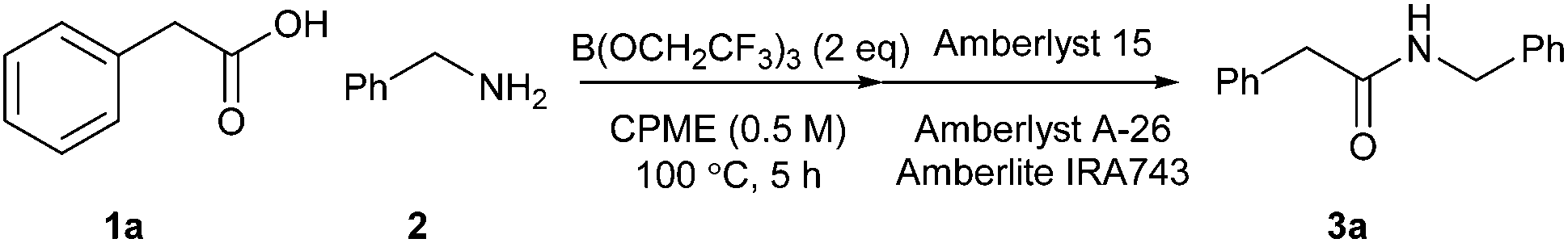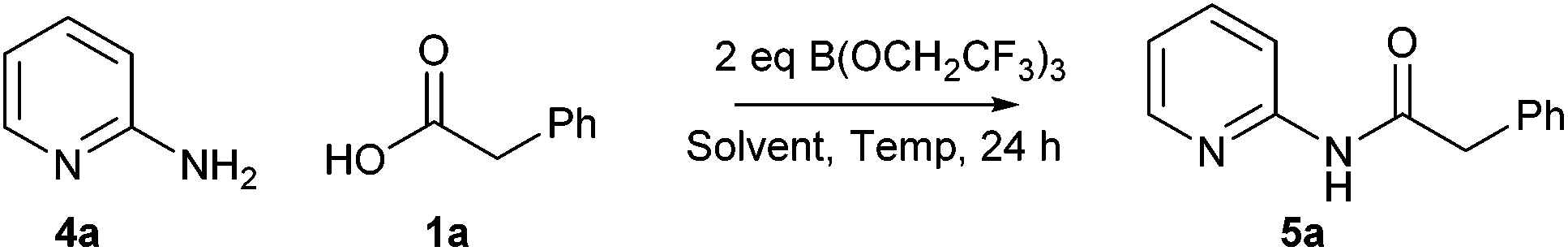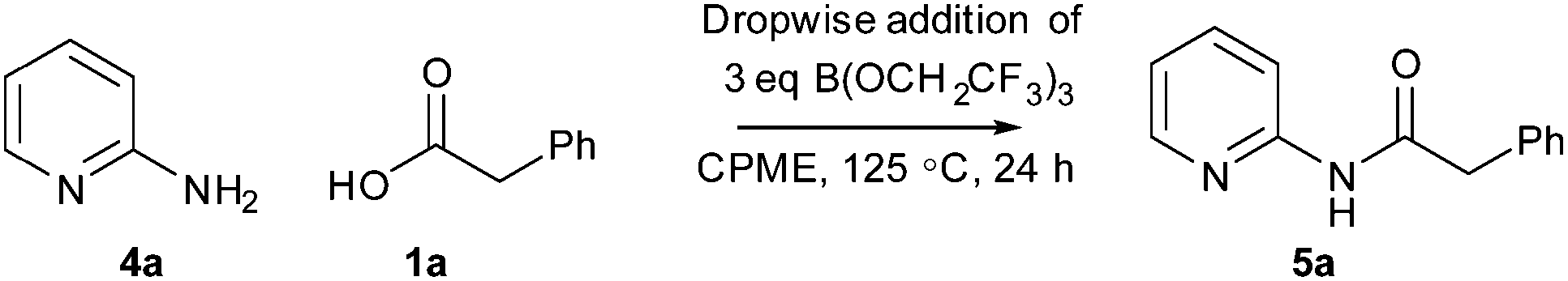DOI:
10.1039/C5OB01801C
(Paper)
Org. Biomol. Chem., 2015,
13, 10888-10894
B(OCH2CF3)3-mediated direct amidation of pharmaceutically relevant building blocks in cyclopentyl methyl ether†
Received
28th August 2015
, Accepted 8th September 2015
First published on 14th September 2015
Abstract
The use of B(OCH2CF3)3 for mediating direct amidation reactions of a wide range of pharmaceutically relevant carboxylic acids and amines is described, including numerous heterocycle-containing examples. An initial screen of solvents for the direct amidation reaction suggested that cyclopentyl methyl ether, a solvent with a very good safety profile suitable for use over a wide temperature range, was an excellent replacement for the previously used solvent acetonitrile. Under these conditions amides could be prepared from 18 of the 21 carboxylic acids and 18 of the 21 amines examined. Further optimisation of one of the low yielding amidation reactions (36% yield) via a design of experiments approach enabled an 84% yield of the amide to be obtained.
Introduction
Amides are highly important in pharmaceuticals and it is well documented that amidation is one of the most commonly used processes in the synthesis of medicinally relevant compounds.1 Amides are typically synthesised either via preparation of a highly reactive acyl derivative (acyl chloride, mixed anhydride, etc.) or via use of a coupling reagent.2 These processes are not without significant drawbacks, however, as they often involve the use of toxic reagents or solvents and lead to the generation of large quantities of waste products. As a consequence of this, there has been considerable interest in the development of alternative methods for achieving direct amidation between carboxylic acids and amines, a process which formally only requires the removal of a molecule of water.3 Notable developments have included the identification of numerous catalysts for mediating amidation reactions under dehydrating conditions (e.g. Dean–Stark water removal or molecular sieves) including systems based around boron,4 group IV metals,5 or other inorganic compounds.6 Progress has also been made on the improvement of direct thermal amidation reactions without a catalyst.7 However, these methods are largely limited to more reactive carboxylic acids and amines (e.g. simple relatively lipophilic alkyl or aryl systems), with very few successful reactions of functionalised compounds being reported. The synthesis of pharmaceutically relevant compounds inherently requires the amidation of such polar functionalised molecules,8 however, with heterocyclic compounds in particular being an essential component of many drug molecules. These highly polar heterocycle-containing acids and amines often show low reactivity in direct amidation reactions (e.g. electron-deficient aminoheterocycles are typically very poor nucleophiles), and the presence of co-ordinating heteroatoms is incompatible with many of the catalytic amidation systems reported to date. In our previous work, we have shown that borate esters9 such as B(OMe)3 and B(OCH2CF3)3 are effective amidation reagents,10,11 and that purification of the amide products can be achieved using a simple filtration work-up in many cases, without any need for chromatography or aqueous work-up.11 In this paper we report new conditions for direct amidation using a simple borate ester that are effective with a wide range of pharmaceutically relevant carboxylic acids and amines.
