
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriazole linker-based trivalent sialic acid inhibitors of adenovirus type 37 infection of human corneal epithelial cells†
Rémi
Caraballo
ab,
Michael
Saleeb
ab,
Johannes
Bauer‡
c,
A. Manuel
Liaci
c,
Naresh
Chandra
bde,
Rickard J.
Storm
bde,
Lars
Frängsmyr
bde,
Weixing
Qian
a,
Thilo
Stehle
cf,
Niklas
Arnberg
bde and
Mikael
Elofsson
*ab
aDepartment of Chemistry, Umeå University, SE90187 Umeå, Sweden. E-mail: mikael.elofsson@chem.umu.se
bUmeå Centre for Microbial Research, Umeå University, SE90187 Umeå, Sweden
cInterfaculty Institute of Biochemistry, University of Tübingen, 72076 Tübingen, Germany
dDepartment of Clinical Microbiology, Division of Virology Umeå University, SE90185 Umeå, Sweden
eLaboratories for Molecular Infection Medicine Sweden, Umeå University, SE-901 87 Umeå, Sweden
fDepartment of Pediatrics, Vanderbilt University School of Medicine, Nashville 37232, TN, USA
First published on 9th July 2015
Abstract
Adenovirus type 37 (Ad37) is one of the principal agents responsible for epidemic keratoconjunctivitis (EKC), a severe ocular infection that remains without any available treatment. Recently, a trivalent sialic acid derivative (ME0322, Angew. Chem. Int. Ed., 2011, 50, 6519) was shown to function as a highly potent inhibitor of Ad37, efficiently preventing the attachment of the virion to the host cells and subsequent infection. Here, new trivalent sialic acid derivatives were designed, synthesized and their inhibitory properties against Ad37 infection of the human corneal epithelial cells were investigated. In comparison to ME0322, the best compound (17a) was found to be over three orders of magnitude more potent in a cell-attachment assay (IC50 = 1.4 nM) and about 140 times more potent in a cell-infection assay (IC50 = 2.9 nM). X-ray crystallographic analysis demonstrated a trivalent binding mode of all compounds to the Ad37 fiber knob. For the most potent compound ophthalmic toxicity in rabbits was investigated and it was concluded that repeated eye administration did not cause any adverse effects.
Introduction
Human adenoviruses (Ads), which belong to the mammalian adenovirus genus, Mastadenovirus, are commonly encountered infectious agents. In humans, Ads are associated with a multitude of clinical symptoms encompassing upper and lower respiratory tract infections, gastroenteritis, hemorrhagic cystitis and ocular diseases such as conjunctivitis and epidemic keratoconjunctivitis (EKC).1 Ads are ubiquitous in nature and new types are continuing to be discovered.2,3 Since the isolation of the first Ads about 60 years ago,4,5 over 60 types that are grouped into seven species (A–G) have been identified. Ad infections are usually self-limited in immunocompetent patients while they can become a serious life-threatening disease in immunocompromised individuals.6,7To date, there are no specific antiviral drugs available for the treatment of Ad infections.8 Ads are obligate intracellular pathogens that are fully dependent on the cellular replication machinery. The selective inhibition of Ad replication by antiviral compounds is therefore difficult to achieve as some of the essential functions of the host cells may also be altered. The acyclic nucleoside analogue cidofovir has been shown effective against EKC-causing Ads. Unfortunately, nephrotoxicity and lacrimal canalicular blockage have been reported after systemic administration.1,8 An alternative approach to the intracellular activity of nucleoside analogues is to block the initial interaction between the virus and host cell and thereby block cell attachment and subsequent entry.9
EKC is a severe and highly contagious ocular infection that is contracted by millions of individuals every year. Among the Ad types responsible for EKC, Ad8, Ad19 and Ad37 remain the principal causative agents of the infection;10 however, new EKC-causing types such as Ad53, Ad54 and Ad56 have been recently isolated from patients.11–13 Common symptoms are keratitis, conjunctivitis, edema, pain, lacrimation, formation of pseudomembranes and decreased vision.14 Because these viruses are spread by contact (e.g. hand to eye contact),14 EKC is frequent in densely populated areas and in medical wards with insufficient hygiene precautions. The infection commonly lasts for up to two weeks; however, some patients continue to suffer from sight impairment for several months, years or even permanently. Whereas other non-EKC causing Ads use CD46 and coxsackie-adenovirus receptor (CAR) as receptors,15 EKC-causing Ads bind via their homotrimeric fiber knobs to sialic acid-containing glycans that are situated on epithelial cells in the cornea and/or conjunctiva.16 The fiber knobs of EKC-causing Ads are located at the most distal part of each of the 12 fibers that protrude from the icosahedral virion. The knobs are highly homologous and the critical sialic acid-interacting residues are thus conserved within the different types of an Ad species.17 Consequently, substances targeting a single type also have the potential to prevent cell attachment by the other types. Recently, glycoproteins with glycans corresponding to the carbohydrate portion of the GD1a gangliosides were shown to function as receptors for the infection of ocular cells by EKC-causing Ads.17 The crystal structure of the Ad37-GD1a complex showed that the terminal sialic acid residues, located on each of the two branches of the GD1a glycan, are accommodated into two out of three carbohydrate recognition sites on top of the Ad37 fiber knob. Thus, inhibition of Ads with natural or synthetic sialic acid derivatives may prevent the virion to attach to, penetrate into and infect new cells. As a result, the infection and its spread would become limited. Importantly and especially in the case of EKC, the poor pharmacologic properties of carbohydrate-based drugs that include rapid serum clearance and poor cellular uptake obtained after systemic administration can be bypassed by the use of a topical mode of administration (e.g. cream, ointment, eye drops) and extracellular targeting of the virus particles.
Efficient sialic acid–based inhibitors of Ad37 infection of human corneal epithelial (HCE) cells have been recently reported.18–21 In order to circumvent the relatively low efficacy of monovalent sialic acid derivatives, the authors took advantage of the trimeric binding site at the Ad37 fiber knob. The use of multivalent sialic acid derivatives or glycoconjugates that can simultaneously bind to several carbohydrate recognition domains per knob considerably improved the inhibitory potency in comparison to the sialic acid monosaccharide. For instance, ME032221 (Fig. 1a), a synthetic trivalent sialic acid derivative was reported as four orders of magnitude more potent than the natural sialic acid monosaccharide. Other successful reports of trivalent glycosides include, for example, the synthesis of glycoclusters targeting the hepatic asialoglycoprotein receptor.22–24
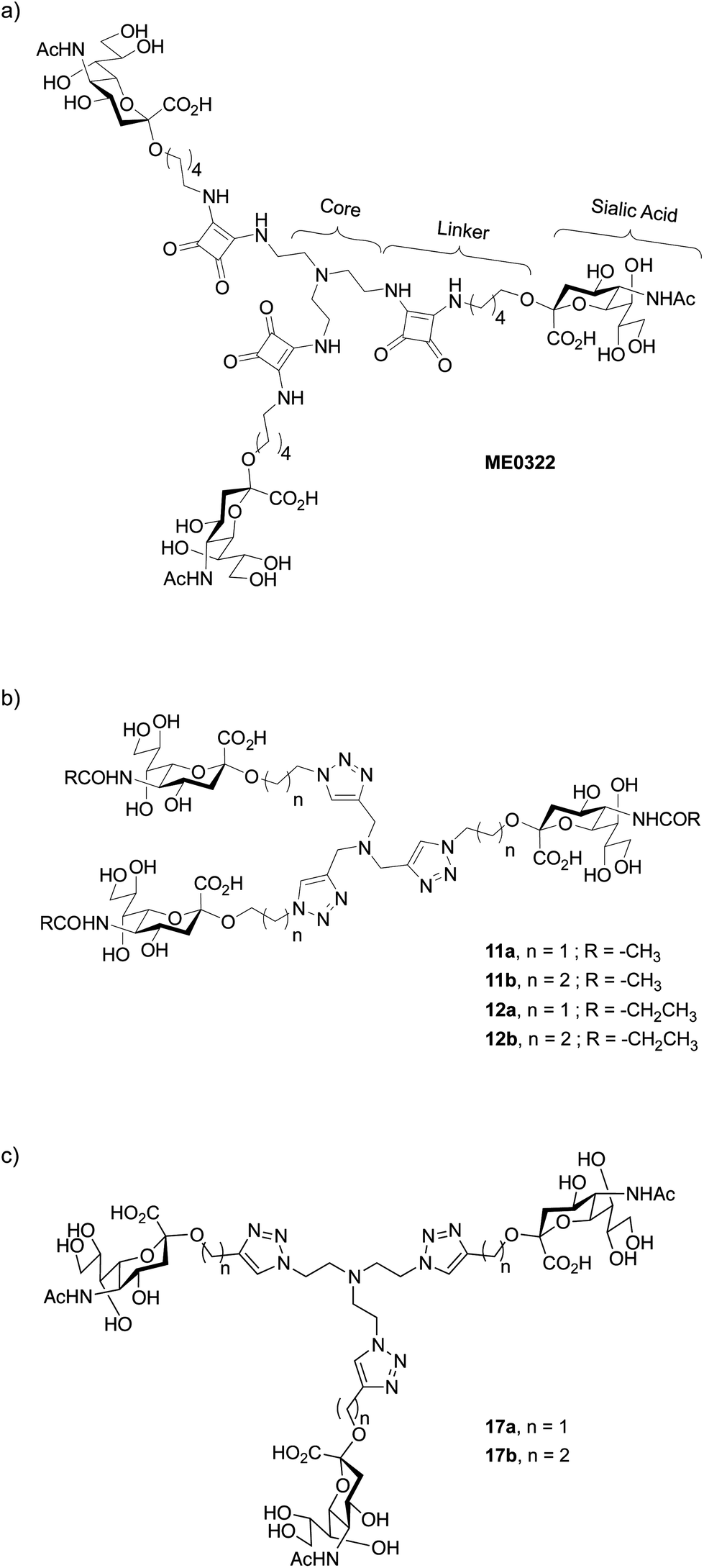 | ||
| Fig. 1 Structure of (a) trivalent sialic acid ME0322,21 a potent inhibitor of Ad37, (b) the first-generation triazole linker-based trivalent sialic acid derivatives and (c) the second-generation triazole linker-based trivalent sialic acid derivatives. | ||
Herein, we report on the design, synthesis and evaluation of new potent trivalent sialic acid inhibitors of Ad37 infection of HCE cells where the trivalent scaffolds were conveniently accessed by “click” chemistry. We probed the beneficial effect of a more compact linker and the influence of a small modification at the N-acetyl moiety on efficacy. Finally, the analysis of the respective crystal structures allowed us to confirm that the trivalent binding mode is engaged by all the inhibitors to the fiber knob as well as to reason on the potency differences.
