
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of analogues of natural products†
Martin E.
Maier
Institut für Organische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: martin.e.maier@uni-tuebingen.de
First published on 23rd March 2015
Abstract
In this article strategies for the design and synthesis of natural product analogues are summarized and illustrated with some selected examples. Proven strategies include diverted total synthesis (DTS), function-oriented synthesis (FOS), biology-oriented synthesis (BIOS), complexity to diversity (CtD), hybrid molecules, and biosynthesis inspired synthesis. The latter includes mutasynthesis, the synthesis of natural products encoded by silent genes, and propionate scanning. Most of the examples from our group fall in the quite general concept of DTS. Thus, in case an efficient strategy to a natural product is at hand, modifications are possible at almost any stage of a synthesis. However, even for compounds of moderate complexity, organic synthesis remains a bottle neck. Unless some method for predicting the biological activity of a designed molecule becomes available, the design and synthesis of natural product analogues will remain what it is now, namely it will largely rely on trial and error.
Introduction
Natural product research represents an important part of our scientific culture. From natural products (NPs) a lot of biology could be learned. Their diverse structures have enriched the collection of known organic molecules. Studies related to their biosynthesis have provided valuable insights into genetics and enzymology. Moreover, many NPs turned out to be useful molecules or lead compounds in the fragrance, crop science or pharmaceutical industries. For the latter this is well documented. Thus, an analysis of the sources of new drugs from 1981–2010 indicated that only 36% of the new chemical entities (NCEs) were discovered without inspiration from a natural product.1 Despite the obvious importance, interest in natural product isolation by the pharmaceutical industry has declined in recent years. For this, several reasons might be responsible. Certainly, natural product isolation is time consuming and elaborate. A big challenge for the discovery of new NPs is dereplication, that is, to recognize and weed out already known substances. In parallel the last decade has seen a downward trend in the NCEs per year from 2001 to 2010. According to a recent hypothesis this might be due to the heavy reliance of the industry on combinatorial chemistry.1 Also, it seems that computational techniques like virtual screening are not yet able to produce good lead compounds. Basically, we are far from the vision that a drug candidate can be produced by rational design on a computer or by virtual screening of the chemical space. We still do not know enough about molecular recognition and what it takes for a ligand to trigger a signal on a receptor. Certainly, drug discovery is much more difficult than to make a plaster of a shoe print. It is rather comparable to horse racing or football betting, where knowledge and experience are advantageous.Therefore it might be worthwhile to reconsider natural products as the inspiration and the starting point of novel drugs.2 Several papers have analyzed differences between NPs and compounds from medicinal chemistry. Thus, classical medicinal chemistry is characterized by a lot of C–N bond formation. In contrast, NP syntheses strongly rely on C–C bond forming reactions. These are often accompanied by creation of OH groups, for example through allylation or aldol reactions. As a consequence, the number of asymmetric centers is higher in NPs. In addition, most of the rings in NPs are partly or completely saturated. With regard to physicochemical or drug-like properties of NPs, an analysis by A. Ganesan of 24 NPs that became approved drugs between 1970 and 2006 revealed that these fall into two categories.3 NPs of the first category adhere to the Lipinski Rule of Five. NPs of the second category just follow the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P < 5 rule and the rule regarding the number of H-bond donors <5. The obvious advantage of NPs over non-natural molecules is that during their biosynthesis they have already interacted with different enzymes and proteins.4 Accordingly, their core scaffolds can be considered privileged structures. In other words, out of all possible small organic molecules NPs only occupy certain areas of chemical space that seems predestined for interaction with proteins.
P < 5 rule and the rule regarding the number of H-bond donors <5. The obvious advantage of NPs over non-natural molecules is that during their biosynthesis they have already interacted with different enzymes and proteins.4 Accordingly, their core scaffolds can be considered privileged structures. In other words, out of all possible small organic molecules NPs only occupy certain areas of chemical space that seems predestined for interaction with proteins.
The analysis and visualization of chemical space is a topic of bioinformatics. Thus, software programmes were devised that are able to generate all theoretically possible molecules following basic rules for covalent bonds and functional groups. For example, the chemical universe database GDB-17 includes all molecules up to a size of 17 atoms considering C, N, O, S and halogens. It contains 166.4 billion molecules.5 In principle this database should contain many NP-like structures. To explore such large collections of molecules, descriptors are calculated for each molecule followed by projecting multidimensional property space in lower dimensions for visualization purposes.6,7 In another instance, a representative universal library spanning the small molecule chemical space was constructed from the known chemical universe by stochastic search, including mutation and crossover for generating new molecules from known ones. This library contains 8.9 million structures.8 All these libraries appear useful for virtual screening and bioinformatics analysis. However, hits would still have to be synthesized in order to find out whether they show biological activity or not.
Because of the reasons mentioned above, NPs appear good starting points for analogue synthesis. Chemistry related to NPs also provides the opportunity to test new reactions and synthetic strategies. In the following we discuss strategies that have been used in the synthesis of NP analogues. Of course only representative examples could be considered, since countless papers deal with the synthesis of NP analogues.
Diverted total synthesis (DTS)
The synthesis of NP analogues is probably as old as the field of total synthesis. In the simplest approach a NP can be converted to an analogue by modifications of its functional groups. In a total synthesis programme, modifications are possible on advanced intermediates. Moreover, total synthesis allows deep seated structural modifications by using modified building blocks in fragment-based synthesis. The term diverted total synthesis (DTS) was suggested by Danishefsky to describe this strategy.9 This includes the preparation of modified NPs as probes for elucidating the mode of action.10 To streamline and simplify the synthesis of NP analogues, attempts have also been made to implement solid-phase strategies.11 Famous cases from the last one or two decades for diverted total synthesis include the epothilone story.12 Two prominent epothilones are sagopilone (2) even though it is not marketed, and the amide analogue 3 (ixempra), which is in clinical use. The development of carfilzomib (5) from epoxomicin (4)13 or the synthesis of the powerful antibiotic eravacycline (7) taking tetracycline (6) as the lead structure (Fig. 1) can also be mentioned.14,15 Related to DTS is the concept of diversity-oriented synthesis.16 While the compound collections obtained by this strategy are typically different from natural products, some examples are known where NP-like structures were obtained.17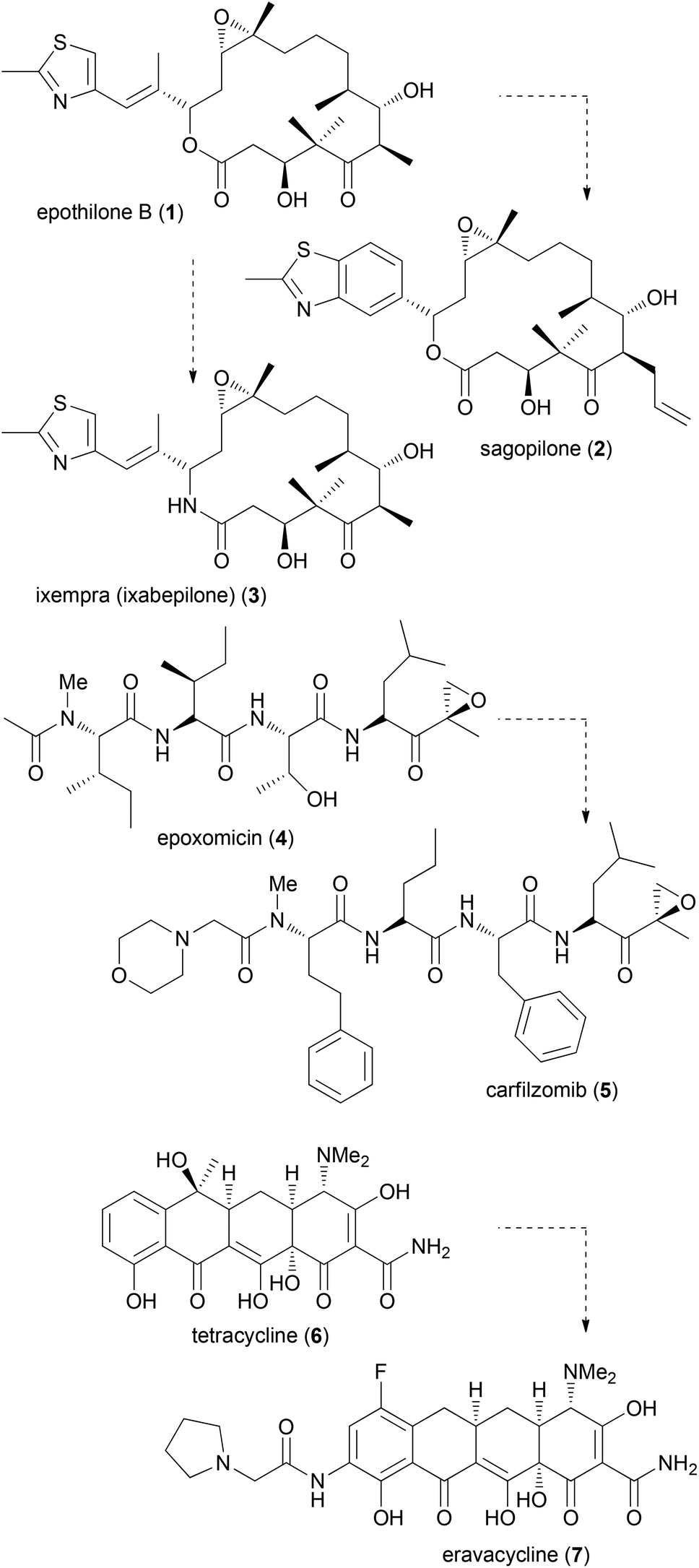 | ||
| Fig. 1 Some recent examples of the synthesis of promising natural product analogues. | ||
Latrunculin analogues
An illustrative case for DTS was put forward by the Fürstner group in connection with the total synthesis of the two marine macrolides latrunculin A (8) and B (9) (Fig. 2).18 The latrunculins were isolated from a sponge collected in the red sea.19 They display a range of biological activities with the dominant one being the disruption of actin polymerization. For this reason the latrunculins are important tools for chemical biology. Key structural features include a 16- or 14-membered macrolactone ring that contains several double bonds. The side chain features an acylthiazolidinone substituent. The keto function of 8 forms a hemiacetal with the 13-OH of the macrolactone ring. Studies on semisynthetic latrunculin analogues revealed that N-alkylated derivatives are inactive.20 Carbonate derivatives of 17-OH (hemiacetal OH) display reduced affinity to actin.21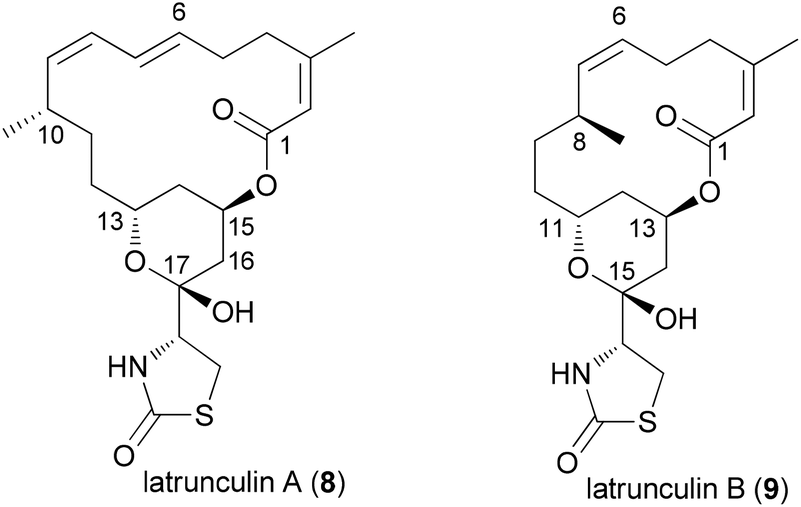 | ||
| Fig. 2 Structures of the two latrunculins. | ||
Due to the convergent synthetic strategy used by the Fürstner group twelve latrunculin analogues could be prepared that addressed the role of the methyl groups in the macrocyclic ring and the configurations in the thiazolidinone ring.18 The general synthetic strategy, exemplified by the synthesis of latrunculin B (9), is shown in Scheme 1.22 An aldol reaction between alkynal 10 and acetylthiazolidinone 11 followed by cleavage of the silyl protecting group and acetalization furnished pyran 12. This key fragment was then connected with several unsaturated carboxylate salts 13, yielding esters like 14. Formation of the macrolactone could be realized by a ring-closing alkyne metathesis (rcam). Routine steps then led to the target molecules.
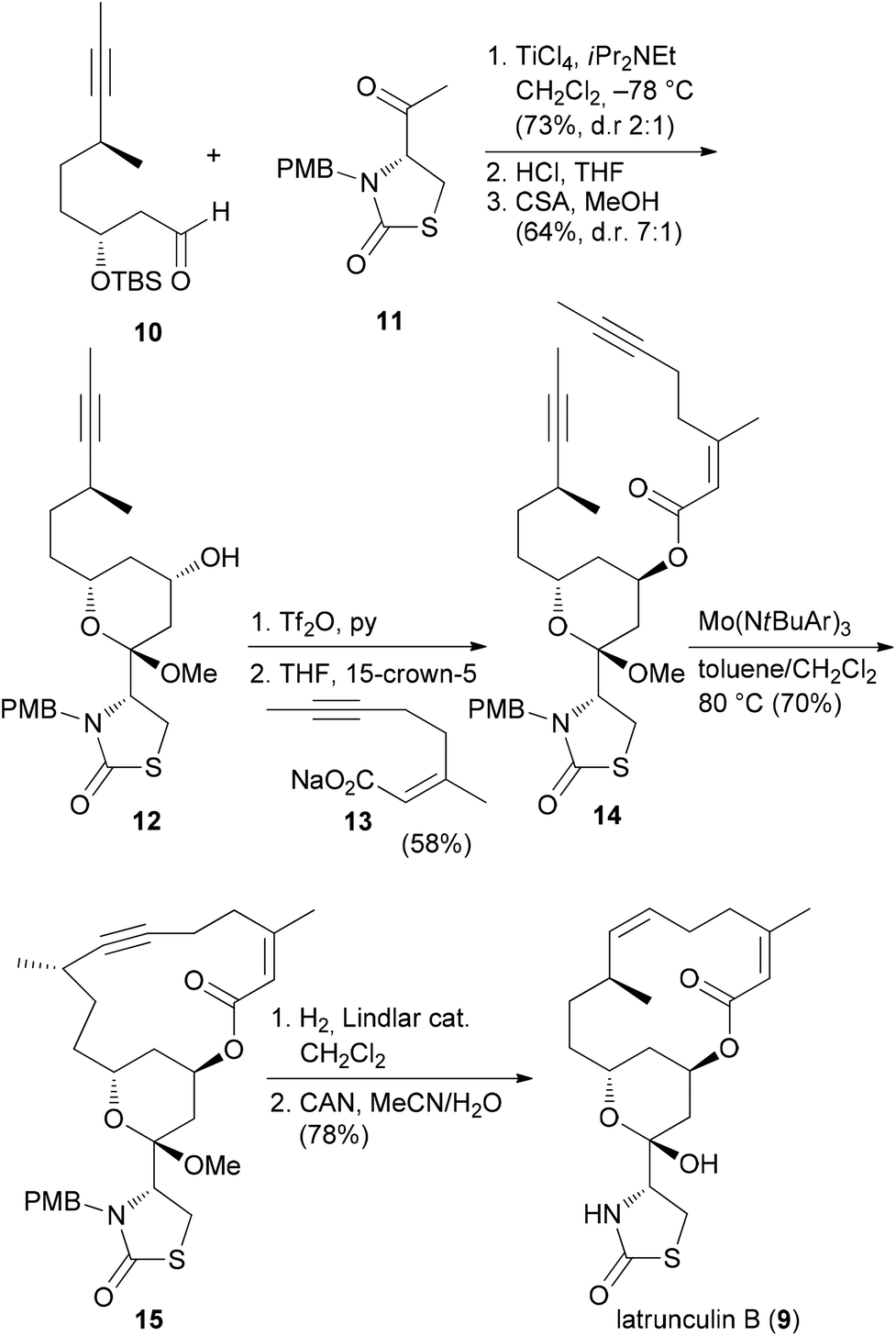 | ||
| Scheme 1 Synthetic strategy for the synthesis of natural and non-natural latrunculins. Ar = 3,5-dimethylphenyl. | ||
The five most active compounds, listed with decreasing microfilament activity, are shown in Fig. 3. While the activity of latrunculin A (8) could not be surpassed, the bis-nor-methyl-latrunculin B analogue 16 turned out to be more active than 9.