Results and discussion
Solvent screen
In our original report of borate-mediated amidation reactions we screened a selection of different solvents for the B(OMe)3-mediated reaction between phenylacetic acid 1a and benzylamine 2 to give amide 3a.10 From this initial screen, acetonitrile was selected as a solvent for further study and this was employed for subsequent amidation reactions with other borate esters from which B(OCH2CF3)3 emerged as the most promising reagent, especially for less reactive substrates. However, no subsequent screen of solvents using B(OCH2CF3)3 was carried out. In general, amidation reactions with less reactive substrates were observed to proceed more effectively at higher temperatures, but using acetonitrile as solvent, this led to the need to carry out reactions of particularly difficult substrates at 100 °C in a sealed tube. Even under these conditions, very unreactive substrates such as the poorly nucleophilic 2-aminopyridine gave <20% yield of the corresponding amide.11 Before challenging our amidation method with a set of highly functionalised acids and amines, we therefore elected to carry out a further solvent screen to determine whether other solvents might be more suitable for carrying out B(OCH2CF3)3-mediated amidation reactions (Scheme 1 and Table 1).12 As in our previous work, we used the reaction of phenylacetic acid and benzylamine as a benchmark to screen the different solvents. The amidation product in this case can easily be isolated rapidly via a filtration work-up, without the need for aqueous work-up or chromatography.
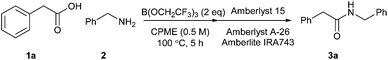 |
| | Scheme 1 Direct amidation between phenylacetic acid and benzylamine. | |
Table 1 Direct amidation between 1a and 2 in a variety of solvents
| Entry |
Solvent |
Temperature |
Yielda |
|
Isolated yields.
|
|
1
|
MeCN
|
80
|
87%
|
| 2 |
DMSO |
80 |
35% |
|
3
|
tert-Amyl methyl ether (TAME)
|
80
|
88%
|
|
4
|
Cyclopentyl methyl ether (CPME)
|
80
|
87%
|
| 5 |
4-Methyltetrahydropyran |
80 |
43% |
| 6 |
Tetrahydrofuran (THF) |
80 |
58% |
| 7 |
2-Methyltetrahydrofuran (2-MeTHF) |
80 |
56% |
| 8 |
tert-Butyl methyl ether (TBME) |
80 |
53% |
| 9 |
Ethyl acetate (EtOAc) |
80 |
50% |
Dimethylsulfoxide (entry 2), selected as it readily dissolves most polar acids/amines, was a poor solvent for the reaction. Ethereal solvents (entries 3–8) proved particularly promising, however, although it should be noted that there was considerable variation in yield within this class of solvent. tert-Amyl methyl ether (TAME, entry 3) and cyclopentyl methyl ether (CPME, entry 4)13 were both excellent solvents for the amidation reaction, whereas the use of 4-methyltetrahydropyran (entry 5) resulted in only a moderate yield of the product. Notably, in this latter solvent immediate precipitation was observed on mixing the acid/amine suggesting that salt formation is particularly favourable. This was not the case in the other solvents examined. Other ether solvents including THF, 2-MeTHF and TBME gave the amide in moderate yield, as did ethyl acetate. The latter example elegantly demonstrates the compatibility of esters with the reaction conditions. We selected CPME for further use as it has a relatively high boiling point (bp 106 °C), providing scope for increasing the reaction temperature when studying the amidation of less-reactive substrates. Preliminary studies suggested that whilst many heterocyclic acids/amines may not readily dissolve in CPME at room temperature, they typically dissolve and react at higher temperatures.
Application to pharmaceutically relevant carboxylic acids and amines
In order to challenge the scope of the B(OCH2CF3)3-mediated direct amidation reactions we studied the reaction of sets of 21 carboxylic acid and 21 amines specifically selected by researchers in the pharmaceutical industry as ‘difficult substrates’ which can often prove incompatible with newly developed synthetic methodology.14 Standard reaction conditions (2 eq. B(OCH2CF3)3, CPME, 100 °C, 5 h) were examined with all of the compounds in the first instance, with the temperature or the reaction time being increased if required for less reactive examples.