Results and discussion
Design strategy for the first-generation triazole linker-based compounds
The design of new trivalent sialic acid conjugates was based on the structural features of ME0322 (Fig. 1). Thus, the compounds were composed of three key building blocks: the core fragment that is necessary for the construction of a trivalent network; the sialic acid residue that is required for the binding to the carbohydrate recognition sites at the Ad37 fiber knob and the linker that is connecting the sialic acid residues to the core fragment.The tertiary amino scaffold that constituted the core fragment in ME0322 and the sialic acid residues that are crucial for the interactions with the target protein were conserved in the new ligands. Contrarily, the linker, which could not be visualized in the crystal structure of ME0322 in complex with the Ad37 fiber knob21 likely due to flexibility and lack of interactions with the protein, was revised. The design efforts were initially focused on both, shortening the original linker with the idea to bring the core fragment closer to the receptor and, introducing a heteroaromatic ring moiety to possibly create new contacts with the fiber knob (Fig. 1b). After analysis of the commercial availability of functionalized tertiary amino building blocks (core fragment) and robust chemical reactions that allow a rapid access to multivalent sialic acid containing structures our choice was triazole-based linkers.25–30 Two linker lengths were investigated to probe an eventual size effect on the potency.
In addition to trivalent sialic acid compounds 11a and 11b, their corresponding N-acetyl modified analogues 12a and 12b were also included in this series (Fig. 1b). Thus, we aimed to further investigate the hydrophobic pocket to which the N-acetyl group of the sialic acid is directed in the Ad37-ME0322 complex.21 Previous studies on a set of multivalent human serum albumin (HSA) conjugates highlighted a detrimental effect of an increased lipophilicity at the N-acetyl group of the sialic acid on the compound potency, although crystallographic investigations suggested that these modifications should readily fit in the hydrophobic fiber knob pocket that accommodate the sialic acid N-acetyl group.31 This loss in potency could eventually be scaffold- and/or linker-dependent as suggested by a better tolerance of an increased lipophilicity during the evaluation of their corresponding monovalent analogues. Therefore, the effect of such modifications on trivalent sialic acid derivatives that have a linker especially designed to better accommodate into the fiber knob cellular receptor should clarify the scaffold- and linker-dependence hypothesis.
Synthesis of the first-generation triazole linker-based compounds
The route to unmodified N-acyl trivalent sialic acids 11a and 11b proved straightforward and could be achieved in eight steps from commercially available chemicals or in five steps from key intermediate 1 (Scheme 1). The synthesis of the sialic acid thiophenyl derivative 1, readily prepared from sialic acid, was performed according to published procedures.32 Sialosides 3a and 3b were then accessed in good conversion by glycosylation of 2-bromoethanol and 3-bromopropan-1-ol respectively, with compound 1. The reactions yielded inseparable mixtures of anomers together with the resulting elimination product that was not further purified at this stage. Bromo derivatives 3a and 3b were readily converted to their azido analogues 5a and 5b. Subsequent O-deacylation using standard Zemplèn conditions afforded anomerically pure 7a and 7b in 40% and 58% yields, respectively, over three steps. Then, compounds 7a and 7b were reacted with tripropargylamine in a copper-catalyzed azide–alkyne cycloaddition reaction (“click” reaction). Thus, methyl esters 9a and 9b were obtained in 51% and 45% yields, respectively. Subsequent saponification provided the final target compounds 11a and 11b in 76% and 41% yields, respectively. The route to N-acyl modified trivalent sialic acids 12a and 12b proved somewhat more challenging. The target compounds could be reached in 13 steps from commercially available sialic acid or in five steps from key intermediate 2 (Scheme 1). The synthesis of compound 2 was performed according to published procedures.31 The following steps were analogous to the above-described synthetic route to compounds 11a and 11b. Thus, the successive glycosylation, azide formation and O-deacylation reactions afforded anomerically pure product 8a and 8b in 46% and 55% yields over three steps, respectively. Subsequent “click” reactions provided the trivalent compounds 10a and 10b in 50% and 34% yields, respectively. Finally, saponification of the methyl esters gave the target products 12a and 12b in good yields.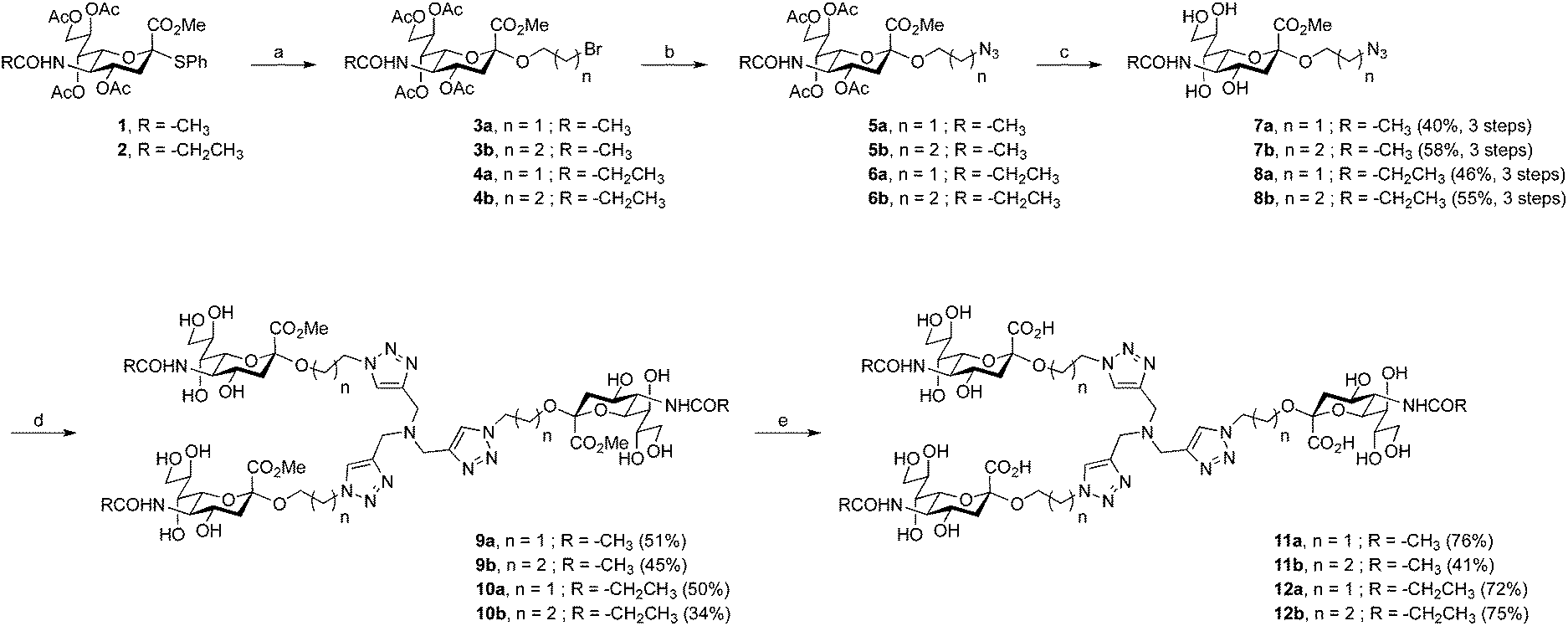 | ||