 | ||
| Fig. 3 The most active analogues prepared by the Fürstner group together with their microfilament disrupting activity (at 10 μM effective concentration). Abbreviations: ± weak effect, + significant effect, ++ strong effect, +++ very strong effect (less than 20% viable cells). | ||
Spongistatin analogues
With highly complex NPs, the production of a larger collection of analogues is a rather elaborate effort. But even with smaller numbers of analogues, useful information regarding SAR can be obtained. Thus, among two analogues prepared, compound 20, which is the E-ring dehydrated version of spongistatin 1 (19), was even more potent than the NP itself (IC50 values are given for HCT116 colon cell lines) (Fig. 4).23 Compound 20 was formed as a by-product in the deprotection of a 35-OTBS ether with aqueous HF-MeCN. This example highlights the role of serendipitous discoveries in chemistry. One should note that already the NP is active in the low picomolar range. So to beat this activity is truly remarkable. In contrast the analogue with a truncated side chain (ending with C46) was much less active (407 nM).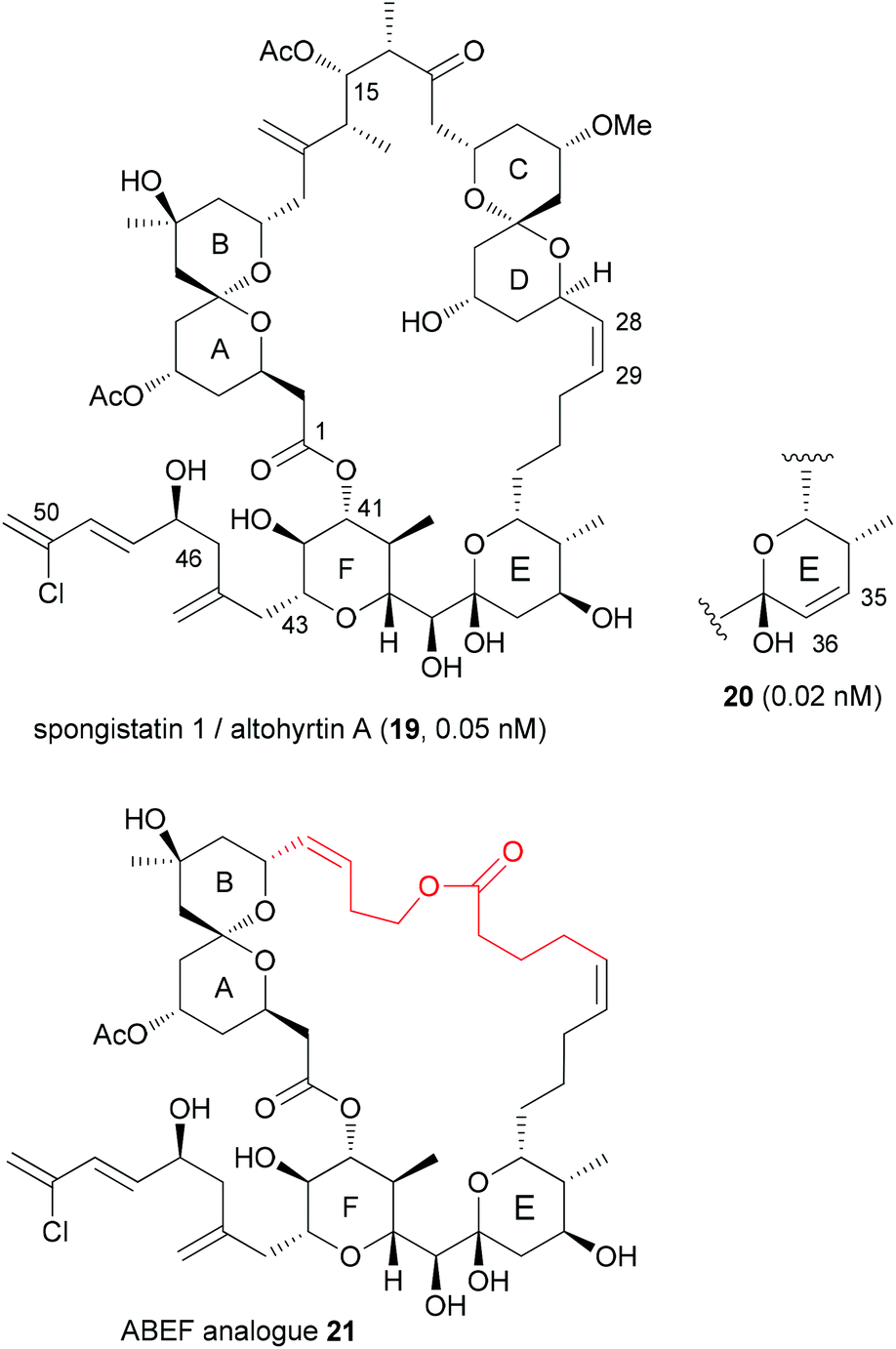 | ||
| Fig. 4 Structure of (+)-spongistatin 1 (19) and the analogues 20 and 21. | ||
The spongistatins are bis(spiroacetal) macrolides, which were isolated from marine sponges. Their potent antitumor activity is due to inhibition of mitosis by binding in the vinca binding region of tubulin.24
Another option for DTS of complex macrolides is to replace the sections of the macrocycle that are not involved in binding to the target proteins with simpler fragments. Based on the hypothesis that the small-molecule recognition domains most likely include the more rigid parts of a macrocycle, Smith, III, et al. designed spongistatin analogues with a simple tether between the ABEF rings (Fig. 4).25 While not as active as the NP lead, the ABEF analogue 21 still displayed nanomolar activity, for example an IC50 of 60.5 nM versus 0.06 nM (for 19) against U937 lymphoma cells.
Pironetin analogues
A common strategy in the design of NP analogues is to truncate or simplify the structure down to the essential elements. In a recent study, Marco et al. prepared four analogues of the NP pironetin (22) (Scheme 2).26 Unlike many molecules that bind to microtubules, pironetin exerts its antitumor activity by binding to α-tubulin and not to β-tubulin. Well known NPs that interact with β-tubulin include colchicine, paclitaxel and the epothilones. The microtubule dynamics can be disturbed either by breaking up microtubules or by stabilization them. Pironetin inhibits the assembly of tubulin to microtubules. It seems that the Lys352 residue of α-tubulin adds to the enoate double bond. In addition, hydrogen bonding of Asn258 to the pyrone carbonyl and the 9-OMe group contributes to the binding. SAR studies on pironetin point to the importance of a 9-OH or 9-OMe group and a 7-OH group. The Marco group removed the alkyl groups at C4, C8 and C10, thus simplifying the syntheses significantly. The terminus of the side chain was varied in the analogues. The synthetic strategy is outlined in Scheme 2. Thus, repetitive Brown allylation reactions came to use for the creation of the three stereocenters. A final acrylate formation, followed by ring-closing metathesis (rcm) and deprotection, delivered the desired analogues. | ||
| Scheme 2 Synthesis of pironetin analogues. | ||
The growth inhibitory activity of the analogues was determined on ovarian cancer cell lines (Table 1). While pironetin was highly potent, the analogues were at least three orders of magnitude less active, with IC50 values in the micromolar range. Cell cycle analysis indicated that analogue 26 shares a common mode of action with pironetin (22), whereas analogues 23–25 possibly work by another mechanism. This study shows that the omission of alkyl groups may cause a reduction of the hydrophobic contribution to binding. Moreover, a loss of conformational control may be detrimental.
| Compound | IC50 [μM] |
|---|---|
| a In this and other tables the biological activity data for the compounds are given without standard deviation. | |
| Pironetin (22) | 0.0062 |
| 23 | 39.7 |
| 24 | 14.1 |
| 25 | 9.2 |
| 26 | 54.7 |
Benzolactone enamide analogues
Our own work regarding the synthesis of NP analogues mainly falls in the category DTS. Thus, in cases where we found an efficient synthetic strategy for a NP, we looked for opportunities in the sequence to branch out towards analogues. An early example from our group addressed apicularen A analogues.27,28 Apicularen A (32) is a prominent member of the benzolactone enamide class of NPs.29 Besides apicularen A, the family comprises the salicylihalamides 31, the oximidines, like 33, and lobatamide C (34) (Fig. 5). Common structural features include a macrocyclic salicylate ring with an enamide side chain on the aliphatic part of the macrolactone.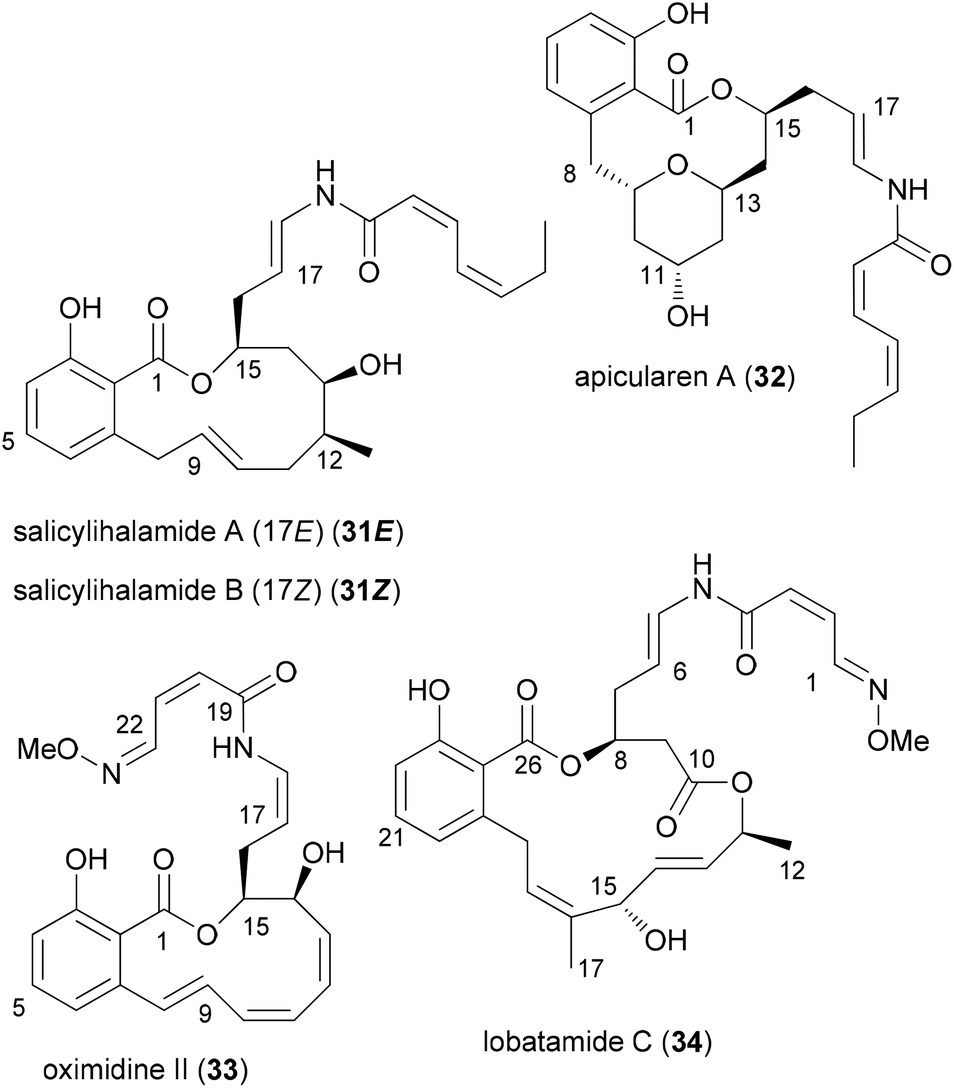 | ||
| Fig. 5 Structures of some prominent benzolactone enamides. | ||
The benzolactone enamides are potent inhibitors of vacuolar (H+)-ATPases. These are multicomponent proton pumps that are responsible for acidification of single membrane intracellular compartments like endosomes and lysosomes.30 Biochemical studies showed that salicylihalamide binds to the V0 domain of the proton pump.31 While most of the total syntheses of apicularen A pass through a trans pyran prior to macrolactonization,32 the strategy we developed features a macrolactonization to a 12-membered macrolactone ring followed by a transannular etherification (Scheme 3).33,34 The precursor 35 does indeed show a striking similarity to the salicylihalamide core structure.
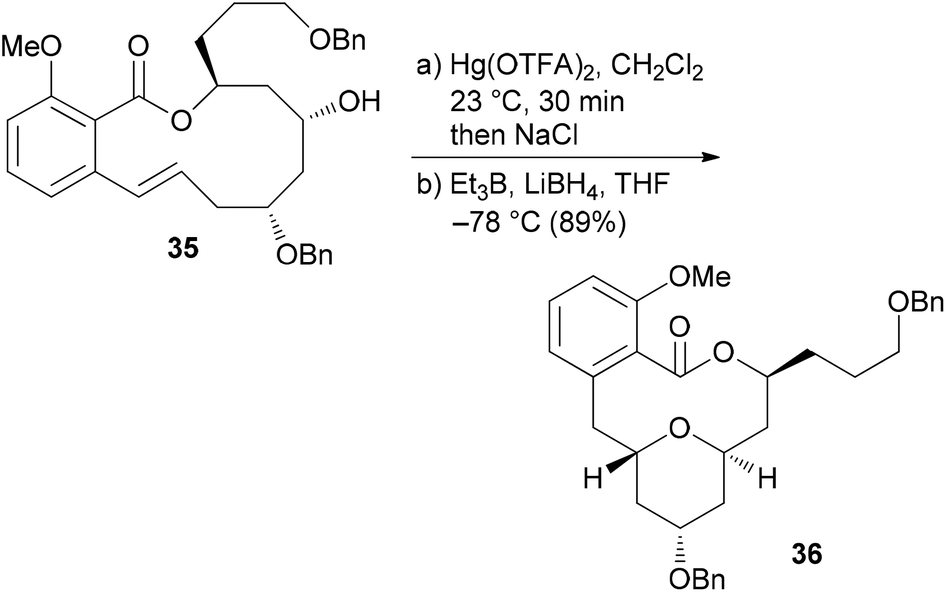 | ||
| Scheme 3 Key transannular cyclization towards the core structure 36 of apicularen A. | ||
From model studies on the transannular cyclization and from the total synthesis campaign several aldehydes could be made available. These were used for attachment of the enamide side chain via formation of the hemiaminal followed by dehydration as shown for the conversion of 37 to 40. In all, five analogues 40, 41, 42, 43 and 44 were prepared (Scheme 4). Out of these, 40 and 41 contain the intact enamide side chain but have changes in the macrolactone core.
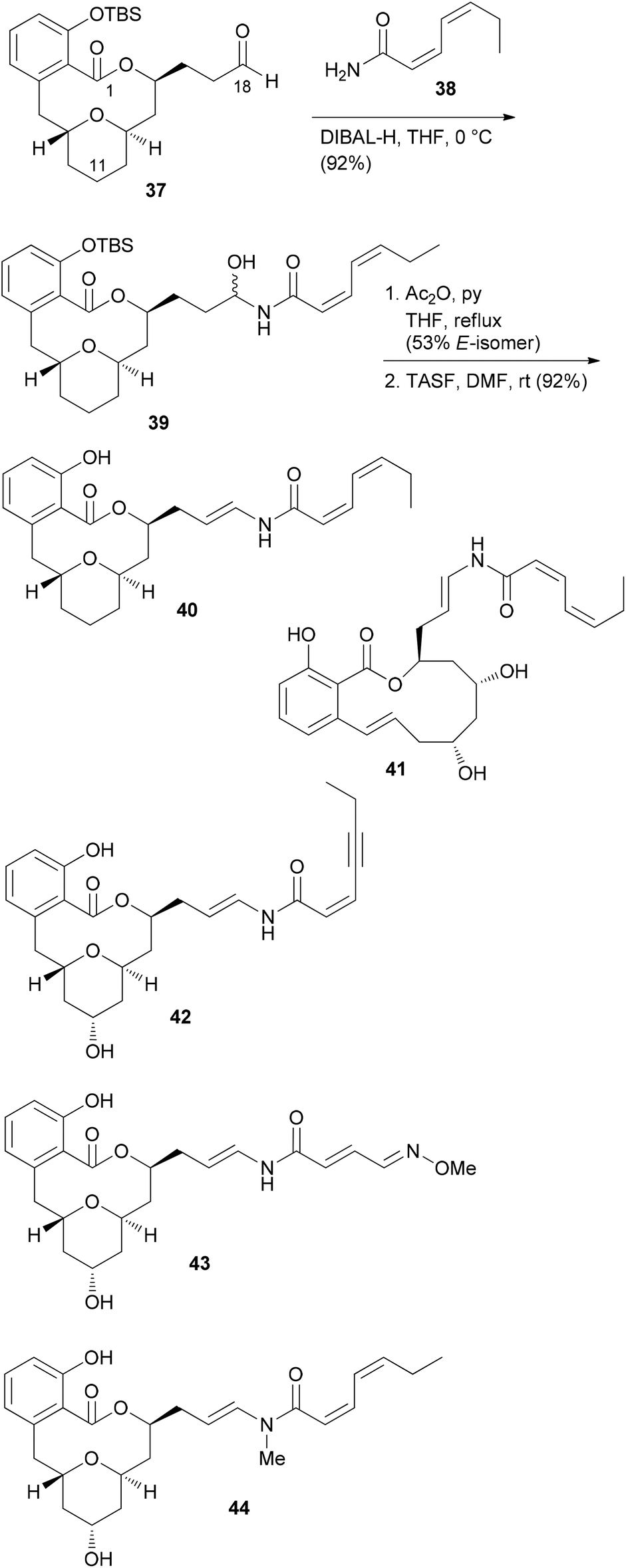 | ||
| Scheme 4 Structures of some apicularen A analogues. TASF = tris(dimethylamino)sulfonium fluoride. | ||
These compounds turned out to be quite active (Table 2). On the other hand, compounds 42, 43 and 44 feature the natural core structure but a different enamide part. These analogues were roughly three orders of magnitude less cytotoxic (Table 2). Other studies concur with these findings.35 Thus, it seems that variations in the acyl part of the enamide are tolerated provided that the chain is neither too short nor too long. On the other hand, the macrolactone part seems to be less sensitive to structural modifications.
| Compound | Cell line | ||
|---|---|---|---|
| L-929 | KB-3-1 | KB-V1 | |
| 32 | 6.8 | 2.3 | 15.9 |
| 40 | 19 | 7.1 | 2.4 |
| 41 | 200 | 180 | 1360 |
| 42 | 4550 | 1820 | 9100 |
| 43 | 1800 | 1130 | 9000 |
| 44 | 770 | 660 | 1980 |
| Sali A (31E) | 57 | 6.8 | 9.1 |
For the other prominent benzolactone enamide, salicylihalamide A (31E), our group contributed with several approaches to the macrolactone core. These strategies are summarized in Scheme 5. The first approach relied on an intramolecular Suzuki coupling of ester 46 to macrolactone 45. Here a diastereoselective hydroboration generated the stereocenter at C12 prior to the coupling step.36 This strategy was also successful in the reverse order. Thus, hydroboration of fragment 48 followed by coupling with vinyl iodide 47 gave rise to seco compound 49. Deprotection to the corresponding hydroxy acid set the stage for an intramolecular Mitsunobu esterification.37 Possibly a more efficient strategy is to close the macrolactone ring by ring-closing metathesis. In this variant the stereocenters at C12 and C13 could be created in an efficient manner by an Evans aldol reaction.38 Using this strategy for the synthesis of the aliphatic sector 51, we prepared macrolactone 45 in very good yield.