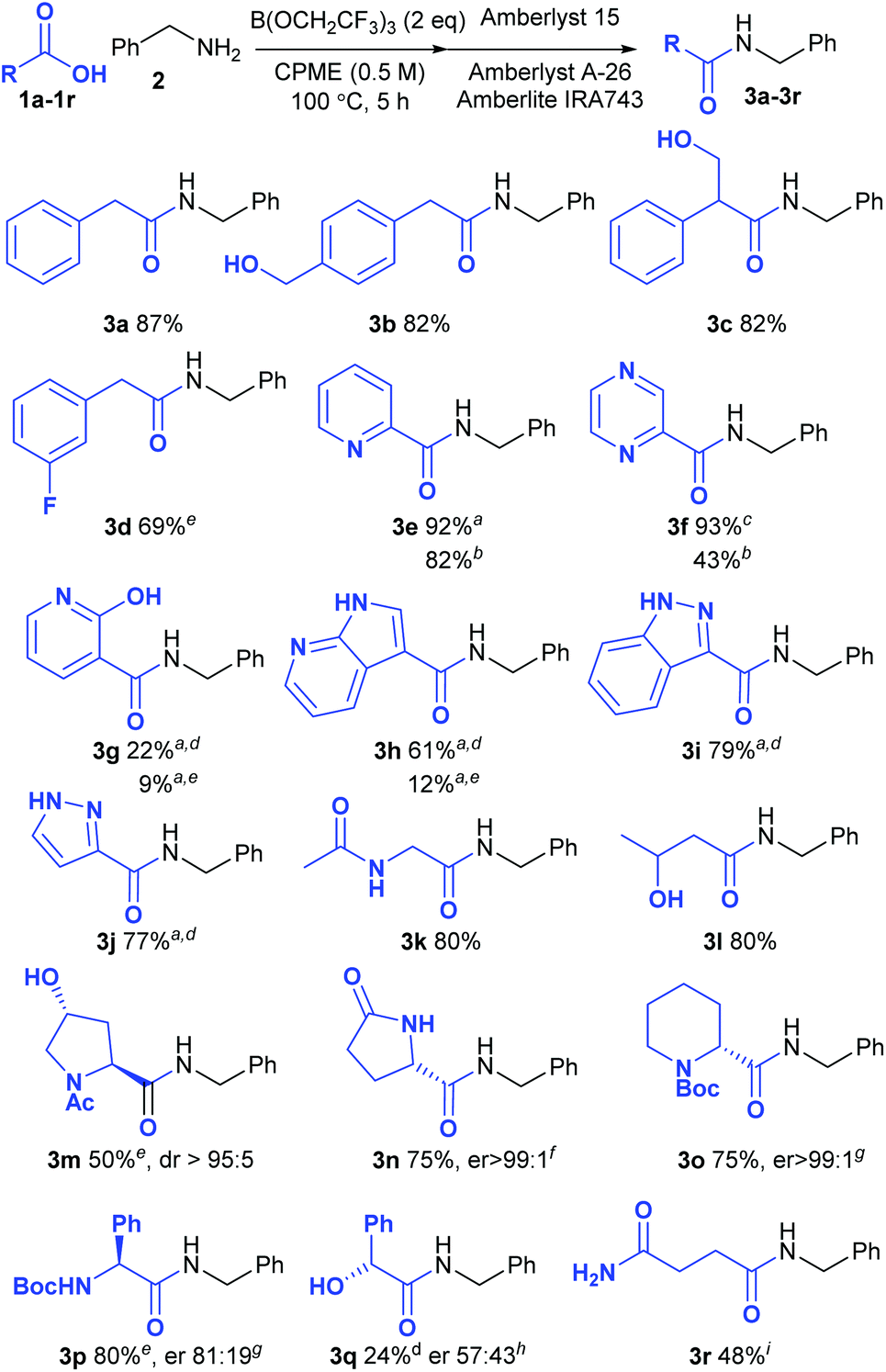
Screen of functionalised carboxylic acids
In order to provide a good comparison of the reactivity, the acids were all examined in direct amidation reactions with benzylamine as the nucleophile (Scheme 2). Pleasingly the standard procedure worked well for the vast majority of carboxylic acids tested (18/21). Unless otherwise indicated, the amide product could be isolated via a simple solid phase filtration procedure without the need for aqueous work-up or chromatography. Substrates containing free alcohols readily gave the corresponding amides in good yield (3b, 3c, 3l). 3-Fluorophenylacetic acid 1d was notably less reactive than the parent compound 1a, giving only 69% of amide 3d. Pleasingly, a wide range of heterocyclic carboxylic acids underwent amidation in excellent yield, including acids containing a pyridine (3e), a pyrazine (3f), a 7-azaindole (3h), an indazole (3i) and a pyrazole (3j), although with the less reactive examples the reaction temperature had to be increased to 125 °C. The 2-pyridone/2-hydroxypyridine substrate 1g gave only a moderate yield of the amide 3g even at this higher reaction temperature, however. With some of the amides containing basic nitrogen heterocycles, purification via the standard solid-phase work-up was not possible due to absorption of the product on the Amberlyst 15 resin. However, in some cases replacement of this strongly acidic resin with Amberlite IRA86 carboxylic acid resin enabled the products to be isolated without chromatography (3e–3f). In other cases, purification by chromatography was necessary (3g–3j). Functionalised aliphatic carboxylic acids also underwent amidation efficiently including substrates containing acetamides (3k, 3l), free hydroxyl groups (3c, 3l, 3m) and a lactam (3n). The amino-acid derivatives N-acetylhydroxyproline (3m), L-pyroglutamic acid (3n), Boc-pipecolic acid (3o) and Boc-phenylglycine (3p) were all good substrates for the amidation with only the latter compound undergoing any significant degree of racemisation under the reaction conditions. Mandelic acid (3q) gave only 24% yield of the amide, however, and underwent almost complete racemisation during the amidation process. The primary amide in succinic acid monoamide 1r was compatible with the reaction conditions, although the resulting amide 3r appeared to undergo cyclisation to the corresponding N-benzylsuccinimide during the solid-phase work-up procedure. As a consequence, this compound was purified by recrystallisation to give the amide 3r in 48% yield.
