
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of bicyclic tetrahydrofurans from linear precursors using manganese(III) acetate†
Anne-Caroline
Chany
,
Léo B.
Marx
and
Jonathan W.
Burton
*
Department of Chemistry, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: jonathan.burton@chem.ox.ac.uk
First published on 13th August 2015
Abstract
We have recently developed methodology based on oxidative radical reactions for the synthesis of [3.3.0]-bicyclic lactones containing both cyclopentanes and γ-lactams along with application of this methodology to the synthesis of natural products and complex molecular architectures. Herein we report an extension of this methodology to the synthesis of oxygen heterocycles including bicyclic bis-lactones.
The tetrahydrofuran (THF) moiety is present in a vast array of biologically active natural products including: the polyketides, the acetogenins, halogenated natural products from Laurencia species, and numerous terpenes.1,2 As a result the development of new methodology for the synthesis of THFs, and related 5-membered oxygen heterocycles, is an on-going and important aspect of modern synthetic organic chemistry.3 Recently we have developed methodology based on oxidative radical reactions4 for the synthesis of [3.3.0]-bicyclic lactones containing both cyclopentanes5 and γ-lactams6 along with application of this methodology to the synthesis of natural products6,7 and complex molecular architectures.8,9 Herein we report an extension of this methodology to the synthesis of 5-membered oxygen heterocycles containing bicyclic bis-lactones with up to four stereocentres. In accord with our previous work,5,6 we proposed that exposure of substrates such as 1 to a transition metal oxidant could generate the corresponding malonyl radical 2, which would undergo 5-exo-trig cyclisation presumably via the pre-transition state assembly 3 in accord with the Beckwith–Houk model of 5-exo-trig radical cyclisation, to give the adduct radical 4 (Fig. 1).10,11 Oxidation of the so formed adduct radical with concomitant C–O bond formation would deliver the dioxocarbenium ion 5 which gives the desired [3.3.0]-bicyclic γ-lactone 6 on hydrolysis. As in our study of the cyclisation of radicals derived from amidomalonates,6 we were aware that malonyl radicals such as 2 are captodative in nature12 and, as such might be prone oxidation to the corresponding oxocarbenium ions prior to cyclisation.
 | ||
| Fig. 1 Proposed mechanism of formation of [3.3.0]-bicyclic γ-lactones. | ||
The initial substrates we selected for study were the terminal and phenyl-substituted olefins 7a and 7c as we had demonstrated that the corresponding substrates, in the all carbon series, could be successfully converted into [3.3.0]-bicyclic γ-lactones under oxidative conditions.5,9 These substrates, and all of the substrates in the paper were prepared by O–H insertion into the corresponding diazo-malonates according to the procedure described by Hatakeyama and co-workers (see ESI†).3d,13