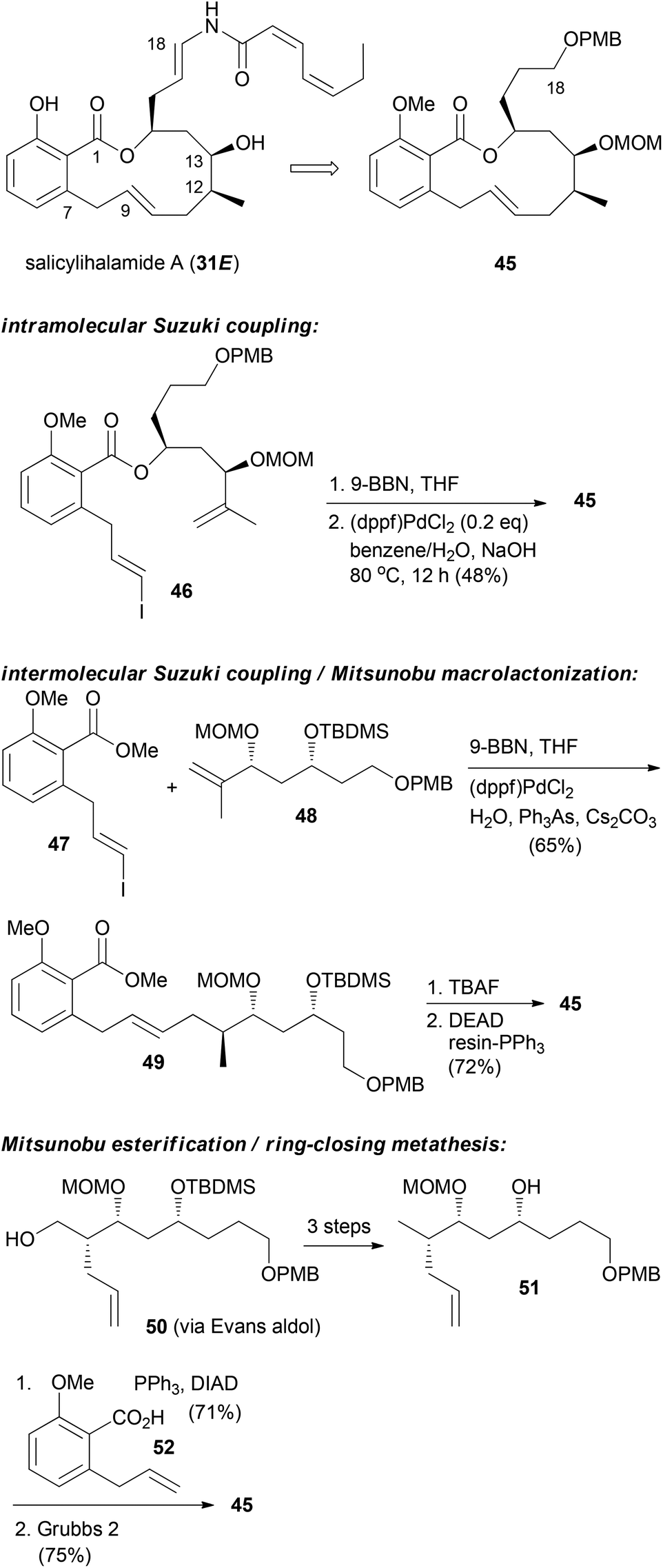 | ||
| Scheme 5 Synthetic strategies developed in our group towards the synthesis of salicylihalamide congeners. | ||
In the case of a terminal alkyne its protection as TIPS derivative 53 was required in order to achieve good yields in the rcm reaction. But the alkynyl group allowed for a formal total synthesis of salicylihalamide A by converting it to a vinyl iodide (Scheme 6).39 In addition, we prepared the pyridine substituted derivative 55 by Sonogashira cross-coupling. In a simpler substrate lacking the C12 and C13 substituents, we used the side chain alkyne for click reactions with a range of azides. This way the triazoles 56 and 57 were obtained (Scheme 6).40 These analogues showed cytotoxicity in the micromolar range, pointing to the crucial role of the enamide side chain in 31E.
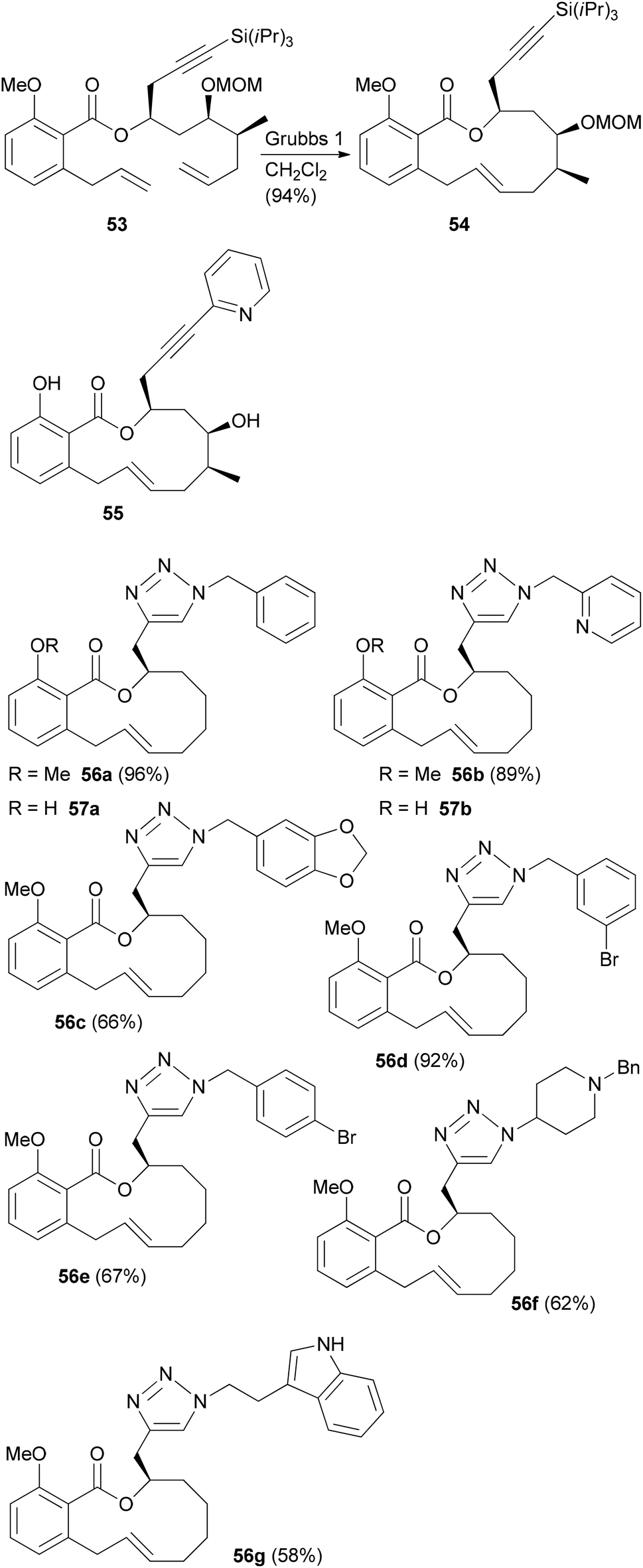 | ||
| Scheme 6 Benzotriazole analogues of salicylihalamide A prepared from an alkyne precursor by a click reaction. | ||
The ring-closing metathesis strategy for the synthesis of salicylihalamide A (31E) has been pioneered by De Brabander et al.41,42 They prepared a range of analogues where the enamides were created by nucleophilic addition to vinyl isocyanates. One of the more promising analogues, called saliphenylhalamide (66, SaliPhe), has been explored as a potential lead for anticancer therapy.43 The methyl ketone 58 was added to aldehyde 59 in a boron-aldol reaction in a non-selective reaction (Scheme 7). The mixture was carried on through the next steps. Silylation and syn-selective reduction was followed by Mitsunobu esterification and ring-closing metathesis. At the stage of the phenol 63 the unwanted diastereomer could be removed by chromatography. The final steps established the enamide via addition of lithium phenylacetylide to the intermediate vinyl isocyanate.
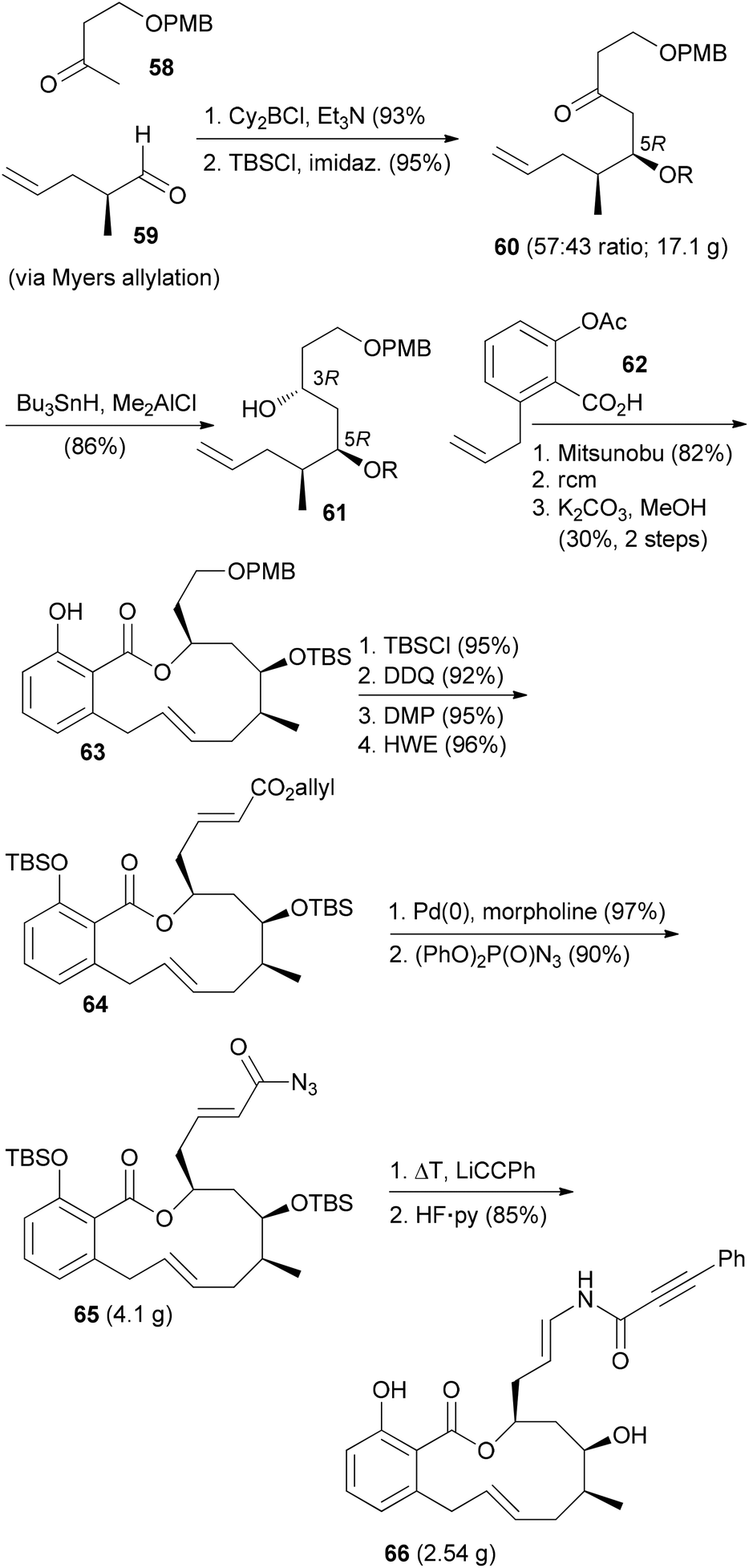 | ||
| Scheme 7 Synthetic route towards saliphenylhalamide 66. | ||
In a recent paper SaliPhe and other V-ATPase inhibitors were tested as antiviral compounds.44 It could be shown that V-ATPase is required for human papillomavirus (HPV) infection. Addition of inhibitors affects uncoating/disassembly but not the process of endocytosis. If the toxicity towards the host could be controlled, the inhibition of V-ATPases might be an interesting antiviral strategy.
Cruentaren A analogues
Unlike the benzolactone enamides, which target V-ATPase, the macrolactone cruentaren A (67) inhibits mitochondrial F-ATPase (Scheme 8). This protein is quite important since it uses a proton gradient to promote the synthesis of ATP. A recent study described that the inhibition of F1F0-ATPase by 67 disrupts the synthase-Hsp90 interaction.45 As Hsp90 is an important chaperone,46 cruentaren A can indirectly cause client protein degradation. Structural features of this NP, which was isolated from the myxobacterium Byssovorax cruenta,47 include a 12-membered lactone with a Z-double bond and an unsaturated side chain amide. With its biological activity and our previous experience in the synthesis of benzolactones, cruentaren A (67) appeared as a promising target. Up to now, four total syntheses for 67 have been published.48–51 Three of them, including our own, rely on a ring-closing alkyne metathesis52 (rcam) to establish the macrocyclic ring. Major differences in these routes are in the preparation of the building blocks, a benzoic acid like 68, and a homopropargylic alcohol such as 69. For example, we prepared fragment 68via an Evans aldol reaction forming the C9–C10 bond (Scheme 9). Here the 10-Me group came from a reduction of a carboxylic function. The other fragment 69, featuring four stereocenters, was constructed by an aldol reaction (C15–C16) and a Marshall–Tamaru reaction. In the latter transformation a chiral allenyl metal was added to an aldehyde generating a 2-substituted butynyl structure. The triple bond was then converted via a hydroboration/reduction step to the hydroxyethyl moiety. The two fragments 68 and 69 were combined to give ester 70 after converting the acid 68 to the corresponding carbonyl imidazolide. Using the Schrock catalyst 71 an excellent yield for the rcam reaction could be realized. The alkyne in the macrolactone ring was kept for the elaboration of the side chain. This prevented an undesired translactonization to the six-membered lactone. Conversion of the advanced intermediate 73 to the propargylic amine 76 was followed by condensation with acid 77. After deprotection steps, a final Lindlar hydrogenation provided cruentaren A (67) (Scheme 8).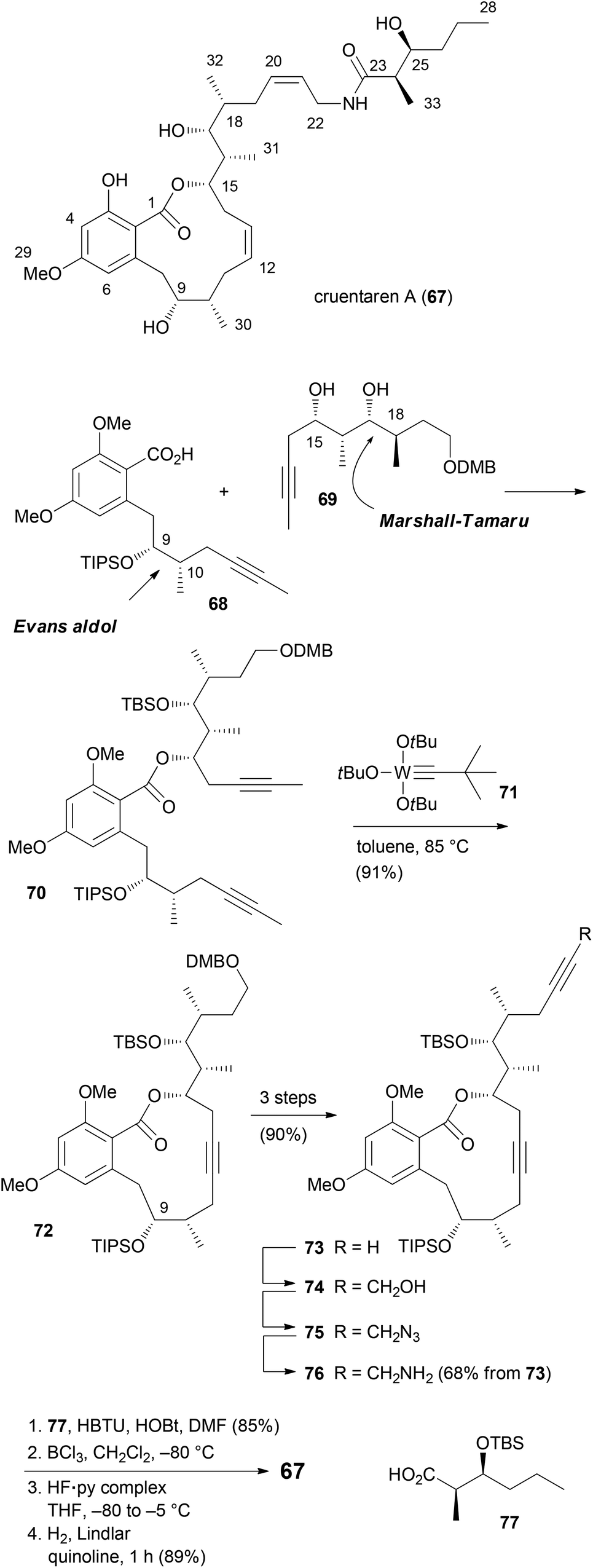 | ||
| Scheme 8 Overview of the synthetic route to cruentaren A (67). | ||
 | ||
| Scheme 9 Summary of the strategies towards the pyran system of neopeltolide, part 1. | ||
The efficient route to amine 76 allowed access to sufficient material and the preparation of several amide analogues (Fig. 6).53 All of them were somewhat less active with 78c being the most potent one. This points to the importance of the hydroxyl group of the amide. Compound 83 with two triple bonds was not very active (IC50 = 11.1 μM). The 3-OH group also significantly contributes to the biological properties as can be seen in the reduced activity of 3-OMe-cruentaren A (IC50 = 28 nM vs. 0.7 nM for 67).
