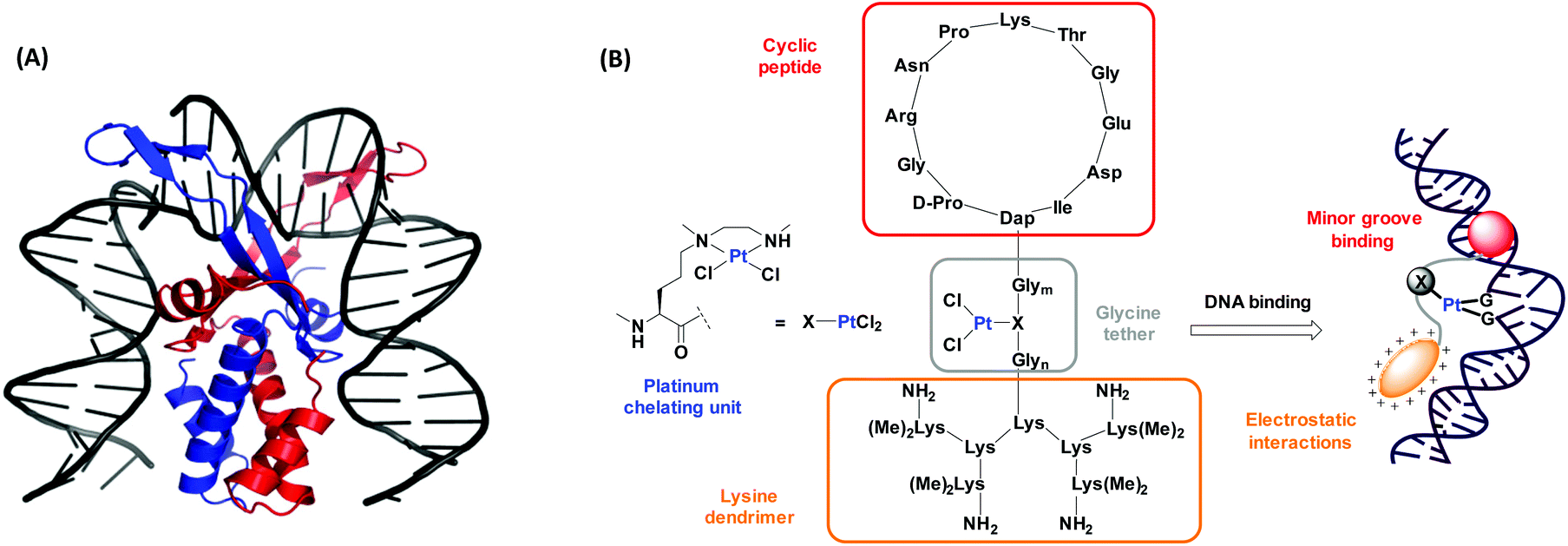 | ||
| Fig. 1 (A) The cocrystal structure of IHF bound to a 35 bp dsDNA fragment consisting of the H′ site of phage λ. The α-subunit is shown in red and the β–subunit in blue. Image reproduced using PyMOL (PDB entry: 1IHF).40 (B) Our model of the platinum complex/peptide chimera mimicking the α-subunit of IHF for targeting DNA by minor groove binding (through the cyclic peptide shown in red) and by electrostatic interactions with the negatively charged DNA (through the lysine dendrimer shown in orange). The IHF mimicking units are attached through a glycine linker wherein, the platinum chelating unit (X) is flexibly inserted to provide a second DNA recognition center. | ||
Results and discussion
The design of the IHF mimicking peptides is based on the model peptide IHF-1 (Fig. 2) as reported earlier.41,42 It has a closer resemblance to the α-subunit of IHF mainly due to its contribution to sequence specific DNA recognition within the minor groove.40 However, for the model peptide IHF-1 only a modest DNA bending effect was observed based on FRET measurements of respectively labeled DNAs.42 The basic structure of the IHF mimicking peptides can be divided into three parts, wherein each part contributes to different kinds of interactions with DNA (Fig. 1B). Firstly, a cyclic peptide unit resembles the minor groove binding loop of the IHF α-subunit and consists of the amino acid residues around the intercalating proline. Secondly, a lysine dendrimer mimics the positively charged body of IHF in order to compensate for electrostatic interactions with the negatively charged DNA phosphate backbone. Thirdly, a glycine linker not only tethers the two main peptide units but also has a functional role by containing a norvaline derived platinum chelating artificial amino acid. The platinated IHF mimics were prepared by placing the platinum metal center at flexible positions within the glycine linker: in close proximity to the cyclic unit (IHF-2), in the middle (IHF-3), and near the dendrimeric unit (IHF-4), respectively. The DNA interaction of the platinated mimics IHF-2/3/4 was compared to that of the platinated dendrimeric peptide P1, cyclic peptide P2 and the unplatinated IHF mimic IHF-1 (Fig. 2). Irrespective of the position of the platinum center within the glycine linker all three mimics IHF-2/3/4 showed equally enhanced binding to oligonucleotide sequences containing a GG site.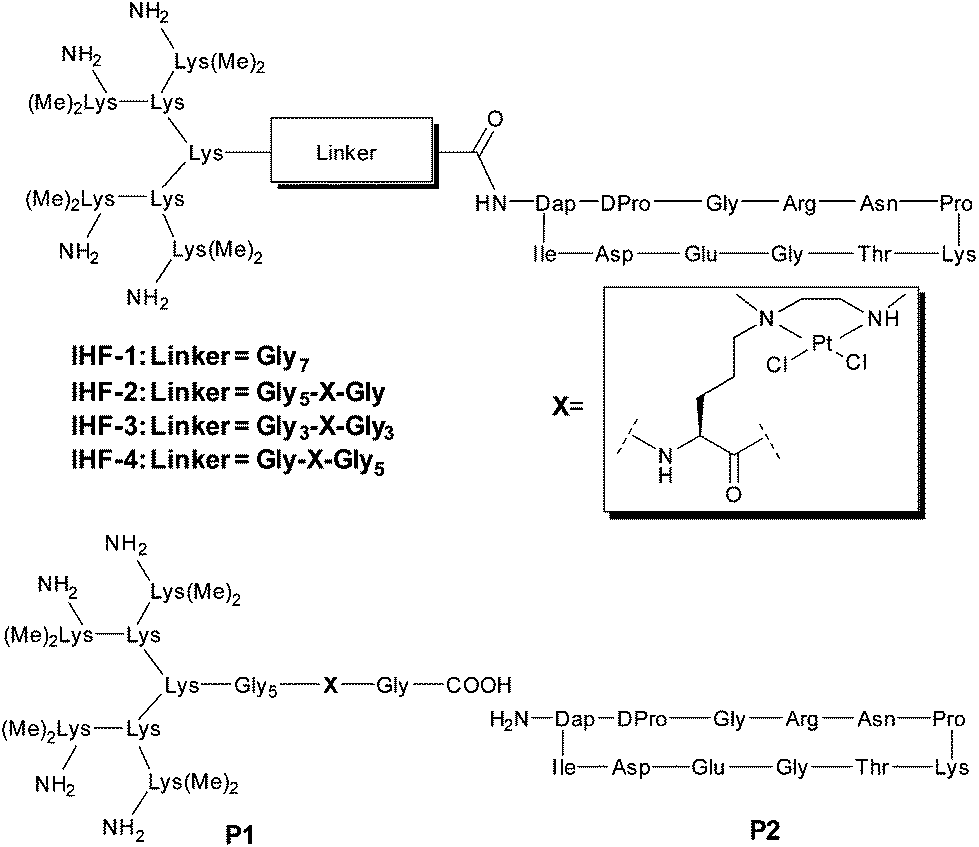 | ||
| Fig. 2 Various IHF mimicking peptides used in this study: IHF-1 is the unplatinated peptide, the mimics IHF-2, IHF-3 and IHF-4 contain a platinum chelating unit at different positions within the glycine linker, P1 is the platinated dendrimeric peptide and P2 is the cyclic peptide mimicking the minor groove intercalating unit. | ||
Synthesis of the platinum chelating amino acid residue