Exposure of the dimethyl malonate 7a to two equivalents of the one electron oxidant manganese(III) acetate14,15 in the presence of one equivalent of copper(II) triflate in acetonitrile at 40 °C, according to our previous work, gave the corresponding [3.3.0]-bicyclic γ-lactone 8a in 81% yield (Table 1, entry 1).‡ The corresponding di-tert-butyl malonate 7b cyclised with similar efficiency (Table 1, entry 2). Using the phenyl-substituted alkenes 7c and 7d gave the corresponding [3.3.0]-bicyclic lactones 8c and 8d in good yield and with high diastereocontrol at the lactone bearing stereocentre (major diastereomer shown, Table 1, entries 3 and 4).§,¶ In keeping with previously reported results in the all carbon series,5 cyclisation of ethyl-substituted alkene 7e gave the product 8e in reduced yield and with lower diastereoselectivity and hence we focussed our attention on substrates bearing a terminal alkene or styrene.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Yield (%) | dr |
| a Combined isolated yield of mixture of diastereomers. b Diastereomeric ratio (dr) refers to the mixture of diastereomers at the lactone stereocentre measured from the crude 1H NMR; major diastereomer shown. | |||||
| 1 | a | Me | H | 81 | — |
| 2 | b | tBu | H | 83 | — |
| 3 | c | Me | Ph | 92a | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1b :1b |
| 4 | d | tBu | Ph | 62a | 16:1b |
| 5 | e | Me | Et | 50a | 2:1b |

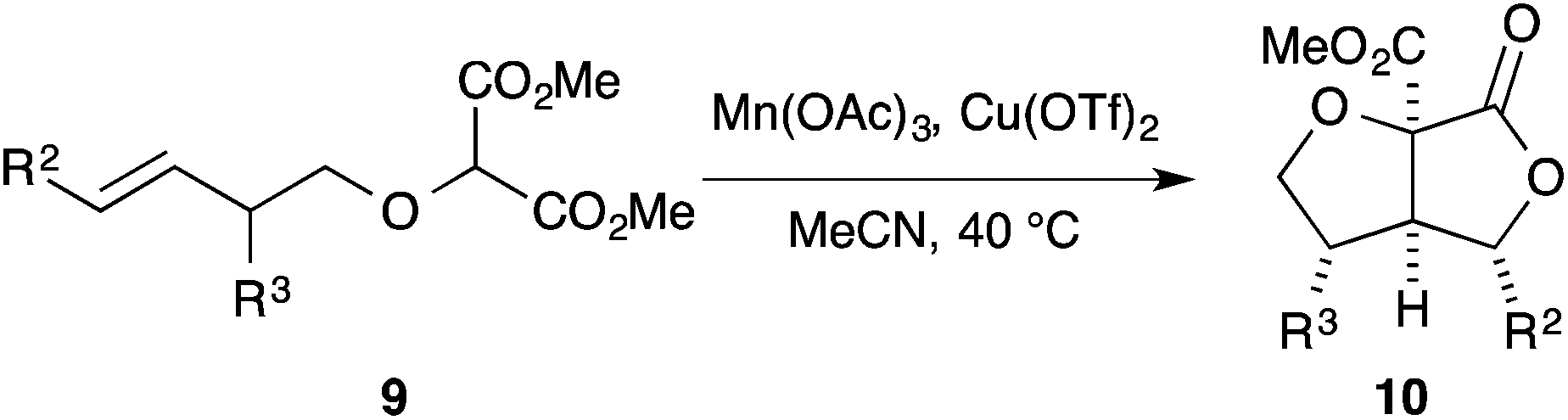
Given the successful cyclisation of substrates 7, we turned our attention to substrates carrying a substituent at the allylic position (Table 2). With relatively small substituents (9, R3 = Me, CH2CO2tBu), the corresponding lactones 10 were formed with moderate diastereocontrol (Table 2, entries 1, 2, 4 and 5).§,¶ In the cyclisation of 5-hexenyl radicals, allylic substituents frequently impart higher levels of diastereocontrol than exhibited above. This is a result of the allylic substituents having a preference for the pseudo-equatorial position in the chair-like transition state so as to minimise both allylic strain and 1,3-diaxial interactions. The modest selectivity exhibited in the cyclisation of substrates 9a,b,d and e is in keeping with the chair-like pre-transition state assembly where R3 occupies a pseudo-equatorial position (Fig. 2, TSmaj). However the preference for R3 to occupy a pseudo-equatorial position is determined by allylic strain;16 if R3 occupies a pseudo-axial position it would suffer only very minimal 1,3-diaxial interactions with one of the lone pairs of the oxygen atom of the forming THF (TSmin).|| With much larger substituents (9c and f), the corresponding lactones 10c,f were formed with high levels of diastereoselectivity (Table 2, entries 3 and 6). In all cases, the [3.3.0]-bicyclic γ-lactones were formed in synthetically useful yields.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | R3 | R2 | Yielda (%) | dr |
|
a Combined isolated yield of mixture of diastereomers; major diastereomer shown.
b Diastereomeric ratio (dr) refers to the mixture of diastereomers at the R3-bearing sterocentre measured from the crude 1H NMR.
c Ratio is given as major diastereomer:sum of minor diastereoisomers; detailed dr = 6.7:1:0.7:0.4.