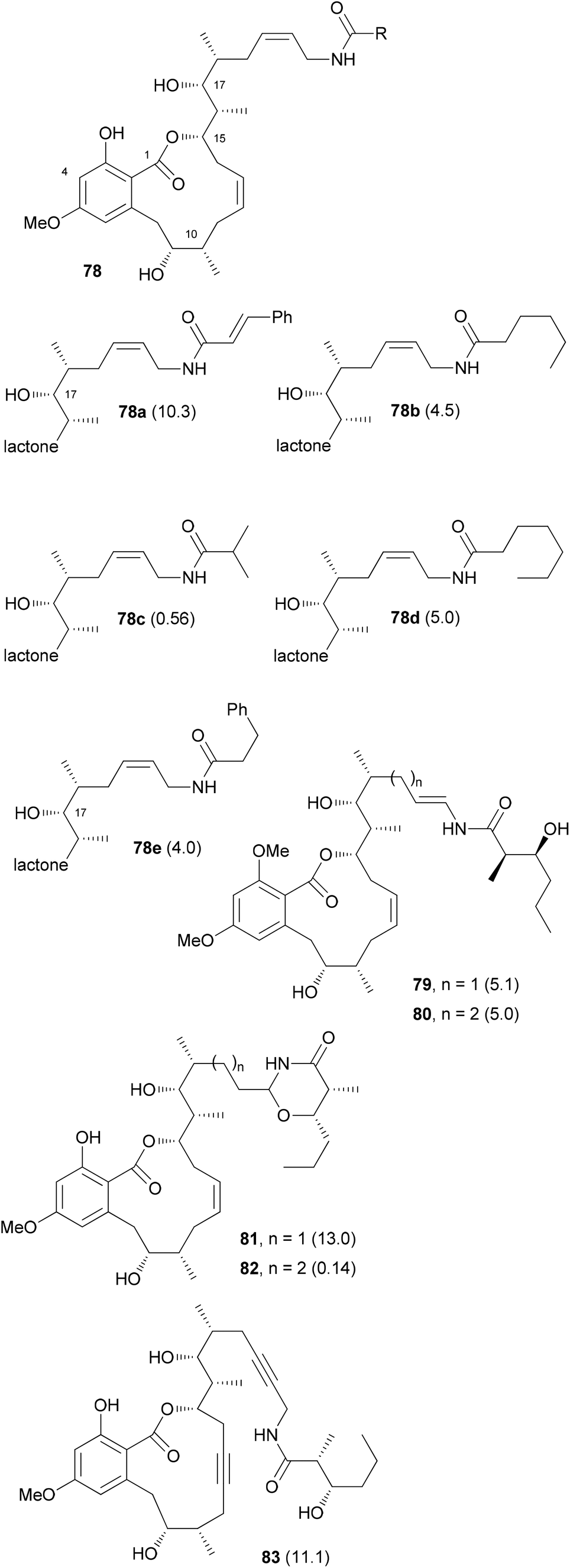 | ||
| Fig. 6 Structures of some cruentaren A analogues. The IC50 values (L929 cell line, μM) are given in brackets after the compound numbers. | ||
In addition to the allylamide derivatives we also wanted to prepare enamide derivatives. Although speculative it might be that the allylamide isomerizes to an enamide that then could form a highly electrophilic acyliminium ion, similar to the benzolactone enamides. It was possible to prepare vinyl iodides by hydrozirconation and iodination. A cross-coupling reaction with the side chain amide was used to create the enamide functionality. Cleavage of the silicon protecting groups and Lindlar reduction of the triple bond furnished analog 79. Unfortunately, the enamide did not survive the demethylation step with BCl3. Rather under these conditions the 1,3-oxazinan-4-ones 81 and 82 were formed, respectively. Out of these, 82 showed good growth inhibitory activity (IC50 = 140 nM). Enamides 79 and 80 themselves were only moderately active (Fig. 6).
The Fürstner group also prepared a range of analogues.54 Among them were stereoisomers of the side chain hydroxy acid and the 9-epimer 87 (Fig. 7). In addition, the 9-deoxy analogue 89 was prepared. Among the prepared analogues 85, 86 and 87 were the most active ones. It seems that a hydroxyl group at C9 is quite important for the activity. Moreover, either a syn configuration in the amide side chain or at least the correct configuration of the secondary hydroxyl group is crucial for activity.
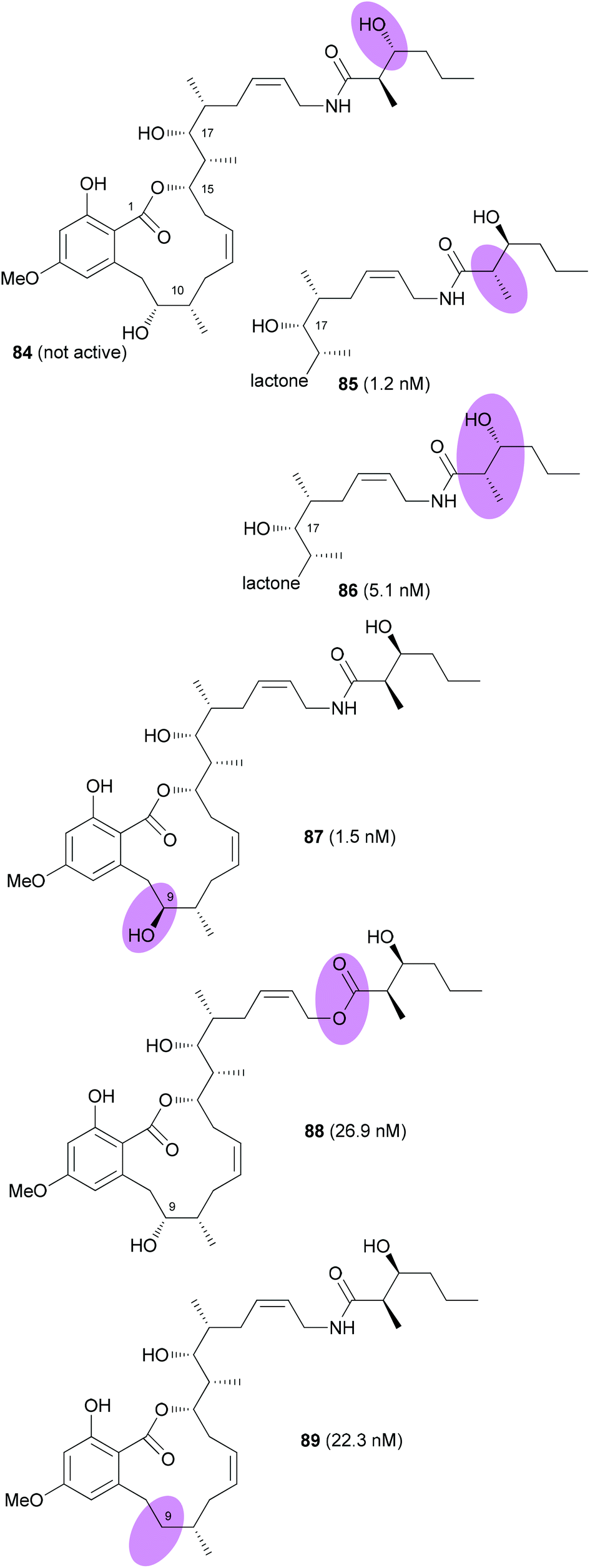 | ||
| Fig. 7 Structures of cruentaren A analogues prepared by the Fürstner group. IC50 values (against L 929 cells) are given in brackets. | ||
Neopeltolide analogues
Neopeltolide (90) (Fig. 8) is another macrolide for which we developed an efficient synthetic route that allowed us to prepare a range of analogues.55 This macrolide was isolated from a sponge Daedalopelta of the Neopeltodae family.56 It exhibits potent cytotoxic activity with IC50 values in the nanomolar range against a number of cancer cell lines. | ||
| Fig. 8 Structure of (+)-neopeltolide (90). | ||
It is certainly not among the most complicated structures but it does pose certain challenges in constructing the macrolactone and the oxazole-containing side chain. So far, ten total syntheses of neopeltolide have been published.57 The first two, by Panek and Scheidt, also corrected the initial published structure that had been misassigned at positions 11 and 13. In addition to the total syntheses at least ten formal total syntheses have been reported in the literature.58 All these papers nicely illustrate the state of the art in the synthesis of 2,6-cis-tetrahydropyrans and the stereoselective generation of stereocenters in 1,3-distance. Schemes 9 and 10 summarize the key steps that have been used in the synthesis of the pyran ring. In several of the syntheses a Prins cyclization came to use. Other strategies feature a hetero-Diels–Alder reaction.
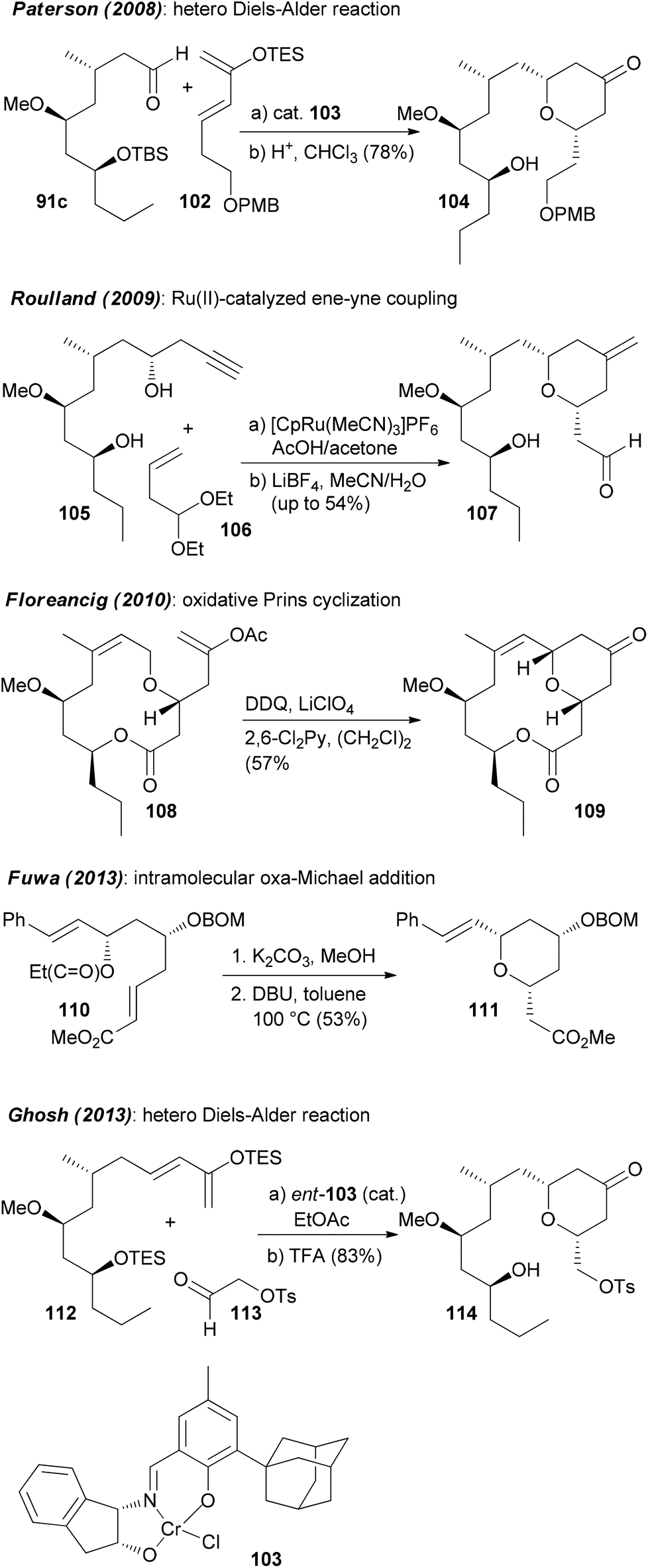 | ||
| Scheme 10 Summary of the strategies towards the pyran system of neopeltolide, part 2. | ||
Regarding the mode of action of neopeltolide a major contribution came from the Kozmin group.57e Using a yeast haploinsufficiency screen the cytochrome bc1 complex was identified as the molecular target of neopeltolide (90) and the related NP leucascandrolide A (115) (Fig. 9). Thus, these compounds interfere with the crucial energy supply chain in a cell that explains their strong toxicity. The fact that leucascandrolide A (115), which has a macrolactone core different from 90, addresses the same cellular target shows that some variations in the macrolactone should be possible without a significant reduction in potency. So far several groups have contributed to SAR studies of 90.59 The key findings are highlighted in Fig. 9. Thus, the pyran as well as the intact oxazole side chain are essential for activity. They basically represent the minimal structural units. In addition, the axial orientation of the ester at C5 significantly contributes to the activity. The region bridging C7 and C3 seems to tolerate changes to some degree, for example, deletion of substituents, dehydrogenation or epimerization at C9 or C11.
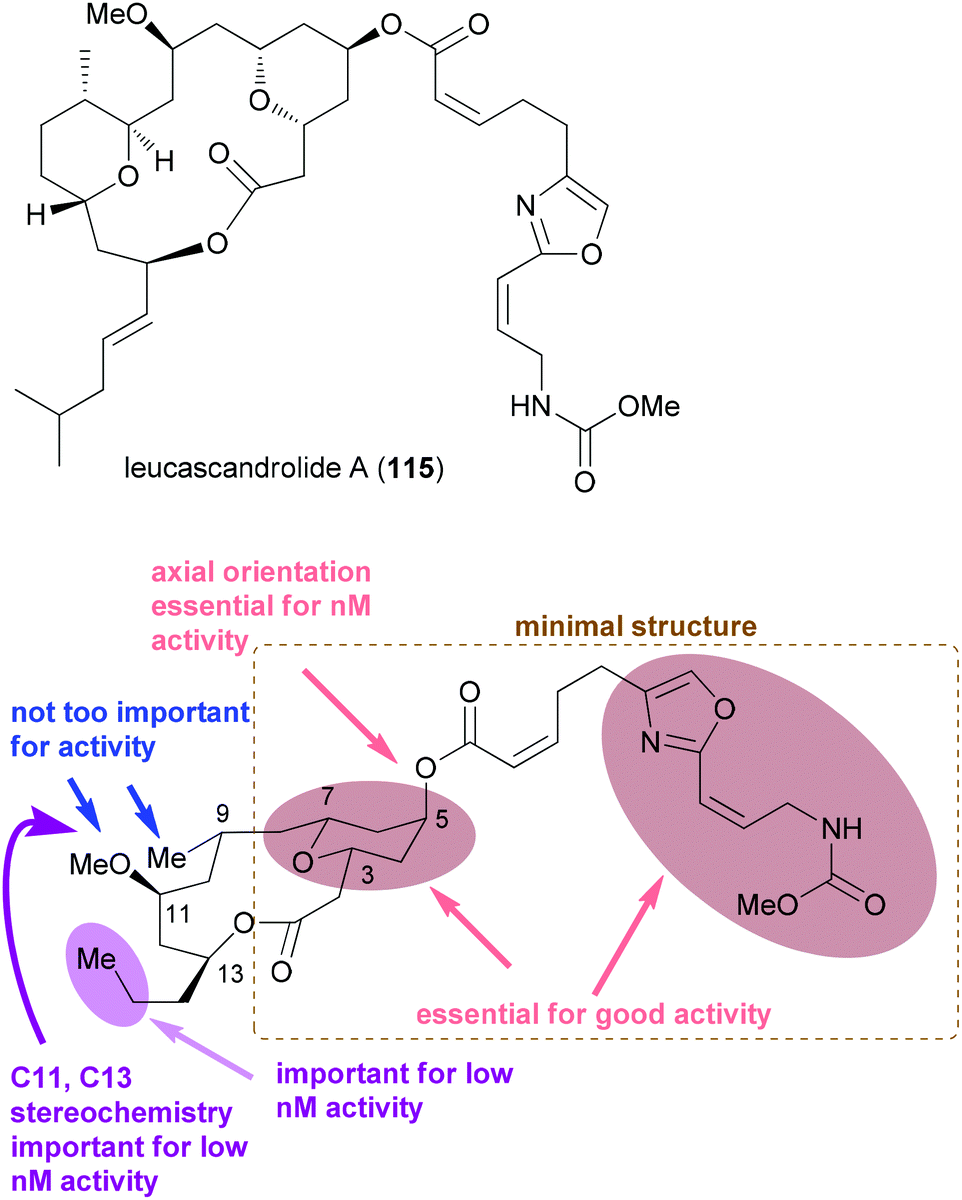 | ||
| Fig. 9 Structure of the related NP leucascandrolide (115) and a summary of the SAR for neopeltolide. The lower part of the figure was adapted from ref. 59d. | ||
Some specific analogues are shown in Fig. 10. The exact binding side in the target cytochrome however is not known. Due to the high toxicity, most likely neopeltolide will remain as a tool for biology. Its potential as drug is probably low owing to the toxicity.
 | ||
| Fig. 10 Representative neopeltolide analogues. The IC50 values were determined on the A549 cell line. Compounds 116–119 (ref. 59d), and compounds 120–122 (ref. 55). | ||
Chondramide A analogues
Among natural products, those which consist of obvious subunits are ideal for the synthesis of analogues. In this regard peptides, cyclic peptides or cyclic depsipeptides can be mentioned. The latter represent a unique class of secondary metabolites that in a way bridge the peptide with the polyketide world.60 They may contain unusual amino acids like D-configured or N-methylated ones. In addition, they incorporate a hydroxyl acid from the polyketide pathway. This building block connects the peptide part giving rise to an ester bond. Some prominent examples include the jasplakinolides61 (123) and the structurally related chondramides (Fig. 11).62 While the jasplakinolides have been isolated from a marine sponge,63 the chondramides were found in myxobacteria. The tripeptide segments of the jasplakinolides and the chondramides are quite similar, containing an L-alanine, an N-methyl-D-tryptophan and an L-β-tyrosine. However, the ring sizes are different since jasplakinolide contains an 8-hydroxy acid, whereas the chondramides feature a 7-hydroxy acid. The lagunamides64 and viequeamide A65 (130) are some recent examples of cyclic depsipeptides. Both show cytotoxic properties.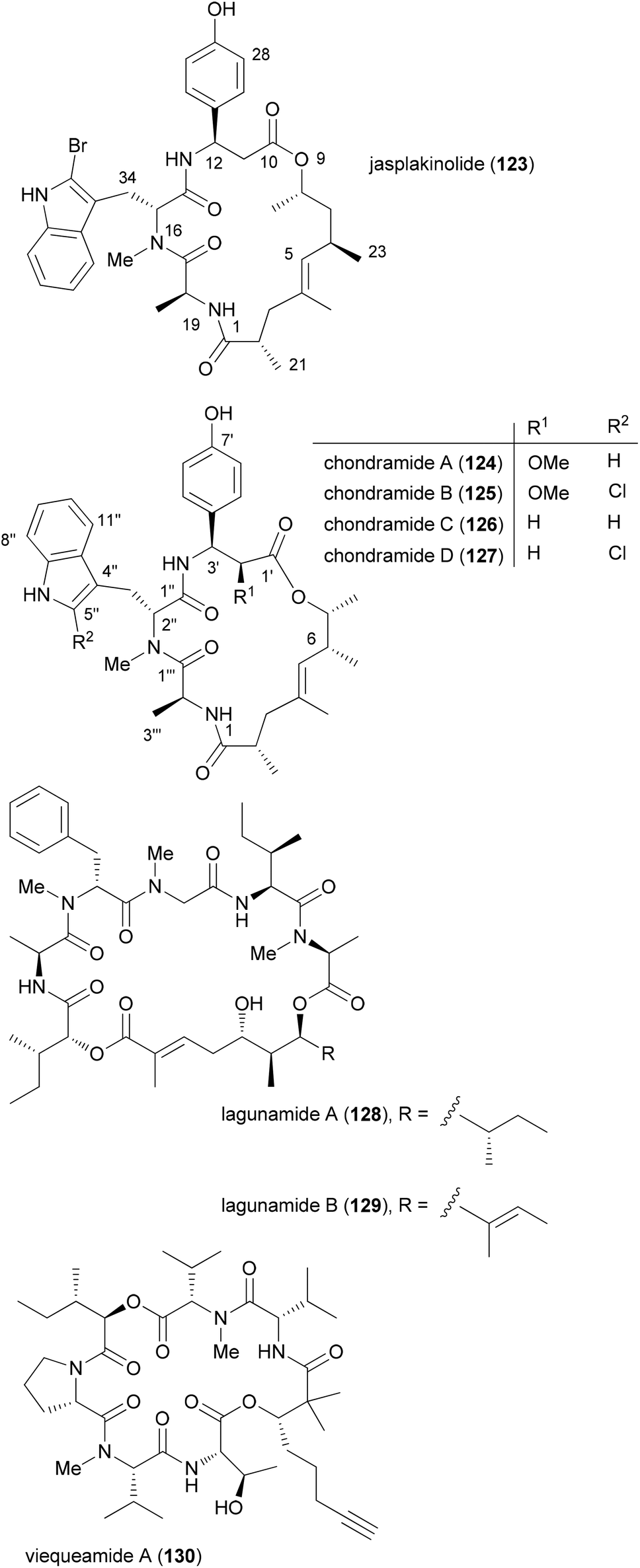 | ||
| Fig. 11 Structures of some cyclodepsipeptides. | ||
For jasplakinolide several total syntheses have been described.66 Most of them close the macrocyclic ring by formation of the ester or an amide bond. The Waldmann group utilized a ring-closing metathesis strategy.67 The first total syntheses of chondramide C were achieved by the Waldmann and Kalesse groups.68,69 They also secured the stereochemistry in the ω-hydroxy acid.