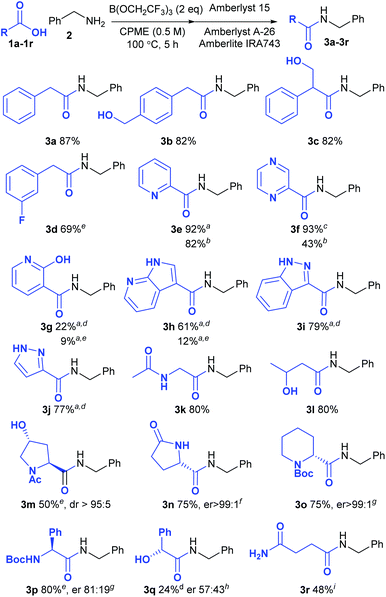 |
| | Scheme 2 Direct amidation reactions of functionalised carboxylic acids; aPurified by column chromatography; bAmberlite IRA86 (CO2H) resin used instead of Amberlyst 15; cAmberlyst 15 resin was not used; d24 h, 125 °C; e24 h; fDetermined using an organic chiral shift reagent;15gDetermined by derivatisation with Marfey's reagent;16hDetermined by HPLC using a chiral stationary phase;17iPurified by recrystallisation; no solid-phase work-up. | |
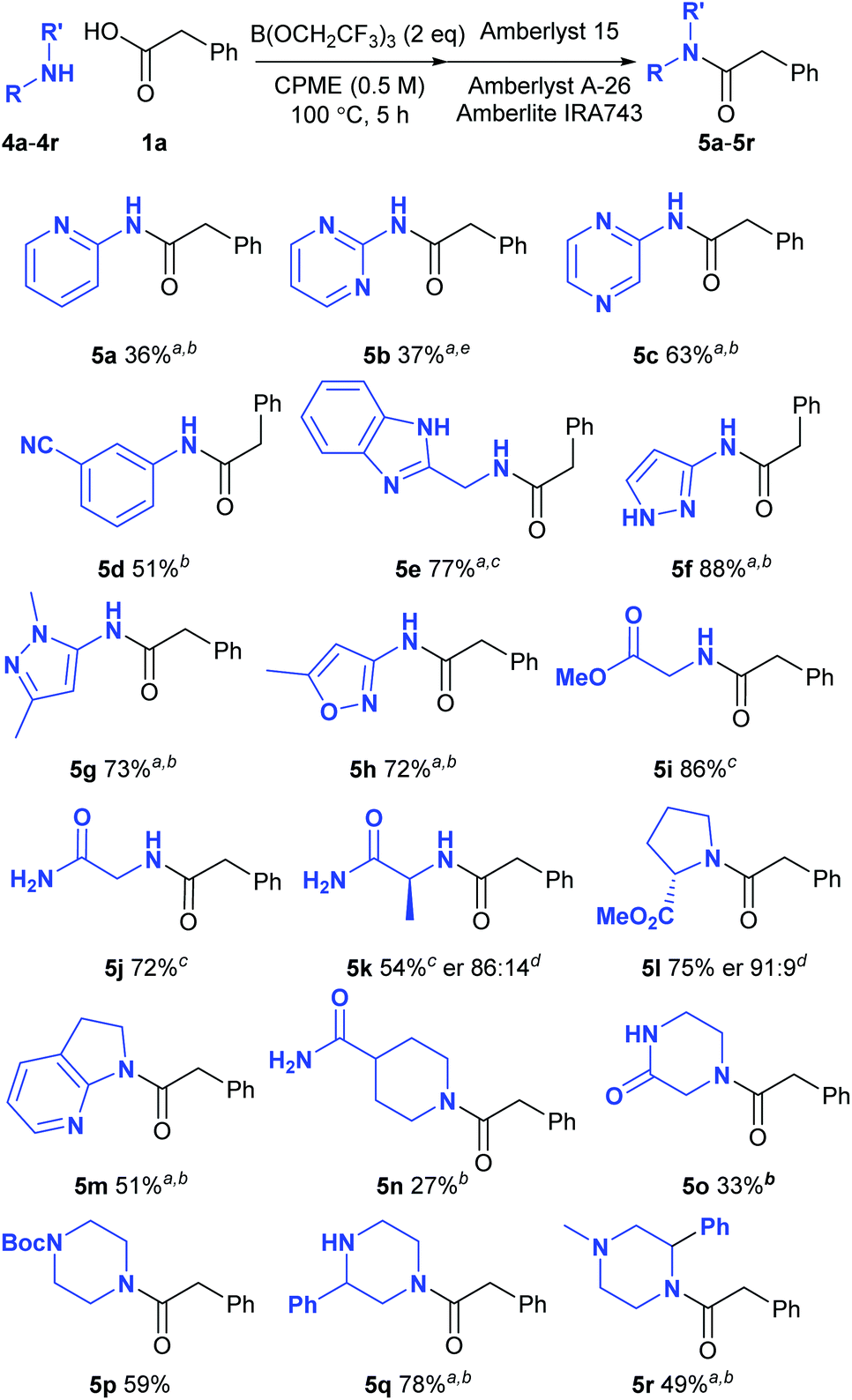
Screen of functionalised amines
We next went on to examine the amidation of a set of pharmaceutically relevant amines (Scheme 3). In a similar fashion to the carboxylic acids above, we chose to study their reaction with a standard reaction partner (phenylacetic acid 1a) in order to provide a good understanding of their comparative reactivity. Heterocyclic amines are notoriously unreactive substrates for amidation reactions as the strongly electron-withdrawing nature of many nitrogen heterocycles makes the adjacent amine an extremely poor nucleophile. Low yields of amide were obtained from 2-aminopyridine (5a) and 2-aminopyrimidine (5b), but the corresponding 2-aminopyrazine underwent amidation effectively (5c). An electron-deficient aniline bearing a cyano group also gave a reasonable yield of the corresponding amide 5d. Pleasingly, amines containing a benzimidazole (5e), a pyrazole (5f–5g) and an oxazole (5h) all gave the amide derivatives in good yield. Relatively unreactive aliphatic amines such as glycine methyl ester (5i), glycinamide (5j), alaninamide (5k) and proline methyl ester (5l) were also fairly good substrates, with some racemisation being observed with the two chiral substrates. Moderate to good yields of amides were obtained from other challenging substrates including a cyclic aminopyridine (5m), an amide-containing piperidine (5n), piperazin-2-one (5o) as well as a series of piperazine derivatives (5p–5r). 2-Phenylpiperazine 4q reacted selectively at the least hindered amine to give amide 5q, with no diamidated product being isolated from the reaction. Amines which were supplied as hydrochloride salts could also be used directly in the amidation reaction, with triethylamine being added to liberate the free amine from the salt (5e, 5i–5k). In a similar manner to the reactions of the carboxylic acids described above, amide products containing basic nitrogen atoms could not be purified via the solid-phase work-up procedure.
