
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnatomy of gold catalysts: facts and myths
Beatrice
Ranieri
a,
Imma
Escofet
a and
Antonio M.
Echavarren
*ab
aInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: aechavarren@iciq.es
bDepartament de Química Analítica i Química Orgànica, Universitat Rovira i Virgili, C/Marcel·li Domingo s/n, 43007 Tarragona, Spain
First published on 19th May 2015
Abstract
This review article covers the main types of gold(I) complexes used as precatalysts under homogeneous conditions in organic synthesis and discusses the different ways of catalyst activation as well as ligand, silver, and anion effects.
1. Introduction
Although a few reports were already published in the thirties on the chlorination of arenes catalysed or mediated by AuCl and AuCl3,1,2 the first important application of gold(I) complexes in homogeneous catalysis, namely the formation of acetals by addition of methanol to alkynes, was reported by the group of Teles less than 20 years ago.3 Since then, homogeneous gold catalysis has seen an almost exponential growth leading to the development of many synthetic transformations4 of considerable importance for the build-up of complex molecular systems.5Although much is known about the basic mechanisms for the activation of unsaturated functional groups, such as alkynes and allenes,6 there is still a need to more clearly understand the mechanisms of reactions catalysed by gold(I) and gold(III) in order to develop more efficient and selective transformations. This is particularly important in the case of enantioselective gold catalysis, an area in which very little rational design has been reported to date. Furthermore, most of the gold-catalysed reactions are carried out by using stable linear dicoordinated chloride gold(I) complexes [LAuCl], where L is a phosphine or other donating ligand, which are precatalysts that have to be activated by chloride abstraction, leading to species that are often formulated as monocoordinated [LAu]+ complexes. However, no structural proof of the existence of these coordinatively unsaturated d12 electron species has yet been obtained.
There is also some confusion regarding the particular role of silver(I) salts, often used as co-catalysts, which has led to invoke the so-called “silver effects” in gold(I) catalysis.
This review article covers, mainly from a personal experience, known facts and discusses some “myths” about ligand and counterion effects7 on gold catalysis, as well as the different activation modes commonly used to generate active species from a stable precatalyst.
2. Gold(I) vs. gold(III) catalysis
Most gold-catalysed reactions rely on the use of stable gold(I) precatalysts, whereas gold(III) complexes have been less explored.8 Switching from gold(I) to gold(III) can have a profound effect, even leading to divergent reaction pathways. This is well exemplified in the reaction of indole 1 with [(PPh3)AuCl] in the presence of AgSbF6, which leads to indolenine-fused cyclobutane 2 by a formal [2 + 2] cycloaddition between the indole and an allene formed in situ, whereas the use of dichloro(pyridine-2-carboxylato)gold(III) gave 3, a product of a [3 + 2] cycloaddition (Scheme 1).9 | ||
| Scheme 1 Reactivity of gold(I) versus gold(III) in the cyclisation of indole 1. | ||
A dramatic effect in the reaction outcome was also found in the cyclisation of alkynyl-substituted indoles 4 (Scheme 2).10 By using gold(I) catalysts, a 7-exo-dig cyclisation led to 5, whereas AuCl3 gave indoloazocines 6 by a formal 8-endo-dig cyclisation. Here, it is important to note that the AuCl also provided indoloazocines 6, although the isolated yields were slightly lower. This suggests that chloride as the ligand, more than the oxidation state of the actual catalytic species, is responsible for this change in the cyclisation mode, which has also been observed in the cycloisomerisation of alkynyl cycloheptatrienes.11
 | ||
| Scheme 2 Formation of seven- and eight-membered indole-fused rings using gold(I) and gold(III). | ||
Divergent cyclisation modes were also observed by treating the alkynyl benzothioamide 7 with gold(I) or gold(III), leading to acyclic (aryl)(heteroaryl)carbene gold complex 8 or mesoionic carbene complex 9 respectively (Scheme 3).12
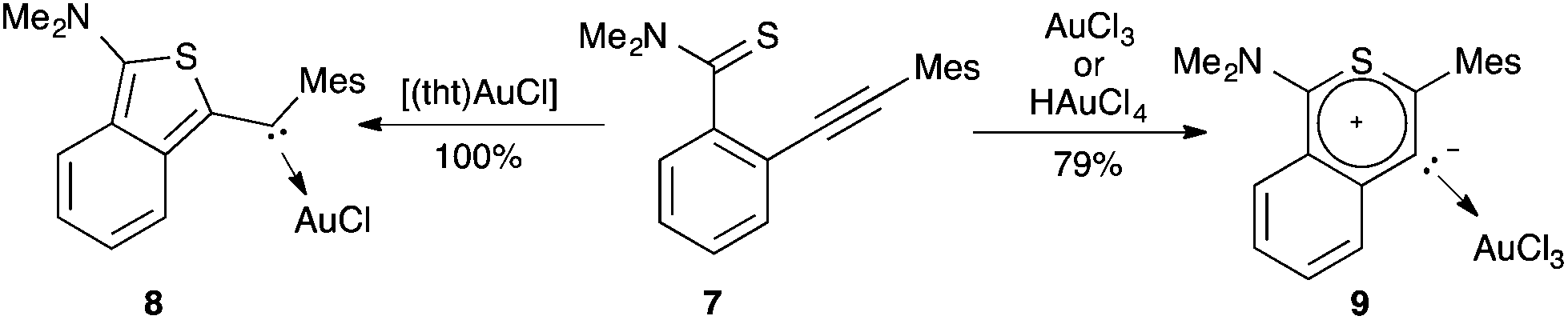 | ||
| Scheme 3 Formation of carbenes promoted by gold(I) and gold(III) by 5-exo-dig or 6-endo-dig ring closure. | ||
The different reactivity found in the reaction between ketene acetal 10 and cinnamaldehyde 11 depending on the oxidation state of the gold catalyst is significant (Scheme 4).13 Thus, whereas NHC-gold(I) 14b activated by AgOTf promotes formation of Mukaiyama aldol product 12 by a 1,2-addition, the new gold(III) complex 15 leads to the products of 1,4-addition 13, in a Mukaiyama–Michael reaction.
 | ||
| Scheme 4 Different reactivities of NHC gold(I) and gold(III) complexes. | ||
In many cases, it is clear that gold(III) salts act as precatalysts that are reduced in situ to gold(I),14 as in the cycloisomerization of α-amino and α-thioallenes 16 to the corresponding 3-pyrrolines and 2,5-dihydrothiophenes 17, which are most likely catalysed by gold(I) (Scheme 5).14b,c The reduction of gold(III) salts to gold(I) takes place with a variety of easily oxidized functional groups, including alcohols15 and can also take place by a photoinduced electron transfer process.16
 | ||
| Scheme 5 Gold(III) as a precatalyst in the cycloisomerization of α-amino and α-thioallenes. | ||
In this context, it may be important to recall that gold(III) salts react with water to form aquo complexes that are Brønsted acids.17 Cationic gold(I) complexes also coordinate with water18 or alcohols19 to form [LAu(ROH)]+ species, or related dinuclear complexes, in which the Lewis acid significantly enhances the Brønsted acidity of ROH.