|
|||||
| 1 | a | Me | H | 73 | 3:1b |
| 2 | b | CH2CO2tBu | H | 76 | 3.6:1b |
| 3 | c | iPr | H | 73 | 22:1b |
| 4 | d | Me | Ph | 92 | 4.6:1b |
| 5 | e | CH2CO2tBu | Ph | 69 | 3.2:1c |
| 6 | f | iPr | Ph | 92 | 28:1 |
 | ||
| Fig. 2 Plausible pre-transition state assemblies for formation of major and minor diastereomers relating to Table 2. | ||
We next moved to investigate cyclisation reactions of homoallylic-substituted substrates 11 (Table 3). Cyclisation of such substrates would give access to THFs carrying 2/5-substituents – positions that are frequently substituted in THF-containing natural products.1,2 Cyclisation of the substrates 11a–d (Table 3, entries 1–4) gave the product bicyclic lactones 12a–d in good yields with pleasing levels of diastereocontrol.§,¶ With the phenyl-substituted alkenes 11e–h the corresponding products 12e–h were formed as a mixture of three diastereomers.§ For all the products 12, the relative configuration of the major diastereromer of the products was in keeping with the Beckwith–Houk model10,11 for 5-hexenyl radical cyclisation. The levels of stereocontrol are in keeping with related cyclisations and examples from our group.5
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | R4 | R2 | Yielda (%) | drb |
|
a Combined isolated yield of mixture of diastereomers; major diastereomer shown.
b Ratio is given as major diastereomer:sum of minor diastereomers.
|
|||||
| 1 | a | Me | H | 96 | 6.1:1 |
| 2 | b | iPr | H | 94 | 5.7:1 |
| 3 | c | Ph | H | 80 | 7.5:1 |
| 4 | d | CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
H | 86 | 5:1 |
| 5 | e | Me | Ph | 66 | 2.8:1 |
| 6 | f | iPr | Ph | 61 | 3.2:1 |
| 7 | g | Ph | Ph | 70 | 2.6:1 |
| 8 | h | CHCH2 |
Ph | 80 | 2.8:1 |
Having investigated the cyclisation of ether substrates we briefly turned to investigate cyclisation of ester substrates 13 (Table 4). Gratifyingly, under our previously optimised conditions efficient cyclisation occurred to give the bicyclic bis-lactones 14 with good levels of diastereocontrol, although it was not always possible to isolate the products in pure form.
Many of the small densely functionalised products formed in the above cyclisation reactions contain differentiated oxygen functional groups which can be independently manipulated (Scheme 1). For example, on treatment of the [3.3.0]-bicyclic γ-lactone 8b with the phenyl selenide anion17 the carboxylic acid 15 is formed in 54% yield. Reduction of the carboxylic acid 15 in the presence of the ester could be readily achieved by initial conversion into the corresponding acid chloride followed by treatment with lithium tri-tert-butoxyaluminum hydride18 giving the alcohol 16. Oxidative elimination from 16 then provided the exo-methylene THF 17 bearing full substitution at C-2.