Since cyclodepsipeptides are still peptide-like, they often modulate protein–protein interactions. The potent antitumor activity of the jasplakinolides and chondramides is a consequence of their stabilizing effect on F-actin filaments.70
In our own work related to the chondramides we initially developed a concise synthesis of the ω-hydroxy acid based on a vinylogous aldol reaction and an asymmetric alkylation.71 Thereafter, we achieved a total synthesis of chondramide A.72 Unlike chondramide C which contains a β-tyrosine, chondramide A features a 2-methoxy-β-tyrosine. Its configuration was determined as (2S,3S). This amino acid derivative could be prepared by a Sharpless dihydroxylation followed by a selective Mitsunobu substitution of the benzylic hydroxyl group with hydrazoic acid (Scheme 11). With the tyrosine derivative 134 and the tryptophan 131 in hand, assembly of the tripeptide segment was started. Thereafter the tripeptide acid derived from 135 was combined with the ω-hydroxy ester 136 by Mitsunobu esterification. The synthesis could be completed with macrolactam formation in the presence of N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate (TBTU) and HOBT in reasonable yield.
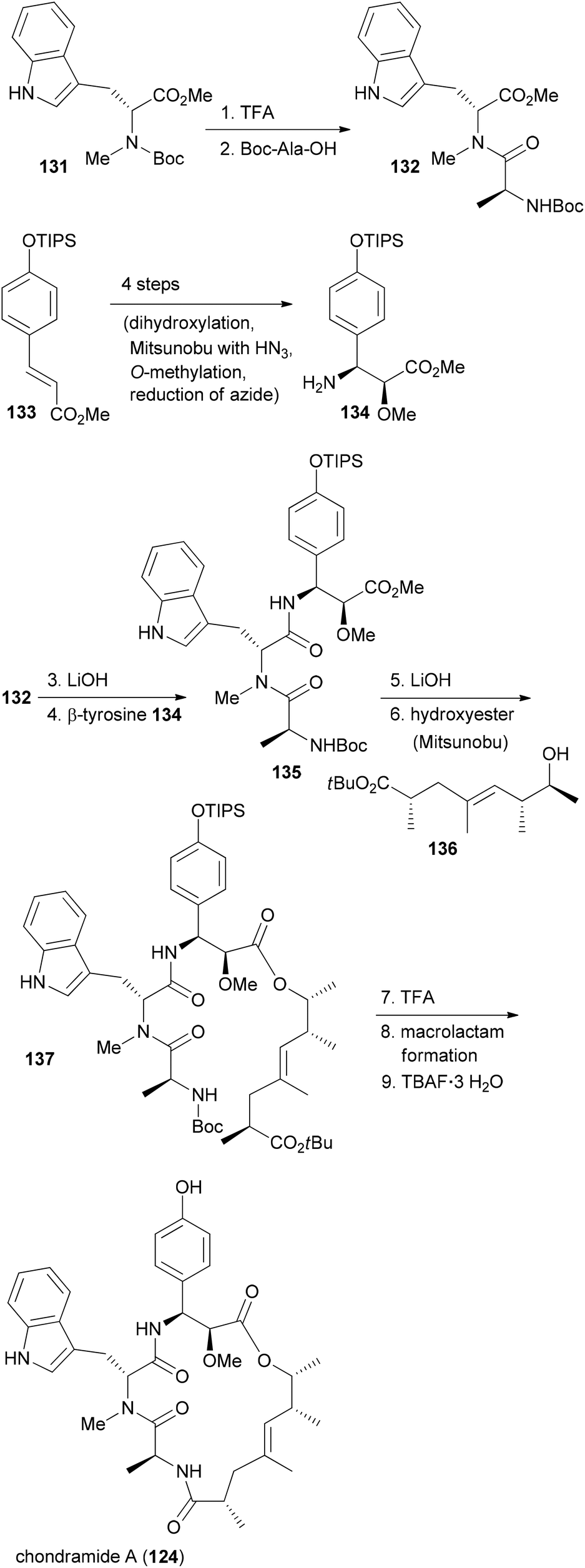 | ||
| Scheme 11 Summary of the synthetic strategy to chondramide A (124). | ||
With efficient routes to the building blocks and a reliable strategy for assembly and ring closure, the synthesis of chondramide A analogues was undertaken.73,74 One reason was to contribute to the SAR. Another reason was to exploit differences in the actin filaments between parasites and mammalian cells. For example, in the regions of contact, sequence alignment indicated residue differences at positions 200, 270 and 272. In Toxoplasma gondii they are G, K and A, whereas human actin has S, M and S at these positions. Altogether, we synthesized ten chondramide A analogues where the phenolic hydroxyl group was replaced with some other substituents. Most of the analogues retained high cytotoxicity in the low nanomolar region. Among the most potent ones were the –CN, –F and –NO2 substituted β-tyrosine analogues. The para-position also tolerates hydroxymethyl and hydroxyethyl substitution. On the other hand replacement of the OH group with a phenyl or carboxamide caused a reduction of activity. Scheme 12 also summarizes the SAR from the contributions of the Waldmann, Kalesse and the Maier group illustrated with jasplakinolide.75
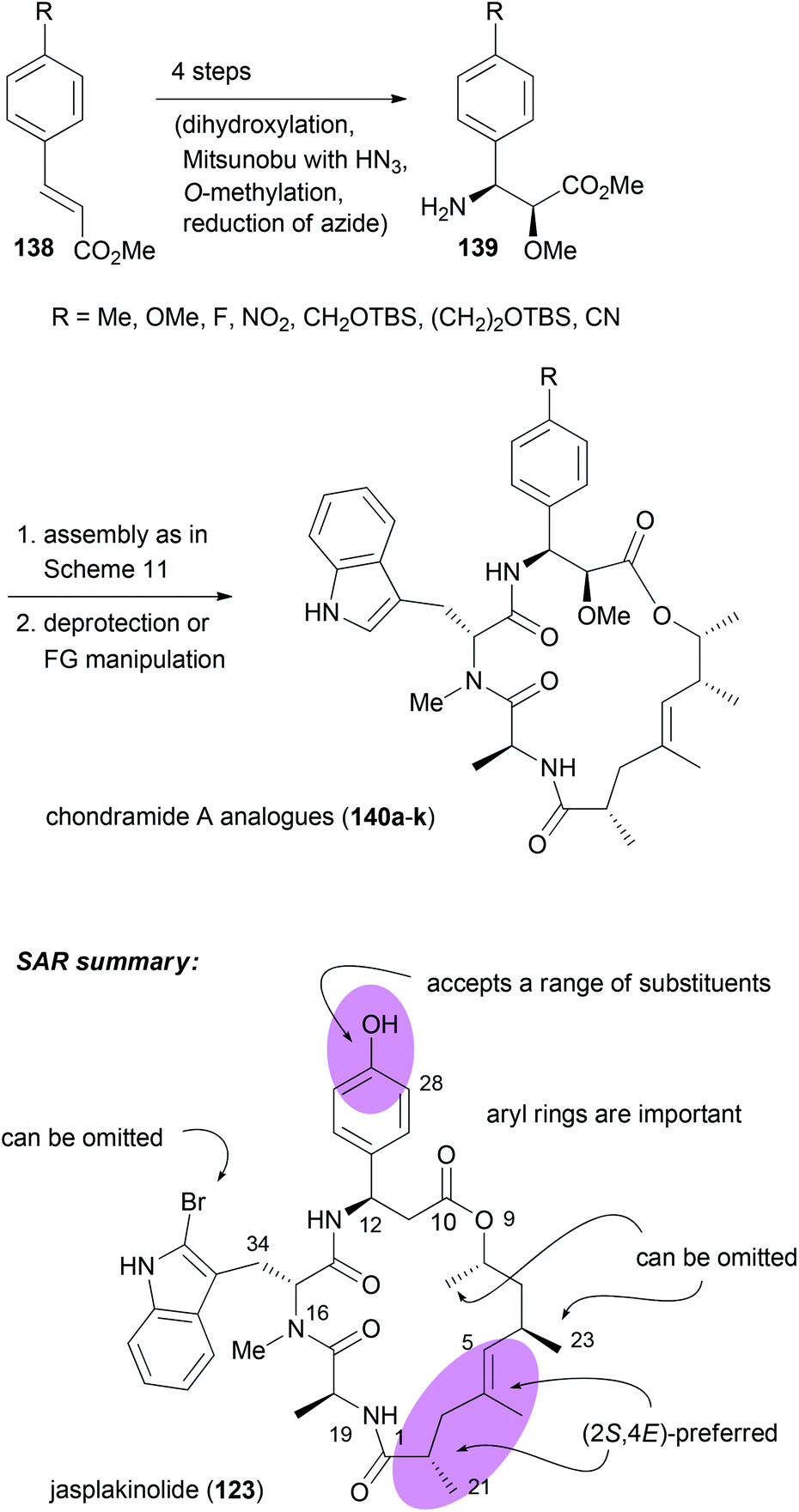 | ||
| Scheme 12 Synthesis of various 2-methoxy-β-tyrosine analogues and their incorporation into the chondramide A system. Summary of the SAR for the chondramide/jasplakinolide class of cyclodepsipeptides illustrated on the structure of jasplakinolide (123). | ||
These chondramide A analogues were indeed able to block parasite invasion into host cells (Table 3).76 Although they lack specificity for parasite vs. host actin other analogues might be able to disturb actin dynamics in parasites without harming the host.
| Compound | R | EC50 (HFF) [μM] | EC50 (T. gondii) [μM] |
|---|---|---|---|
| 124 | OH | 0.109 | 0.51 |
| 140b | Me | 0.027 | 0.53 |
| 140c | OMe | 0.066 | 0.69 |
| 140d | F | 0.029 | 0.32 |
| 140e | NO2 | 0.040 | 0.61 |
| 140f | NH2 | 0.057 | 0.66 |
| 140g | CH2OH | 0.052 | 0.74 |
| 140h | (CH2)2OH | 0.059 | 0.60 |
| 140i | CN | 0.027 | 0.38 |
| 140j | CONH2 | 1.424 | >45 |
| 140k | Ph | 0.213 | 1.34 |
Pladienolide B
The relevance of NPs stems also from the fact that occasionally compounds are isolated that lead to the discovery of biological processes which offer new opportunities in the treatment of diseases. Recent examples include NPs that bind to components of the spliceosome. In retrospect this is not too surprising since the spliceosome is responsible for the correct removal of introns from mRNA precursors in eukaryotic cells.77 The spliceosome is assembled from five small nuclear ribonucleoproteins (snRNP; U1, U2, U4, U5, U6) and associated protein factors. The intron removal requires sequence recognition. This is guided by certain motifs in the introns like branch sequences and polypyrimidine regions. It seems that the U2 snRNP has a key role in that it pairs with the branch sequence. This snRNP itself is composed of the U2 snRNA, U2 snRNP specific proteins (sm proteins) and two splicing factors SF3a and SF3b. The latter is a multiprotein complex made up of splicing associated proteins (SAP) 155, 145 and 130. These are also known as SF3b1, which seems to be the target of pladienolide B,78 SF3b2 and SF3b3. The exons can be joined in various ways as a result of alternative splicing. Thus, one gene can code for several proteins. Erroneous splicing is a key feature of cancer cells. Accordingly, inhibition of splicing appears as a promising anticancer strategy, at least for some types of cancer.So far several natural products are known to inhibit the splicing process (Fig. 12). They include polyketides of the pladienolide family, like the natural products pladienolide B (141), pladienolide D (142)79 and FD-895 (144).80 Molecules like GEX1A (herboxidiene) (145)81 have a side chain similar to the one in pladienolide B. Another group of spliceosome inhibitors represented by FR901464 (146)82 features two pyran rings. A related compound is thailanstatin (148).83 The Eisai company, which had discovered the pladienolides, early on described the structure of the carbamate derivative E7107 (143). This compound is also active indicating that modifications at C7 are tolerated. It even advanced to human trials.84,85 A dose-limiting study revealed common side effects of antitumor compounds, for example, nausea, vomiting and diarrhoea. However, since some patients also suffered from bilateral optic neuritis (visual disturbance due to inflammation of the optic nerve) the human trials were discontinued.
 | ||
| Fig. 12 Structures of some important spliceosome inhibitors. | ||
In order to discover improved and simpler SF3b inhibitors, SAR studies are clearly important. For FR901464 a range of analogues have been prepared by the groups of Koide and Webb. For example on meayamycin (149), an analogue that has two methyl groups at C1, SAR studies revealed the importance of the (Z)-pentenamide side chain with the allylic acetate, the methyl groups on the pyran ring, the diene region and the spiro epoxide (Fig. 13).86 Changes or omissions on these parts produced much less active compounds. Related analogues, called sudemycins, were developed by Webb et al.87 These analogues also contained the pharmacophore common to FR901464 and the pladienolides, namely the spiro epoxide at a certain distance to the allylic acetate.88 In some cell lines these analogues achieved IC50 values in the low micromolar range.
 | ||
| Fig. 13 Key structural feature important for the activity of FR901464 and analogues inspired by FR901464. | ||
Since pladienolide appears to be more challenging from a synthetic point of view, fewer analogues are known. A simplified pladienolide analogue 154 was made by Webb et al.89 It lacks the methyl groups at C10, C15 and C22 as well as the 3-OH (Fig. 14). While this compound turned out to be essentially inactive, the more hydrophobic compound 153 (21-O-tert-butyldimethylsilyl) was at least moderately active (IC50 10.7 μM, SK-MEL-2, melanoma cell line). This result clearly underscores the role of methyl groups as contributors to hydrophobic binding and as conformational control elements. Research by Chandrasekhar showed that aryl analogues of pladienolide B, like 155, with a truncated side chain are also active.90 However, this seems not to be in line with the pharmacophore model of Webb. Compound 156 with a truncated side chain lacking the epoxide and with the wrong configuration at C7 was also completely inactive.91 Pladienolide D analogue 157 lacking the 6-OH turned out to be quite active.92
 | ||
| Fig. 14 Pladienolide B analogues and SAR information gathered from them. Compounds 153 and 154 (ref. 89), compound 155 (ref. 90), compound 156 (ref. 91), compound 157 (ref. 92), compounds 158 and 159 (ref. 93), and compounds 160 and 161 (ref. 94). | ||
In the course of the elucidation of the side chain stereochemistry of FD-895 several diastereomers were prepared.93 With 17-epi derivative 158 an analogue was found that was almost 20 times more active than FD-895 itself. As can be seen with the less active cyclopropyl analogue 159, the epoxide does contribute to the biological activity. Studies on pladienolide–herboxidiene hybrid compounds highlighted the importance of the methyl substituent in the pyran ring and the role of the hydroxyl group in the side chain.94 However, one should say that the findings partly contradict themselves. Thus whereas 160 with a free hydroxyl group is inactive, the free hydroxyl group of 161 seems to be crucial.
Focusing on the synthesis of pladienolides, the most challenging seems to be creation of building blocks that contain the C6–C7 diol and the formation of the macrolactone. One strategy utilized by Kotake is the asymmetric dihydroxylation of (Z)-alkene 162.95 An esterification of the derived alkenoic acid 164 with alkenol 165 provided substrate 166 for a ring-closing metathesis (Scheme 13). The Burkart group first prepared monoprotected diol 169 by a Brown allylation of aldehyde 168. Thereafter the tertiary alcohol at C6 was created via addition of MeMgBr to the alkoxyketone derived from 169. As before, the ring was closed via rcm (Scheme 13).