3. Gold(I) precatalysts
In the early years of gold catalysis, gold(I) as well as gold(III) were mainly used as the chloride salts. However, it was soon realised that ligands play a fundamental role in modulating the reactivity of gold(I) catalysts in the activation of alkynes, allenes, and alkenes.4–6 In many cases, the careful choice of the ligand is essential to achieve the desired reactivity.The vast majority of gold(I) complexes show a linear dicoordination,20 although higher coordination numbers (3 or 4) are also possible.21 In general, gold(I) complexes with donating ligands that are sterically hindered are the most useful catalysts. Although many phosphine–gold(I) complexes have been prepared,22 probably the most successful are complexes of commercially available bulky dialkyl biphenyl phosphines, a type of ligands that were initially developed for Pd-catalysed C–C and C–X cross-coupling reactions.23 Thus, gold(I) complexes such as 18a–d24 are very common precatalysts that need to be activated by chloride abstraction with a silver salt to generate active catalysts. Cationic complexes 19a–c are obtained as stable white salts and are often more convenient catalysts since reactions can be carried out in the absence of silver(I) salts.25 These complexes bear acetonitrile as a relatively weak coordinating ligand, which has to be replaced by the reactive substrate via π-coordination to enter the productive catalytic cycles. Closely related complexes 20a–c with a weakly coordinating bis(trifluoromethanesulfonyl)amide ligand show similar reactivity (Fig. 1).26
 | ||
| Fig. 1 Gold(I) complexes 18–20 with dialkyl biphenyl phosphine ligands. | ||
The possible existence of a stabilizing arene–gold(I) interaction in complexes 18–20 was studied by comparing the structure of complex 19c with isoleptic silver(I) and copper(I) complexes 21a–b (Fig. 2).27 As expected, the metal–ligand distances decrease in the order Ag > Au > Cu. More interesting is the very significant difference that was observed in the P–M–NCMe angles, from an almost linear coordination for gold(I) complex 19c (173.1°) to more bent structures for silver and copper complexes 21a (168.0°) and 21b (148.8°). Moreover, the closest metal–arene distance for 19c was much larger (3.04 Å) than that in 21a (2.89 Å) or 21b (2.48 Å), the latest two values being characteristic of significant metal–arene interactions. Additional theoretical studies confirmed that in the case of gold(I) complexes of type 19, the metal centre does not interact significantly with the closest arene ring.27 Complex 22 and related anthracene complexes show the closest Au–arene distances between 2.96 and 3.246 Å,28 much longer than those found in well-characterized arene–gold(I) complexes. Very weak interactions were observed in a gold(I) complex related to 19c but with a pyrimidinium betaine at the ortho position.29
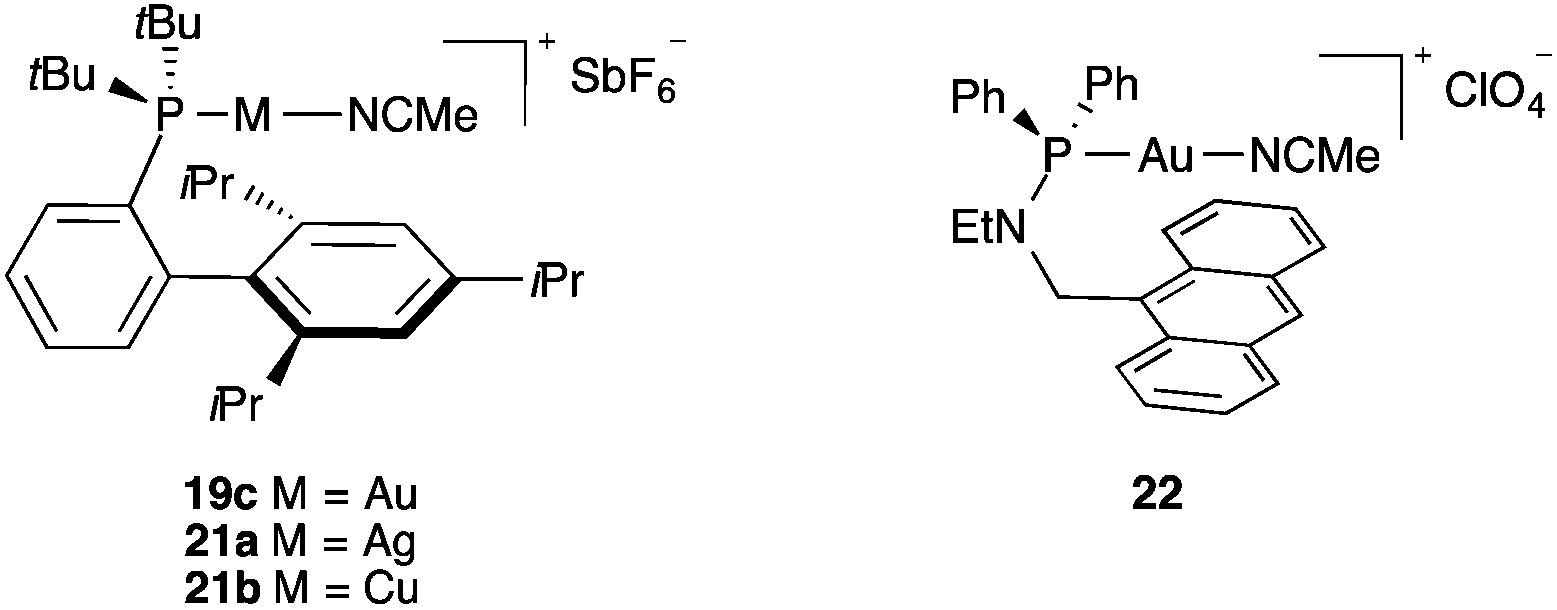 | ||
| Fig. 2 Isoleptic gold(I), silver(I) and copper(I) complexes 19c, and 21a–b and complex 22. | ||
Well-characterized gold(I)–arene complexes have been found to have the usual linear coordination around gold(I). Thus, cationic η1/ η2 complexes of type 23 were prepared by exchange of the acetonitrile ligand in complexes of type 19 in an aromatic solvent and were shown to have the shortest Au–arene distances (2.20–2.24 Å) (Fig. 3).25,27 A similar benzene complex 24 with a sterically demanding cyclic (alkyl)(amino)carbene showed slightly longer Au–arene distances (2.32–2.34 Å).30
 | ||
| Fig. 3 Arene-gold(I) complexes. | ||
The highest electrophilicity is achieved with gold(I) complexes such as 25a–b with less donating phosphite ligands (Fig. 4).31 Although less commonly employed, these complexes proved to be far superior to phosphine and NHC-derived gold catalysts for the activation of the less reactive substrates and for enhancing the carbocationic character of the reactive intermediates in the addition of C-nucleophiles of 1,6-enynes32 and in other processes.33–37 Other related highly electrophilic complexes have also been developed.38,39
 | ||
| Fig. 4 Phosphite gold(I) complexes. | ||
A variety of chiral mono- and dinuclear phosphine, phosphite, and phosphoramidite gold complexes 26–34 have been developed for the enantioselective activation of alkynes and allenes (Fig. 5).40–54
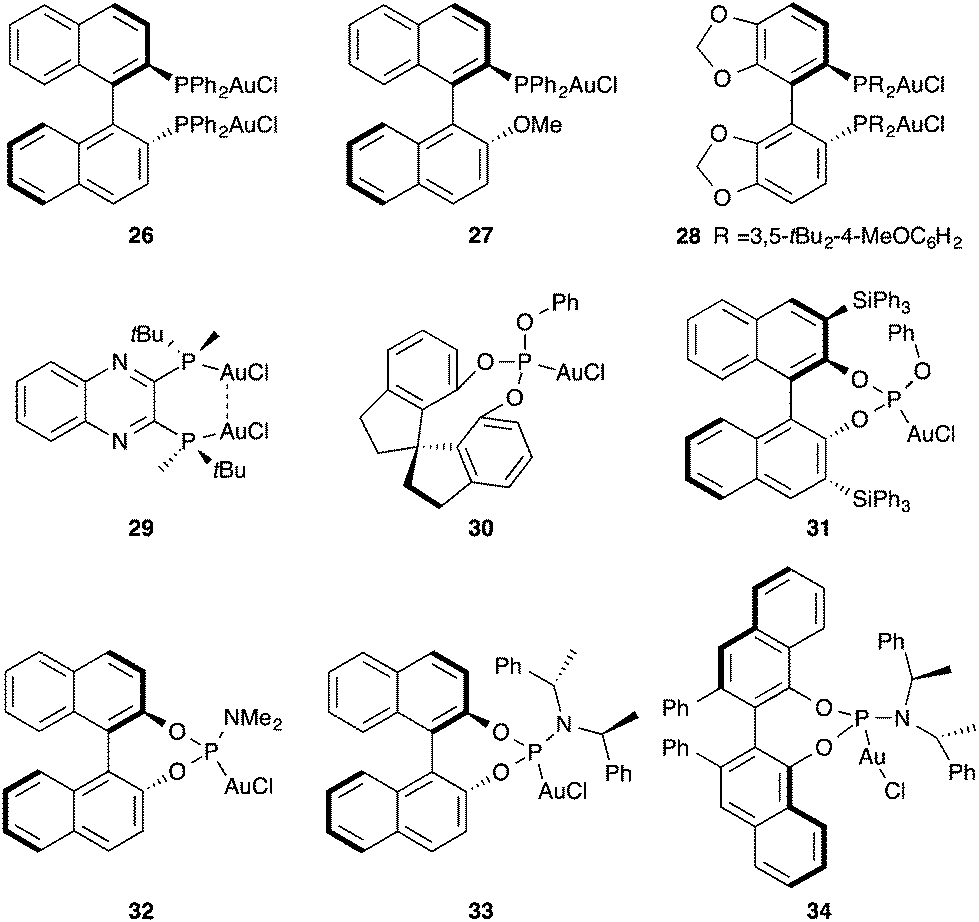 | ||
| Fig. 5 Representative chiral gold(I) complexes used in enantioselective catalysis. | ||
Another important class of gold(I) catalysts bears N-heterocyclic carbenes (NHC), which are two-electron donor bulky ligands with strong σ-donating properties. Gold(I) complexes with highly donating NHC ligands are usually less electrophilic than those with less-electron-donating phosphine ligands, which makes these catalysts more selective in many transformations.55,56 Complexes of type [(NHC)AuCl] (14) can be prepared from the free carbenes57 or via the corresponding silver(I) complexes by transmetalation58 (Scheme 6). Recently a more direct method of synthesis of complexes 14 has been developed from imidazolium salts, which bypasses the preparation of free carbenes by using K2CO3 as the base.59
 | ||
| Scheme 6 Preparation of complexes [(NHC)AuCl]. | ||
By using these procedures, many stable cationic NHC complexes have been prepared.55,60 In particular, cationic complexes such as 35a–b,32 and IPr or IMes neutral complexes with NTf2 ligands 36a–b61 have found widespread applications in homogeneous gold(I)-catalysis (Fig. 6).62 It is important to recall that the first application of NHC-gold(I) complexes in catalysis was reported in 2003 by Herrmann and co-workers, who showed that one of these complexes was moderately active in the hydration of 3-hexyne to form ketone 3-hexanone.63
 | ||
| Fig. 6 Representative N-heterocyclic carbene gold(I) complexes. | ||
Complexes 37a–b with nitrogen acyclic gold(I) carbenes (NAC), readily prepared by the addition of amines to isocyanide gold(I) complexes, have also been prepared and used in catalysis.64,65 Rotational barriers around the NAC-gold(I) bond have been used to determine the extent of π-back donation of a series of X and L ligands in [(NAC)AuX] and [(NAC)AuL]+X− complexes.66 Chiral complexes 38–41 have also been applied for several gold(I)-catalysed enantioselective transformations (Fig. 7).67–70
 | ||
| Fig. 7 Acyclic N-heterocyclic carbene gold(I) complexes. | ||
An interesting class of chiral gold(I) carbene complexes is represented by 42a–b with axially chiral triazoloisoquinolin-3-ylidene ligands, which have been applied in the enantioselective [2 + 2] cycloaddition of allenamides with dienes (Fig. 8).71 Similarly, other axially chiral NHC-Au(I) complexes have been developed and applied to the asymmetric acetoxycyclization and symmetric oxidative rearrangement of 1,6-enynes.72
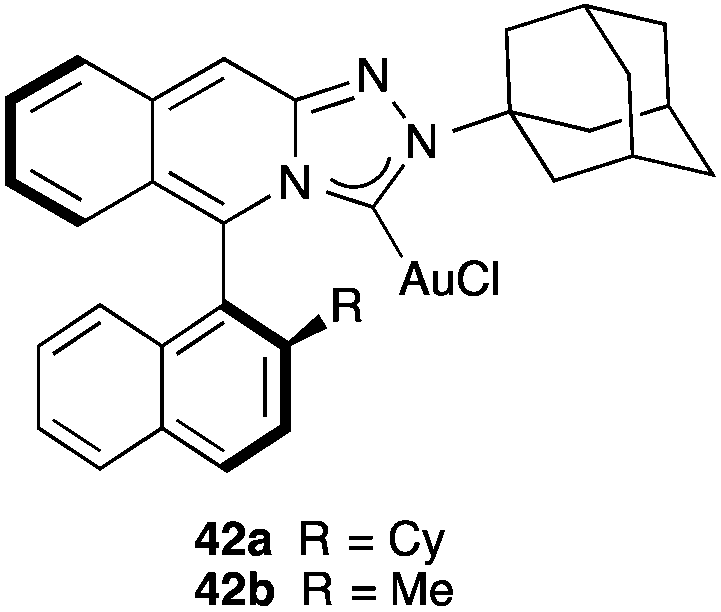 | ||
| Fig. 8 Chiral N-heterocyclic carbene gold(I) complex. | ||
There are numerous instances in which the selectivity of the reaction can be controlled by the ligand on gold.4 In a particularly illustrative example, the attack of indole to 1,6-enyne 43 was found to give two adducts 44 and 45 in ratios dependent on the gold(I) complex used as the catalyst (Scheme 7).32 Adduct 44 became the major product when phosphine gold(I) complex 18b was used, whereas 45 was the major one with complex 35a with IPr as the ligand. This different reactivity can be explained by the attack of indole onto the carbene centre of intermediate 46, which has a carbene-like character due to the higher electron-donating ability of the NHC ligand, while the corresponding intermediate with a phosphine ligand formed using catalyst 18b possesses a more carbocationic-like character, favouring attack at the benzylic position.