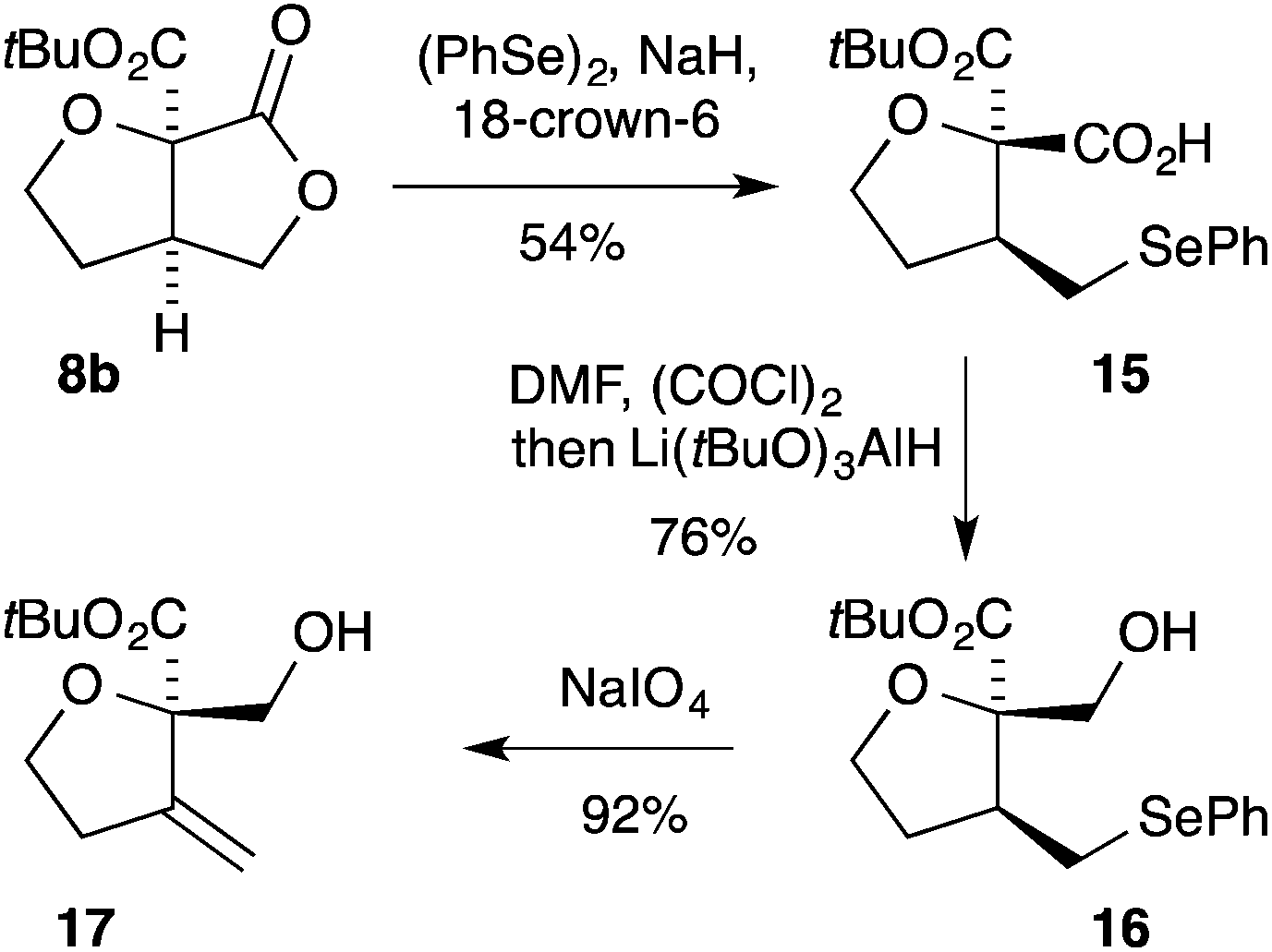 | ||
| Scheme 1 Synthetic manipulations of lactone 8b. | ||
Conclusions
In summary, we have reported a mild and operationally simple synthesis of bicyclic THFs and bicyclic bis-lactones in synthetically useful yields and with good levels of diastereocontrol. Application of this methodology to the synthesis of natural products and related targets is on-going.Acknowledgements
We thank the European Commission (FP7-PEOPLE-2012-IEF-329876) and the EPSRC for financial support. We thank the analytical sections of our Department for their excellent support.Notes and references
- A. Bermejo, B. Figadere, M. C. Zafra-Polo, I. Barrachina, E. Estornell and D. Cortes, Nat. Prod. Rep., 2005, 22, 269–303 RSC.
- A. Lorente, J. Lamariano-Merketegi, F. Albericio and M. Alvarez, Chem. Rev., 2013, 113, 4567–4610 CrossRef CAS PubMed.
- For reviews on the synthesis of THFs see: (a) J. P. Wolfe and M. B. Hay, Tetrahedron, 2007, 63, 261–290 CrossRef CAS PubMed; (b) G. Jalce, X. Franck and B. Figadère, Tetrahedron: Asymmetry, 2009, 20, 2537–2581 CrossRef CAS PubMed; (c) J. D. Rainier, Top. Heterocycl. Chem., 2014, 35, 1–41 CAS. For a recent natural product synthesis featuring cyclisation of an alkoxy malonate that is relevant to this work see: (d) F. Urabe, S. Nagashima, K. Takahashi, J. Ishihara and S. Hatakeyama, J. Org. Chem., 2013, 78, 3847–3857 CrossRef CAS PubMed.