The amino acid residue to be introduced into the sequence of the IHF mimicking peptides was modified to contain an ethylenediamine ligand as side chain functionality for platinum coordination (Scheme 1). N,N′-Dimethylethylenediamine 1 was allyloxycarbonyl (Alloc) protected to yield N-allyloxycarbonyl-N,N′-dimethylethylenediamine 2. The platinum chelating artificial amino acid was obtained by reduction of the commercially available butyloxycarbonyl (Boc) protected amino acid derivative, N-Boc-glutamic acid benzyl ester with sodium borohydride followed by bromination to afford 3.43 Nucleophilic substitution of the brominated compound 3 with 2 led to the formation of 4 in a good yield. Removal of Boc and benzyl protecting groups released the amine and the carboxylic acid functionalities, respectively. The platinum chelating amino acid 5 was obtained by 9-fluorenylmethyloxycarbonyl (Fmoc) protection of the free amino group and was further employed for the synthesis of 7a–c (Scheme S1 in the ESI†) using solid phase peptide synthesis (SPPS). | ||
| Scheme 1 Synthesis of the platinum chelating amino acid building block 5. | ||
Synthesis of the IHF mimicking platinum complex/peptide chimera
The synthesis of the platinated IHF mimics IHF-2/3/4 started from connecting the cyclic unit 641 with the peptides 7a–c followed by a series of synthetic steps (Scheme 2). All the reaction intermediates were purified by RP-HPLC and characterized by HR-MS. The cyclic unit 6 was obtained as a free amine following a previously reported procedure.41 The peptides 7a–c containing the lysine dendrimer unit along with different glycine linkers were prepared by Fmoc based manual SPPS starting from Fmoc-Gly-NovaSyn® TGT resin (Scheme S1 in the ESI†). All the amino acids within the glycine linker including the platinum chelating building block 5 were sequentially coupled on the resin. The first and second dendrimer generations were obtained by coupling Fmoc–Lys(Fmoc)–OH. The third generation dendrimer was prepared by coupling Boc–Lys(Me)2–OH. Cleavage of the peptide from the resin under mild cleavage conditions utilizing a solution of 30% 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) in dichloromethane yielded the dendrimeric peptides 7a–c. | ||
| Scheme 2 Synthesis of the platinated IHF mimicking peptides IHF-2/3/4. | ||
Connection of the two main peptide units 6 and 7a–c using N,N′-diisopropylcarbodiamide (DIC), N-methylmorpholine (NMM) and 1-hydroxy-7-azabenzotriazole (HOAt) yielded peptides 8a–c. Palladium catalyzed deprotection of the Alloc group in the presence of a borane dimethylamine complex afforded 9a–c.44 Subsequent platination reaction yielding peptides 10a–c was followed by complete deprotection of the side chain protecting groups using 95% trifluoroacetic acid (TFA) leading to the formation of the final platinated IHF mimicking peptides IHF-2/3/4. For comparative studies the dendrimeric peptide P1 without the cyclic recognition unit was prepared in an analogous manner starting from peptide 7c (Scheme S1 in the ESI†). Deprotection of the Alloc group followed by platination and removal of the side chain protecting groups afforded the dendrimeric peptide P1. The cyclic peptide P2 was obtained by treatment of 6 with 95% TFA (Scheme S2 in the ESI†). The peptides P1 and P2 were further employed as controls in DNA binding studies.