 | ||
| Scheme 7 Change in the reaction outcome due to different ligands. | ||
Triazole gold(I) complexes 47–49 have recently been introduced as thermally stable catalysts73 applied for a variety of synthetic applications74 (Fig. 9).
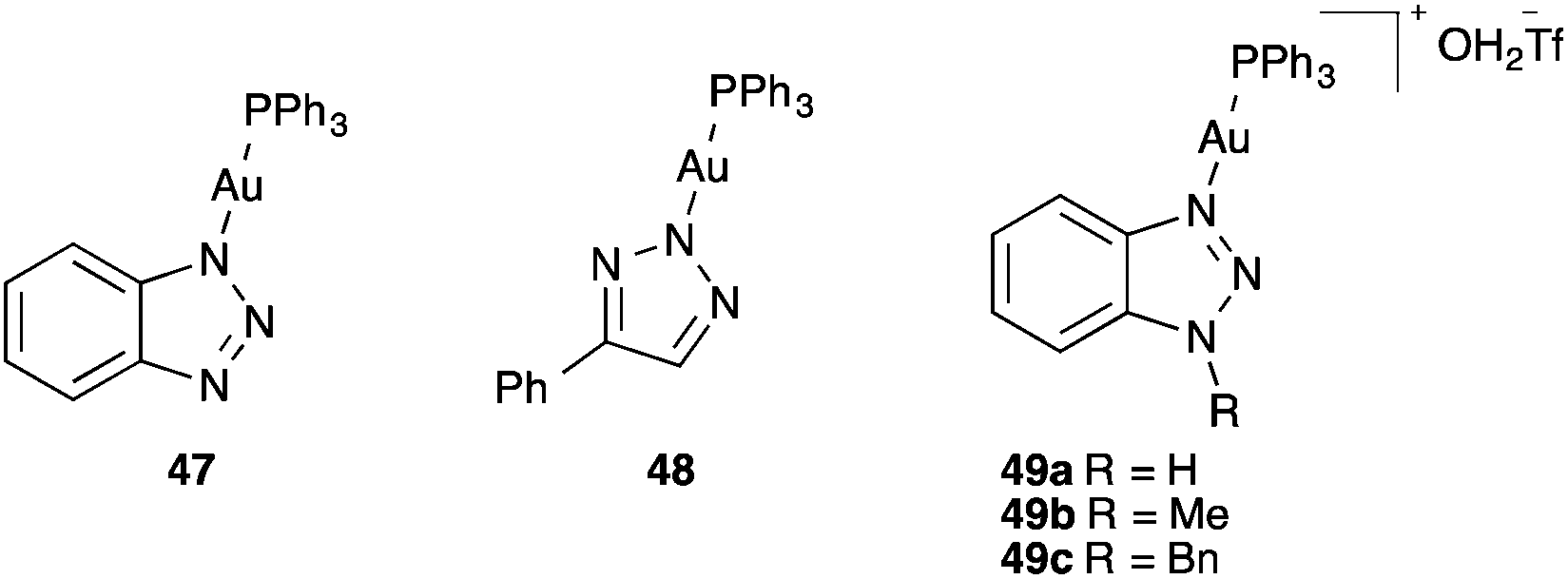 | ||
| Fig. 9 Triazole gold(I) complexes. | ||
A multipurpose gold(I) precatalyst [Au(tmbn)2](SbF6) 50 has been developed with the electron-donating nitrile 2,4,6-trimethylmethoxybenzonitrile as a ligand (tmbn) (Scheme 8).75 A variety of active catalysts [LAu(tmbn)]SbF6 and [(L–L)Au2 (tmbn)2](SbF6) were readily prepared from air-stable precursor 50 by simple ligand substitution with mono- and bidentate ligands. Recently the (η2-alkyne) complex η2-alkyne gold(I) complex [(Ph3P)Au(tBuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CtBu)]SbF6, has been used as a catalyst in some gold-catalysed transformations.76
CtBu)]SbF6, has been used as a catalyst in some gold-catalysed transformations.76
 | ||
| Scheme 8 Precatalyst [Au(tmbn)2](SbF6) and generation of active catalysts [LAu(tmbn)]SbF6. | ||
All the above-mentioned precatalysts for the activation of alkynes and other unsaturated functional groups are linear dicoordinated chloride gold(I) complexes [LAuCl]. An interesting different class of tricoordinated gold(I) complexes 51a–b with o-carborane diphosphines has been reported by the group of Bourissou (Fig. 10).77 Although the application of these complexes for the activation of alkynes has not been reported, these complexes undergo facile oxidative addition with aryl iodides in the presence of GaCl3 or AgNTf2,77a a reaction that does not take place with linear complexes [LAuCl].78 Bulky P,N-ligands such as 4-(2-(diadamanthylphosphanyl)phenyl)morpholine (Mor-Dalphos) have also been proposed to act as bidentate ligands in certain gold(I)-catalysed reactions.79
 | ||
| Fig. 10 Tricoordinated gold(I) complexes with o-carborane diphosphines. | ||
Complexes 52a–b have been shown to be more catalytically active than Johnphos gold(I) complex 19b in the intramolecular [4 + 2] cycloaddition of arylalkynes with alkenes (Fig. 11).80 These complexes have a Z-type boryl ligand, although the Au–B bond distances in the solid state (3.55–3.62 Å) indicate that this interaction is weak. Complex 53 with another Z-type boryl ligand is also catalytically active in the cyclisation of enynes.81 In this last case, presumably a stronger interaction exists, as shown in the parent neutral complex with the chloride ligand, which displays a much shorter Au–B bond distance (2.34 Å).