- For our early work on the formation of THFs using radical cyclisations see: D. G. Hulcoop, H. M. Sheldrake and J. W. Burton, Org. Biomol. Chem., 2004, 2, 965–967 CAS.
- L. H. Powell, P. H. Docherty, D. G. Hulcoop, P. D. Kemmitt and J. W. Burton, Chem. Commun., 2008, 2559–2561 RSC.
- A. W. J. Logan, S. J. Sprague, R. W. Foster, L. B. Marx, V. Garzya, M. S. Hallside, A. L. Thompson and J. W. Burton, Org. Lett., 2014, 16, 4078–4081 CrossRef CAS PubMed.
- J. J. Davies, T. M. Krulle and J. W. Burton, Org. Lett., 2010, 12, 2738–2741 CrossRef CAS PubMed.
- D. G. Hulcoop and J. W. Burton, Chem. Commun., 2005, 4687–4689 RSC.
- A. W. J. Logan, J. S. Parker, M. S. Hallside and J. W. Burton, Org. Lett., 2012, 14, 2940–2943 CrossRef CAS PubMed.
- A. L. J. Beckwith and C. H. Schiesser, Tetrahedron Lett., 1985, 26, 373–376 CrossRef CAS.
- K. N. Houk, M. N. Paddonrow, D. C. Spellmeyer, N. G. Rondan and S. Nagase, J. Org. Chem., 1986, 51, 2874–2879 CrossRef CAS.
- For a review on the captodative effect see: L. Stella and J. N. Harvey, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, WILEY-VCH, Weinheim, 2001, vol. 1 Search PubMed.
- K. Takahashi, M. Midori, K. Kawano, J. Ishihara and S. Hatakeyama, Angew. Chem., Int. Ed., 2008, 47, 6244–6246 CrossRef CAS PubMed.
- For reviews of manganese(III) acetate in organic synthesis see: B. B. Snider, Chem. Rev., 1996, 96, 339–363 CrossRef CAS PubMed; G. G. Melikyan, Org. React., 1997, 49, 427–675 Search PubMed; A. S. Demir and M. Emrullahoglu, Curr. Org. Synth., 2007, 4, 321–351 CrossRef; J. W. Burton, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, John Wiley & Sons Ltd, Chichester, UK, 2012, pp. 901–942 RSC; M. Mondal and U. Bora, RSC Adv., 2013, 3, 18716–18754 RSC.
- For a review of the mechanisms of manganese(III) acetate-mediated reactions see: B. B. Snider, Tetrahedron, 2009, 65, 10738–10744 CrossRef CAS PubMed.
- For a review see: R. W. Hoffmann, Chem. Rev., 1989, 89, 1841–1860 CrossRef CAS.
- V. Rodeschini, J.-G. Boiteau, P. Van de Weghe, C. Tarnus and J. Eustache, J. Org. Chem., 2004, 69, 357–373 CrossRef CAS PubMed.
- H. C. Brown and R. F. McFarlin, J. Am. Chem. Soc., 1958, 80, 5372–5376 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterisation data and NMR spectra of new compounds. See DOI: 10.1039/c5ob01091h |
| ‡ Control experiments indicated that both manganese(III) acetate and copper(II) triflate were required for efficient product formation – see ESI† for details. |
| § In some cases other components were present in the crude reaction mixtures that may be other diastereomers but these components could not be characterised. |
| ¶ The relative configuration of the major diastereomer of the product lactones was assigned on the basis of 1H NMR nOe experiments or by analogy – see ESI† for details. The relative configuration of a small number of the minor diastereomers was also assigned by 1H NMR nOe experiments. |
| || Boat-like transition states are also possible. |
| This journal is © The Royal Society of Chemistry 2015 |