Plasmid DNA interaction studies
The interaction of the platinated IHF mimicking peptides with DNA can interrupt the structural organization of DNA by means of three simultaneous forces acting in synergy: (1) interaction of the cyclic peptide with the DNA minor groove; (2) electrostatic attraction between the positively charged dendrimeric unit of the IHF mimic and the negatively charged phosphate backbone of DNA; (3) covalent modification of DNA by the platinum unit, known to distort DNA's structure due to a local unwinding of the duplex.45 Gel mobility shift assays utilizing native agarose gel electrophoresis were performed in order to study the conformational changes induced in the circular plasmid DNA upon interaction with various synthetic peptides (Fig. 3). The supercoiled form of the plasmid has a more compact structure and hence migrates faster in comparison with the relaxed form. Unwinding of the plasmid DNA affects the degree of supercoiling influencing its migration pattern on the gel; an increasing amount of platinated DNA is expected to be visualized by appearance of a slower migrating band.46 The ratio of the nucleotide (CDNA) to the peptide derivate (Cpeptide) concentration (rf = Cpeptide/CDNA) was varied in the range of 0–0.15 and the peptides were incubated for 2 h at 37 °C in 10 mM phosphate buffer, pH = 5.8.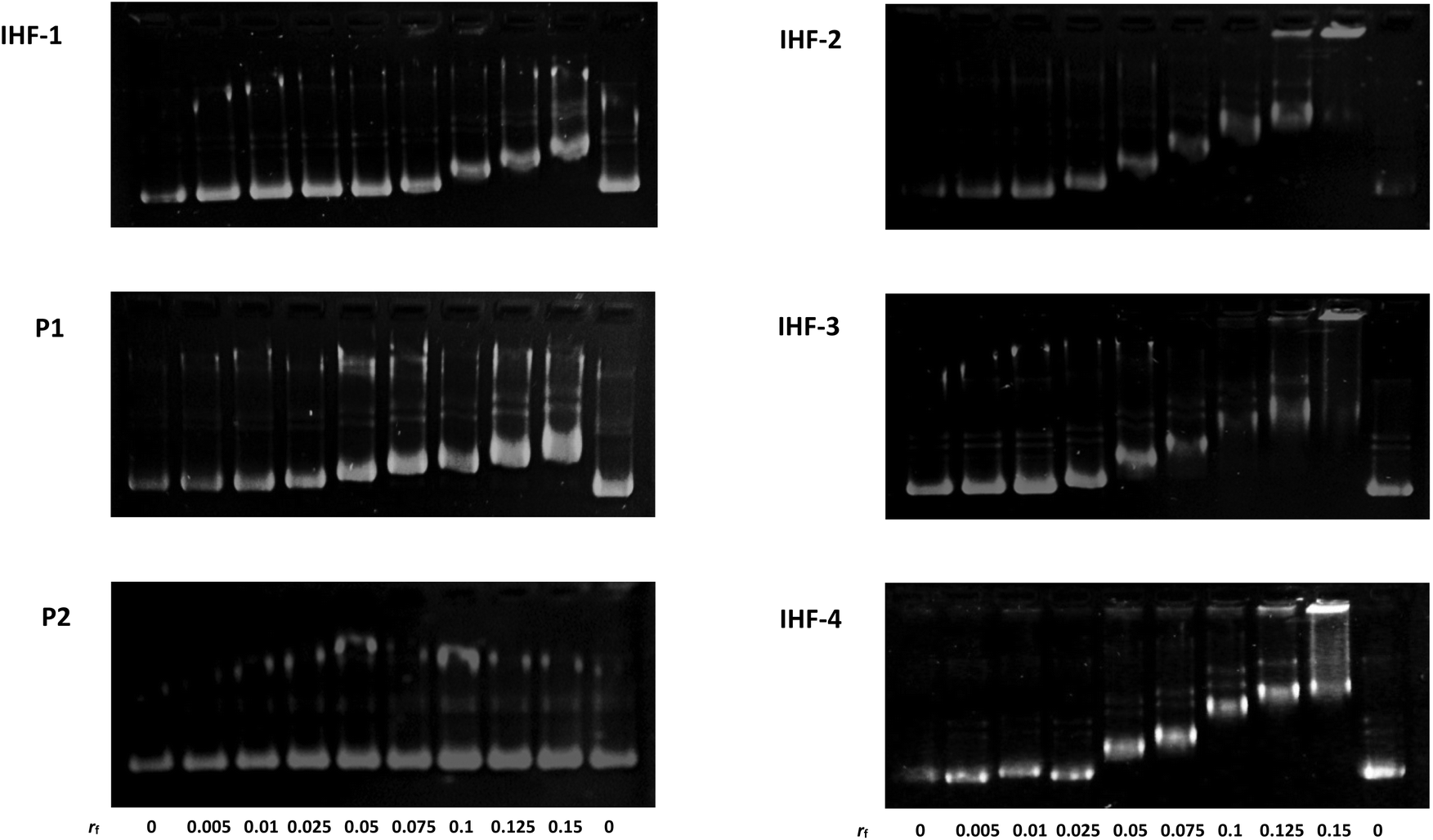 | ||
| Fig. 3 Agarose gel electrophoresis to study the interactions of the IHF mimicking peptides IHF-1/2/3/4, the platinated lysine dendrimer P1 and the cyclic peptide P2 with pUC18 plasmid DNA (incubation time for 2 h at 37 °C, with rf varying from 0 to 0.15, 10 mM Na2HPO4/NaH2PO4, pH 5.8, [pUC18] = 0.2 mM nucleobases). | ||
In this study the interaction of the IHF mimicking peptides IHF-1/2/3/4, the platinated lysine dendrimer P1, and the non-platinated cyclic peptide P2 was tested with the negatively supercoiled pUC18 plasmid. The platinated IHF mimics IHF-2/3/4 interact with DNA via both covalent as well as non-covalent interactions whereas IHF-1 can interact with DNA only through non-covalent interactions. All three platinated mimics IHF-2/3/4 led to the formation of slower migrating bands already at a lower rf value of 0.05 compared to the unplatinated IHF mimic IHF-1 which only had an effect on the plasmid DNA migration pattern at an rf around 0.1. This is a first clue that the presence of the platinum chelating unit in the IHF mimics results in enhanced DNA binding and unwinding.
The platinated dendrimeric peptide P1 induces a shift starting at an rf of 0.075, whereas the cyclic peptide P2 does not show an efficient gel mobility shift in the plasmid DNA migration pattern. The cyclic peptide P2 had no influence on the migration properties of the supercoiled plasmid due to lack of electrostatic forces for having sufficient interaction with the negatively charged DNA, unless it is connected to the positively charged body of the dendrimeric peptide. The effect is emphasized when comparing P2 with IHF-1. This is an indication that the cyclic peptide P2 does not affect plasmid unwinding and that only incorporation within the IHF mimics brings it in close proximity to the interaction center, facilitating DNA binding.
Moreover, on comparing the platinated compound P1 with its platinum-IHF analogues, IHF-2/3/4, it is obvious that the platinated IHF mimics induce a higher DNA unwinding compared to the platinated lysine dendrimer. Hence, the electrostatic interactions of the dendrimeric peptide with the plasmid DNA solely are not enough to distort its structure, but it influences the effect of the whole IHF mimic. Therefore, the connection of the two IHF mimicking peptide units and platination are necessary to have an optimum interaction, and furthermore, structural distortion of DNA.
Binding studies with the IHF consensus sequence
Native and denaturing polyacrylamide gel electrophoresis were utilized to investigate the interactions of the IHF mimicking peptides with 34-mer DNA duplexes consisting of the IHF consensus sequence. Native gels allow visualization of the binding events resulting from both covalent and non-covalent interactions whereas denaturing gels assist in specifically visualizing covalent linkages between the peptide and the DNA. The following two DNA sequences were applied in this study: (3′-ATTTTTTCGTAAC ATAGTTAAACAACGTTGCT-5′) and (3′-ATTTTTTCGTAACGAATAGTTAAACAACGTTGCT-5′). In the former sequence, adjacent guanine residues (GG site) were introduced due to their greater tendency for platination. The oligomers consisted of a fluorescent 6-carboxyfluorescein label at the 5′ end for visualization and were annealed with their complementary DNA to give duplexes D1 and D2, respectively. The peptides were incubated with the duplexes for 17 h at 37 °C in 10 mM Na2HPO4/NaH2PO4, 100 mM NaClO4, pH = 5.8 prior to electrophoresis. Since in polyacrylamide gels, larger molecules tend to have a slower migration pattern,41,47–49 the binding of the peptide to the DNA was monitored by appearance of a slower migrating band compared to the unbound DNA.