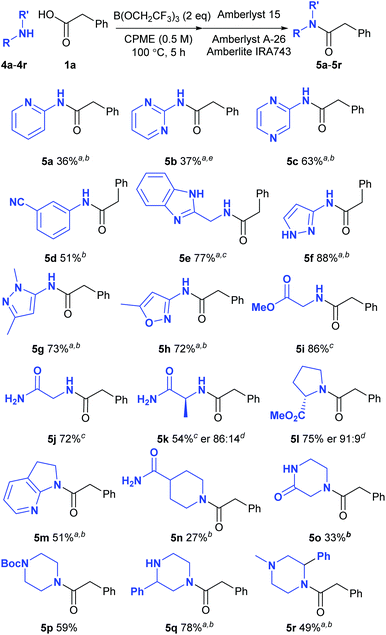 |
| | Scheme 3 Direct amidation reactions of functionalised amines; aPurified by column chromatography; b125 °C, 24 h; cAmine hydrochloride salt was used; Et3N was added to the reaction; dDetermined by HPLC using a chiral stationary phase;17e24 h. | |
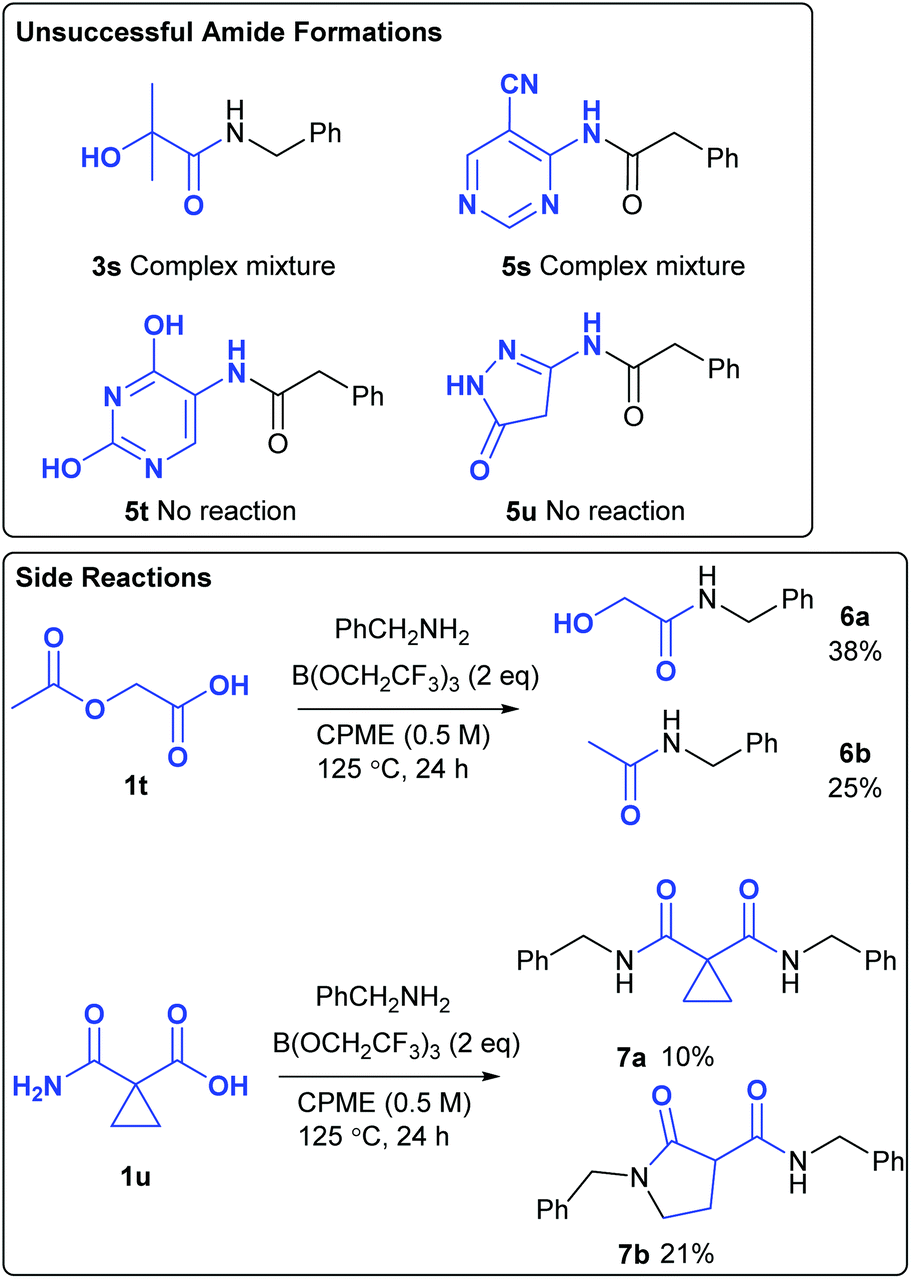
Unsuccessful amidation reactions
Amide 3s, derived from 2-hydroxyisobutyric acid could not be obtained from the amidation reaction, with a complex mixture of products being formed (Scheme 4). Attempted formation of amide 5s also gave a complex mixture of products due to numerous side-reactions. The formation of amides 5t and 5u was unsuccessful as the corresponding amines 4t–4u were insoluble in the reaction mixture. Although acetoxyacetic acid 1t did undergo amidation of the carboxylic acid group, this was accompanied by amidation of the acetoxy group to give amides 6a/6b in moderate overall yield. Given the fact that the borate-mediated amidation reaction is compatible with esters (Table 1, entry 9), this suggests that reaction of this acetate group is probably assisted by co-ordination of the adjacent amide or carboxylic acid group to the boron atom. Cyclopropyl malonate derivative 1u gave a mixture of products 7a and 7b in moderate overall yield. Thus, direct amidation of the carboxylic acid competes with transamidation of the primary amide as well as ring opening of the highly activated cyclopropane. Heating cyclopropane 7a on its own or in the presence of B(OCH2CF3)3 with/without benzylamine did not result in the formation of 7b, suggesting that these products were formed via divergent competing reaction pathways in the original amidation reaction.
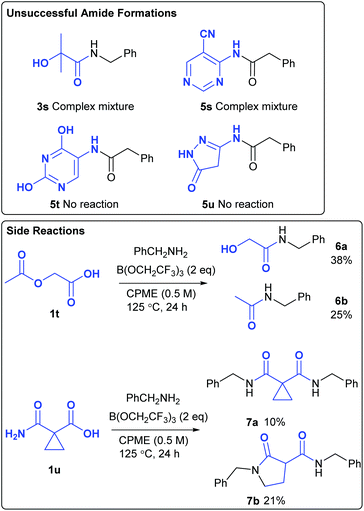 |
| | Scheme 4 Unsuccessful amidation reactions. | |
Design of Experiments optimisation of the reaction of 2-aminopyridine with phenylacetic acid
In order to further demonstrate the utility of this amidation method, we sought to take one of the less efficient amidation reactions and optimise the procedure to give good yields of the amide product. We selected the reaction between 2-aminopyridine 4a and phenylacetic acid 1a to give amide 5a as this reaction gave only a moderate yield of product (Scheme 5 and Table 2). Indeed, under our original conditions (MeCN, 100 °C), only a 12% yield of the amide was obtained (entry 1).11 As can be seen above, switching the solvent to CPME and raising the temperature led to a significant improvement, giving a 36% yield of the amide (entry 2). In an initial investigation, we explored whether dropwise addition of one or more reagents to the reaction could lead to improved yields of the product. Dropwise addition of the borate reagent to a mixture of acid/amine led to a small improvement in the yield (entry 3, 39%), whilst dropwise addition of a mixture of acid/borate to the amine gave a similar yield (entry 4, 40%). Interestingly, a significant improvement in yield (entry 5, 60%) was observed by addition of the borate reagent dropwise to a mixture of acid, amine and triethylamine suggesting that keeping the borate concentration low and the overall reaction mixture basic is advantageous.