 | ||
| Fig. 11 Tricoordinated gold(I) complexes with o-carborane diphosphines. | ||
Interestingly, a hexanuclear gold(I) cluster 54 supported by only four phosphine ligands has also been found to be able to activate a variety of alkynyl substrates (Fig. 12).80
 | ||
| Fig. 12 Catalytically active hexanuclear gold(I) cluster. | ||
4. Ligand substitution and activation of gold(I) complexes
Linear dicoordinated gold(I) complexes [LAuX] undergo ligand substitutions by associative mechanisms,82–88 an aspect that is often overlooked in mechanistic discussions. The alternative dissociative processes would generate a coordinatively unsaturated [LAu]+ cation, which is unlikely under mild conditions. Thus, for example, high energies of activation (78–82 kcal mol−1) have been calculated for chloride dissociation in a series of [(NHC)AuCl] complexes.89In general, commonly used linear dicoordinated chloride gold(I) complexes [LAuCl] are poorly reactive in reactions towards unsaturated substrates and require activation by a silver(I) salt to form insoluble AgCl.90 There are however instances in which direct dissociation of a chloride anion has been claimed to take place from [LAuCl], although neither structural nor kinetic evidence has yet been provided. Thus, a recent report describes the hydration of terminal alkynes with [(IPr)AuCl] and related complex catalysts which takes place at 110–120 °C for 6–12 h in MeOH–H2O.91 However, it is important to remember that the hydration and the analogous alkoxycyclisation of alkynes take place under much milder conditions for complexes [LAuX] with less strongly coordinating anions such as tosylates, triflates, or triflimides.92–94
It is also somewhat puzzling that ammonium salt-tagged [(NHC)AuCl] complexes 55 (Fig. 13), which are water soluble, are catalytically active in the cyclisation of hydroxy allenes despite the presence of a strongly coordinating chloride anion as the ligand.95 At very high concentrations inside self-assembled nanospheres, [LAuCl] complexes have also been found to catalyse the intramolecular addition of alcohol to an allene to form a dihydrofuran and the single-cleavage rearrangement of a 1,6-enyne.96
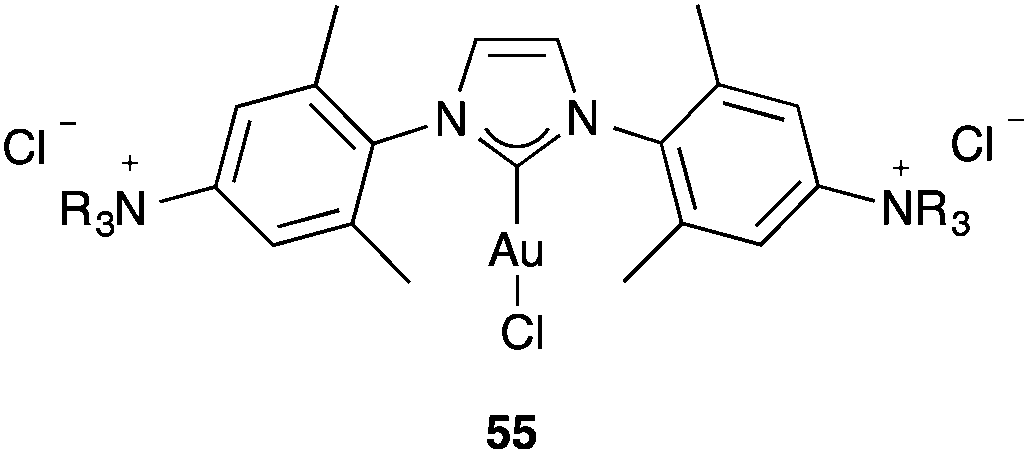 | ||
| Fig. 13 Ammonium salt-tagged NHC-gold(I) complexes. | ||
Although the structure of most complexes used in homogeneous gold(I) catalysis has been determined by X-ray diffraction, still some uncertainties remain on the structure of the actual catalytically active species that are generated in situ by chloride abstraction using different silver salts. This is particularly important in the case of chiral binuclear complexes, where the mono- or dicationic species can actually lead to very different enantioselectivities.41
Even in a very crowded environment, complex 56 with a very bulky NHC ligand (IPr**) reacts with AgNTf2 to form neutral species 57 with a covalently bound triflimide ligand (Scheme 9).97 However, a coordinatively unsaturated species [LAu]+BArF−4 was proposed to form upon treatment of complex 56 with NaBArF−4 although no structural proof was given for the resulting cationic species.98
 | ||
| Scheme 9 Complex 56 with very bulky IPr** ligand and complex 57. | ||
In this regard, it is important to mention the structure of dicationic digold(I) complex 58, which was shown to have a covalent structure with the tetrafluoroborate anions bound to the gold(I) centres (Au–F bond distances of approximately 2.1 Å) (Fig. 14).53 The same structure was retained in solution for 58 and the corresponding bis-perchlorate complex according to DOSY 1H NMR experiments. This result is rather significant, considering that in the series [R3PAu–X], tetrafluoroborate was found to have the lowest dissociation energy: CF3CO2− > Cl− > NO3− > TsO− > TfO− > BF4−,99 which points to related covalent structures for many species that are often formulated as ionic species [LAu]+X− without structural proof.
 | ||
| Fig. 14 Structure of the dicationic complex with tetrafluoroborate anions. | ||
Ligand exchange in well-characterized η2-alkyne-,100,101 η2-alkene-,102–104 η2-(1,3-diene)-,105 and η2-allene-gold(I) complexes106,107 takes place by associative mechanisms. Equilibrium constants for ligand substitution in several synthetically important phosphine and NHC gold(I) complexes have been determined in solution.108 In solution, both alkenes and alkynes coordinate to gold(I), although alkynes are always selectively activated under homogeneous conditions. This high selectivity for alkynes (alkynophilicity) has mainly a kinetic origin, driven by the very low LUMO of the coordinated triple carbon–carbon bond.109 In general, alkynes coordinate more tightly to gold(I) than do alkenes as shown in the equilibrium between complexes 59 and 60 (Scheme 10), although the differences in binding affinities are low.100a,110
 | ||
| Scheme 10 Ligand substitution in dialkyl biphenyl phosphine gold(I) complexes. | ||
In catalytic cycloisomerizations of 1,6-enynes, the kinetic data are also consistent with associative ligand substitution between [LAu(product)]+ and [LAu(substrate)]+ which is the rate-determining step.111
Cationic gold(I) species have also been generated by protonolysis of the Au–Me bond in [LAu(Me)] complexes with a strong acid such as methanesulfonic acid, fluoroboric acid, trifluoroacetic acid, or phosphotungstic acid.112,113 However, it is important to mention that the strong Brønsted acids are not innocent in the presence of alkynes and other unsaturated functional groups and could therefore lead to undesired side reactions. As an alternative, monomeric gold(I) hydroxide complex [(IPr)AuOH] 61, obtained by treating the neutral complex [(IPr)AuCl] 14b with KOH,114 can be activated in the presence of Brønsted acids to form cationic dinuclear hydroxygold(I) complex 62 (Scheme 11).
 | ||
| Scheme 11 Preparation and activation of [(IPr)AuOH] precatalyst. | ||
5. On the silver effect
Treatment of [LAuCl] with a stoichiometric amount of a non- or weakly coordinating silver salt AgX is the standard procedure for the in situ formation of the active catalyst. In some instances using a high loading of AgX was beneficial.115It is also important to mention that silver salts such as AgOTf used as the chloride abstractors can react with 1,2-dichloroethane, a commonly used solvent in gold(I) catalysis, leading to the formation of triflic acid, which could be the actual catalyst (hidden Brønsted acid catalysis).116
Furthermore, silver(I) salts are not totally innocent in gold catalysis. One of the first examples was found in the context of the intramolecular hydroarylation of 63 catalysed by gold(I) to give rise to 2-H-chromene 64 (Scheme 12).117 When 63 was treated with [(Ph3P)AuCl] and HBF4, product 64 was formed exclusively. However, by using [(Ph3P)AuCl] and AgSbF6, dimeric 65 was isolated along with 64. This [2 + 2] cycloaddition was found to be a silver(I)-catalysed process.
 | ||
| Scheme 12 Gold(I)-catalysed formation of 2-H-chromenes and the corresponding dimer. | ||
Interestingly, different enantioselectivities were obtained in the cycloisomerization of enallene 66 depending on the manner of activation of the chiral precatalyst (Scheme 13).118 When the reaction was carried out by using isolated [(R)-3,5-xylyl-binap(AuOTf)2], product 67 was obtained with much lower enantioselectivity than when the catalyst was generated in situ from [(R)-3,5-xylyl-binap(AuCl)2] and AgOTf.
 | ||
| Scheme 13 The effect of silver(I) on catalytic enantioselective cycloisomerization of enallenes. | ||
Several inconsistencies in the experimental results depending on the different procedures used for the activation of neutral gold complexes [LAuCl] by silver salts led to the suggestion of the existence of “silver effects” in gold(I) catalysis.119 In same cases, it was proposed that the nature of the particular catalytically active species generated in situ by mixing a gold catalyst [LAuCl] and AgX depended on whether or not the resulting mixture was filtered through Celite.
An evaluation of this problem by using complex 18b as a representative phosphine gold(I) complex concluded that most of so-called “silver effects” could be attributed to the ready formation of dinuclear chloride-bridged species such as 68, which were characterized by X-ray diffraction with different X anions (SbF4−, BF4−, OTf−, NTf2−) (Scheme 14).120 Presumably, an associative ligand substitution reaction initiated by coordination of silver(I) to the chloride ligand of [LAuCl] to form transient [LAuCl⋯Ag]+, followed by addition of a second molecule of [LAuCl] as the incoming ligand leads to [LAuClAuL]+ and AgCl. These dinuclear complexes 68 were readily formed regardless of the counterion when a silver salt AgX was added in the absence of the coordinating substrate in non-coordinating solvents like CH2Cl2, and were found to be considerably less catalytically active than cationic complexes [LAu(NCMe)]X towards several transformations of substituted alkynes. On the other hand, when the silver salt was added last, by premixing [LAuCl] with the substrate, formation of dinuclear chloride-bridged species such as 68 could be minimized.
 | ||
| Scheme 14 Formation of the chloride-bridged complexes in a non-coordinating solvent. | ||
Interestingly, by mixing complex 18b with AgSbF6 followed by filtration through Celite, complex 69 that shows an additional coordination of silver(I) with the aryl rings could be isolated and characterized by X-ray diffraction (Fig. 15).121
 | ||
| Fig. 15 Structure of an Au–Ag–Au triangular motif. | ||
Formation of the chloride-bridged species in a gold(I) catalysed reaction was also found in the cycloisomerisation of 1,6-enynes such as 70 in the presence of complex 72 with a secondary phosphine oxide ligand (Scheme 15).122 The yield of 71 was significantly increased by using an excess of AgSbF6, which minimized the formation of less reactive [LAuClAuL]+ species, which were detected by ESI mass spectrometry.
 | ||
| Scheme 15 Effect of the amount of silver salts on the cyclisation of enynes by the SPO-gold(I) complex. | ||
The routine use of highly hygroscopic silver salts as halide scavengers can introduce significant amounts of water in the reaction mixtures leading to unwanted hydration of alkynes and hydrolysis reactions. In addition, water reacts with gold(I) complexes to form bridged [LAu(OH)AuL]+ such as 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 120 and related complexes108,114c,e or trinuclear oxonium cations 74108,123 (Fig. 16).