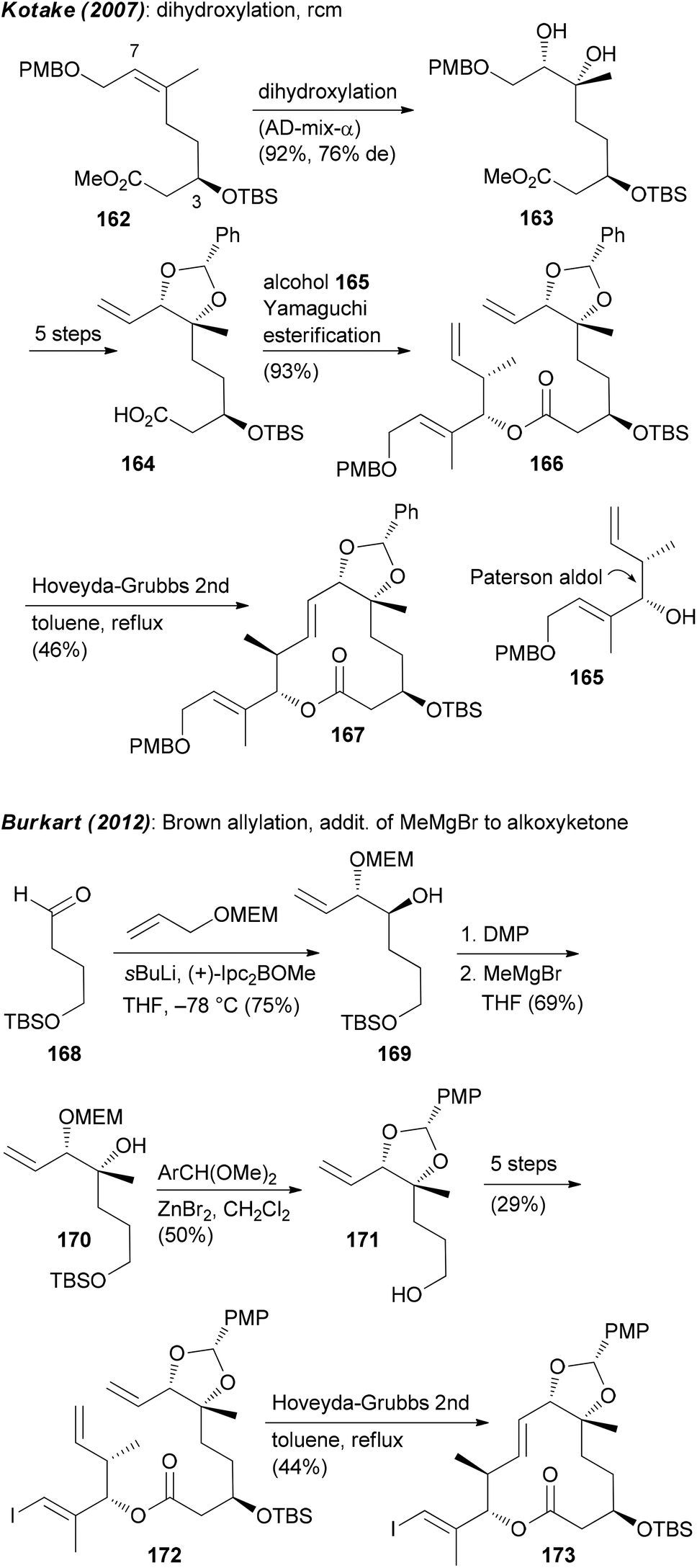 | ||
| Scheme 13 Synthetic strategies to create building blocks containing the C6,C7-diol. | ||
The chelation controlled Grignard addition to an α-alkoxyketone was also used by Ghosh et al.96 However, here the synthesis of the required ketone started with divinylmethanol 174. Opening of the derived epoxide 175 with the dienolate of 176 and asymmetric reduction of the keto ester 177 gave diol derivative 178 (Scheme 14). Oxidation and reaction with the Grignard reagent led to enoate 179. This was further elaborated to macrolactone 181.
 | ||
| Scheme 14 Other strategies to create building blocks containing the C6,C7-diol. | ||
A very simple and clever way to set up the C6–C7 region was put forward by Chandrasekhar.90 Thus, a Sharpless asymmetric epoxidation (SAE) led to epoxide 182 (Scheme 15). Its hydrolytic opening followed by protection of the diol gave compound 183. As can be seen, further elaboration of 183 to acid 185 did require a number of steps. In this work also an rcm strategy came to use for the creation of the macrolactone.
 | ||
| Scheme 15 Unexpected synthesis of 7-epi-pladienolide B (199) due to Mitsunobu acetylation under retention of configuration. MNBA = 2-methyl-6-nitrobenzoic anhydride. | ||
Our own approach to the pladienolide core was different but not without problems. Since most of the rcm approaches gave the macrolactone only in moderate yield we opted for a classical macrolactonization approach. We also recognized the match of the stereocenter in (R)-(−)-linalool with that of C6 in the pladienolides. To combine the subunits for the seco acid, a HWE reaction was planned followed by reduction of the enone. Accordingly, (R)-(−)-linalool was converted to aldehyde 186 which upon an acetate aldol reaction furnished hydroxy acid derivative 187 (Scheme 15). Manipulation of the carboxylic function and the double bond delivered aldehyde 188. Addition of lithiated phosphonate 189 and oxidation gave rise to keto phosphonate 190. Under optimized conditions phosphonate 190 underwent a HWE reaction with aldehyde 191 providing enone 192. Cyclization of the derived seco acid 193 with the Shiina reagent97 afforded macrocyclic enone 194 in good yield. Unfortunately, a reduction of the enone under Luche conditions gave the 7-epi-alcohol 195. In the course of the synthesis of pladienolide B analogue 156 (Scheme 15), after a Stille cross-coupling with phenyltributylstannane, the C7 allylic alcohol was acetylated under Mitsunobu conditions.91 However, as we later found out, the Mitsunobu reaction had occurred with retention of configuration. Thus, acetylation of 195 either under Mitsunobu or classical acetylation conditions gave the same allylic acetate 196. Cleavage of the MEM and TBS protecting groups delivered core structure 197. A final Stille cross-coupling with vinylstannane 198 furnished pladienolide B analogue 199.98 Its 1H NMR spectrum showed distinct differences to that from pladienolide B (141).
Since it was not possible to reduce the enone in the macrolactone 194 in the desired way, attempts were made to reduce the enone function on an acyclic precursor. After some trials we found that the chelation controlled reduction of enone 192 delivered the desired allylic alcohol 200 as the major diastereomer (Scheme 16). In a similar manner to that for the 7-epi analogue, ester 201 was converted to macrolactone 204 and then to pladienolide B (141). The biological evaluation of both compounds revealed a dramatic effect of the configuration at C7. Thus, whereas pladienolide B was highly active (IC50 = 7.5 nM, L929 mouse fibroblast cells), the 7-epi-epimer 199 was devoid of any activity.98 This analogue showed that an acetate or carbamate at C7 with the correct configuration is an essential part of the pharmacophore.
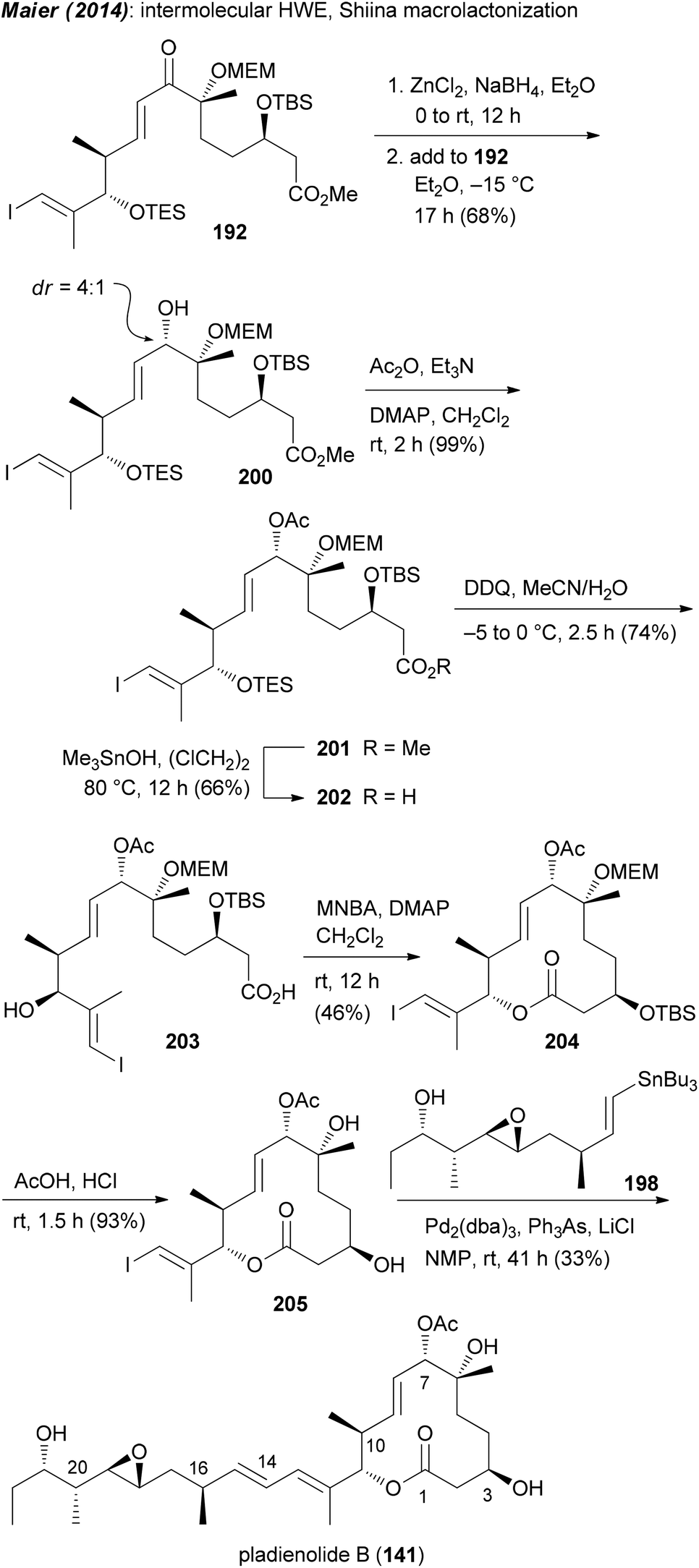 | ||
| Scheme 16 Synthesis of pladienolide B (141) via chelation controlled reduction of enone 192. | ||
Function-oriented synthesis (FOS)
The idea of function-oriented synthesis is to circumvent some inherent disadvantages of biologically active NPs. Thus, often they are difficult to obtain from natural sources, in particular if they originate from the marine environment. If their structures are highly complex, they are generally not amenable to total synthesis in larger amounts. Also, in many cases they are not suitable for direct use in humans before some chemical modifications. The ultimate goal of FOS is the design of simpler analogues of NPs where the activity is optimized towards the intended application. At the same time the analogues should allow for straightforward syntheses.99 The concept of FOS is kind of obvious and trivial. It is similar to DTS but the emphasis here is on function. The analogue of a FOS programme might be much simpler and structurally much different from the NP. The Wender group demonstrated this strategy with the synthesis of several bryostatin analogues. The bryostatins comprise 20 marine macrolides, which were isolated from the bryozoan Bugula neritina.100 They are polyketides featuring three pyran rings and several stereocenters. They essentially have the same macrocyclic core structure but most of them differ in the substituents at C7 and C20.The bryostatins are characterized by a range of biological activities, but prevalent is their ability to modulate the activity of protein kinase C. This leads to inhibition of cell growth and angiogenesis. So far several syntheses of bryostatins were reported.101 The shortest one, the synthesis of bryostatin 7 (207), requires 36 steps in total (TS) with 20 steps for the longest linear sequence (LLS) (Scheme 17).102 Based on the notion that the relevant pharmacophoric regions include the upper lipophilic A and B rings plus the polar carbonyl and hydroxyl groups at C1, C19 and C26, respectively, the Wender group synthesized many highly potent but simpler bryostatin analogues. Among the most potent derivatives was macrolactone 210 (Scheme 17). Esterification of acid 209 with alcohol 208 followed by macrocyclization via intramolecular acetal formation gave analogue 210 (32 TS), (17 LLS).103 The design of these analogues was probably inspired by the good availability of the lower fragment 208.104 This allowed for connection with various simplified upper fragments.105
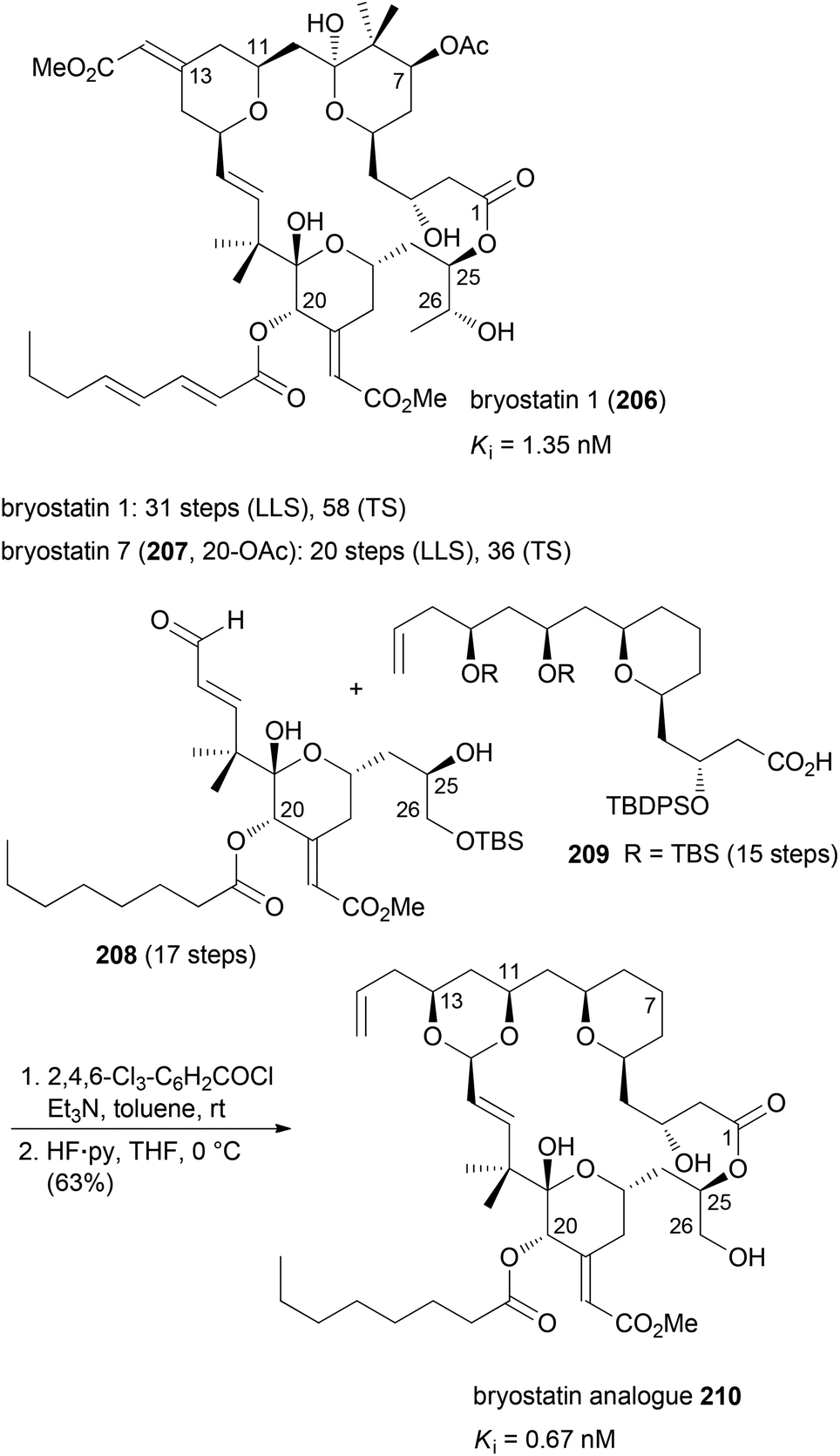 | ||
| Scheme 17 Synthesis of bryostatin analogue 210 from the lower fragment 208 and the diol derivative 209. | ||
Although not extensively covered in this account, one should mention the discovery of eribulin mesylate whose design was inspired by the marine-derived natural product halichondrin B. This rather complex polyketide is a potent antitumor agent which works by inhibition of microtubule dynamics. In the course of synthetic studies using a convergent approach it was discovered that the biological activity resides in the right part of the molecule. Among simplified macrolactone, macrocyclic ether, macrolactam and macrocyclic ketone analogues, the latter was found to be the best compromise.106,107 Eribulin mesylate (216) has been approved for clinical use in 2010 in the USA (Scheme 18). Key fragments were prepared using a chiron approach starting with D-glucuronolactone (211) and D-(−)-gulonolactone (212). In addition, epoxide 215 served as a starting material. First the C0–C1 bond was formed by addition of the anion of 213 to the aldehyde derived from 214. The macrocyclic ring was closed between C13–C14 using a Nozaki–Hiyama–Kishi (NHK) reaction. Altogether, three key bond formations relied on the NHK coupling (C19–C20, C26–C27, C13–C14). This case nicely underscores the high level of organic synthesis in some pharmaceutical companies.