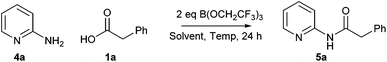 |
| | Scheme 5 Reaction of 2-aminopyridine 4a and phenylacetic acid 1a. | |
Table 2 Study of the effect of different experimental procedures on the yield of amide 5a
| Entry |
Conditions |
Yielda |
|
Isolated yields; NMR yields in parentheses.
|
| 1 |
MeCN, 100 °C |
12% |
| 2 |
CPME, 125 °C |
36% |
| 3 |
CPME, 125 °C, dropwise addition of borate |
(39%) |
| 4 |
CPME, 125 °C, dropwise addition of borate/acid |
(40%) |
| 5 |
CPME, 125 °C, 1 eq. Et3N, dropwise addition of borate |
(60%) |
With this information in hand, we then went on to carry out a ‘Design of Experiments’ optimisation of this reaction using the conditions where a solution of borate was added dropwise to a mixture of amine/acid/Et3N as our starting point. ‘Design of Experiments’ is a statistical approach to reaction optimisation which enables the variation of multiple factors simultaneously in order to evaluate the important parameters in a reaction using a relatively small number of experiments.18 For the DoE optimisation, seven different continuous variables were chosen for the study, and two solvents were evaluated: CPME and propionitrile (EtCN), the latter being chosen as a higher boiling point equivalent to MeCN. The parameters were investigated via a Resolution IV design consisting of 16 experiments plus two centre points. The factors studied and the ranges covered are shown in Table 3.
Table 3 Factors and ranges covered in the ‘Design of Experiments’ optimisation of the formation of amide 5aa
| Variable |
Low |
High |
|
For full details of the Design of Experiments study, see the ESI.
|
| (eq. amine 4a)-(eq. acid 1a) |
−2 |
+2 |
| eq. B(OCH2CF3)3 |
0.5 |
3 |
| eq. Et3N |
0.1 |
3 |
| Solvent volume in borate solution |
0.5 mL |
2 mL |
| Solvent volume in reaction vessel |
0.5 mL |
2 mL |
| Addition rate (syringe pump) |
0.254 mL h−1 |
13 mL h−1 |
| Temperature |
80 °C |
125 °C |
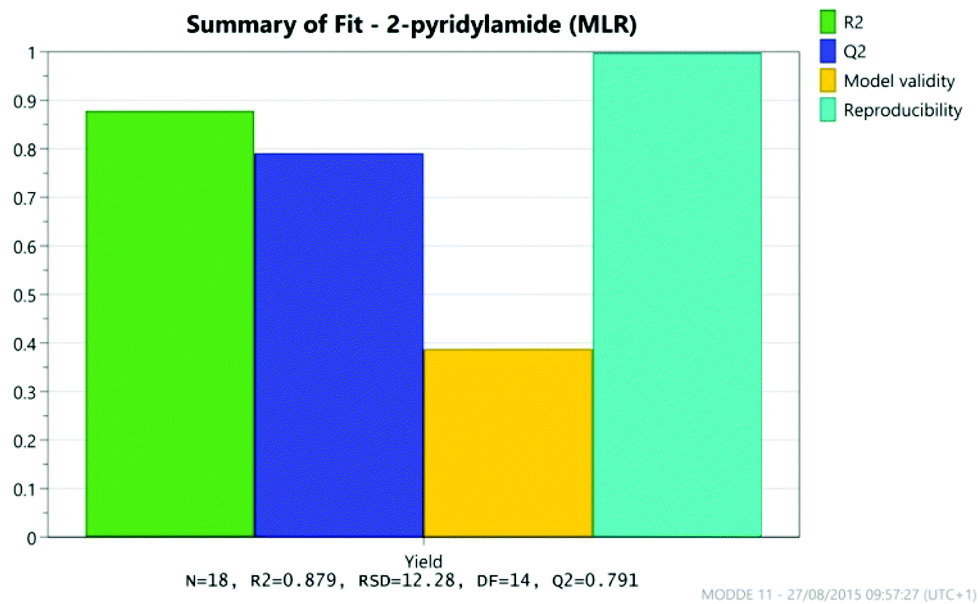
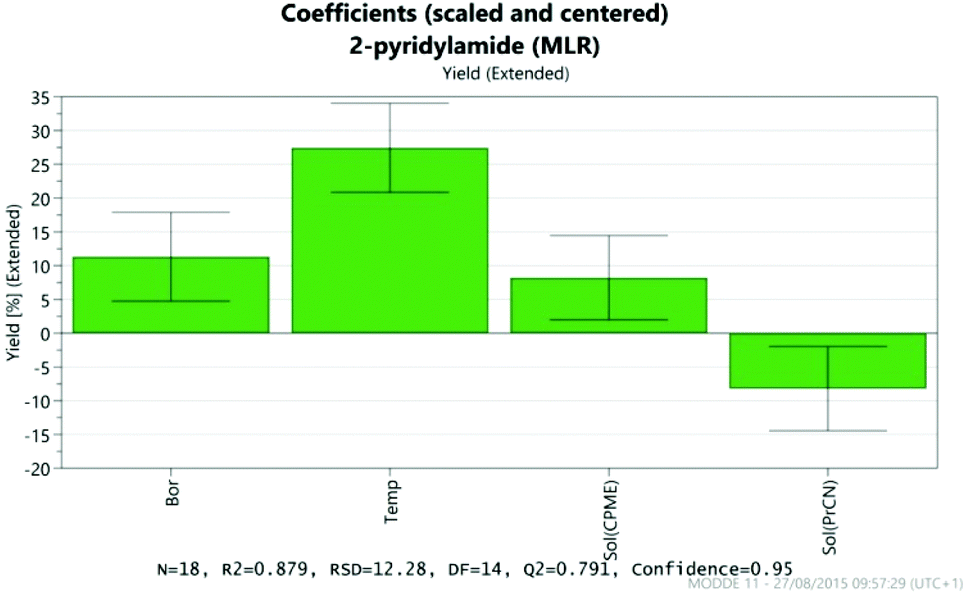
The results were analysed using MODDE 10 software,19 and a model was generated to fit the data using multiple linear regression (MLR) which provided a good fit to the data (Fig. 1). The important factors in the reaction are shown in the coefficient plot in Fig. 2. By far the most significant factor in the reaction was temperature with the higher temperature giving, on average, a ∼27% increase in the reaction yield. The addition of more borate reagent was also beneficial to the outcome, as was the use of CPME as solvent rather than EtCN. None of the other factors were found to be significant across the ranges explored and there were no significant interactions between the factors. The factor (amine-acid) explored the use of an excess (3 eq.) of either the amine or the acid relative to the other component. The design suggested that there was no particular benefit in using either an excess of the amine or the acid, though it should be noted that the highest yields in the DoE study were obtained with either an excess of acid or amine at high temperature and with high borate loading in CPME. The centre points of the DoE which employed equimolar quantities of acid and amine gave considerably lower yields. In further studies we observed that an excess of one reagent or the other was important in order to obtain a good isolated yield of the amide (vide infra, Table 4).