Scheme 1 Synthesis of 11a, 11b, 12a and 12b. Reagents and conditions: (a) i: molecular sieves 3 Å, 2-bromoethanol or 3-bromopropan-1-ol, CH3CN/CH2Cl2 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), rt, 2 h, ii: AgOTf, IBr, −73 °C, 4.5 h, iii: DIPEA, −73 °C, 30 min. (b) NaN3, TBAI, DMSO, rt, 6 h. (c) i: NaOMe, MeOH, rt, 3 h, ii: H+ ion exchange resin. (d) Tripropargylamine, CuSO4, sodium ascorbate, THF/H2O (1:1), 50 °C, 3 h then rt, 18 h. (e) i: LiOH, MeOH, rt, 9 h, ii: H+ ion exchange resin. :2), rt, 2 h, ii: AgOTf, IBr, −73 °C, 4.5 h, iii: DIPEA, −73 °C, 30 min. (b) NaN3, TBAI, DMSO, rt, 6 h. (c) i: NaOMe, MeOH, rt, 3 h, ii: H+ ion exchange resin. (d) Tripropargylamine, CuSO4, sodium ascorbate, THF/H2O (1:1), 50 °C, 3 h then rt, 18 h. (e) i: LiOH, MeOH, rt, 9 h, ii: H+ ion exchange resin. | ||
Biological evaluation of the first-generation triazole linker-based compounds
Cell-binding assays using 35S-labeled virions were performed to investigate the efficiency of compounds 11a, 11b, 12a and 12b to prevent the attachment of Ad37 virions to HCE cells (Fig. 2a). The assays were carried out essentially as previously described but with minor modifications16,33 and, ME0322, sialic acid and GD1a glycan were used as reference compounds.17,21 In brief, 35S-labeled Ad37 virions were mixed with or without the trivalent sialic acid derivatives, GD1a glycan and sialic acid at various concentrations. The mixtures were incubated with HCE cells and unbound virions were then washed away. Finally, the cell-associated radioactivity was measured by using a scintillation counter.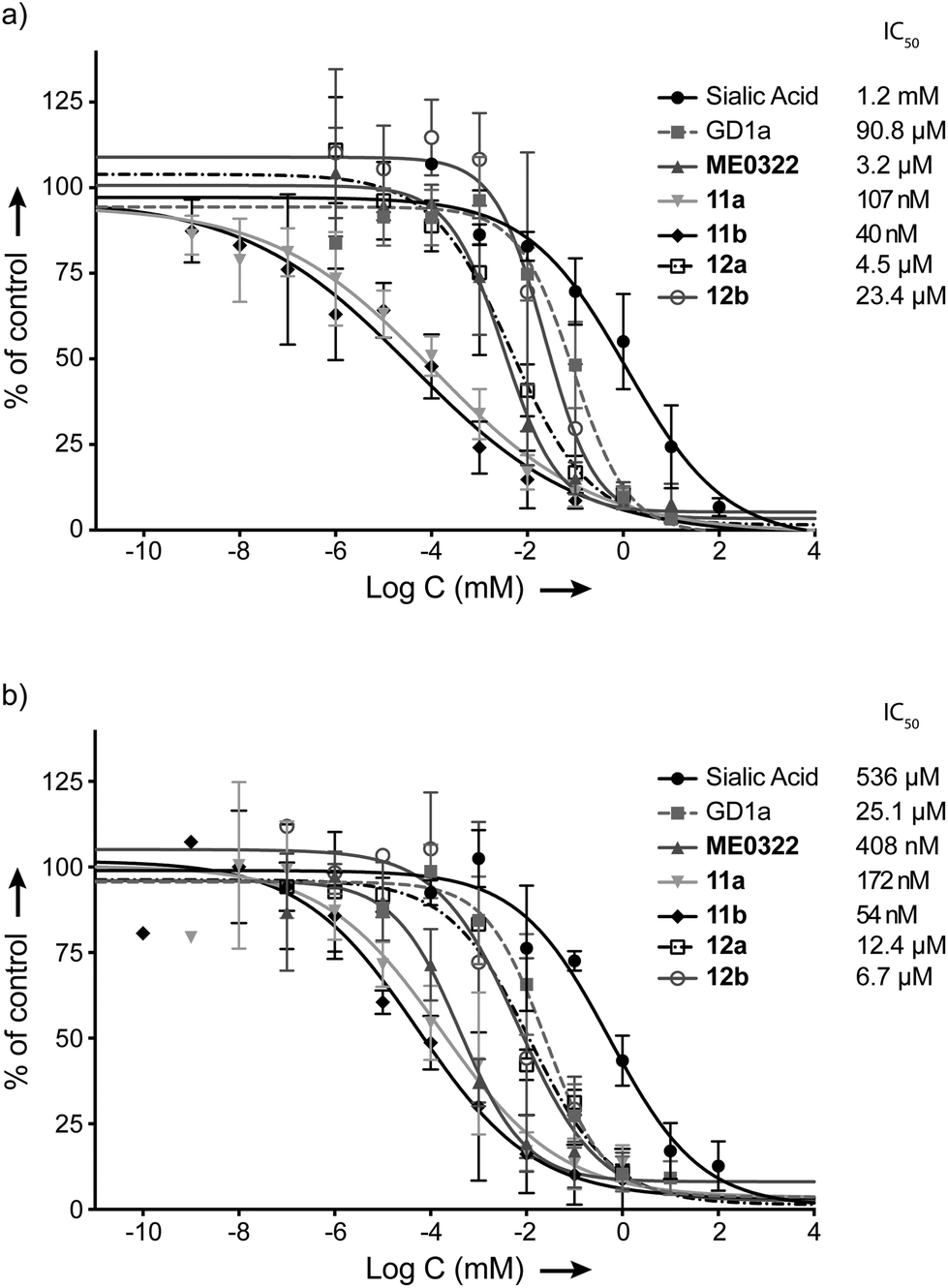 | ||
| Fig. 2 Effect of the set of trivalent sialic acid derivatives 11a–b and 12a–b on Ad37 binding to and infection of HCE cells. (a) Virion binding in the presence of inhibitors at different concentrations. (b) Infection at different concentrations of the inhibitors. Data are presented as % of control that is the value obtained in the absence of inhibitor. | ||
The attachment of Ad37 virions to HCE cells was dramatically hindered in the presence of the triazole linker-based compounds (Fig. 2a). Indeed, the new trivalent sialic acid derivatives were considerably more potent than the sialic acid monosaccharide (IC50 = 1.2 mM) and bivalent GD1a glycan (IC50 = 90.8 μM). Compounds 11a and 11b were found as the most efficient compounds to prevent Ad37 virions from HCE cells attachment with IC50 values of 107 nM and 40 nM, respectively. The previously described compound, ME0322, proved to be less potent (IC50 = 3.2 μM). The N-acyl modified derivatives 12a and 12b completed the series with IC50 values of 4.5 μM and 23.4 μM, respectively.
A correlation between the linker length and the ligand potency could not be demonstrated. Thus, reduction of the linker size in the N-acetyl series (11avs. 11b) resulted in a 2.7 times decrease in binding potency. Contrarily, the same transformation in the N-acyl modified series (12avs. 12b) proved 5.2 times more beneficial. These results suggest that some degree of variation of the linker length is tolerated. However, increasing the lipophilicity of the ligands at the N-acyl moiety was clearly not beneficial to the binding to Ad37 virions (11avs. 12a and 11bvs. 12b). Even though these data are in agreement with previous investigations;31 herein, the drop in potency is not as pronounced. Indeed, while N-acyl modified multivalent HSA conjugates were very poorly effective, compounds 12a and 12b prevent the attachment of Ad37 virions to HCE cells rather efficiently. Therefore, the loss in binding potency cannot only be attributed to an increased lipophilicity and/or bulkiness at the N-acyl moiety and, the nature of the multivalent scaffold is most likely to play an important role.
Infection experiments were then performed to further evaluate our set of compounds (Fig. 2b). The assays were carried out essentially as previously described but with minor modifications.16,33 In brief, unlabeled virions were mixed with or without the trivalent sialic acid derivatives, GD1a glycan or sialic acid at various concentrations. These mixtures were then added to HCE cells and incubated at +4 °C. Unbound virions were washed away, the resulting mixtures were incubated at +37 °C and a synchronized infection – all virions enter the cells simultaneously – was then obtained. After 44 h of infection, the cells were rinsed, fixed, incubated with rabbit polyclonal anti-Ad37 antibodies (that recognize viral capsid proteins) prior to being washed and stained. Finally, the cells were washed and the number of infected cells was quantified by immunofluorescence microscopy.
Hence, the trends from the cell-binding assays were confirmed in the infection experiments (Fig. 2a and b). Compounds 11a and 11b prevented infection of HCE cells by Ad37 virions most efficiently with IC50 values of 172 nM and 54 nM, respectively. The half maximal inhibitory concentration of ME0322 that had previously been estimated to 380 nM21 was herein calculated to 408 nM. Compounds 12a and 12b were evaluated as the least potent trivalent sialic acid derivatives with IC50 values of 12.4 μM and 6.7 μM, respectively. The GD1a glycan (IC50 = 25.1 μM) and sialic acid (IC50 = 536 μM) completed the series.