120 and related complexes108,114c,e or trinuclear oxonium cations 74108,123 (Fig. 16).
 | ||
| Fig. 16 Hydroxo-bridged complex 73 and trigold(I) oxonium complexes 74. | ||
More drastic changes can take place in other cases in the presence of silver salts. Thus, upon mixing gold(I) carbene complex 14a with AgBF4 in a non-coordinating solvent, cationic bis-NHC gold(I) complex 75 was formed (Scheme 16).124
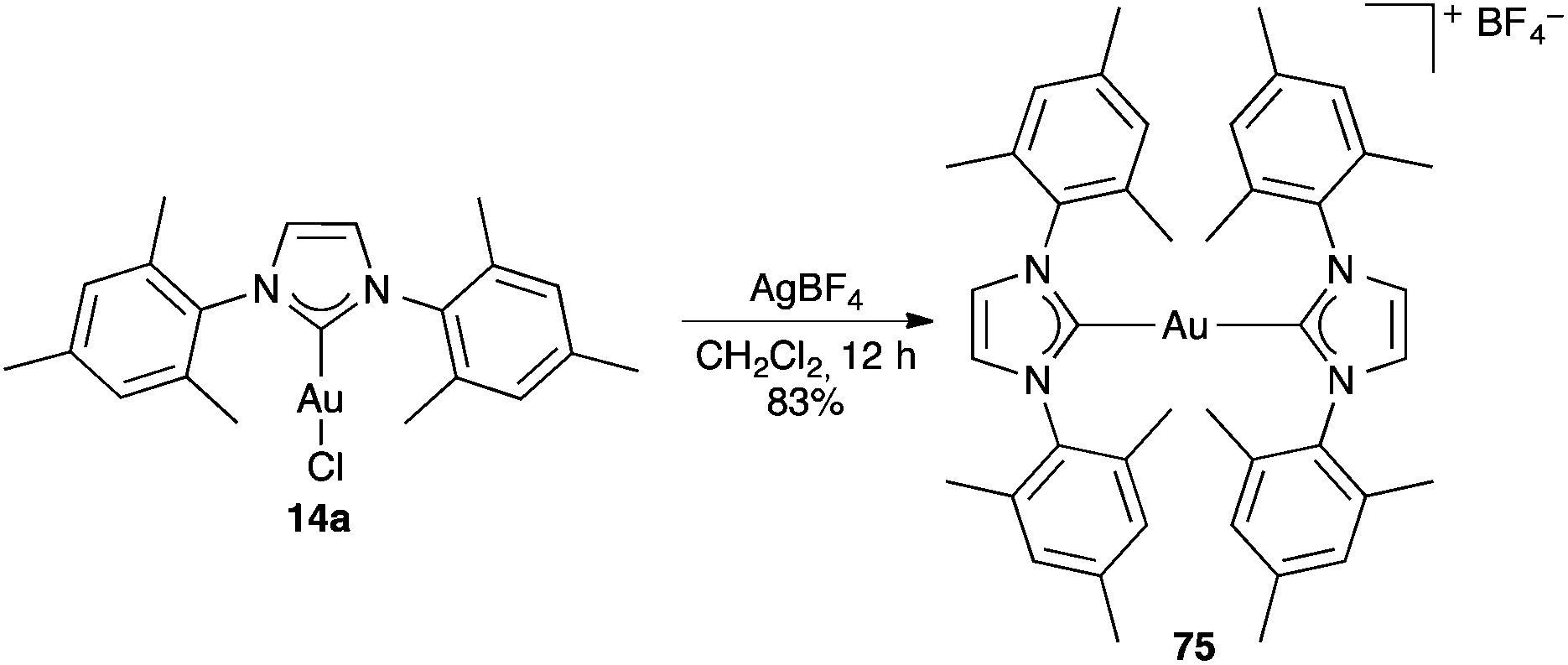 | ||
| Scheme 16 Formation of bis(IMes) gold(I) complex. | ||
As an alternative to silver(I), copper(II) salts, such as Cu(OTf)2, can also be used for the chloride abstraction of gold(I) complexes.125 Similarly, several Lewis acids of the transition and main group metals can be used for the generation of active species from [LAuCl] complexes.126
6. Digold complexes
Although digold(I) complexes have been known since the mid seventies,127 their relevance in homogeneous gold(I) catalysis has only been recognized recently.128–130 In this section, we will focus only on the findings regarding their role in gold(I)-catalysed transformations.The first evidence of the dual activation of alkynes via σ,π-activation was found in the context of the cycloisomerisation of 1,5-allenynes catalysed by [(Ph3PAu)3O]BF4 (Scheme 17).131 The experimental and computational data for the cyclisation of allenynes 76 to form 77 supported a mechanism in which the coordination of a cationic fragment [(Ph3P)Au]+ to the gold acetylide in complex 78 promoted a 5-endo-dig cyclisation via gem-diaurated species 79 and 80.
 | ||
| Scheme 17 Proposed role of gem-diaurated species in the cycloisomerization of 1,5-allenynes. | ||
The involvement of related σ,π-digold(I) species was proposed in the cycloisomerization of diynes.132 Although similar species were observed by mass spectrometry under electrospray ionization, their real involvement in the cycloisomerization of 1,6-enynes was questioned.133 Other alkenyl-86,134–137 and aryl138–140 digold complexes with Au2C three-centre-two-electron bonds have been observed.141
σ,π-Digold(I) alkyne digold(I) complexes 83 are easily formed by the in situ deprotonation of terminal alkynes in 81via gold(I) alkynyl complexes 82 (Scheme 18).100b,133,142–144 Complexes of type 83 have been identified as species out of the catalytic cycle in the cycloisomerization of 1,6-enynes,133 the intermolecular [2 + 2 + 2] cycloaddition of alkynes with oxoalkenes,143 as well as in the [2 + 2] cycloaddition of alkynes with alkenes.145
 | ||
| Scheme 18 Formation of digold-alkyne complexes. | ||
Importantly, gold(I) binds more strongly to gold(I)-acetylides than to free alkynes,146 which explains the very low reactivity of digold(I) complexes 83 in the activation of substrates containing alkyne functionalities under catalytic conditions.143,145 Formation of unproductive σ,π-digold(I) alkyne digold(I) complexes in intermolecular reactions of alkynes with alkenes and related processes could be minimized by changing the counterion of the cationic catalyst from SbF6− to the softer anion BArF−4.143,144
Although σ,π-digold(I) alkyne complexes 83 are unproductive dead-ends in reactions of alkynes with alkenes, they are excellent catalysts in reactions of diynes in which one of the alkynes is a terminal one, by allowing the simultaneous formation of a σ-alkynyl gold(I) species and a π-alkyne gold(I) that react with each others in a dual-gold(I) catalysed process.147–149 This apparent contradiction can be explained by the Janus-like character of complexes 83, which contain, in addition to the usual Lewis acid, a basic gold acetylide. Thus, the annulation of o-dialkyne derivatives 84 to form indeno[2,1-a]indenes 85 proceeds via σ,π-digold(I) alkyne complexes 86, which give rise to gold(I) vinylidenes 87 that cyclise to 88 (Scheme 19).136b
 | ||
| Scheme 19 Annulation via σ,π-digold(I) alkyne complexes to form indeno[2,1-a]indenes. | ||
7. Anion effects
The awareness of the effect of the counterion on gold(I) catalysis has experienced an outbreak recently.7,150 A particular illustrative example is the reaction of gold(I)-catalysed ring expansion alkynyl aziridines such as 89 to form pyrroles by a tandem cyclisation-opening/Wagner–Meerwein sequence.151 When the reaction was performed in the presence of AgOTf, product 90 was obtained, whereas using a catalyst with a sufficiently basic counterion such as tosylate, proton elimination and atom transfer took place to give 91 (Scheme 20). | ||
| Scheme 20 Role of counterions in the ring expansion of alkynyl aziridines. | ||
The anion effect was also studied in the gold(I)-catalysed reaction of diyne 92 to give rise to naphthalenes 93 and 94 using σ,π-digold(I) alkyne complexes 95 with different anions (Scheme 21).152 A fast transformation was observed for TfO−, although full conversion could not be obtained and, whereas the complex with PF6− as the anion gave almost full conversion after 1 h, the corresponding complex with SbF6− gave less than 20% conversion after 8 h.
 | ||
| Scheme 21 Counterion effect in the formation of naphthalenes by σ,π-digold(I) alkyne complexes. | ||
Not surprisingly, ion pairing is responsible for some of the counterion effects. Based on NMR experiments and DTF calculations, the position of the BF4− anion in systems [(Ph3P)Au(4-Me-styrene)] 96 and [IPrAu(4-Me-styrene)] 97 was studied by 19F–1H HOESY NMR spectroscopy combined with theoretical modelling (Fig. 17).153 These results demonstrate that the counterion is placed away from the gold(I) site, although the specific localization strongly depends on the neutral ligand (phosphine or NHC).