ATAGTTAAACAACGTTGCT-5′) and (3′-ATTTTTTCGTAACGAATAGTTAAACAACGTTGCT-5′). In the former sequence, adjacent guanine residues (GG site) were introduced due to their greater tendency for platination. The oligomers consisted of a fluorescent 6-carboxyfluorescein label at the 5′ end for visualization and were annealed with their complementary DNA to give duplexes D1 and D2, respectively. The peptides were incubated with the duplexes for 17 h at 37 °C in 10 mM Na2HPO4/NaH2PO4, 100 mM NaClO4, pH = 5.8 prior to electrophoresis. Since in polyacrylamide gels, larger molecules tend to have a slower migration pattern,41,47–49 the binding of the peptide to the DNA was monitored by appearance of a slower migrating band compared to the unbound DNA.
In both native and denaturing gels, the platinated IHF mimics exhibit stronger binding to duplex D1 containing a GG site (Fig. 4a and b) compared to duplex D2 without a GG site (Fig. 4c and d). On the contrary, the unplatinated peptide IHF-1 (lane 2) does not bind to either of the two sequences indicating a crucial role of the platinum unit in enhanced DNA interaction. Changing the position of the GG site within a given DNA sequence doesn't seem to influence the binding of the platinum-IHF mimics (see ESI Fig. S1†). This reveals their lack of sequence specificity for targeting a particular DNA.
 | ||
| Fig. 4 Polyacrylamide gel electrophoresis assay to study the interactions of the IHF mimicking peptides with the 34 mer DNA duplex D1, consisting of a GG site and the duplex D2, excluding the GG site. The samples were incubated for 17 h at 37 °C in 10 mM Na2HPO4/NaH2PO4, 100 mM NaClO4, pH 5.8. (a) and (b) Lanes 1–11 contain 0.5 μM DNA duplex D1. Lanes 2, 3, 4, 5, 6 contain 10 μM of IHF-1, IHF-2, IHF-3, IHF-4, P1 respectively. Lanes 7, 8, 9, 10 and 11 contain an additional 50 μM cisplatin to IHF-1, IHF-2, IHF-3, IHF-4, P1 respectively. (c) and (d) Lanes 1–7 contain 0.5 μM DNA duplex D2. Lanes 2, 3, 4, 5, 6, 7 contain 10 μM of IHF-1, IHF-2, IHF-3, IHF-4, P1, P2 respectively. (a) and (c) Performed under non-denaturing conditions (20% PAA, 1× TBE). (b) and (d) Performed under denaturing conditions (20% PAA, 7 M urea, 1× TBE). | ||
The interaction of the IHF mimics with duplex D1 containing the GG site was studied in detail both in the absence (lanes 1–6) and in the presence (lanes 7–11) of cisplatin (Fig. 4a and b). All platinated peptides induced a shift in the migration pattern of DNA (lanes 3–6 and lanes 7–11). In the absence of cisplatin (lanes 3–6), the first product band with the highest intensity corresponds to the mono-peptide–DNA adduct, whereas the other less intense bands with further gel shifts correspond to various higher order adducts. The native gel reveals (Fig. 4a), a comparable amount of peptide–DNA adduct formation with platinum-IHF mimics, IHF-2/3/4 in the presence and absence of cisplatin (lanes 3–5 & 7–10, 50% average yield). The denaturing gel indicates (Fig. 4b) a vast difference in the formation of the peptide–DNA adduct with IHF-2/3/4 in the absence of cisplatin (lanes 3–5, 40% average yield) when compared to that in the presence of five times excess of cisplatin (lanes 7–10, 15% average yield). These differences point towards the ability of platinum-IHF mimics to non-covalently interact with the cisplatin modified DNA, which can still have a synergistic effect on DNA interactions. Additionally, covalent binding of platinum-IHF mimics to the GG site containing duplex D1 even in the presence of five times excess of cisplatin clearly indicates a competition between the two for the same binding site.
The native gel indicates (Fig. 4a) the platinated dendrimeric peptide P1 led to nearly complete disappearance of the unbound DNA band due to the formation of multiple adducts, most of which were below the level of detection. Moreover, the denaturing gel displays (Fig. 4b) a much weaker covalent binding of P1 (lane 6, 20%; lane 11, 10%) both in the absence and presence of cisplatin compared to IHF-2/3/4. These results further validate the design of the IHF mimicking platinum complex/peptide chimera which include the minor groove binding cyclic peptide, a positively charged lysine dendrimer for electrostatic interactions and a platinum unit for covalent linkage with DNA.