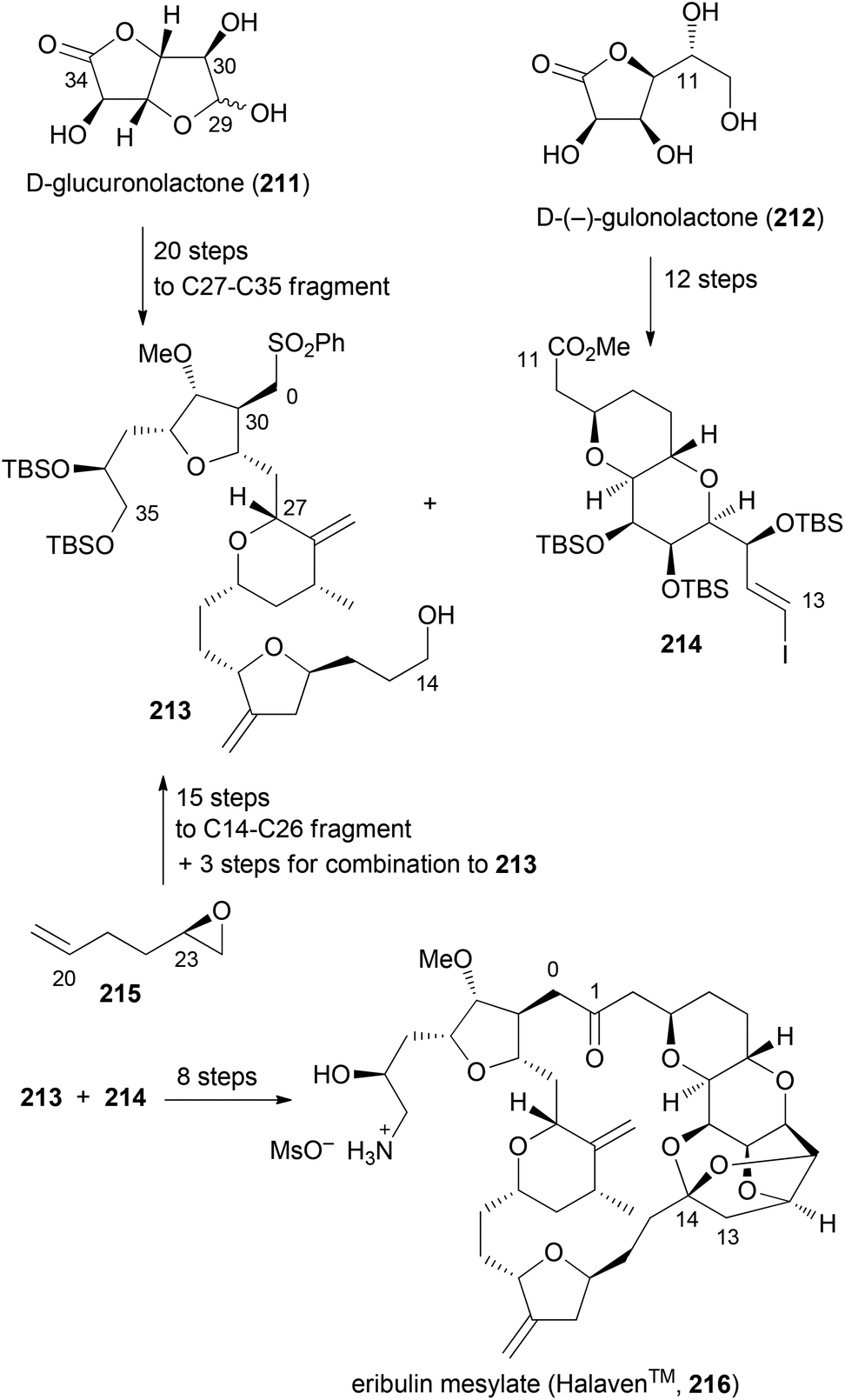 | ||
| Scheme 18 Synthesis of eribulin mesylate (216) on the production scale. | ||
Taking the benzolactone hypothemycin (218) as a lead, the Winssinger group designed simpler mimetics that react irreversibly with certain kinases that have an exposed cysteine in the so-called DFG binding motif. Thus, analogues like 218 feature the resorcyclic acid part as a hinge binder and an electrophilic trap to address the cysteine (Fig. 15).108
 | ||
| Fig. 15 Design of affinity-based probes for kinases with cysteine in the binding region. The cellular inhibition of EGFR autophosphorylation in A431 cells after stimulation with EGF is given for the most active compound 218. | ||
Biology-oriented synthesis (BIOS)
The principle of BIOS is to use scaffolds contained in NPs as starting points for the synthesis of compound libraries.109 This concept is based on the notion that NP scaffolds and possibly substructures of NP core structures define privileged structures. In other words, evolutionarily formed NP scaffolds match highly conserved subfolds of ligand sites.For the selection of suitable NP-like scaffolds the Waldmann group conceived two complementary approaches. The first is a hierarchical structural organization of scaffolds contained in NPs. After removal of side chains that do not contain rings, in each truncation step one peripheral ring is removed according to certain guidelines. This way, parent–child relationships are generated with the parent being the simpler structure. This leads to tree-like branches of NP scaffolds. One such branch is shown in Fig. 16. Representative NPs for each scaffold include digitoxigenin (219), the aglycon of the cardiac glycoside digitoxin, dehydroepiandrosterone (DHEA, 220), the tetradecahydrophenanthrene-type diterpene klysimplexin T110 (221), nakamurol A111 (222), and gabosine A112 (223).
 | ||
| Fig. 16 Example of a branch from the NP scaffold tree (left column). The parent is always the simpler structure in a chain. Representative NPs for each scaffold are shown in the right column. | ||
If one argues that a parent (simpler) scaffold to a child scaffold which contains an active representative might also show some activity if properly functionalized, then a parent scaffold might form the basis of a compound library. Of particular interest are branches that contain gaps, meaning core structures in a sequence that do not have a corresponding NP. Software to visualize the hierarchical organization of NPs is freely available.113 Of particular use is this kind of clustering if biological activity data are available for molecules, representing scaffolds in the branches. In this case, so-called virtual scaffolds located between a complex and a simpler one for both of which compounds are known with related activity should be ideally suited for library development.
A second approach put forward by the Waldmann group starts with proteins. In this protein structure similarity clustering (PSSC), protein folds are clustered according to their similarity. The hypothesis is that a ligand for a member of a protein cluster might also bind to other members of a cluster.114 Therefore, it should be possible to use such NP scaffolds, their parents or grand parents for library construction to address members of a protein cluster that might be relevant for medicinal application.
Recent examples from the Waldmann group are still based on the BIOS approach but at the same time are substantially chemistry driven.115,116 This means that substructures of NPs are used as guidelines which can be constructed or functionalized by novel and simple chemistry. For example, taking the tetrahydroisoquinoline NP noscapine (224) as a lead, a compound collection of tetrahydroisoquinolines 226 was prepared by reacting iminium ions 225 with acetylene nucleophiles (Scheme 19). Not surprisingly, several derivatives were found that, like 227, interfere with microtubule polymerization. In the assays performed, alkyne 227 showed the highest activity.
 | ||
| Scheme 19 Synthesis of a tetrahydroisoquinoline collection. | ||
If a class of NPs comprises a large number of members they also should be promising leads for analogue syntheses. For example, iridoids are widespread in nature and display a range of biological activities.117 The Waldmann group utilized an asymmetric [3 + 2]-cycloaddition between azomethine ylides, generated from glycine imines 229, and pyranones 230 (Scheme 20). The latter are available from furfuryl alcohols via oxidative rearrangement. In the presence of a Cu(I) salt, the ligand 231 and DBU, the cycloaddition proceeded with good dia- and enantioselectivities.118 Out of a collection of 115 derivatives, nine were able to inhibit Wnt signalling with an IC50 below 10 μM. The Wnt pathway plays a role in cell differentiation and stem-cell renewal. Other representatives from this collection turned out to be inhibitors of the hedgehog signalling pathway.
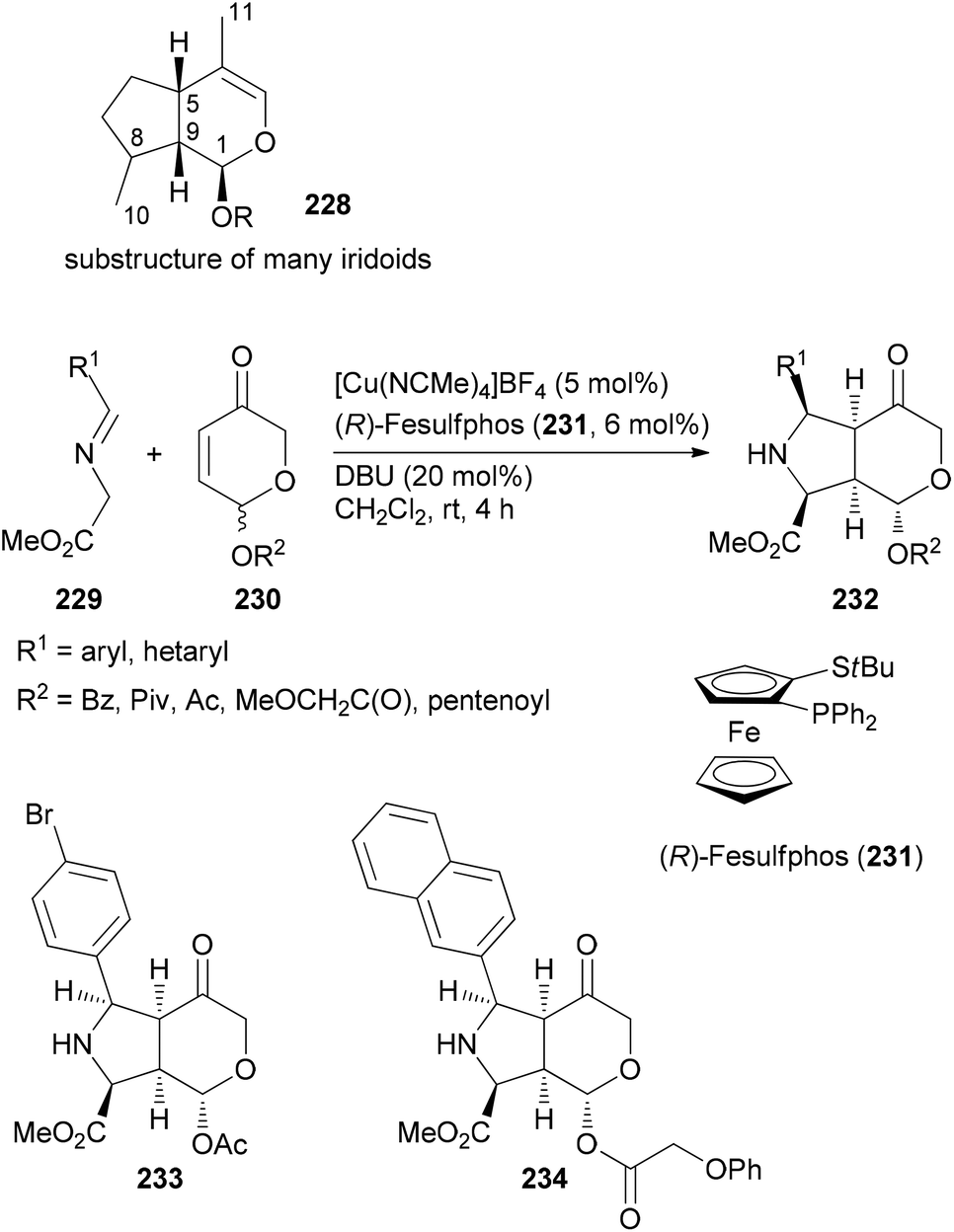 | ||
| Scheme 20 Synthesis of a library of fused heterocycles 232 inspired by the structure of iridoids. | ||
Complexity to diversity (CtD)
Due to the fact that NPs have unique shapes, ring systems and diverse functional groups they are in principle interesting starting points for the preparation of NP analogues. The problem is to selectively address the various functional groups of a NP in order to avoid the formation of complex mixtures. According to Hergenrother, who coined the term complexity to diversity (CtD) one can distinguish two categories.119 One strategy entails reactions on a single functional group to generate a new structure. A classical singular example is the preparation of ansa-steroids by a domino Diels–Alder/retro-Diels–Alder reaction between ergosterol derivatives and propynal.120,121 For example, steroid 235 could be opened to macrocycle 237 utilizing this concept (Scheme 21).122 A second strategy addresses several functional groups on a NP to create new derivatives.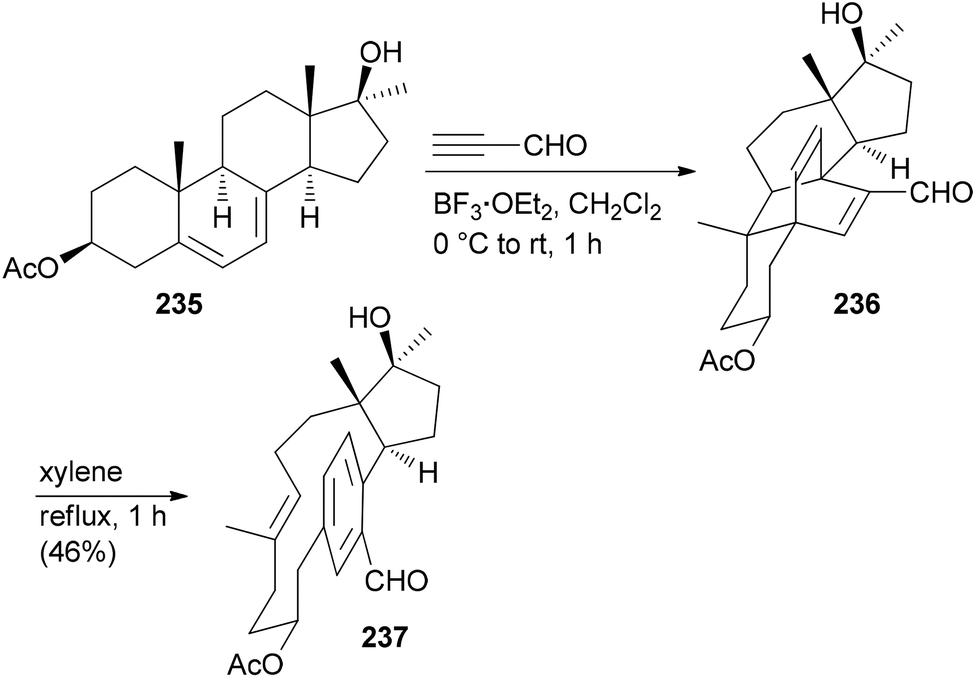 | ||
| Scheme 21 Synthesis of macrocyclic paracyclophane 237 from diene 235via a Diels–Alder/retro-Diels–Alder sequence. | ||
Selective modifications of the diterpene abietic acid (238) enabled the construction of 84 different compounds.123 The initial challenge is to differentiate the two double bonds and the carboxylic group. This is possible by dihydroxylation. From diol 240 a new ring system can be obtained by oxidative cleavage followed by intramolecular aldol condensation (Scheme 22).
 | ||
| Scheme 22 Synthesis of tricyclic compound 242 by the ring distortion strategy. | ||
Manipulation of the B-ring is possible after protection of the diol in 240 as a cyclic carbonate. Ring expansion was illustrated with cyclopropanation followed by electrocyclic ring opening induced by AgBF4 in MeOH (Scheme 23). Ring system 245 resulted from dihydroxylation of the double bond, oxidative cleavage and intramolecular aldol reaction.
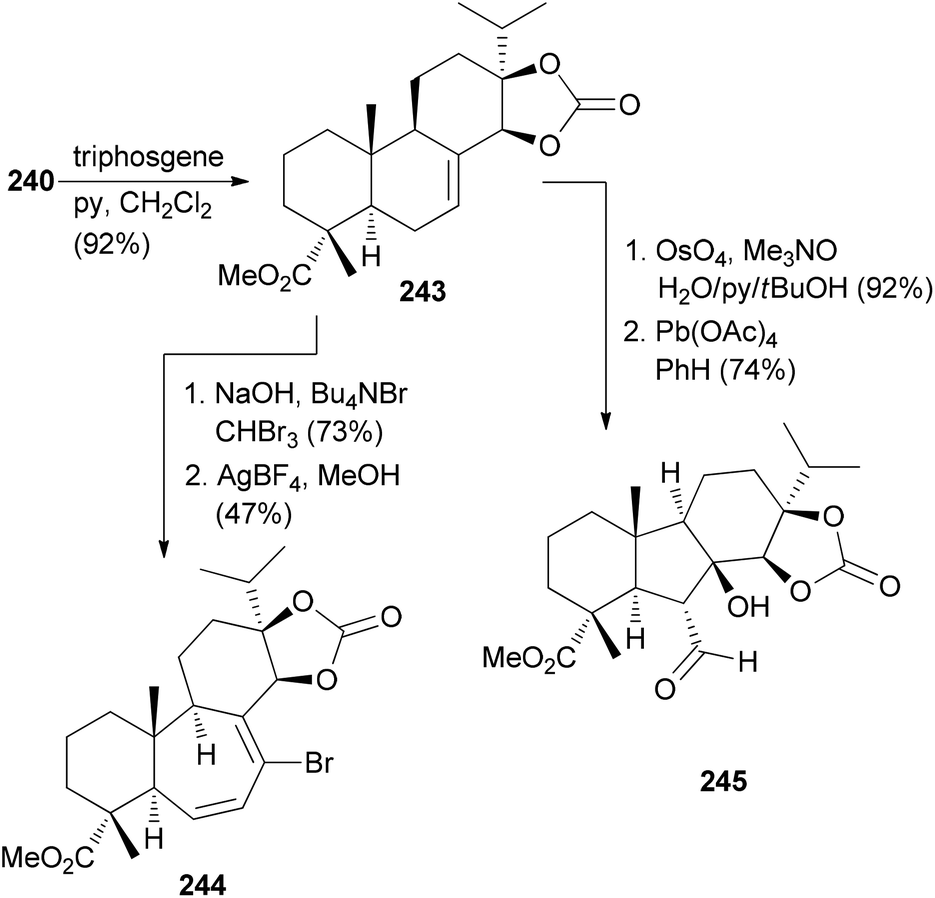 | ||
| Scheme 23 Conversion of alkene 240 to compounds 244 and 245. | ||
The CtD strategy can be applied to all kinds of natural products. For example, the alkaloid quinine (246) was also subjected to controlled structural changes (Scheme 24).124 Thus, treatment of 246 with thionochloroformate resulted in ring cleavage between N1 and C2. The putative intermediate thiocarbonate rearranged to the polycyclic S-alkyl thiocarbonate 247 whose structure could be confirmed by X-ray crystallography. On the other hand, acid induced Hoffmann elimination led to piperidine 248. After protection of the amine, Petasis methylenation of the keto function followed by ring-closing metathesis resulted in octahydroisoquinoline derivative 250 (Scheme 24). Further unique structures were accessible by Grignard additions to the quinoline ring of 246.