 | ||
| Fig. 17 Key 19F–1H HOESY contacts in complexes 97·BF4 (left) and 98·BF4 (right). | ||
The impact of the anion on the enantioselectivity of gold(I) catalysed reactions has also been investigated.154,155 This effect was best illustrated in the enantioselective gold(I) catalysed cyclisation of allenols such as 98 to give tetrahydrofuran 99 using achiral complexes [LAuCl] activated by silver(I) binol phosphate salts (Scheme 22) and in other related transformations of allenyl sulfonamides.154,156 Complexes [(L–L)(AuX)2] and [(L–L)Au2XCl] (L–L = bidentate phosphine) were also active in the enantioselective cyclisation of hydroxy- and sulfonamidoallenes.157 Gold complexes combined with chiral phosphoric acids have also been used in other enantioselective transformations.158 Interestingly, in the gold(I)-catalysed enantioselective hydroammination by intramolecular addition of tosylamides to allenes, the best results were obtained when using AgOPNB (PNB = p-nitrobenzoate), presumably by generating the monocationic gold complex [((R)-BINAP)Au2PNB]+.156b
 | ||
| Scheme 22 Enantioselective cyclisation of allenols with gold(I) catalysts in the presence of chiral phosphate anions. | ||
The above transformations were suggested to proceed in the presence of an ion pair [LAu]+X− with a chiral phosphate anion. However, the actual complexes 101a–b, prepared from the dimeric phosphate silver(I) salts 100a–b, are very robust covalent species both in the solid-state and in solution. (Scheme 23)159 Notably, whereas these complexes catalyse the cyclisation of allenol 98 to form tetrahydrofuran 99, they were surprisingly inactive in the cyclisation of several 1,6-enynes. However, the catalytic activity could be restored upon addition of [Ag(NCMe)2]+SbF6− or by performing the reaction in the presence of a protic solvent like MeOH. Presumably, in both cases, the associative ligand substitution step generates cationic complexes [(Ph3P)Au(S)]+ (S = MeCN or MeOH), which are catalytically active. These results show that in order to extend the chiral counterion concept to gold(I)-catalysed activation of alkynes, anionic ligands less basic than phosphates would have to be used.
 | ||
| Scheme 23 Synthesis of well-characterized chiral gold(I)-phosphate complexes. | ||
8. Conclusions
Gold(I) complexes emerged as powerful catalysts for the selective electrophilic activation of multiple bonds under mild conditions. Several aspects have to be taken into account in order to accomplish a gold(I)-catalysed reaction. Thus, although ancillary ligands tune the electronic and steric properties of the gold(I) complex, it is important to remember that precatalysts enter catalytic cycles by associative ligand substitution, which may be rate determining. Anions and anionic ligands also play a significant role in determining the reaction outcome and the enantioselectivity when using chiral catalysts.In the case of gold(III) precatalysts, uncertainties still exist regarding the actual structure and even the oxidation state of the actual catalytic species since the presence of highly donating chloride as the ligand, more than the oxidation state, seems to be responsible for the observed selectivities.
It is difficult and, in many cases, wrong to overgeneralise in gold(I) catalysis since different substrates require different modes of activation. Thus, although seemingly contradictory, σ,π-digold(I) alkyne species are both dead-ends in reactions of alkynes with alkenes and catalysts in reactions of diynes bearing terminal acetylenes, which are mechanistically very different processes. Similarly, phosphine gold(I) phosphates are good catalysts for the activation of allenes, but very poor ones in cyclizations of enynes.
Finally, particularly in the reactions of the less reactive substrates, premixing the neutral gold complexes [LAuCl] with silver salts leads to the formation of significant amounts of chloride-bridged dinuclear species [LAuClAuL]+ that are poor catalysts. In these cases, premixing [LAuCl] with the substrate, followed by addition of silver salts, minimizes the formation of the unreactive dinuclear complexes. The use of silver-free, cationic [LAu(S)]+X− is often the alternative of choice in gold(I) catalysis.
Acknowledgements
We thank MINECO (project CTQ2013-42106-P and Severo Ochoa Excellence Accreditation 2014-2018 (SEV-2013-0319), the European Research Council (advanced grant no. 321066), the AGAUR (2014 SGR 818), the Marie Curie program (postdoctoral fellowship 658256-MSCA-IF-EF-ST to B.R.), and the ICIQ Foundation.References
-
(a) M. S. Kharasch and H. S. Isbell, J. Am. Chem. Soc., 1931, 53, 3053–3059 CrossRef CAS
; (b) M. S. Kharasch and T. M. Beck, J. Am. Chem. Soc., 1934, 56, 2057–2060 CrossRef CAS
- W. Schwemberger and W. Gordon, Chem. Zentralbl., 1935, 106, 514–514 Search PubMed
- J. H. Teles, S. Brode and M. Chabanas, Angew. Chem., Int. Ed., 1998, 37, 1415–1418 CrossRef CAS
-
(a) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed
- R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115 DOI:10.1021/cr500691k
-
(a) C. Obradors and A. M. Echavarren, Chem. Commun., 2014, 50, 16–28 RSC
- A very recent review on counterion effects in homogeneous gold catalysis: M. Jia and M. Bandini, ACS Catal., 2015, 5, 1638–1652 CrossRef CAS
-
(a) D. Aguilar, M. Contel, R. Navarro and E. P. Urriolabeitia, Organometallics, 2007, 26, 4604–4611 CrossRef CAS
-
(a) L. Zhang, J. Am. Chem. Soc., 2005, 127, 16804–16805 CrossRef CAS PubMed
-
(a) C. Ferrer and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 1105–1110 CrossRef CAS PubMed
- P. R. McGonigal, C. de León, Y. Wang, A. Homs, C. R. Solorio-Alvarado and A. M. Echavarren, Angew. Chem., Int. Ed., 2012, 51, 13093–13096 CrossRef CAS PubMed
- G. Ung, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 758–761 CrossRef CAS PubMed
- C.-Y. Wu, T. Horibe, C. B. Jacobsen and F. D. Toste, Nature, 2015, 517, 449–454 CrossRef CAS PubMed
-
(a) N. Morita and N. Krause, Org. Lett., 2004, 6, 4121–4123 CrossRef CAS PubMed
- B. Pal, P. K. Sen and K. K. S. Gupta, J. Phys. Org. Chem., 2001, 14, 284–294 CrossRef CAS PubMed
-
(a) C. D. McTiernan, E. I. Alarcon, G. L. Hallett-Tapley, J. Murillo-Lopez, R. Arratia-Perez, J. C. Netto-Ferreira and J. C. Scaiano, Photochem. Photobiol. Sci., 2014, 13, 149–153 RSC
- W. Robb, Inorg. Chem., 1967, 6, 382–386 CrossRef CAS
- C. H. Chen, C. D. Wang, Y. F. Hsieh and R. S. Liu, Org. Biomol. Chem., 2014, 12, 9831–9836 CAS
- O. Kanno, W. Kuriyama, Z. J. Wang and F. D. Toste, Angew. Chem., Int. Ed., 2011, 50, 9919–9922 CrossRef CAS PubMed
- P. Schwerdtfeger, H. L. Herman and H. Schmidbaur, Inorg. Chem., 2003, 42, 1334–1342 CrossRef CAS PubMed
- M. C. Gimeno and A. Laguna, Chem. Rev., 1997, 97, 511–522 CrossRef CAS PubMed
- Lead references:
(a) A. K. Hashmi, A. Loos, S. Doherty, J. G. Knight, K. J. Robson and F. Rominger, Adv. Synth. Catal., 2011, 353, 749–759 CrossRef CAS PubMed
-
(a) S. Kaye, J. M. Fox, F. A. Hicks and S. L. Buchwald, Adv. Synth. Catal., 2001, 343, 789–794 CrossRef CAS
- C. Nieto-Oberhuber, S. López and A. M. Echavarren, J. Am. Chem. Soc., 2005, 127, 6178–6179 CrossRef CAS PubMed
- E. Herrero-Gómez, C. Nieto-Oberhuber, S. López, J. Benet-Buchholz and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 5455–5459 CrossRef PubMed
-
(a) N. Mézailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133–4136 CrossRef PubMed
- P. Pérez-Galán, N. Delpont, E. Herrero-Gómez, F. Maseras and A. M. Echavarren, Chem. – Eur. J., 2010, 16, 5324–5332 CrossRef PubMed
-