Design strategy for the second-generation triazole linker-based compounds
A second round of linker engineering (Fig. 1c), where the connecting “click” strategy was reversed with respect to the first generation of ligands (Fig. 1b), was performed. The linker lengths of 11a and 11b were preserved in compounds 17a and 17b, respectively; however, the triazole ring was moved closer to the sialic acid residues. Therefore, we could not only investigate the influence of the triazole ring orientation but also probe if additional interactions with the fiber knob could be achieved.Synthesis of the second-generation triazole linker-based compounds
Compounds 17a and 17b were prepared in four steps from key intermediate 1 (Scheme 2). The synthesis of anomerically pure sialosides 14a and 14b (48% and 31% yields over two steps, respectively) followed the above-described glycosylation and O-deacylation procedures, albeit the use of the appropriate alcohol derivatives in the first step. Compounds 14a and 14b were then reacted with tris (2-azidoethyl)amine (15) in a “click” reaction. The core building block 15 was synthesized in two steps from commercially available triethanolamine. First, tris (2-chloroethyl)amine was obtained according to published procedure34 and the chloro derivative was then readily converted to its azido analogue 15. Compound 15 was judged as potentially explosive and was therefore kept in solution at all times. The methyl esters 16a and 16b were obtained in 76% and 53% yields, respectively. Subsequent saponification provided the final target compounds 17a and 17b in quantitative and 88% yields, respectively.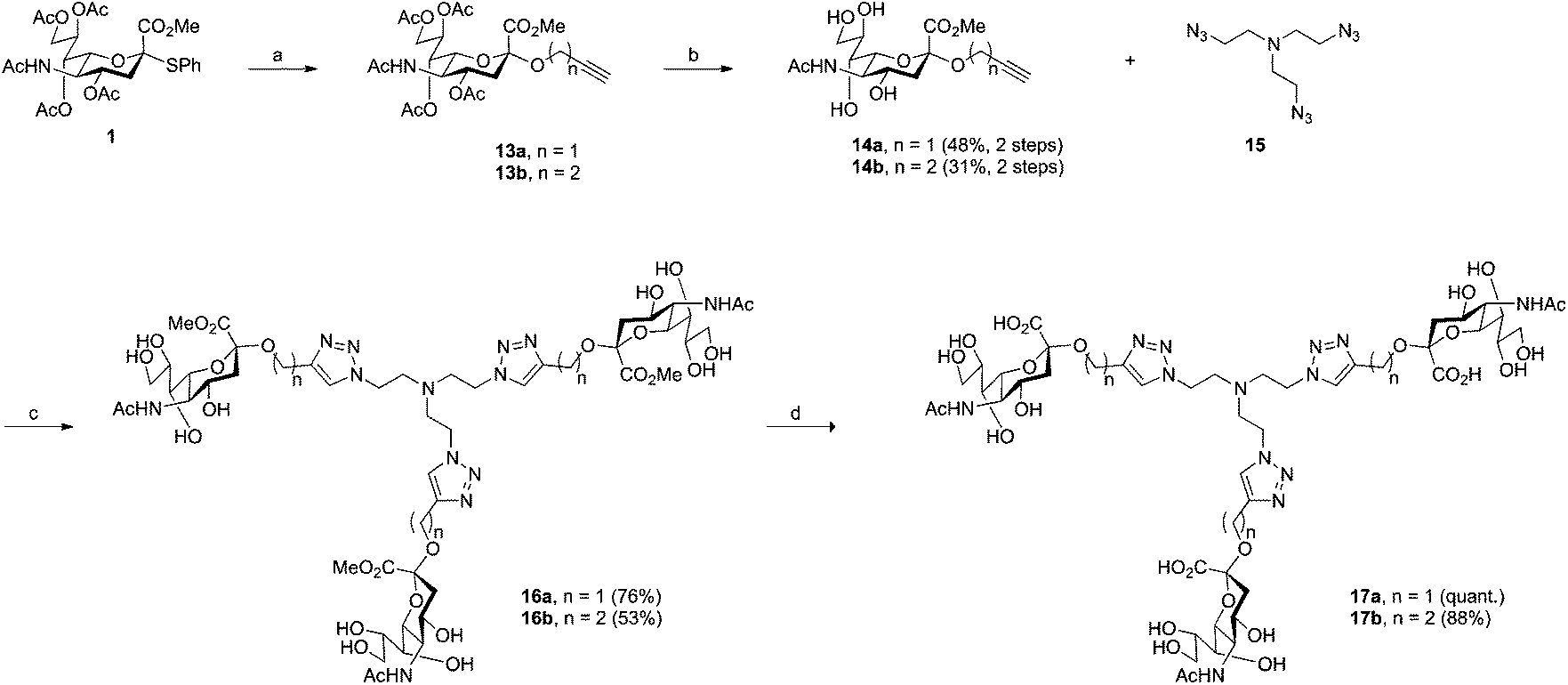 | ||
| Scheme 2 Synthesis of 17a and 17b. Reagents and conditions: (a) i: molecular sieves 3 Å, propargyl alcohol or 3-butyn-1-ol, CH3CN/CH2Cl2 (3:2), rt, 2 h, ii: AgOTf, IBr, −73 °C, 4.5 h, iii: DIPEA, −73 °C, 30 min. (b) i: NaOMe, MeOH, rt, 3 h, ii: H+ ion exchange resin. (c) CuSO4, sodium ascorbate, THF/H2O (1:1), 50 °C, 3 h then rt, 18 h. (d) i: LiOH, MeOH, rt, 9 h, ii: H+ ion exchange resin. | ||
Biological evaluation of the second-generation triazole linker-based compounds
The compounds were subsequently investigated in cell-binding assays (Fig. 3a). The linker optimization strategy proved successful and a 76 times increase in potency was observed for 17a (IC50 = 1.4 nM) with respect to 11a. Contrarily, 17b (IC50 = 376 nM) was evaluated about 9 times less efficient than 11b to prevent the attachment of Ad37 virions to HCE cells and 17a was over two orders of magnitude more potent than 17b. These results contrast our earlier observations where a clear effect of the linker length on the ligand potency could not be evidenced. A tentative explanation for the potency of 17a could be that the position of the triazole ring adjacent to the sialic acid residue might result in additional interactions with the fiber knob. These potentially favorable contacts would be harder to achieve for 11a, 17b and 11b, where the triazole ring is situated further away from the sialic acid residue by one or two methylene groups, respectively. The reasons for the relatively low efficiency of 17b in comparison to 11a and 11b to prevent Ad37 virions attachment to HCE cells however remain unclear.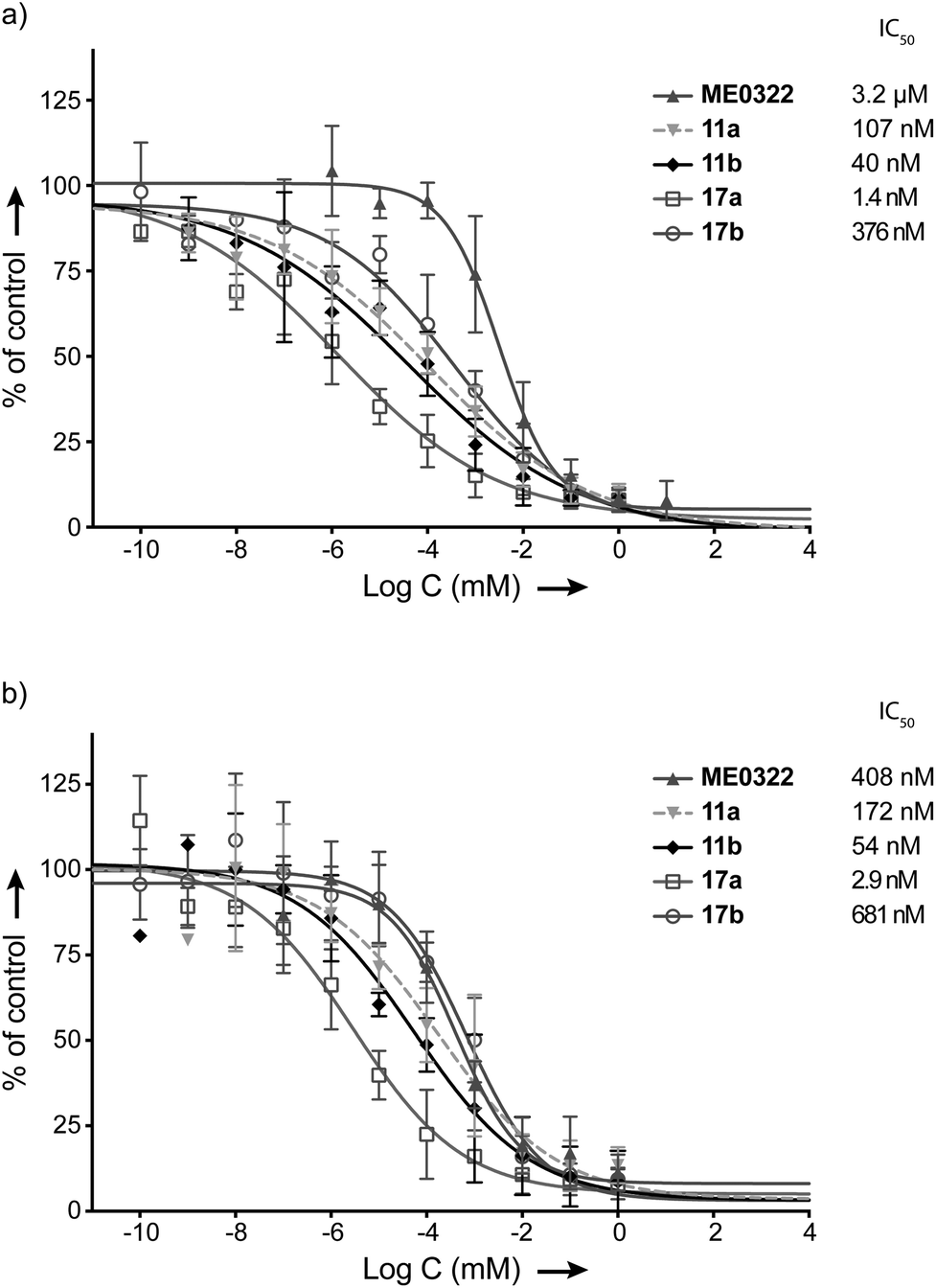 | ||
| Fig. 3 Effect of the set of trivalent sialic acid derivatives (17a–b) on Ad37 binding to and infection of HCE cells. (a) Virion binding in the presence of inhibitors at different concentrations. (b) Infection at different concentrations of the inhibitors. Data are presented as % of control that is the value obtained in the absence of inhibitor. | ||
Compounds 17a and 17b were then subjected to cell-infection assays. The results were in agreement with the cell-binding assays and 17a (IC50 = 2.9 nM) was confirmed as the best inhibitor against Ad37 infection to HCE cells. Interestingly, the optimization strategy entirely based on the linker re-design provided an inhibitor over 140 times more potent than the initial lead compound (17avs. ME0322). Compound 17b also proved efficient against Ad37 infection to HCE cells, despite a relatively low potency in relation to 17a.