Thermal melting analysis
The effects of the IHF mimicking peptides on the thermal stability of the 34-mer duplex D1 containing a GG-site within the IHF consensus sequence was analyzed by monitoring temperature dependent UV absorbance (Fig. 5). The observed melting profile of the DNA duplex is a net result of its interactions with the peptidic scaffold and the platinum unit. The platinum complexes distort the structure of DNA by forming covalent crosslinks and depending on the type of crosslinking they either enhance or reduce the stability of the duplex.50,51 On the other hand, binding of the positively charged DNA ligands apparently enhances the duplex stability.52 Prior to measurement the peptides were pre-incubated with the duplex for 17 h at 37 °C. Thermal denaturation and subsequent renaturation processes were studied by heating from 20 to 95 °C and cooling from 95 to 10 °C, respectively. The melting temperatures (Tm) for each cycle were obtained by calculating the maxima of the first derivative of the heating and the cooling curves (Table 1). | ||
| Fig. 5 UV melting profile of duplex D1 studied after pre-incubation with the IHF mimicking peptides IHF-1/2, P1, P2 as well as DNA alone for 17 h at 37 °C (1 μM of DNA with 5 μM of peptides) in 10 mM Na2HPO4/NaH2PO4, pH = 5.8. The melting profiles of IHF-2/3/4 were found to be similar and the profile for only IHF-2 has been used for representation. | ||
| Peptidea | Denaturation (20–95) °C | Renaturation (95–10) °C | ||
|---|---|---|---|---|
| T m (°C) | ΔTm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (°C) b (°C) |
T m (°C) | ΔTmb (°C) |
|
| a The peptides were pre-incubated with the duplex D1 for 17 h at 37 °C in 10 mM phosphate buffer, pH = 5.8 where the concentration of the DNA was 1 μM and the concentration of the peptide was 5 μM. b ΔTm represents Tm (in the presence of peptide) – Tm (control DNA) for the heating and the cooling curve, respectively. | ||||
| Control | 53.6 ± 0.5 | — | 52.7 ± 0.1 | — |
| IHF-1 | 65.6 ± 0.1 | +12.0 | 66.0 ± 0.1 | +13.3 |
| IHF-2 | 61.5 ± 1.5 | +7.9 | 50.2 ± 0.9 | −2.5 |
| IHF-3 | 62.1 ± 0.9 | +8.5 | 50.0 ± 0.1 | −2.7 |
| IHF-4 | 59.6 ± 0.6 | +6.0 | 50.6 ± 0.6 | −2.1 |
| P1 | 57.9 ± 0.3 | +4.3 | 51.1 ± 0.7 | −1.6 |
| P2 | 54.1 ± 0.1 | +0.5 | 53.9 ± 0.1 | +1.2 |
In line with the reversibility of melting curves, the denaturation and renaturation cycles were nearly superimposable for the duplex D1 (1 μM, 10 mM Na2HPO4/NaH2PO4, pH = 5.8) in the absence of peptides as well as in the presence of unplatinated peptides, IHF-1 and cyclic peptide P2. While the unplatinated IHF mimic IHF-1 led to a sharp increase in melting temperature (ΔTm ≥ 12 °C) of duplex D1, the cyclic peptide P2 had a mild stabilization effect (ΔTm = 1 °C). This indicates that while IHF-1 stabilizes the duplex by electrostatic interactions with its negatively charged phosphate backbone, the cyclic peptide P2 can only interact weakly with the duplex in the absence of the positively charged lysine dendrimer. The platinated mimics IHF-2/3/4 and the dendrimeric peptide P1, however, did not exhibit reversibility of the melting profile for the heating and cooling cycle. In the heating cycle, though less in comparison to the unplatinated mimic IHF-1, all the platinated IHF mimics (ΔTm = 6–8 °C) and the dendrimeric peptide P1 (ΔTm = 4 °C) indicate a net stabilizing effect on the duplex. At elevated temperatures single stranded DNA obtained after denaturation of the duplex is readily accessible for reaction with the platinum complex/chimera, triggering the formation of a covalent linkage between the platinated peptides and DNA. This hinders the renaturation process of the duplex and as a result, a net decrease in the melting temperature is observed.
Experimental section
General
All purchased chemical reagents were of analytical grade and were utilized as received from the supplier. All solvents were of the highest commercial grade available. All the amino acid derivatives, coupling reagents and resins utilized in solid peptide synthesis were purchased from NovaBiochem (Darmstadt, Germany), GL Biochem (Shanghai, China), ABCR (Karlsruhe, Germany), Bachem (Bubendorf, Switzerland), IRIS Biotech (Marktredwitz, Germany), Merck (Darmstadt, Germany) and VWR (Darmstadt, Germany). All other chemical reagents were obtained from Carl Roth GmbH (Karlsruhe, Germany), Fisher Scientific GmbH (Nidderau, Germany), Alfa Aesar (Karlsruhe, Germany), Bachem (Bubendorf, Switzerland), TCI (Eschborn, Germany), NovaBiochem (Darmstadt, Germany), Sigma Aldrich (Taufkirchen, Germany) and Acros Organic (Geel, Belgium). DNA oligomers used for binding experiments were supplied by Biomers (Ulm, Germany). Ultrapure water was obtained utilizing the water purification system “Simplicity” (Millipore, Bedford, UK). The artificial amino acid residues N-tert-butoxycarbonyl-δ-bromo-norvaline benzyl ester and Nα-Boc-Nε,Nε-dimethyl-L-lysine were prepared by following literature known procedures.43,53 Glassware utilized for performing reactions under an argon atmosphere were flame dried prior to usage. Thin layer chromatography (TLC) was performed using Merck aluminium backed plates coated with silica gel F254. Purification by flash chromatography was performed using Merck silica gel 60. The compounds were visualized under UV light or by staining with ninhydrin solution followed by charring.1H and 13C NMR spectra were recorded on a Varian Unity 300 or Varian Inova 500 spectrometer. The chemical shifts (δ) reported in parts per million (ppm) denote a downfield shift relative to trimethylsilane (TMS), taken as the internal standard. Signal multiplicities were abbreviated as: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; b, broad. The coupling constants (J) were reported in hertz (Hz). ESI mass spectra were measured on a Finnigan instrument (type LCQ or TSQ 7000) and high-resolution mass spectra were measured on Bruker spectrometers (types Apex-Q IV 7 T, microTOF API). Fmoc solid phase peptide synthesis was performed manually utilizing standard protocol and coupling reagents 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b] pyridinium 3-oxid hexafluorophosphate (HATU) and 1-hydroxy azabenzotriazole (HOAt). HPLC was carried out on Amersham Pharmacia Biotech systems (Äkta basic, Pump type P-900, variable wavelength detector, GE Healthcare, London, UK). A linear gradient of A (0.1% TFA in H2O) to B (0.1% TFA in MeCN/H2O, 8/2, (v/v)) or to B′ (0.1% TFA in MeCN/H2O, 9/1, (v/v)) was applied and the UV absorption was monitored at 215 nm. Peptides were analyzed and purified utilizing a Macherey-Nagel Nucleodur 100 column, RP-C18, 250 × 21.0 mm, 5 μm, with a flow rate of 10 mL min−1 or a Macherey-Nagel Nucleodur 100 column, RP-C18, 250 × 21.0 mm, 5 μm, with a flow rate of 3 mL min−1. The concentration of the oligonucleotides was determined by utilizing the extinction co-efficient at 260 nm and measuring absorbance using a Nanodrop ND-2000c spectrometer.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.28–5.12 (m, 2-H, CHCH2), 4.55–4.50 (m, 2-H, –OCH2), 3.35 (t, 3JH–H = 6.6 Hz, 2-H, N–CH2), 2.90 (s, 3-H, N–CH3), 2.70 (t, 3JH–H = 6.6 Hz, 2-H, NH–CH2), 2.39 (s, 3-H, NH–CH3), 1.36 (sbr, 1-H, NH). 13C-NMR (125 MHz, CDCl3): δ (ppm) = 156.1 (CO), 133.0 (–CHCH2), 117.1 (–CHCH2), 66.0 (O–CH2), 49.7 (NHCH2), 48.6 (NCH2), 36.4 (NHCH3), 35.2 (NCH3). MS (ESI): m/z = 173.1 [M + H]+, 195.1 [M + Na]+, 345.2 [2M + H]+, 367.2 [2M + Na]+. HRMS (ESI) = calculated for [C8H17N2O2]+ ([M + H]+) = 173.1285, found = 173.1286.