(a) F.-B. Xu, Q.-S. Li, L.-Z. Wu, X.-B. Leng, Z.-C. Li, X.-S. Zeng, Y. L. Chow and Z.-Z. Zhang, Organometallics, 2003, 22, 633–640 CrossRef CAS
- L. Noël-Duchesneau, N. Lugan, G. Lavigne, A. Labande and V. César, Eur. J. Inorg. Chem., 2015, 1752–1758 CrossRef PubMed
- V. Lavallo, G. D. Frey, S. Kousar, B. Donnadieu and G. Bertrand, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13569–13573 CrossRef CAS PubMed
- S. López, E. Herrero-Gómez, P. Pérez-Galán, C. Nieto-Oberhuber and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 6029–6032 CrossRef PubMed
-
(a) C. H. M. Amijs, C. Ferrer and A. M. Echavarren, Chem. Commun., 2007, 698–700 RSC
-
(a) S. Pascual, C. Bour, P. de Mendoza and A. M. Echavarren, Beilstein J. Org. Chem., 2011, 7, 1520–1525 CrossRef CAS PubMed
-
(a) H. Faustino, I. Alonso, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2013, 52, 6526–6530 CrossRef CAS PubMed
-
(a) B. Bolte and F. Gagosz, J. Am. Chem. Soc., 2011, 133, 7696–7699 CrossRef CAS PubMed
- D. Pflästerer, E. Rettenmeier, S. Schneider, E. de Las Heras Ruiz, M. Rudolph and A. S. K. Hashmi, Chem. – Eur. J., 2014, 20, 6752–6755 CrossRef PubMed
- C. Winter and N. Krause, Angew. Chem., Int. Ed., 2009, 51, 6339–6342 CrossRef PubMed
- M. C. Blanco Jaimes, F. Rominger, M. M. Pereira, R. M. B. Carrilho, S. A. C. Carabineiro and A. S. K. Hashmi, Chem. Commun., 2014, 50, 4937–4940 RSC
- G. Henrion, T. E. J. Chavas, X. Le Goff and F. Gagosz, Angew. Chem., Int. Ed., 2013, 52, 6277–6282 CrossRef CAS PubMed
- Reviews on enantioselective gold catalysis:
(a)
N. Huguet and A. M. Echavarren, Asymmetric Synthesis II, ed. M. Christmann and S. Bräse, Wiley-VCH Verlag, 2012, Chapter 26, pp. 205–212 Search PubMed
- M. P. Muñoz, J. Adrio, J. C. Carretero and A. M. Echavarren, Organometallics, 2005, 24, 1293–1300 CrossRef
-
(a) M. J. Johansson, D. J. Gorin, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 18002–18003 CrossRef CAS PubMed
-
(a) C. Liu and R. A. Widenhoefer, Org. Lett., 2007, 9, 1935–1938 CrossRef CAS PubMed
- C.-M. Chao, D. Beltrami, P. Y. Toullec and V. Michelet, Chem. Commun., 2009, 6988–6990 RSC
- A. Martínez, P. García-García, M. A. Fernández-Rodríguez, F. Rodríguez and R. Sanz, Angew. Chem., Int. Ed., 2010, 49, 4633–4637 CrossRef PubMed
- I. Alonso, B. Trillo, F. López, S. Montserrat, G. Ujaque, L. Castedo, A. Lledós and J. L. Mascareñas, J. Am. Chem. Soc., 2009, 131, 13020–13030 CrossRef CAS PubMed
- S. Suárez-Pantiga, C. Hernández-Díaz, E. Rubio and J. M. González, Angew. Chem., Int. Ed., 2012, 51, 11552–11555 CrossRef PubMed
- L.-I. Rodríguez, T. Roth, J. L. Fillol, H. Wadepohl and L. H. Gade, Chem. – Eur. J., 2012, 18, 3721–3728 CrossRef PubMed
- S. Handa and L. M. Slaughter, Angew. Chem., Int. Ed., 2012, 51, 2912–2915 CrossRef CAS PubMed
- E. M. Barreiro, D. F. D. Broggini, L. A. Adrio, A. J. P. White, R. Schwenk, A. Togni and K. K. Hii, Organometallics, 2012, 31, 3745–3754 CrossRef CAS
- N. Delpont, I. Escofet, P. Pérez-Galán, D. Spiegl, M. Raducan, C. Bour, R. Sinisi and A. M. Echavarren, Catal. Sci. Technol., 2013, 3, 3007–3012 CAS
- E. M. Barreiro, E. V. Boltukhina, A. J. P. White and K. K. M. Hii, Chem. – Eur. J., 2015, 21, 2686–2690 CrossRef CAS PubMed
- M.-A. Abadie, X. Trivelli, F. Medina, F. Capet, P. Roussel, F. Agbossou-Niedercorn and C. Michon, ChemCatChem, 2014, 6, 2235–2239 CrossRef CAS PubMed
-
(a) H. Teller, S. Flügge, R. Goddard and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 1949 CrossRef CAS PubMed
-
(a) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776–1782 RSC
- K. Molawi, N. Delpont and A. M. Echavarren, Angew. Chem., Int. Ed., 2010, 49, 3517–3519 CrossRef CAS PubMed
- P. de Frémont, N. M. Scott, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 2411–2418 CrossRef
- H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972–975 CrossRef CAS
-
(a) R. Visbal, A. Laguna and M. C. Gimeno, Chem. Commun., 2013, 49, 5642–5644 RSC
- P. de Frémont, E. D. Stevens, M. R. Fructos, M. M. Díaz-Requejo, P. J. Pérez and S. P. Nolan, Chem. Commun., 2006, 2045–2047 RSC
- L. Ricard and F. Gagosz, Organometallics, 2007, 26, 4704–4707 CrossRef CAS
-
(a) Y. Peng, M. Yu and L. Zhang, Org. Lett., 2008, 10, 5187–5190 CrossRef CAS PubMed
- S. K. Schneider, W. A. Herrmann and E. Herdtweck, Z. Anorg. Allg. Chem., 2003, 629, 2363–2370 CrossRef CAS PubMed
-
(a) C. Bartolomé, M. Carrasco-Rando, S. Coco, C. Cordovilla, J. M. Martín-Alvarez and P. Espinet, Inorg. Chem., 2008, 47, 1616–1624 CrossRef PubMed
- A. S. K. Hashmi, T. H. C. Lothschütz and F. Rominger, Adv. Synth. Catal., 2010, 352, 1315–1337 CrossRef CAS PubMed
- G. Ciancaleoni, L. Biasiolo, G. Bistoni, A. Macchioni, F. Tarantelli, D. Zuccaccia and L. Belpassi, Chem. – Eur. J., 2014, 21, 2467–2473 CrossRef PubMed
- C. Bartolomé, D. García–Cuadrado, Z. Ramiro and P. Espinet, Inorg. Chem., 2010, 49, 9758–9764 CrossRef PubMed
-
(a) L. M. Slaughter, ACS Catal., 2012, 2, 1802–1816 CrossRef CAS
- Y.-M. Wang, C. N. Kuzniewski, V. Rauniyar, C. Hoong and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 12972–12975 CrossRef CAS PubMed
- H. G. Raubenheimer, Angew. Chem., Int. Ed., 2012, 51, 5042–5044 CrossRef CAS PubMed
- J. Francos, F. Grande-Carmona, H. Faustino, J. Iglesias-Sigüenza, E. Díez, I. Alonso, R. Fernańdez, J. M. Lassaletta, F. Loṕez and J. L. Mascareñas, J. Am. Chem. Soc., 2012, 134, 14322–14325 CrossRef CAS PubMed
- W. Wang, J. Yang, F. Wang and M. Shi, Organometallics, 2011, 30, 3859–3869 CrossRef CAS
-
(a) H. Duan, S. Sengupta, J. L. Petersen, N. G. Akhmedov and X. Shi, J. Am. Chem. Soc., 2009, 131, 12100–12102 CrossRef CAS PubMed
-
(a) Y. Chen, W. Yan, N. G. Akhmedov and X. Shi, Org. Lett., 2010, 12, 344–347 CrossRef CAS PubMed
- M. Raducan, C. Rodríguez-Escrich, X. C. Cambeiro, E. C. Escudero-Adán, M. A. Pericas and A. M. Echavarren, Chem. Commun., 2011, 47, 4893–4895 RSC
- D. Zhu, X. Cao and B. Yu, Org. Chem. Front., 2015, 2, 360–365 RSC
-
(a) M. Joost, A. Zeineddine, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654–14657 CrossRef CAS PubMed
- M. Livendahl, C. Goehry, F. Maseras and A. M. Echavarren, Chem. Commun., 2014, 50, 1533–1536 RSC
-
(a) K. Ji, Y. Zhao and L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 6508–6512 CrossRef CAS PubMed
- E. S. Smirnova and A. M. Echavarren, Angew. Chem., Int. Ed., 2013, 52, 9023–9026 CrossRef CAS PubMed
- F. Inagaki, C. Matsumoto, Y. Okada, N. Maruyama and C. Mukai, Angew. Chem., Int. Ed., 2014, 54, 818–822 CrossRef PubMed
- S. Komiya, T. A. Albright, R. Hoffmann and J. K. Kochi, J. Am. Chem. Soc., 1976, 98, 7255–7265 CrossRef CAS
- C. B. Colburn, W. E. Hill, C. A. Mcauliffe and R. V. D. Parish, J. Chem. Soc., Chem. Commun., 1979, 218–219 RSC
- P. N. Dickson, A. Wehrli and G. Geier, Inorg. Chem., 1988, 27, 2921–2925 CrossRef CAS
- S. Komiya, M. Iwata, T. Sone and A. Fukuoka, J. Chem. Soc., Chem. Commun., 1992, 1109–1110 RSC
- A. Zhdanko and M. E. Maier, Organometallics, 2013, 32, 2000–2006 CrossRef CAS
- Y. Xi, Q. Wang, Y. Su, M. Li and X. Shi, Chem. Commun., 2014, 50, 2158–2160 RSC
- R. E. M. Brooner, B. D. Robertson and R. A. Widenhoefer, Organometallics, 2014, 33, 6466–6473 CrossRef CAS
- A. Couce-Rios, G. Kovács, G. Ujaque and A. Lledós, ACS Catal., 2015, 5, 815–829 CrossRef CAS
- M. Preisenberger, A. Schier and H. Schmidbaur, J. Chem. Soc., Dalton Trans., 1999, 1645–1650 RSC
- F. Li, N. Wang, L. Lu and G. Zhu, J. Org. Chem., 2015, 80, 3538–3546 CrossRef CAS PubMed
-
(a) L. Biasiolo, M. Trinchillo, P. Belanzoni, L. Belpassi, V. Busico, G. Ciancaleoni, A. D'Amora, A. Macchioni, F. Tarantelli and D. Zuccaccia, Chem. – Eur. J., 2014, 20, 14594–14598 CrossRef CAS PubMed