Surface plasmon resonance (SPR) experiments of selected trivalent inhibitors
Finally, 11a, 11b, 17a, 17b and ME0322 were investigated in surface plasmon resonance (SPR) experiments and their respective binding affinities (KD) for immobilized Ad37 fiber knobs were determined (see Experimental section for further details). Due to the fast on and off rates, the association and dissociation constants could not be determined. SPR data corroborated well the trends from both cell-binding and cell-infection assays and the five compounds proved to interact with the Ad37 fiber knob. Thus, 17a (KD = 10.3 ± 1.4 μM, n = 3) was confirmed to best interact with the Ad37 fiber knob, followed by 11b (KD = 72.8 ± 3.3 μM, n = 3), 11a (KD = 78.6 ± 2.7 μM, n = 3), ME0322 (KD = 135.4 ± 17.9 μM, n = 3) and 17b (KD = 163.4 ± 12.3 μM, n = 3) (see ESI† for SPR curve tracing). It is also worth noting the influence of the Ad37 fiber knob construct on the KD values. Indeed, while histidine-free fiber knobs were used during our assays, histidine-tagged proteins were previously utilized thus providing a different KD value for ME0322 (KD value previously evaluated at 14 μM).21 This clearly underlines the need of an internal reference during the SPR experiments.Crystallography
Crystal structures of Ad37 in complex with our new trivalent sialic acid conjugates were obtained by soaking (using 2 mM 12a) or co-crystallization (for 11a, 11b, 12b, 17a and 17b) using previously reported methods.§17,21 In all complex structures (Fig. 4, S11 and Table S2†), electron density for the entire conjugate was visible thus allowing for unambiguous placement of the ligands. A simultaneous binding of the three sialic acid residues from a single ligand to the same fiber knob was evidenced, and most sialic acid/Ad37 contacts observed in previous complexes17,21,35 were retained within the different complex structures (Fig. 4 and S12†). Lys345, Pro317 and Tyr312 were confirmed as key contributors to ligand binding and directly interact with the sialic acid moiety, whereas the Ser344/sialic acid contact occurs via water-mediated hydrogen bonds. Additional interactions from the linker and/or core fragment with the fiber knob could however not be observed and the conformations and flexibility of the linker varied significantly from one Ad37-ligand complex to another (Fig. 4 and 5a). Although the hypothesis of favorable direct interactions between the triazole ring of 17a and the fiber knob protein could thus not be verified, there may still exist long-range electrostatic interactions that favor binding. However, the analysis of the Ad37-17b complex highlighted an additional internal order of 17b that could eventually explain its lower potency. Indeed, in the complex the sialic acid residues of 17b were rotated sideward and the linker formed a bell-like shape, possibly due to a staggered arrangement of the triazole rings (Fig. 4f and magenta structure in Fig. 5a). This ordered and more compact conformation could then directly affect the potency of the ligand.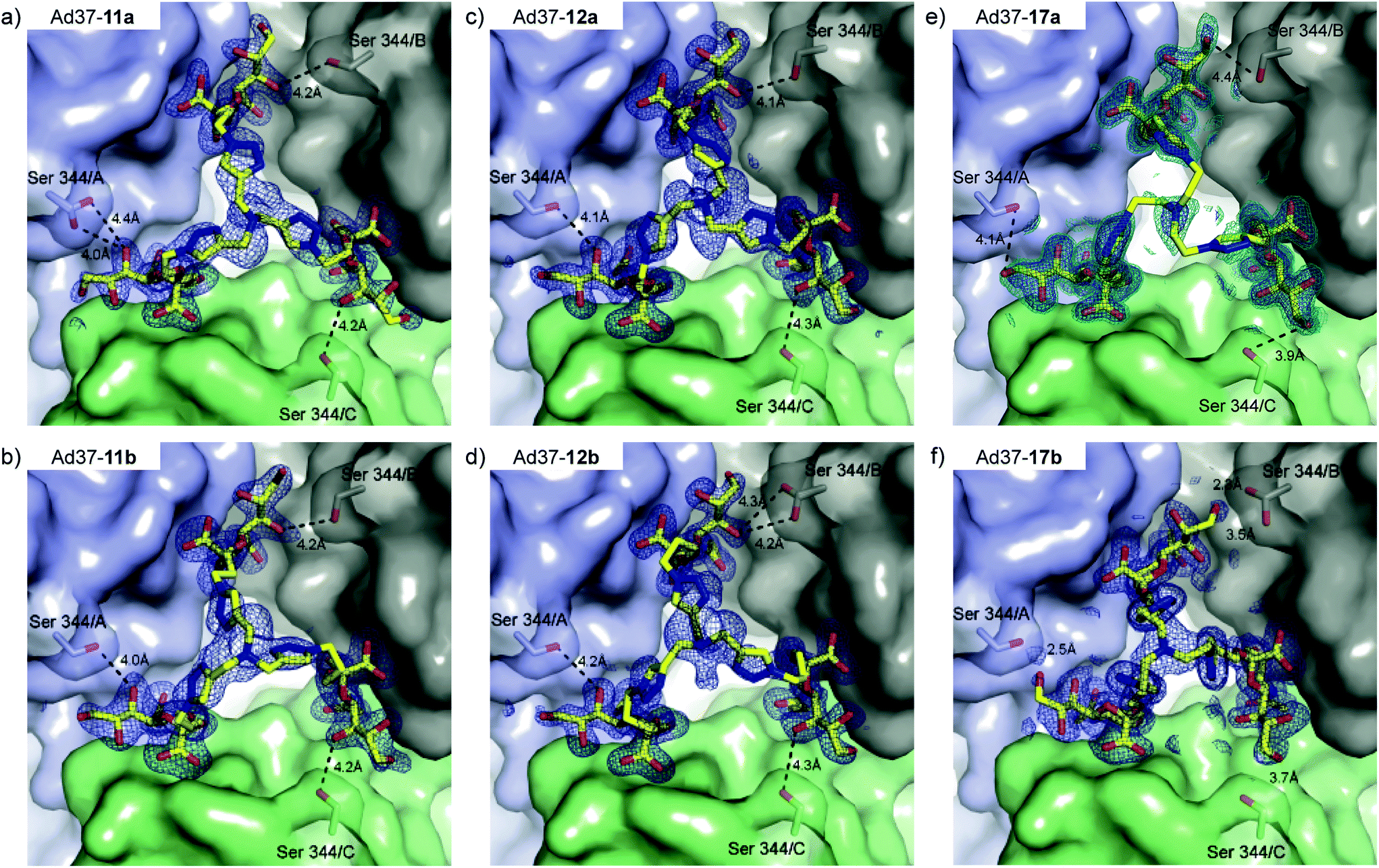 | ||
| Fig. 4 Binding of 11a, 11b, 12a, 12b, 17a and 17b to the Ad37 fiber knob (top view, a–f). Simulated annealing omit difference electron density maps for 11a (a, 2.0 Å resolution, PDB ID 4K6T), 11b (b, 1.9 Å resolution, PDB ID 4K6U), 12a (c, 1.5 Å resolution, PDB ID 4K6W), 12b (d, 1.5 Å resolution, PBD ID 4K6V), 17a (e, 1.4 Å resolution, 4XQA) and 17b (f, 1.6 Å resolution, 4XQB) were contoured at 3σ and shown with a radius of 2.4 Å around the ligand (dark blue). For 17a (e), an additional map at 2σ shows an ordered density for the ligand base (cyan). One inhibitor molecule is simultaneously bound with its three terminal sialic acid residues. Serine 344 (Ser344) is within the van-der-Waals radius of the sialic acid moieties and only 17b (f) is capable of forming hydrogen bonds with this residue. Black dashed lines in a–e indicate distances to Ser344. | ||
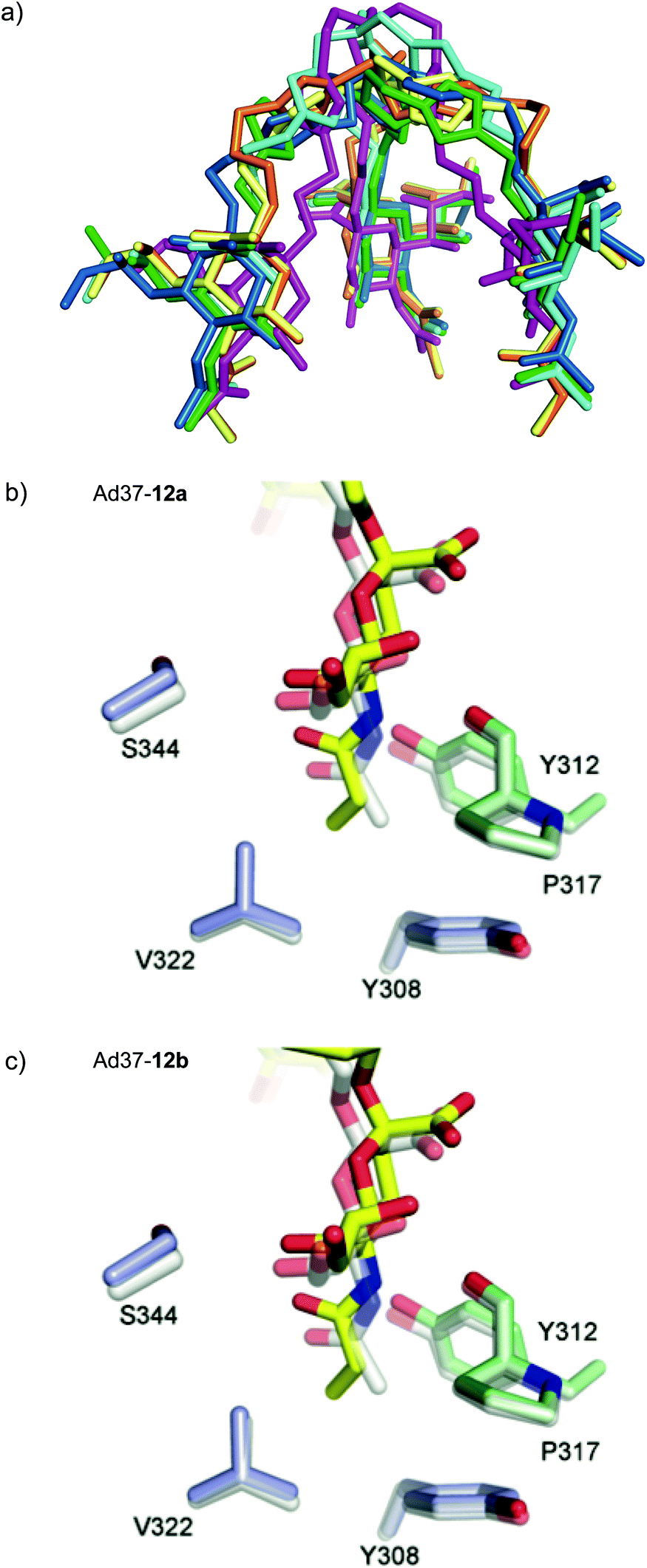 | ||
| Fig. 5 (a) Superposition of the inhibitor compounds (11a = blue, 11b = cyan, 12a = yellow, 12b = orange, 17a = green, 17b = magenta). The corresponding protein chains were aligned in PyMOL. 17b possesses additional internal order, its sialic acid moieties are rotated sideward and its linker forms a bell-like structure; (b) and (c) hydrophobic interactions of 12a (b) and 12b (c). For the two compounds, the additional methyl group of the propionic acid group is facing away from Y308 and V322, pushing the whole ligand up. | ||
Crystal structures of the N-acyl modified compounds 12a and 12b showed an overall binding mode similar to their corresponding unmodified N-acyl analogues (Fig. 4a–d, respectively). Moving the analysis to the sialic acid binding site, the Ad37-12a and Ad37-12b complexes provided important information (Fig. 5b and c, respectively). The additional methyl group in 12a and 12b that is oriented away from Tyr308 and Val322 pushes the entire ligand slightly upwards. This unfavorable interaction most likely causes the drop in inhibitory potency for the N-acyl modified series of compounds.
Ophthalmic toxicity of compound 17a in rabbit