CH2), 5.30–5.05 (m, 4-H, CHCH2, CH2–Ph), 4.49 (dt, 2–H, 4JH–H = 1.5 Hz, 3JH–H = 5.4 Hz, CH2-Alloc), 4.10–3.97 (m, 1-H, α-CH), 3.27 (t, 3JH–H = 6.9 Hz, 2-H, CO–NCH2), 2.83 (s, 3-H, CO–NCH3), 2.39 (t, 3JH–H = 6.75 Hz, 2-H, N–CH2), 2.27 (t, 3JH–H = 6.9 Hz, 2-H, δ-CH2), 2.12 (s, 3-H, NCH3), 1.72–1.55 (m, 2-H, β-CH2), 1.46–1.40 (m, 2-H, γ-CH2), 1.38 (s, 8-H, tBoc-CH3), 1.29 (s, 1-H, tBoc-CH3, rotamer). 13C-NMR (125 MHz, DMSO-d6): δ (ppm) = 172.4 (CO-OBn), 155.5 (CO-Boc), 155.1 (CO-Alloc), 136.0 (Phipso), 133.5 (–CH2–CHCH2), 128.3, 127.9, 127.7 (Ph), 116.6 (–CH2–CHCH2), 78.1 [C(CH3)3], 65.6 (CH2–Ph), 64.9 (–CH2–CHCH2), 56.5 (δ-CH2), 54.4 (NCH2), 53.5 (α-CH), 46.1 (CO–NH2), 41.6 (NCH3), 33.9 (CO–NCH3), 28.4 (β-CH2), 28.1 [C(CH3)3], 23.0 (γ-CH2). MS (ESI): m/z = 478.3 [M + H]+, 476.3 [M − H]−. HRMS (ESI) = calculated for [C25H40N3O6]+ ([M + H]+) = 478.2912, found = 478.2917.
CH2), 5.17–5.13 (m, 1-H, CH2), 4.50–4.48 (m, 2-H, CH2-Alloc), 4.31–4.24 (m, 2-H, CH2-Fmoc), 4.23–4.19 (m, 1-H, CH-Fmoc), 3.92–3.88 (m, 1-H, α-CH), 3.29 (t, 3JH–H = 6.6 Hz, 2-H, CONCH2), 2.83 (sbr, 3-H, CONCH3), 2.44 (t, 3JH–H = 6.6 Hz, 2-H, NCH2), 2.33 (sbr, 2-H, δ–CH2), 2.16 (s, 3-H, NCH3), 1.74–1.68 (m, 1-H, β-CH2), 1.62–1.56 (m, 1-H, β-CH2), 1.49–1.41 (m, 2-H, γ-CH2). 13C-NMR (125 MHz, DMSO-d6): δ (ppm) = 174.2 (COOH), 155.8 (CO-Fmoc), 155.1 (CO-Alloc), 143.7 (C6-Fmoc), 140.7 (C5-Fmoc), 133.52 (CHAlloc), 127.5 (C3-Fmoc), 127.0 (C2-Fmoc), 125.2 (C1-Fmoc), 120.0 (C4-Fmoc), 116.7 (CH2-Alloc), 65.5 (CH2-Alloc), 65.0 (CH2-Fmoc), 56.7 (δ-CH2), 54.7 (NCH2), 54.1 (α-CH), 46.7 (CH-Fmoc), 45.8 (CONCH2), 41.5 (NCH3), 33.9 (CONCH3), 29.1 (β-CH2), 23.0 (γ-CH2). MS (ESI): m/z = 510.3 [M + H]+, 532.3 [M + Na]+, 548.2 [M + K]+, 1019.5 [2M + H]+. HRMS (ESI) = calculated for [C28H36N3O6]+ ([M + H]+) = 510.2599, found = 510.2601.
:1 ratio with its complementary strand 5′-TAAAAAAGCATTGCCTATCAATTTGTTGCAACGA-3′ in 10 mM phosphate buffer (Na2HPO4/NaH2PO4, pH = 5.8). The two strands were annealed by heating at 95 °C for 5 min followed by slow cooling to room temperature for 30 min to form duplex D1. Thereafter, the peptides were mixed with the duplex D1 such that the final concentration of the peptide was 5 μM and that of the duplex was 1 μM in 10 mM phosphate buffer (Na2HPO4/NaH2PO4, pH = 5.8). The peptides were pre-incubated for 17 h at 37 °C with the duplex D1 prior to measurement. All experiments were performed in at least as triplicates on independent occasions in 1 cm quartz cuvettes. A Cary 100 UV/Vis spectrophotometer (Varian) equipped with a multiple cell holder and a Peltier temperature controller was utilized to measure the absorbance versus temperature profiles at 250 nm. The data points were taken after every 0.5 °C at a heating/cooling rate of 0.7 °C min−1 during the cycles. The Tm values are given as the calculated maximum of the first derivative of the curves.
CH2), 5.28–5.12 (m, 2-H, CHCH2), 4.55–4.50 (m, 2-H, –OCH2), 3.35 (t, 3JH–H = 6.6 Hz, 2-H, N–CH2), 2.90 (s, 3-H, N–CH3), 2.70 (t, 3JH–H = 6.6 Hz, 2-H, NH–CH2), 2.39 (s, 3-H, NH–CH3), 1.36 (sbr, 1-H, NH). 13C-NMR (125 MHz, CDCl3): δ (ppm) = 156.1 (CO), 133.0 (–CHCH2), 117.1 (–CHCH2), 66.0 (O–CH2), 49.7 (NHCH2), 48.6 (NCH2), 36.4 (NHCH3), 35.2 (NCH3). MS (ESI): m/z = 173.1 [M + H]+, 195.1 [M + Na]+, 345.2 [2M + H]+, 367.2 [2M + Na]+. HRMS (ESI) = calculated for [C8H17N2O2]+ ([M + H]+) = 173.1285, found = 173.1286.