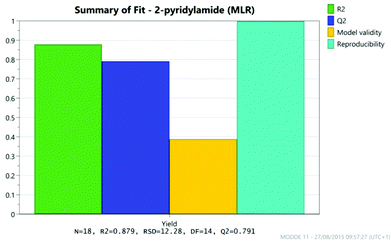 |
| | Fig. 1 Model fit for DoE. | |
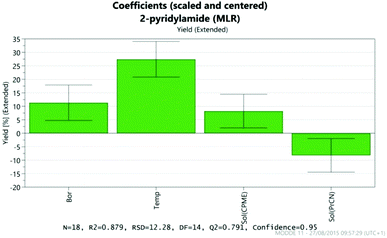 |
| | Fig. 2 Coefficient plot showing the most important factors in the amidation reaction. | |
Table 4 ‘Optimised’ methods for the formation of amide 5a
| Entry |
Conditions |
Yielda |
|
Isolated yields; 1 eq. 4a and 1 eq. 1a used unless otherwise stated.
|
| 1 |
3 eq. Et3N |
58% |
| 2 |
3 eq. 4a, 0.1 eq. Et3N |
84% |
| 3 |
3 eq. 1a; 3 eq. Et3N |
83% |
| 4 |
3 eq. Et3N, Bu2O, 140 °C |
74% |
Following on from the DoE study, we then determined the isolated yield of amide 5a obtained under various sets of ‘optimised’ conditions (Scheme 6 and Table 4). With equimolar equivalents of acid and amine, the best isolated yield obtained was 58% using three equivalents of triethylamine (entry 1). However, with an excess of either the amine (entry 2) or the acid and triethylamine (entry 3) >80% isolated yield of amide 5a was obtained. The ability to carry out the amidation reaction of a poorly reactive substrate with either an excess of the acid or the amine provides flexibility in cases where one of the coupling partners is particularly valuable. Furthermore, switching the solvent to dibutyl ether (Bu2O), which has similar properties to CPME but a higher boiling point, enabled a 74% isolated yield to be obtained using equimolar quantities of amine 4a and acid 1a at 140 °C. This correlates well with the prediction of the DoE model that reaction temperature is by far the most important factor affecting the yield of amide obtained.
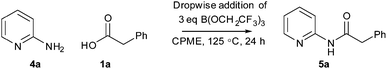 |
| | Scheme 6 Reaction of 2-aminopyridine 4a and phenylacetic acid 1a to give amide 5a under optimised conditions. | |
Conclusions
An improved procedure has been developed for direct amidation reactions with B(OCH2CF3)3 using CPME as the solvent. The method was successfully applied to a wide variety of pharmaceutically relevant carboxylic acids and amines with only 6 of the 42 substrates failing to undergo the desired amidation process. More than half of the amides could be isolated via a simple filtration work-up without the need for chromatographic purification. Furthermore, it was demonstrated that optimisation of the amidation protocol for one of the lower yielding substrates enabled the isolated yield of the amide to be increased from 36% to 84%.
Acknowledgements
We would like to thank Pfizer and the Engineering and Physical Sciences Research Council (EPSRC) for providing a CASE award to VK, and the University College London Department of Chemistry for providing a studentship to RML. We would also like to acknowledge GlaxoSmithKline for providing the carboxylic acid and amine sets, the EPSRC national mass spectrometry facility in Swansea for analytical services, and the EPSRC Dial-a-Molecule Network for supporting the Design of Experiments studies.
Notes and references
-
(a) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC;
(b) L. Amarnath, I. Andrews, R. Bandichhor, A. Bhattacharya, P. Dunn, J. Hayler, W. Hinkley, N. Holub, D. Hughes, L. Humphreys, B. Kaptein, H. Krishnen, K. Lorenz, S. Mathew, G. Nagaraju, T. Rammeloo, P. Richardson, L. Wang, A. Wells and T. White, Org. Process Res. Dev., 2012, 16, 535–544 CrossRef;
(c) K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC;
(d) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
-
(a) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC;
(b) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS PubMed.