To examine if the most potent compound (17a) can be used for topical treatments of eye infections, ophthalmic toxicity was studied in rabbits. Six male New Zealand white rabbits were divided into two groups of three and to each eye 40 μL of 1 mg mL−1 of 17a in 0.9% aq. NaCl or vehicle alone was administered topically. In total, each eye received 48 administrations over a period of seven days. Before the first administration, day 2, and at day 8 the animals were subjected to body weight recordings, slit lamp examinations, intraocular pressure, and pachymetry measurements under local anesthesia. The variations in body weights noted are common among rabbits and are probably not related to the treatment. The data showed that intraocular pressures and corneal thicknesses were within normal limits throughout the study period. The results of the slit lamp examinations show that all eyes were of normal status before start of the study. At the examinations at days 2 and 8 no signs of influence of the test or control items were noted on the cornea, the depth of the anterior chamber, the iris or the lens. No signs of irritation of the test or control item on surrounding tissues were noted. We conclude that compound 17a did not cause irritation in ocular tissues after repeated administration during seven days. In addition, repeated administration did not cause changes in body weights, intraocular pressure, or corneal thickness that would indicate a toxic reaction to the compound.Conclusions
Two generations of new trivalent sialic acid derivatives have been designed, synthesized and evaluated against Ad37 infection of HCE cells. The design of these Ad37 inhibitors was based on the structural features of ME0322, a previously characterized trivalent Ad37 inhibitor, as well as on robust chemical reactions allowing the rapid access to the target compounds. Thus, in this study we set out to improve the potency of ME0322 by revising the linker strategy. First-generation ligands 11a and 11b efficiently prevented the attachment and infection of Ad37 virions to HCE cells while second-generation compound 17a was determined as the most potent inhibitor of Ad37 infection of HCE cells. In addition, the original lead potency was greatly improved. Co-crystallization of the trivalent sialic acids in complex with the Ad37 fiber knob allowed the unambiguous placement of the ligands and therefore confirmed a one-to-one binding mode between the compounds and the Ad37 fiber knob. However, ligand–receptor interactions originating from the linker and/or core fragment were not observed.In this study, we also explored the effect of an increased lipophilicity at the N-acyl moiety (12a and 12b). The trivalent ligands, albeit a great inhibitory potency, were less efficient in preventing Ad37 infection of HCE cells compared to their unmodified analogues (11a and 11b). The analysis of the Ad37-inhibitor structures evidenced an unfavorable interaction between the additional methyl group and the target protein that could explain the lower potency of these compounds.
In conclusion, we have synthesized highly potent inhibitors against Ad37 infection to HCE cells. Compound 17a, the most potent inhibitor, was accessed using a straightforward synthetic route. Some advantages of such drugs are that (i) they can be used for topical treatment, which would overcome systemic treatment related challenges such as rapid serum clearance and poor cellular uptake of carbohydrate based drugs, (ii) their mechanism of action is on the extracellular level whereas most other antiviral drugs act intracellularly (minimizing side effects), and (iii) the active molecule is a normal carbohydrate (also minimizing side effects). Our investigation of ophthalmic toxicity in rabbits show that this type of trivalent sialic acids are well tolerated and thus have the potential to be developed into novel treatments of viral eye infections.
Experimental section
General chemical procedures
1H NMR and 13C NMR spectra were recorded with a Bruker DRX-400 spectrometer at 400 MHz and 100 MHz respectively. NMR experiments were conducted at 298 K in CDCl3 (residual solvent peak = 7.26 ppm (δH)), CD3OD (residual solvent peak = 3.31 ppm (δH) and 49.00 ppm (δC)) and D2O (residual solvent peak = 4.79 ppm (δH)). LCMS was carried out with a Waters LC system equipped with an Xterra C18 column (50 × 19 mm, 5 μm, 125 Å), eluted with a linear gradient of CH3CN in water, both of which contained formic acid (0.2%). A flow rate of 1.5 mL min−1 was used and detection was performed at 214 nm. Mass spectra were obtained on a Water micromass ZQ 2000 using positive and negative electrospray ionization. HRMS was performed using a Bruker MicroTOF II mass spectrometer with electrospray ionization (ES+); Tune Mix ESI solution was used for the calibration. Semi-preparative HPLC separations were performed on a Gilson system HPLC, using a Nucleodur C-18 column HTEC 5 μm (VP 250/21) with a flow rate 20 mL min−1, detection at 214 nm and eluent system: A. aq. 0.005% HCOOH, and B. 0.005% HCOOH in CH3CN. Column chromatography was performed on silica gel (Merck, 60 Å, 70–230 mesh ASTM). Thin Layer Chromatography (TLC) were performed on Silica gel 60 F254 (Merck) with detection under UV light and/or development with 5% H2SO4 in EtOH and heat. Optical rotations were measured with a Perkin-Elmer 343 polarimeter at 20 °C. Organic solvents were dried using a Glass Contour Solvent Systems (SG Water USA) except CH3CN and MeOH that were dried over molecular sieves 3 Å. All commercial reagents were used as received. ME0322 was synthesized according to published procedure.21 All target compounds were ≥95% pure according to HPLC UV-traces. Statistics were calculated using GraphPad Prism (GraphPad Software, Inc, La Jolla, CA).Synthetic procedures
:2; 35 mL mmol−1) at room temperature and under nitrogen atmosphere. 2-Bromoethanol, 3-bromopropan-1-ol, propargyl alcohol or 3-butyn-1-ol (4.5 equiv.) was added and the mixture was stirred for 2 h. The reaction was protected from light and a solution of silver triflate (2.0 equiv.) in CH3CN was added. The mixture was cooled to −73 °C (−70 °C < t < −75 °C) and IBr (1.4 equiv., 1 M in CH2Cl2) was added. The reaction was allowed to proceed for 4.5 h at −73 °C. After completion, DIPEA (6.0 equiv.) was added. The reaction mixture was stirred for a further 30 min at −73 °C and then allowed to warm to room temperature. The mixture was filtered through a Celite® pad, washed with CH2Cl2 or CH3CN and the solvents concentrated to dryness.