CH2), 5.30–5.05 (m, 4-H, CHCH2, CH2–Ph), 4.49 (dt, 2–H, 4JH–H = 1.5 Hz, 3JH–H = 5.4 Hz, CH2-Alloc), 4.10–3.97 (m, 1-H, α-CH), 3.27 (t, 3JH–H = 6.9 Hz, 2-H, CO–NCH2), 2.83 (s, 3-H, CO–NCH3), 2.39 (t, 3JH–H = 6.75 Hz, 2-H, N–CH2), 2.27 (t, 3JH–H = 6.9 Hz, 2-H, δ-CH2), 2.12 (s, 3-H, NCH3), 1.72–1.55 (m, 2-H, β-CH2), 1.46–1.40 (m, 2-H, γ-CH2), 1.38 (s, 8-H, tBoc-CH3), 1.29 (s, 1-H, tBoc-CH3, rotamer). 13C-NMR (125 MHz, DMSO-d6): δ (ppm) = 172.4 (CO-OBn), 155.5 (CO-Boc), 155.1 (CO-Alloc), 136.0 (Phipso), 133.5 (–CH2–CHCH2), 128.3, 127.9, 127.7 (Ph), 116.6 (–CH2–CHCH2), 78.1 [C(CH3)3], 65.6 (CH2–Ph), 64.9 (–CH2–CHCH2), 56.5 (δ-CH2), 54.4 (NCH2), 53.5 (α-CH), 46.1 (CO–NH2), 41.6 (NCH3), 33.9 (CO–NCH3), 28.4 (β-CH2), 28.1 [C(CH3)3], 23.0 (γ-CH2). MS (ESI): m/z = 478.3 [M + H]+, 476.3 [M − H]−. HRMS (ESI) = calculated for [C25H40N3O6]+ ([M + H]+) = 478.2912, found = 478.2917.
CH2), 5.17–5.13 (m, 1-H, CH2), 4.50–4.48 (m, 2-H, CH2-Alloc), 4.31–4.24 (m, 2-H, CH2-Fmoc), 4.23–4.19 (m, 1-H, CH-Fmoc), 3.92–3.88 (m, 1-H, α-CH), 3.29 (t, 3JH–H = 6.6 Hz, 2-H, CONCH2), 2.83 (sbr, 3-H, CONCH3), 2.44 (t, 3JH–H = 6.6 Hz, 2-H, NCH2), 2.33 (sbr, 2-H, δ–CH2), 2.16 (s, 3-H, NCH3), 1.74–1.68 (m, 1-H, β-CH2), 1.62–1.56 (m, 1-H, β-CH2), 1.49–1.41 (m, 2-H, γ-CH2). 13C-NMR (125 MHz, DMSO-d6): δ (ppm) = 174.2 (COOH), 155.8 (CO-Fmoc), 155.1 (CO-Alloc), 143.7 (C6-Fmoc), 140.7 (C5-Fmoc), 133.52 (CHAlloc), 127.5 (C3-Fmoc), 127.0 (C2-Fmoc), 125.2 (C1-Fmoc), 120.0 (C4-Fmoc), 116.7 (CH2-Alloc), 65.5 (CH2-Alloc), 65.0 (CH2-Fmoc), 56.7 (δ-CH2), 54.7 (NCH2), 54.1 (α-CH), 46.7 (CH-Fmoc), 45.8 (CONCH2), 41.5 (NCH3), 33.9 (CONCH3), 29.1 (β-CH2), 23.0 (γ-CH2). MS (ESI): m/z = 510.3 [M + H]+, 532.3 [M + Na]+, 548.2 [M + K]+, 1019.5 [2M + H]+. HRMS (ESI) = calculated for [C28H36N3O6]+ ([M + H]+) = 510.2599, found = 510.2601.
:1 ratio with its complementary strand 5′-TAAAAAAGCATTGCCTATCAATTTGTTGCAACGA-3′ in 10 mM phosphate buffer (Na2HPO4/NaH2PO4, pH = 5.8). The two strands were annealed by heating at 95 °C for 5 min followed by slow cooling to room temperature for 30 min to form duplex D1. Thereafter, the peptides were mixed with the duplex D1 such that the final concentration of the peptide was 5 μM and that of the duplex was 1 μM in 10 mM phosphate buffer (Na2HPO4/NaH2PO4, pH = 5.8). The peptides were pre-incubated for 17 h at 37 °C with the duplex D1 prior to measurement. All experiments were performed in at least as triplicates on independent occasions in 1 cm quartz cuvettes. A Cary 100 UV/Vis spectrophotometer (Varian) equipped with a multiple cell holder and a Peltier temperature controller was utilized to measure the absorbance versus temperature profiles at 250 nm. The data points were taken after every 0.5 °C at a heating/cooling rate of 0.7 °C min−1 during the cycles. The Tm values are given as the calculated maximum of the first derivative of the curves.
Conclusions
In this work, we have presented a combinatorial chemistry approach to design peptide based platinum metal complexes mimicking the sequence specific DNA binding protein, IHF. The synthesis of platinum complex/peptide chimera was achieved by tethering a platinum chelating unit to a model peptide mimicking IHF. Gel mobility shift assays indicate the importance of synergistic interactions resulting from the minor groove binding cyclic peptide, the positively charged lysine dendrimer and the platinum unit, combined together for efficient DNA targeting. However, establishing elements of sequence specific recognition is desirable and can be achieved by further optimizing the design of biomimetic model peptides. Utilizing such DNA targeting peptide designs offers the possibility to develop a novel library of platinum based hybrid chemotherapeutic agents possessing enhanced DNA interaction properties.Acknowledgements
Generous support from Deutsche Forschungsgemeinschaft (DFG, IRTG 1422) is gratefully acknowledged. Furthermore, we are also grateful for financial support from the Swedish Cancer Foundation, Swedish Research Council and FLÄK (Research School in Pharmaceutical Science, Lund University).References
- S. E. Sherman, D. Gibson, A. H.-J. Wang and S. J. Lippard, Science, 1985, 230, 412 CAS.
- A. Eastman, Biochemistry, 1986, 13, 3912 CrossRef.
- J. Reedjik, Pure Appl. Chem., 1987, 59, 181 Search PubMed.
- P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573 CrossRef CAS PubMed.
- M. H. Werner, J. R. Huth, A. M. Gronenborn and G. M. Clore, Cell, 1995, 81, 705 CrossRef CAS.
- C. S. Chow, C. M. Barnes and S. J. Lippard, Biochemistry, 1995, 34, 2956 CrossRef CAS.