-
(a) R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 7453–7465 CrossRef CAS PubMed;
(b) H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714–2742 RSC;
(c) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC;
(d) R. García-Álvarez, P. Crochet and V. Cadierno, Green Chem., 2013, 15, 46–66 RSC;
(e) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
-
(a) N. Gernigon, R. M. Al-Zoubi and D. G. Hall, J. Org. Chem., 2012, 77, 8386–8400 CrossRef CAS PubMed;
(b) K. Arnold, B. Davies, R. L. Giles, C. Grosjean, G. E. Smith and A. Whiting, Adv. Synth. Catal., 2006, 348, 813–820 CrossRef CAS PubMed;
(c) K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196–4197 CrossRef CAS;
(d) R. M. Al-Zoubi, O. Marion and D. G. Hall, Angew. Chem., Int. Ed., 2008, 47, 2876–2879 CrossRef CAS PubMed;
(e) K. Arnold, A. S. Batsanov, B. Davies and A. Whiting, Green Chem., 2008, 10, 124–134 RSC;
(f) T. Maki, K. Ishihara and H. Yamamoto, Tetrahedron, 2007, 63, 8645–8657 CrossRef CAS PubMed;
(g) R. Yamashita, A. Sakakura and K. Ishihara, Org. Lett., 2013, 15, 3654–3657 CrossRef CAS PubMed;
(h) P. Tang, Org. Synth., 2005, 81, 262–272 CrossRef CAS;
(i) K. Arnold, B. Davies, D. Herault and A. Whiting, Angew. Chem., Int. Ed., 2008, 47, 2673–2676 CrossRef CAS PubMed;
(j) S. Fatemi, N. Gernigon and D. G. Hall, Green Chem., 2015, 17, 4016–4028 RSC;
(k) T. M. El Dine, W. Erb, Y. Berhault, J. Rouden and J. Blanchet, J. Org. Chem., 2015, 80, 4532–4544 CrossRef PubMed;
(l) E. K. W. Tam, Rita, L. Y. Liu and A. Chen, Eur. J. Org. Chem., 2015, 1100–1107 CrossRef CAS PubMed.
-
(a) H. Lundberg, F. Tinnis and H. Adolfsson, Chem. – Eur. J., 2012, 18, 3822–3826 CrossRef CAS PubMed;
(b) H. Lundberg and H. Adolfsson, ACS Catal., 2015, 5, 3271–3277 CrossRef CAS;
(c) H. Lundberg, F. Tinnis and H. Adolfsson, Synlett, 2012, 2201–2204 CAS;
(d) C. L. Allen, A. R. Chhatwal and J. M. J. Williams, Chem. Commun., 2012, 48, 666–668 RSC;
(e) L. Y. Shteinberg, S. A. Kondratov and S. M. Shein, Zh. Org. Khim., 1988, 24, 1968–1972 CAS.
-
(a) Y. Terada, N. Ieda, K. Komura and Y. Sugi, Synthesis, 2008, 2318–2320 CAS;
(b) A. Ojeda-Porras, A. Hernández-Santana and D. Gamba-Sánchez, Green Chem., 2015, 17, 3157–3163 RSC;
(c) For amidation of esters using a simple inorganic base, see: N. Caldwell, C. Jamieson, I. Simpson and A. J. B. Watson, Chem. Commun., 2015, 51, 9495–9498 RSC.
-
(a) H. Charville, D. Jackson, G. Hodges and A. Whiting, Chem. Commun., 2010, 46, 1813–1823 RSC;
(b) C. Grosjean, J. Parker, C. Thirsk and A. R. Wright, Org. Process Res. Dev., 2012, 16, 781–787 CrossRef CAS;
(c) L. J. Gooßen, D. M. Ohlmann and P. P. Lange, Synthesis, 2009, 160–164 CrossRef;
(d) H. Charville, D. A. Jackson, G. Hodges, A. Whiting and M. R. Wilson, Eur. J. Org. Chem., 2011, 5981–5990 CrossRef CAS PubMed;
(e) See also ref. 5d.
- A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114–1122 CrossRef CAS PubMed.
-
(a) A. Pelter, T. E. Levitt and P. Nelson, Tetrahedron, 1970, 26, 1539–1544 CrossRef CAS;
(b)
L. Banfi, E. Narisano, R. Riva and D. S. Matteson, Trimethyl borate, in e-EROS Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2007 Search PubMed;
(c)
T. D. Sheppard, Tris(2,2,2-trifluoroethoxy)boron in e-EROS Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2014 Search PubMed.
- P. Starkov and T. D. Sheppard, Org. Biomol. Chem., 2011, 9, 1320–1323 CAS.
- R. M. Lanigan, P. Starkov and T. D. Sheppard, J. Org. Chem., 2013, 78, 4512–4523 CrossRef CAS PubMed.
-
(a) D. S. MacMillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. B. Watson, Green Chem., 2013, 15, 596–600 RSC;
(b) H. E. Eastman, C. Jamieson and A. J. B. Watson, Aldrichimica Acta, 2015, 48, 51–55 Search PubMed;
(c) D. Prat, A. Wells, J. Hayler, H. Sneddon, R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 45 10.1039/c5gc01008j.
- K. Watanabe, N. Yamagiwa and Y. Torisawa, Org. Process Res. Dev., 2007, 11, 251–258 CrossRef CAS.
- The compound
sets of acids and amines were selected from the range of commercially available compounds by researchers at GlaxoSmithKline, Stevenage, UK. The compounds chosen include a diverse set of functional groups that may be incompatible with reactions of the core functional group (carboxylic acid or amine).
- W. H. Pirkle, D. L. Sikkenga and M. S. Pavlin, J. Org. Chem., 1977, 42, 384–387 CrossRef CAS.
-
(a) P. Marfey, Carlsberg Res. Commun., 1984, 49, 591 CrossRef CAS;
(b) R. Bhushan and H. Brückner, Amino Acids, 2004, 27, 231 CrossRef CAS PubMed.
- For full details and HPLC traces, please see the ESI.†.
-
(a) P. M. Murray, S. N. G. Tyler and J. D. Moseley, Org. Process Res. Dev., 2013, 17, 40–46 CrossRef CAS;
(b) S. A. Weissman and N. G. Anderson, Org. Process Res. Dev., 2015 DOI:10.1021/op500169m, in press;
(c) R. Leardi, Anal. Chim. Acta, 2009, 652, 161–172 CrossRef CAS PubMed
R. Carlson and J. E. Carlson, Design and Optimization in Organic Synthesis, in Data Handling in Science and Technology, Elsevier, Amsterdam, 2nd edn, 2005, vol. 24 Search PubMed.
- The DoE optimisation was analysed using MODDE 10 software, available from Umetrics; http://www.umetrics.com/products/modde (accessed 24th August 2015).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, ‘Design of Experiments’ reaction optimisation data, and 1H and 13C NMR spectra. See DOI: 10.1039/c5ob01801c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a