:1, 81 mL mmol−1) was successively added tripropargylamine (1.0 equiv.), CuSO4 (0.9 equiv.) and sodium ascorbate (0.9 equiv.). The reaction was allowed to proceed at 50 °C for 3–3.5 h and at room temperature for a further 18 h. After complete consumption of the starting azide, THF was evaporated under vacuum and the crude was freeze-dried. The crude solid was dissolved in DMSO and purified by HPLC (A: aq. 0.005% HCOOH in H2O, B: aq. 0.005% HCOOH in CH3CN, organic phase gradient 7% to 25%). The collected compound-containing fractions were freeze-dried to afford pure product.
:1, 81 mL mmol−1) was successively added alkyne derivative (4.5 equiv.), sodium ascorbate (0.9 equiv.) and copper(II) sulfate (0.9 equiv.). The reaction was allowed to proceed at 50 °C for 3–3.5 h and at room temperature for a further 18 h. After complete consumption of the starting azide, THF was evaporated under vacuum and the residue was diluted with distilled water and then purified with preparative HPLC (A: aq. 0.005% HCOOH in H2O, B: 0.005% HCOOH in CH3CN, organic phase gradient 5% to 20%/30 min.). The collected compound-containing fractions were freeze-dried to afford pure product.
Cell-binding assay
The assay was carried out essentially as described previously but with minor modifications.16,3335S-labeled Ad37 virions (5 × 108 per well) were mixed with or without the trivalent sialic acid derivatives, GD1a glycan or sialic acid at various concentrations in binding buffer (50 μL; BB: Dulbecco's modified eagle's medium containing 1% BSA (Roche AB, Stockholm, Sweden) and HEPES (20 mM, EuroClone, Milan, Italy), pH 7.5). The mixtures were then added to HCE cells prepelleted (1 × 105 per well) in a 96-well microplate. After re-suspension, the mixtures were incubated at +4 °C for 1 h. Finally, unbound virions were washed away with BB and the cell-associated radioactivity was counted by using a Wallac 1409 scintillation counter.Infection assay
The assay was carried out essentially as described previously with minor modification.16,33 6 × 106 non-labeled virions were added to serum free growth media (50 μL), with or without trivalent sialic acid derivatives, GD1a glycan or sialic acid at various concentrations. The resulting mixtures were then added to monolayers of HCE cells in a 96-well plate (3 × 104 cells per well) and incubated at +4 °C. After 1 h, unbound virions were washed away with serum free growth media. Cells were then incubated with growth media containing 1% fetal bovine serum (FBS) at +37 °C. After 44 h of infection, the cells were rinsed with PBS, fixed with cold (−20 °C) 99% methanol for 10 min and incubated with rabbit polyclonal anti-Ad37 antibodies diluted 1:100 in PBS (pH 7.4) at room temperature. After 1 h, the cells were washed in PBS and incubated with swine anti-rabbit-IgG Alexa flour 647 secondary antibodies diluted 1:250 in PBS for 1 h at room temperature. Finally, the cells were washed in PBS and the number of infected cells was quantified in TROPHOS Plate RUNNER (immunofluorescence microscope).
Surface plasmon resonance (SPR)
The affinity measurements were performed using a surface plasmon resonance BIAcore T200 instrument. Ad37 knob proteins were covalently coupled to a CM5 sensorchip using the amine coupling kit (GE Healthcare), to a concentration of 14–15 ng mm−2 (∼15000 RU). Binding of the trivalent sialic acid conjugates ME0322, 11a, 11b, 17a and 17b to the immobilized knob was performed in 10 mM HEPES, 0.15 M NaCl and 0.05% P20 pH 7.4 (1× HBS-EP+, GE Healthcare). The concentrations of trivalent sialic acid used were 400, 200, 100, 50 (twice), 25, 12.5 (twice), 6.25, 3.125, 1.56 and 0.78 μM. The experiment was performed three times for ME0322, 11a, 11b, 17a and 17b. The binding affinities (KDs) were calculated using BIAcore T200 evaluation software.
Protein production and structure determination
Expression and purification of Ad37 fiber knob protein were essentially carried out as described previously.17 For co-crystallization Ad37 fiber knob trimers were concentrated to 13.0–14.4 mg mL−1 and then incubated with a 1.3 fold excess of the trivalent sialic acid conjugates 11a, 11b, 12b, 17a or 17b. Co-crystals of Ad37-inhibitor complexes were grown following the approach described previously.21 For complex production of Ad37-12a, Ad37 fiber knobs were soaked for 2 h in reservoir solution containing 2 mM conjugate 12a. Crystals were cryo-protected by using 29% (wt/vol) polyethylene glycol 8000, 50 mM zinc acetate, and 100 mM HEPES (pH 6.9–7.2), then flash frozen in liquid nitrogen, followed by data collection on beamlines X06SA and X06DA at the SLS (Villigen, Switzerland) as well as MX-14-1 at BESSY (Berlin, Germany). Diffraction data were recorded with a Pilatus 6M (for 11a, 11b, 17a and 17b) and a Pilatus 2M-F (for 12a and 12b) pixel detector and processed with the XDS-software.36 The structures of Ad37 in complex with 11a, 11b, 12a, and 12b were solved first by molecular replacement using Phaser37 in CCP438 and the native Ad37 knob trimer (pdb-code: 1uxe35) as the search model. For the structures of Ad37 in complex with 17a and 17b, Molrep39 was used for the same purpose. All conjugates were unambiguously placed in Fco-crystallized/soaked − Fnative difference Fourier maps, incorporated into the model, and refined with restraints from either the Refmac540 or the PHENIX41 monomer library and the PRODRG2 server42 (for the ligand). Structural refinement was carried out by alternating rounds of model building in Coot43 and restrained refinement including a combination of isotropic B-factor refinement and the transition-libration-screw method (TLS)44 with Refmac5 or PHENIX (for compound 17b). In the case of 17a, high resolution allowed for an anisotropic refinement of B-factors for all atoms in PHENIX. For TLS refinement, each protomer in the asymmetric unit was attributed to one TLS group (for Ad37-12a two TLS groups per protomer were used). In the case of 17b, PHENIX automatically assigned TLS groups. Waters were located with ARP/wARP45 solvent in CCP4. Simulated annealing was carried out with PHENIX. The final models had excellent geometry. All figures were prepared with PyMOL.46 Statistics on data collection and refinement are given in Table S1† and the simulated annealing omit difference electron density maps for conjugates 11a, 11b, 12a, 12b, 17a and 17b are given in Fig. 4.Ophthalmic toxicity in rabbit
The experiment was performed in agreement with the EMEA-guideline for local tolerance testing of medicinal products.47 Compound 17a was dissolved in 0.9% aq. NaCl to a final concentration of 1 mg mL−1. Six male New Zealand white rabbits were obtained and were allowed to acclimatize before start of the study. The animals were divided into two groups of three. The compound (1 mg mL−1 in 0.9% aq. NaCl) or vehicle (0.9% aq. NaCl) was administered topically in a volume of 40 μL hourly (4 administrations during Saturday and Sunday and 8 administrations during weekdays; 5 × 8 + 2 × 4 = 48 administrations in total) in both eyes during seven days. Before the first administration, day 2, and at day 8 the animals were subjected to body weight recordings, slit lamp examinations, intraocular pressure, and pachymetry measurements under local anesthesia. The study was performed at Adlego Biomedical AB (Uppsala, Sweden) with approval of the local animal ethics committee in Stockholm (N169/14).Acknowledgements
Author contributions: The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. R. C., M. S. and W. Q. synthesized the compounds. J. B. and A. M. L. performed the crystallography experiments. N. C. and R. J. S. performed the cell-based assays. L. F. did the SPR experiments. M. E., N. A., T. S. and R. C. designed the experiments and analyzed the data together with J. B., A. M. L., N. C. and L. F.Funding sources: Swedish Research Council: Dnr: 521-2010-3078 and 621-2010-4746; Knut och Alice Wallenbergs Stiftelse: Dnr: 2009.0009; Stiftelsen för Strategisk Forskning: Dnr: F06-0011; Torsten Söderbergs Stiftelse: Dnr: M4/11; Deutsche Forschungsgemeinschaft SFB 685.