- A. Leyva and A. Corma, J. Org. Chem., 2009, 74, 2067–2074 CrossRef CAS PubMed
- J. A. Goodwin and A. Aponick, Chem. Commun., 2015, 51, 8730–8741 RSC
- K. Belger and N. Krause, Eur. J. Org. Chem., 2015, 220–225 CrossRef CAS PubMed
- R. Gramage-Doria, J. Hessels, S. H. A. M. Leenders, O. Tröppner, M. Dürr, I. Ivanović Burmazović and J. N. H. Reek, Angew. Chem., Int. Ed., 2014, 53, 13380–13384 CrossRef CAS PubMed
- S. G. Weber, D. Zahner, F. Rominger and B. F. Straub, ChemCatChem, 2013, 5, 2330–2335 CrossRef CAS PubMed
- S. G. Weber, D. Zahner, F. Rominger and B. F. Straub, Chem. Commun., 2012, 48, 11325–11327 RSC
- G. Kovàcs, G. Ujaque and A. Lledós, J. Am. Chem. Soc., 2008, 130, 853–864 CrossRef PubMed
-
(a) D. Zuccaccia, L. Belpassi, L. Rocchigiani, F. Tarantelli and A. Macchioni, Inorg. Chem., 2010, 49, 3080–3082 CrossRef CAS PubMed
-
(a) T. J. Brown and R. A. Widenhoefer, J. Organomet. Chem., 2011, 696, 1216–1220 CrossRef CAS PubMed
-
(a) T. J. Brown, M. G. Dickens and R. A. Widenhoefer, Chem. Commun., 2009, 6451–6453 RSC
- T. N. Hooper, M. Green and C. A. Russell, Chem. Commun., 2010, 46, 2313–2315 RSC
- Y. Zhu, C. S. Day and A. C. Jones, Organometallics, 2012, 31, 7332–7335 CrossRef CAS
- R. E. M. Brooner and R. A. Widenhoefer, Organometallics, 2011, 30, 3182–3193 CrossRef CAS
- T. J. Brown, A. Sugie, M. G. D. Leed and R. A. Widenhoefer, Chem. – Eur. J., 2012, 18, 6959–6971 CrossRef CAS PubMed
- T. J. Brown, A. Sugie, M. G. Dickens and R. A. Widenhoefer, Organometallics, 2010, 29, 4207–4209 CrossRef CAS
- A. Zhdanko, M. Ströbele and M. E. Maier, Chem. – Eur. J., 2012, 18, 14732–14744 CrossRef CAS PubMed
- M. García-Mota, N. Cabello, F. Maseras, A. M. Echavarren, J. Pérez-Ramírez and N. López, ChemPhysChem, 2008, 11, 1624–1629 CrossRef PubMed
- L. Jašíková and J. Roithová, Organometallics, 2012, 31, 1935–1942 CrossRef
-
(a) C. Nieto-Oberhuber, S. López, M. P. Muñoz, D. J. Cárdenas, E. Buñuel, C. Nevado and A. M. Echavarren, Angew. Chem., Int. Ed., 2005, 44, 6146–6148 CrossRef CAS PubMed
-
(a) C. Nieto-Oberhuber, M. P. Muñoz, E. Buñuel, C. Nevado, D. J. Cárdenas and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 2402–2406 CrossRef CAS PubMed
-
(a) E. Mizushima, T. Hayashi and M. Tanaka, Org. Lett., 2003, 5, 3349–3352 CrossRef CAS PubMed
-
(a) S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742–2744 RSC
- H. Li and R. A. Widenhoefer, Org. Lett., 2009, 11, 2671–2674 CrossRef CAS PubMed
- T. T. Dang, F. Boeck and L. Hintermann, J. Org. Chem., 2011, 76, 9353–9361 CrossRef CAS PubMed
- C. Nevado and A. M. Echavarren, Chem. – Eur. J., 2005, 11, 3155–3164 CrossRef CAS PubMed
- N. A. Tarselli, A. R. Chianese, S. J. Lee and M. R. Gagné, Angew. Chem., Int. Ed., 2007, 46, 6670–6673 CrossRef PubMed
- D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012–9019 CrossRef CAS PubMed
- A. Homs, I. Escofet and A. M. Echavarren, Org. Lett., 2013, 15, 5782–5785 CrossRef CAS PubMed
- Y. Zhu, C. S. Day, L. Zhang, K. J. Hauser and A. C. Jones, Chem. – Eur. J., 2013, 19, 12264–12271 CrossRef CAS PubMed
- F. Schröder, C. Tugny, E. Salanouve, H. Clavier, L. Giordano, D. Moraleda, Y. Gimbert, V. Mouries-Mansuy, J.-P. Goddardand and L. Fensterbank, Organometallics, 2014, 33, 4051–4056 CrossRef
- A. N. Nesmeyanov, E. G. Perevalova, Y. T. Struchkov, M. Y. Antipin, K. I. Grandberg and V. P. Dyadhenko, J. Organomet. Chem., 1980, 201, 343–349 CrossRef CAS
- D. Canseco-Gonzalez, A. Petronilho, H. Mueller-Bunz, K. Ohmatsu, T. Ooi and M. Albrecht, J. Am. Chem. Soc., 2013, 135, 13193–13203 CrossRef CAS PubMed
- A. Guérinot, F. Weizhen, M. Sircoglou, C. Bour, S. Bezzenin-Lafollée and V. Gandon, Angew. Chem., Int. Ed., 2013, 52, 5848–5852 CrossRef PubMed
- W. Fang, M. Presset, A. Guérinot, C. Bour, S. Bezzenine-Lafollée and V. Gandon, Chem. – Eur. J., 2014, 20, 5439–5446 CrossRef CAS PubMed
-
(a) A. N. Nesmeyanov, E. G. Perevalova, K. I. Grandberg, D. A. Lemenovskii, T. V. Baukova and O. B. Afanassova, J. Organomet. Chem., 1974, 65, 131–144 CrossRef CAS
- I. Braun, A. M. Asiri and A. S. K. Hashmi, ACS Catal., 2013, 3, 1902–1907 CrossRef CAS
- A. Gómez-Suárez and S. P. Nolan, Angew. Chem., Int. Ed., 2012, 51, 8156–8159 CrossRef PubMed
-
(a) G. Seidel, C. W. Lehmann and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 8466–8470 CrossRef CAS PubMed
- P. H.-Y. Cheong, P. Morganelli, M. R. Luzung, K. N. Houk and F. D. Toste, J. Am. Chem. Soc., 2008, 130, 4517–4526 CrossRef CAS PubMed
- Y. Odabachian, X. F. Le Goff and F. Gagosz, Chem. – Eur. J., 2009, 15, 8966–8970 CrossRef CAS PubMed
- A. Simonneau, F. Jaroschik, D. Lesage, M. Karanik, R. Guillot, M. Malacria, J.-C. Tabet, J.-P. Goddard, L. Fensterbank, V. Gandon and Y. Gimbert, Chem. Sci., 2011, 2, 2417–2422 RSC
-
(a) D. Weber and M. R. Gagné, Org. Lett., 2009, 11, 4962–4965 CrossRef CAS PubMed
- G. Seidel, C. W. Lehmann and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 8466–8470 CrossRef CAS PubMed
-
(a) A. S. K. Hashmi, I. Braun, P. Nösel, J. Schädlich, M. Wieteck, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 4456–4460 CrossRef CAS PubMed
-
(a) A. Zhdanko and M. E. Maier, Chem. – Eur. J., 2013, 19, 3932–3942 CrossRef CAS PubMed
- D. Weber, T. D. Jones, L. L. Adduci and M. R. Gagné, Angew. Chem., Int. Ed., 2012, 51, 2452–2456 CrossRef CAS PubMed
- Y. Chen, M. Chen and Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6181–6186 CrossRef CAS PubMed
- J. E. Heckler, M. Zeller, A. D. Hunter and T. G. Gray, Angew. Chem., Int. Ed., 2012, 51, 5924–5928 CrossRef CAS PubMed
- A. Gómez–Suárez and S. P. Nolan, Angew. Chem., Int. Ed., 2012, 51, 8156–8159 CrossRef PubMed
- A. Grirrane, H. Garcia, A. Corma and E. Álvarez, ACS Catal., 2011, 1, 1647–1653 CrossRef CAS
- C. Obradors and A. M. Echavarren, Chem. – Eur. J., 2013, 19, 3547–3551 CrossRef CAS PubMed
- G. Mazzone, N. Russo and E. Sicilia, J. Chem. Theory Comput., 2015, 11, 581–590 CrossRef CAS
- A. Homs, C. Obradors, D. Leboeuf and A. M. Echavarren, Adv. Synth. Catal., 2014, 356, 221–228 CrossRef CAS PubMed
- L. Jašíková and J. Roithová, Organometallics, 2013, 32, 7025–7033 CrossRef
-
(a) L. Ye, Y. Wang, D. H. Aue and L. Zhang, J. Am. Chem. Soc., 2012, 134, 31–34 CrossRef CAS PubMed
- D. D. Vachhani, M. Galli, J. Jacobs, L. Van Meervelt and E. V. Van der Eycken, Chem. Commun., 2013, 49, 7171–7173 RSC
-
(a) A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organometallics, 2012, 31, 644–661 CrossRef CAS
-
(a) G. Lemière, V. Gandon, N. Agenet, J.-P. Goddard, A. de Kozak, C. Aubert, L. Fensterbank and M. Malacria, Angew. Chem., Int. Ed., 2006, 45, 7596–7599 CrossRef PubMed
- P. W. Davies and N. Martin, Org. Lett., 2009, 11, 2293–2296 CrossRef CAS PubMed
- A. S. K. Hashmi, T. Lauterbach, P. Nösel, M. H. Vilhelmsen, M. Rudolph and F. Rominger, Chem. – Eur. J., 2013, 19, 1058–1065 CrossRef CAS PubMed
-
(a) A. Macchioni, Chem. Rev., 2005, 105, 2039–2073 CrossRef CAS PubMed
- G. L. Hamilton, E. J. Kang, M. Mba and F. D. Toste, Science, 2007, 317, 496–499 CrossRef CAS PubMed
-
(a) K. Aikawa, M. Kojima and K. Mikami, Angew. Chem., Int. Ed., 2009, 121, 6189–6193 CrossRef PubMed
-
(a) R. L. LaLonde, B. D. Sherry, E. J. Kang and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 2452–2453 CrossRef CAS PubMed
-
(a) K. Aikawa, M. Kojima and K. Mikami, Angew. Chem., Int. Ed., 2009, 48, 6073–6077 CrossRef CAS PubMed
-
(a) X.-Y. Liu and C.-M. Che, Org. Lett., 2009, 11, 4204–4207 CrossRef CAS PubMed
- M. Raducan, M. Moreno, C. Bour and A. M. Echavarren, Chem. Commun., 2012, 48, 52–54 RSC
| This journal is © The Royal Society of Chemistry 2015 |