- U. M. Ohndorf, M. A. Rould, Q. He, C. O. Pabo and S. J. Lippard, Nature, 1999, 399, 708 CrossRef CAS PubMed.
- D. E. Fisher, Cell, 1994, 78, 539 CrossRef CAS.
- J. C. Huang, D. B. Zamble, J. T. Reardon, S. J. Lippard and A. Sancar, Proc. Natl, Acad. Sci. U. S. A., 1994, 91, 10394 CrossRef CAS.
- J. Zlatanova, J. Yaneva and S. H. Leuba, FASEB J., 1998, 12, 791 CAS.
- M. Kartalou and J. M. Essigmann, Mutat. Res., 2001, 478, 1 CrossRef CAS.
- A. Mandic, J. Hansson, S. Lider and M. C. Shoshan, J. Biol. Chem., 2003, 278, 9100 CrossRef CAS PubMed.
- F. Yu, J. Megyesi and P. M. Price, Am. J. Physiol., 2008, 295, F44 CrossRef CAS PubMed.
- T. Gianferrara, I. Bratsos and E. Alessio, Dalton Trans., 2009, 7588 RSC.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364 CrossRef CAS PubMed.
- S. L. Bruhn, J. H. Toney and S. J. Lippard, Prog. Inorg. Chem., 1990, 38, 477 CrossRef CAS PubMed.
- K. Wang, J. F. Lu and R. C. Li, Coord. Chem. Rev., 1996, 151, 53 CrossRef CAS.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467 CrossRef CAS PubMed.
- P. Jordan and M. Carmo-Fonseca, Cell. Mol. Life Sci., 2000, 57, 1229 CrossRef CAS.
- S. R. McWhinney, R. M. Goldberg and H. L. McLeod, Mol. Cancer Ther., 2009, 8, 10 CrossRef CAS PubMed.
- I. Arany and R. L. Safirstein, Semin. Nephrol., 2003, 23, 460 CrossRef CAS.
- J. T. Hartmann, L. M. Fels, S. Knop, H. Stolt, L. Kanz and C. Bokemeyer, Invest. New Drugs, 2000, 18, 281 CrossRef CAS.
- J. T. Hartmann and H. P. Lipp, Expert Opin. Pharmacother., 2003, 4, 889 CrossRef CAS PubMed.
- G. Giaccone, Drugs, 2000, 59, 9 CrossRef CAS.
- M. A. Fuertes, C. Alonso and J. M. Perez, Chem. Rev., 2003, 103, 645 CrossRef CAS PubMed.
- R. Hamzavi, T. Happ, K. Weitershaus and N. Metzler-Nolte, J. Organomet. Chem., 2004, 689, 4745 CrossRef CAS PubMed.
- F. Noor, A. Wüstholz, R. Kinscherf and N. Metzler-Nolte, Angew. Chem., Int. Ed., 2005, 45, 2429 CrossRef PubMed.
- G. Dirscherl and B. König, Eur. J. Org. Chem., 2008, 597 CrossRef CAS PubMed.
- A. C. Komor and J. K. Barton, Chem. Commun., 2013, 45, 3617 RSC.
- C. Borghouts, C. Kunz and B. Groner, J. Pept. Sci., 2005, 11, 713 CrossRef CAS PubMed.
- L. Crespo, G. Sanclimens, M. Pons, E. Giralt, M. Royo and F. Albericio, Chem. Rev., 2005, 105, 1663 CrossRef CAS PubMed.
- H. Cai, M.-S. Chen, Z.-Y. Sun, Y.-F. Zhao, H. Kunz and Y.-M. Li, Angew. Chem., Int. Ed., 2013, 52, 6106 CrossRef CAS PubMed.
- M. S. Damian, H. K. Hedman, S. K. C. Elmroth and U. Diederichsen, Eur. J. Org. Chem., 2010, 6161 CrossRef CAS PubMed.
- S. Van Zutphen and J. Reedjik, Coord. Chem. Rev., 2005, 249, 2845 CrossRef CAS PubMed.
- N. Goosen and P. Van de Putte, Mol. Microbiol., 1995, 16, 1 CrossRef CAS PubMed.
- N. Goosen and P. Van de Putte, Mol. Microbiol., 1996, 6, 2557 Search PubMed.
- D. I. Friedman, Cell, 1988, 55, 545 CrossRef CAS.
- H. Nash, in Regulation of Gene Expression in Escherichia Coli, eds. E. C. C. Lin and A. S. Lynch, R. G. Landes Company, Austin, Texas, 1996, p. 149 Search PubMed.
- P. A. Rice, S.-W. Yang, K. Mizuuchi and H. A. Nash, Cell, 1996, 87, 1295 CrossRef CAS.
- E. K. Liebler and U. Diederichsen, Org. Lett., 2004, 6, 2893 CrossRef CAS PubMed.
- S. Scholz, E. K. Liebler, B. Eickmann, H.-J. Fritz and U. Diederichsen, Amino Acids, 2012, 43, 289 CrossRef CAS PubMed.
- U. Diederichsen, D. Weicherding and N. Diezemann, Org. Biomol. Chem., 2005, 3, 1058 CAS.
- D. Fernández-Forner, G. Casala, E. Navarro, H. Ryder and F. Albericio, Tetrahedron Lett., 2001, 42, 4471 CrossRef.
- S. F. Bellon, J. H. Coleman and S. J. Lippard, Biochemistry, 1991, 30, 8026 CrossRef CAS.
- G. L. Cohen, W. R. Bauer, J. K. Barton and S. J. Lippard, Science, 1979, 203, 1014 CAS.
- L. S. Lerman and H. L. Frisch, Biopolymers, 1982, 21, 995 CrossRef CAS PubMed.
- M. Leng, Biophys. Chem., 1990, 35, 155 CrossRef CAS.
- S. F. Bellon and S. J. Lippard, Biophys. Chem., 1990, 35, 179 CrossRef CAS.
- C. Hofr and V. Brabec, Biopolymers, 2005, 77, 222 CrossRef CAS PubMed.
- N. Poklar, D. S. Pitch, S. J. Lippard, E. A. Redding, S. U. Dunham and K. J. Breslauer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 7606 CrossRef CAS.
- M. C. Olmsted, J. P. Bond, C. F. Anderson and M. T. Record, Biophys. J., 1995, 68, 634 CrossRef CAS.
- R. M. Hughes, M. L. Benshoff and M. L. Waters, Chem. – Eur. J., 2007, 13, 5753 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob01885d |
| This journal is © The Royal Society of Chemistry 2015 |