Notes and references
- W. S. M. Wold and M. S. Horwitz, in Fields Virology, ed. D. M. Knipe, P. M. Howley, D. E. Griffin, A. M. Martin, R. A. Lamb, B. Roizman and S. E. Straus, Lippincott, Williams and Wilkins, Philadelphia, PA, 5 edn, 2007, vol. 2, pp. 2395–2436 Search PubMed.
- H. Ishiko, Y. Shimada, T. Konno, A. Hayashi, T. Ohguchi, Y. Tagawa, K. Aoki, S. Ohno and S. Yamazaki, J. Clin. Microbiol., 2008, 46, 2002–2008 CrossRef CAS PubMed.
- M. Walsh, A. Chintakuntlawar, C. Robinson, I. Madisch, B. z. Harrach, N. Hudson, D. Schnurr, A. Heim, J. Chodosh, D. Seto and M. Jones, PLoS One, 2009, 4, e5635 Search PubMed.
- M. R. Hilleman and J. H. Werner, Proc. Soc. Exp. Biol. Med., 1954, 85, 183–188 CrossRef CAS PubMed.
- W. P. Rowe, R. J. Huebner, L. K. Gilmore, R. H. Parrott and T. G. Ward, Proc. Soc. Exp. Biol. Med., 1953, 84, 570–573 CrossRef CAS PubMed.
- J. De Jong, A. Wermenbol, M. Verweij-Uijterwaal, K. Slaterus, P. Wertheim-Van Dillen, G. Van Doornum, S. Khoo and J. Hierholzer, J. Clin. Microbiol., 1999, 37, 3940–3945 CAS.
- J. R. Klinger, M. P. Sanchez, L. A. Curtin and D. M. M. B, Am. J. Respir. Crit., 1998, 157, 645–649 CrossRef CAS PubMed.
- P. Kinchington, E. Romanowski and Y. Jerold Gordon, J. Antimicrob. Chemother., 2005, 55, 424–429 CrossRef CAS PubMed.
- E. De Clercq, Nat. Rev. Drug Discovery, 2002, 1, 13–25 CrossRef CAS PubMed.
- K. Aoki and Y. Tagawa, Int. Ophthalmol. Clin., 2002, 42, 49–54 CrossRef PubMed.
- G. Huang, W. Yao, W. Yu, L. Mao, H. Sun, W. Yao, J. Tian, L. Wang, Z. Bo, Z. Zhu, Y. Zhang, Z. Zhao and W. Xu, PLoS One, 2014, 9, e110781 Search PubMed.
- H. Ishiko, Y. Shimada, T. Konno, A. Hayashi, T. Ohguchi, Y. Tagawa, K. Aoki, S. Ohno and S. Yamazaki, J. Clin. Microbiol., 2008, 46, 2002–2008 CrossRef CAS PubMed.
- H. Kaneko, K. Aoki, S. Ishida, S. Ohno, N. Kitaichi, H. Ishiko, F. Tsuguto, Y. Ikeda, M. Nakamura, G. Gonzalez, K. O. Koyanagi, H. Watanabe and T. Suzutani, J. Gen. Virol., 2011, 92, 1251–1259 CrossRef CAS PubMed.
- M. Azar, D. Dhaliwal, K. Bower, R. Kowalski and Y. Gordon, Am. J. Ophthalmol., 1996, 121, 711–712 CrossRef CAS PubMed.
- N. Arnberg, Trends Pharmacol. Sci., 2012, 33, 442–448 CrossRef CAS PubMed.
- N. Arnberg, K. Edlund, A. Kidd and G. Wadell, J. Virol., 2000, 74, 42–48 CrossRef CAS PubMed.
- E. Nilsson, R. Storm, J. Bauer, S. Johansson, A. Lookene, J. Ångström, M. Hedenström, T. Eriksson, L. Frängsmyr, S. Rinaldi, H. Willison, F. Pedrosa Domellöf, T. Stehle and N. Arnberg, Nat. Med., 2011, 17, 105–109 CrossRef CAS PubMed.
- K. Aplander, M. Marttila, S. Manner, N. Arnberg, O. Sterner and U. Ellervik, J. Med. Chem., 2011, 54, 6670–6675 CrossRef CAS PubMed.
- S. Johansson, N. Arnberg, M. Elofsson, G. Wadell and J. Kihlberg, ChemBioChem, 2005, 6, 358–364 CrossRef CAS PubMed.
- S. Johansson, E. Nilsson, M. Elofsson, N. Ahlskog, J. Kihlberg and N. Arnberg, Antiviral Res., 2007, 73, 92–100 CrossRef CAS PubMed.
- S. Spjut, W. Qian, J. Bauer, R. Storm, L. Frängsmyr, T. Stehle, N. Arnberg and M. Elofsson, Angew. Chem., Int. Ed., 2011, 50, 6519–6521 CrossRef CAS PubMed.
- E. A. L. Biessen, D. M. Beuting, H. C. P. F. Roelen, G. A. van de Marel, J. H. Van Boom and T. J. C. Van Berkel, J. Med. Chem., 1995, 38, 1538–1546 CrossRef CAS PubMed.
- D. T. Connolly, R. R. Townsend, K. Kawaguchi, W. R. Bell and Y. C. Lee, J. Biol. Chem., 1982, 257, 939–945 CAS.
- R. T. Lee, P. Lin and Y. C. Lee, Biochemistry, 1984, 23, 4255–4261 CrossRef CAS PubMed.
- S. Andre, D. V. Jarikote, D. Yan, L. Vincenz, G.-N. Wang, H. Kaltner, P. V. Murphy and H.-J. Gabius, Bioorg. Med. Chem. Lett., 2012, 22, 313–318 CrossRef CAS PubMed.
- I. Azcune, E. Balentova, M. Sagartzazu-Aizpurua, S. J. Ignacio, J. I. Miranda, R. M. Fratila and J. M. Aizpurua, Eur. J. Org. Chem., 2013, 2434–2444 CrossRef CAS.
- V. Cendret, M. Francois-Heude, A. Mendez-Ardoy, V. Moreau, F. J. M. Garcia and F. Djedaini-Pilard, Chem. Commun., 2012, 48, 3733–3735 RSC.
- I. S. MacPherson, J. S. Temme, S. Habeshian, K. Felczak, K. Pankiewicz, L. Hedstrom and I. J. Krauss, Angew. Chem., Int. Ed., 2011, 50, 11238–11242 CrossRef CAS.
- V. Percec, P. Leowanawat, H.-J. Sun, O. Kulikov, C. D. Nusbaum, T. M. Tran, A. Bertin, D. A. Wilson, M. Peterca, S. Zhang, N. P. Kamat, K. Vargo, D. Moock, E. D. Johnston, D. A. Hammer, D. J. Pochan, Y. Chen, Y. M. Chabre, T. C. Shiao, M. Bergeron-Brlek, S. Andre, R. Roy, H.-J. Gabius and P. A. Heiney, J. Am. Chem. Soc., 2013, 135, 9055–9077 CrossRef CAS PubMed.
- F. Pertici and R. J. Pieters, Chem. Commun., 2012, 48, 4008–4010 RSC.
- S. Johansson, E. Nilsson, W. Qian, D. Guilligay, T. Crepin, S. Cusack, N. Arnberg and M. Elofsson, J. Med. Chem., 2009, 52, 3666–3678 CrossRef CAS PubMed.
- A. Marra and P. Sinay, Carbohydr. Res., 1989, 187, 35–42 CrossRef CAS.
- N. Arnberg, A. Kidd, K. Edlund, F. Olfat and G. Wadell, J. Virol., 2000, 74, 7691–7693 CrossRef CAS PubMed.
- M. Sun, C.-Y. Hong and C.-Y. Pan, J. Am. Chem. Soc., 2012, 134, 20581–20584 CrossRef CAS PubMed.
- W. P. Burmeister, D. Guilligay, S. Cusack, G. Wadell and N. Arnberg, J. Virol., 2004, 78, 7727–7736 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. J. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- A. Vagin and A. Teplyakov, J. Appl. Crystallogr., 1997, 30, 1022–1025 CrossRef CAS.
- G. N. Murshudov, P. Shubak, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- A. W. Schuttelkopf and D. M. F. van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. D. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- P. D. Painter and E. A. Merritt, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 439–450 Search PubMed.
- V. S. Lamzin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1993, 49, 129–147 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, LLC.
- Committee for Proprietary Medicinal Products (CPMP). Note for guidance on non-clinical local tolerance testing of medicinal products. CPMP/SWP/2145/00. 1 March 2001. London, The European Agency for the Evaluation of Medicinal Products.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, characterization and copies of NMR spectra for isolated intermediates and target compounds; SPR data; X-ray crystallographic tables. See DOI: 10.1039/c5ob01025j |
| ‡ Current address: Centre for Molecular Medicine Norway, University of Oslo, PO Box 1137 Blindern, 0318 Oslo, Norway. |
| § Atomic coordinates and structure factors have been deposited with the Protein Data Bank under Accession Codes 4K6T (Ad37-11a), 4K6U (Ad37-11b), 4K6W (Ad37-12a), 4K6V (Ad37-12b), 4XQA (Ad37-17a) and 4XQB (Ad37-17b). |
| This journal is © The Royal Society of Chemistry 2015 |