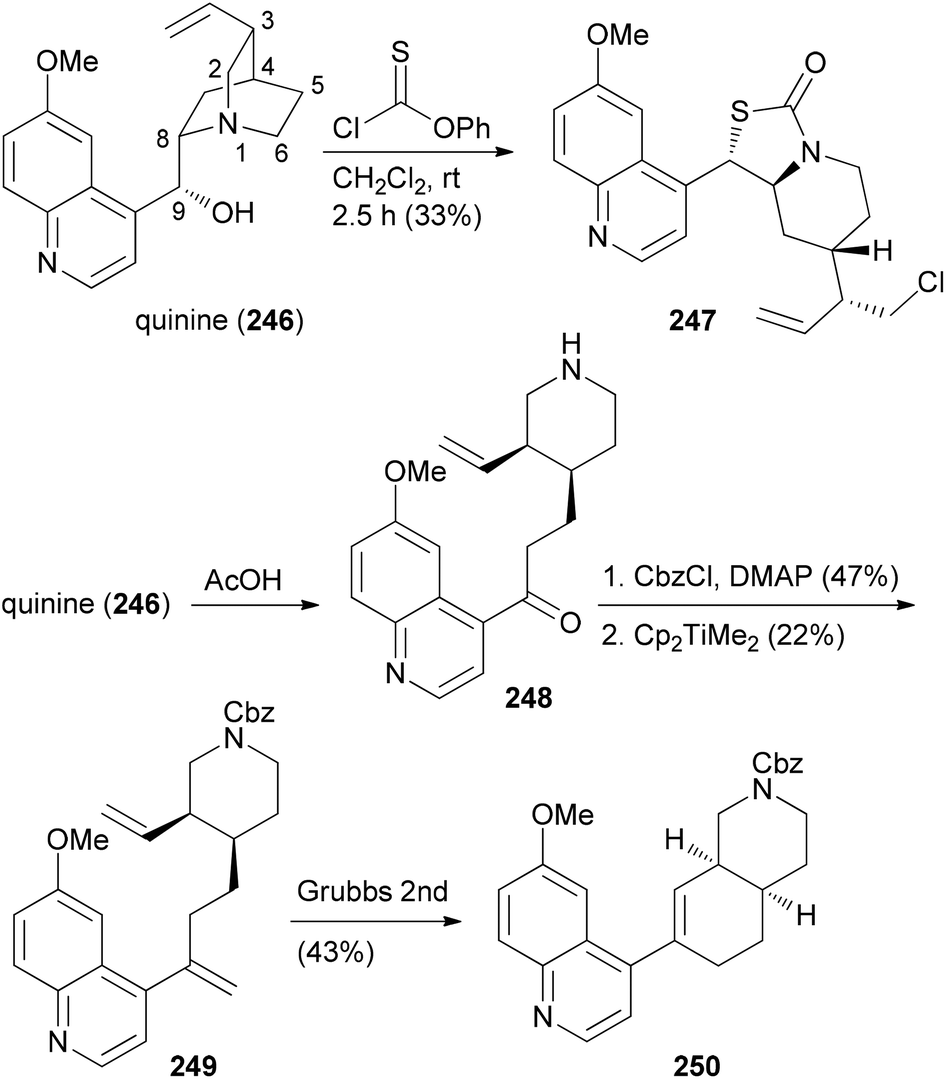 | ||
| Scheme 24 CtD strategies applied to the alkaloid quinine (246). | ||
Several related functional groups in a molecule like an enone or epoxide can be engaged in a domino sequence with a bis-nucleophile to furnish derivatives with increased structural complexity. An illustrative case is the remodeling of fumagillol (251) (Scheme 25). In the presence of the Lewis acid La(OTf)3 and an arylamine a 5-exo-bis-epoxide opening takes place to give annulated pyrrolidine derivatives 252.125 A subsequent reaction of the bicyclic compounds 252 with DDQ (1 equiv.) in the presence of a nucleophile led to a stereoselective addition resulting in complex NP analogues 253. In this example, a terpene is transformed to alkaloid-like compounds.
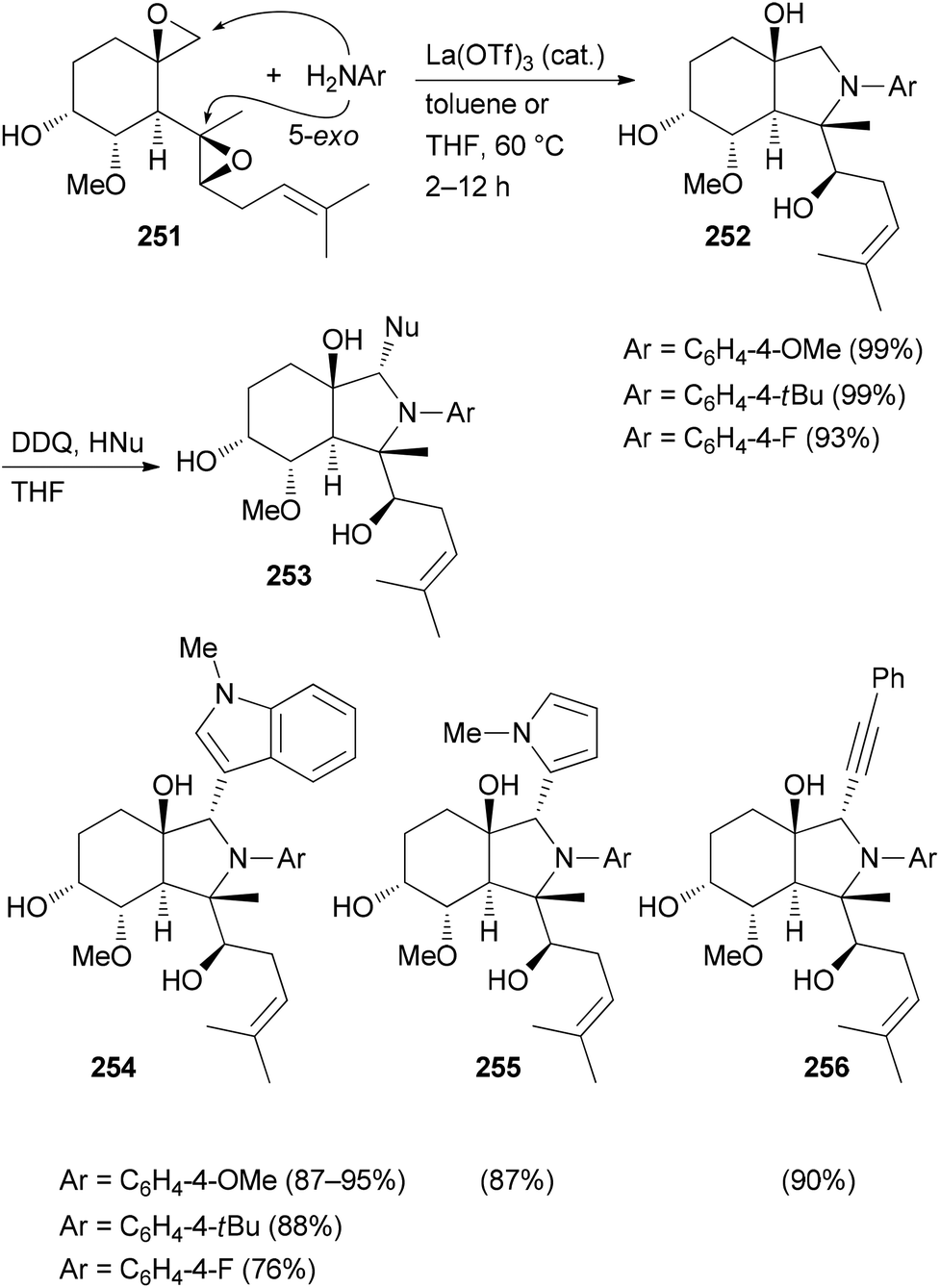 | ||
| Scheme 25 CtD-type manipulations on fumagillol (251). | ||
The CtD strategy appears to be promising and worthwhile for easily available natural products. However, so far it seems that no useful biological activity has been reported for any of the CtD derived NP analogues.
Hybrid molecules
The principle of this concept is to combine partial or whole structures of molecules to create new, possibly more active, molecular entities.126 It is a quite simple and, in certain ways, naive approach. But in biology this concept is widespread. This is very often the case when the subunits of a molecule undertake different functions, for example redox processes or target binding, like DNA recognition. Thus, in the anthracycline antibiotic daunorubicin (257), three functional units can be identified (Fig. 17): a DNA intercalating domain, a domain interacting with the enzyme topoisomerase II, and finally, the sugar part that binds to the minor groove. In fact, the aglycon has 70–100 times lower antitumor activity than daunorubicin. A more complicated structure which can be considered a hybrid molecule is calicheamicin γI1.127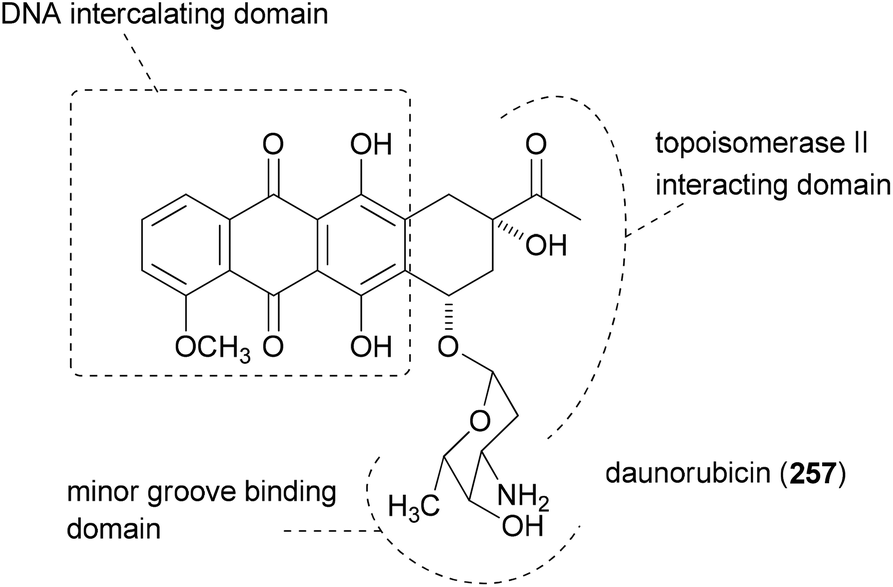 | ||
| Fig. 17 Structure of daunorubicin with its three functional subunits. | ||
The many natural products that are made up of identical parts may also be classified as hybrid molecules. Some prominent examples, including rhizopodin (258), blepharocalyxin D (259), incarvilleatone (260), blennolide G (261), cephalostatin 1 (262) and chaetocin A (263), are shown in Fig. 18.
 | ||
| Fig. 18 Some examples of dimeric natural products. In cases where the subunits are less obvious they are colored. | ||
The dopamine derivative aspongamide A (265) is an example of a trimeric natural product. It is made up of three N-acetyl-dopamine (264) molecules (Fig. 19). This compound was isolated from the insect Aspongopus chinensis, which is found in southern China.128 Dried bodies of this insect are used as traditional medicine for the treatment of pain, stomach problems and kidney diseases. This insect was investigated with the aim to find natural products that are active against chronic kidney disease (CKD). This disease is a consequence of diabetes. The trimer 265 turned out to inhibit smad3 phosphorylation, which plays a role in pathogenesis of CKD.
 | ||
| Fig. 19 Aspongamide A (259) as an example of a trimeric natural product. | ||
The use of hybrid molecules is one of the pillars of bioconjugation. For example, target identification of a bioactive molecule often relies on constructs where the small molecule is linked via a T-piece adapter to a photoreactive group and biotin.129 In these cases it is known from the outset that the biological activity of such constructs is usually reduced, but the purpose of course is different. One such hybrid molecule is depicted in Fig. 20. It contains the microbial product GEX1A (herboxidiene, 145). This NP was originally isolated as a herbicide.130 Later it turned out to also have antitumor activity in vivo. Its side chain is quite similar to that of the polyketide pladienolide B (141), a compound that also displays potent antitumor activity. As pladienolide B targets the SF3b1 subunit of the spliceosome, it is not too surprising that construct 267 led to the discovery that GEX1A also targets a subunit of SF3b.131 In particular, the development of click reactions and chemical ligation methods facilitated the construction of hybrid molecules.
 | ||
| Fig. 20 Hybrid 261 that led to the discovery that GEXA1A targets the spliceosome. | ||
According to Meunier three categories of hybrid molecules can be distinguished (Fig. 21).132 It might be that both parts of the hybrids act on one target. This does not need to be the same binding pocket.
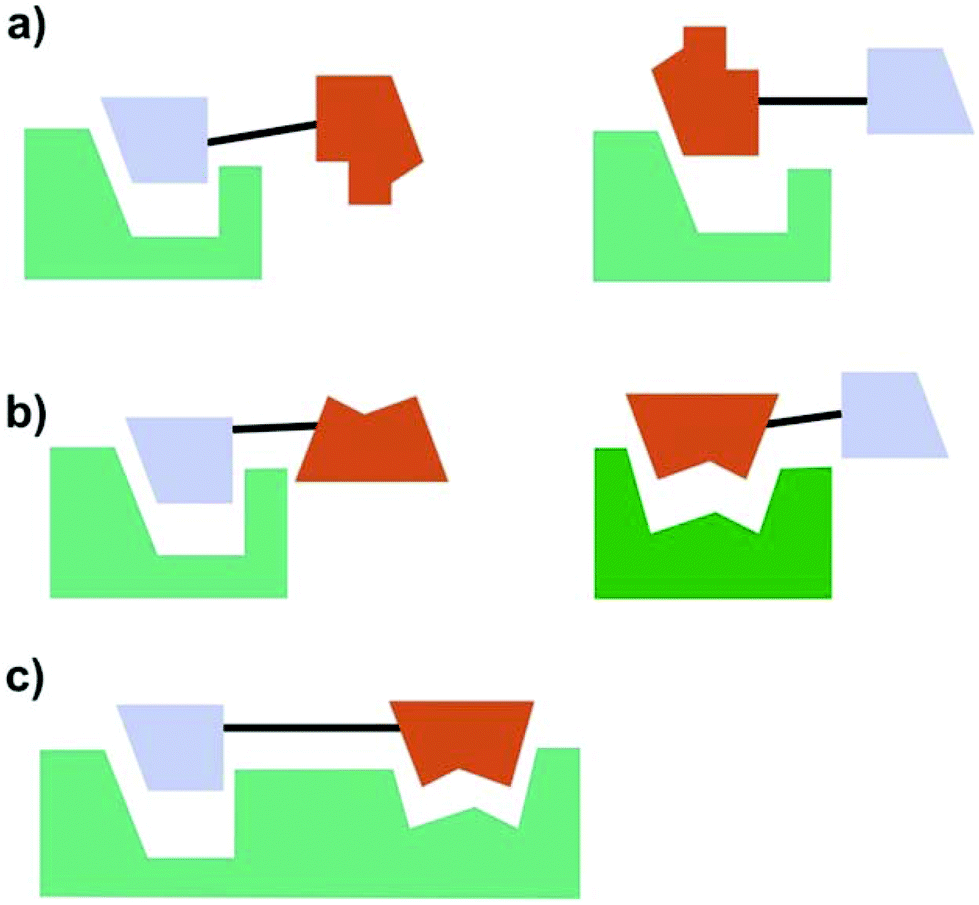 | ||
| Fig. 21 Categories of hybrid molecules according to their mode of action. (a) Both parts act on the same target (not necessarily on the same site); (b) each part acts on a different target; (c) both parts bind to one target at closely related binding sites. | ||
A case in point is the so-called trioxaquines that contain an aminoquinoline and a 1,2,4-trioxane. The latter is inspired by the natural product artemisinin (268) which is a powerful antimalarial drug. The conjugate PA1103/SAR116242 (270) and related molecules are considered “double-edged sword” drugs (Fig. 22). Thus, the aminoquinoline sector can stack with heme, whereas the trioxane part can alkylate heme after reductive activation. Indeed these hybrids are active on chloroquine-resistant strains of Plasmodium falciparum. Compound 270 was eventually selected as an antimalarial drug candidate for further development.133
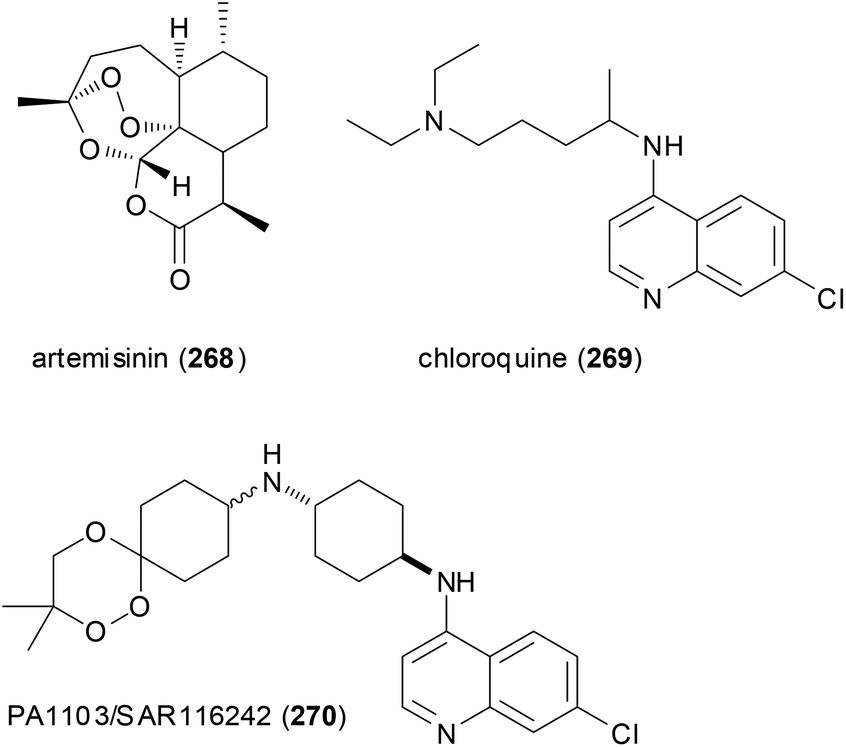 | ||
| Fig. 22 Trioxaquines like 270 are patterned after artemisinin (268) and chloroquine (269). Both parts act on heme of the parasite. | ||
Hybrid molecules of the second category act with each subunit on different and non-related targets. If the linker is cleaved in the biological system, the hybrids are like dual prodrugs. The parts of the hybrid molecule may also act at the same time on two connected targets, as illustrated with category (c). Examples include fragment based drug discovery134 or DNA ligands, like the above-mentioned daunorubicin. If the exact mode of action of a hybrid is not known, it is of course difficult to categorize it according to Fig. 21.
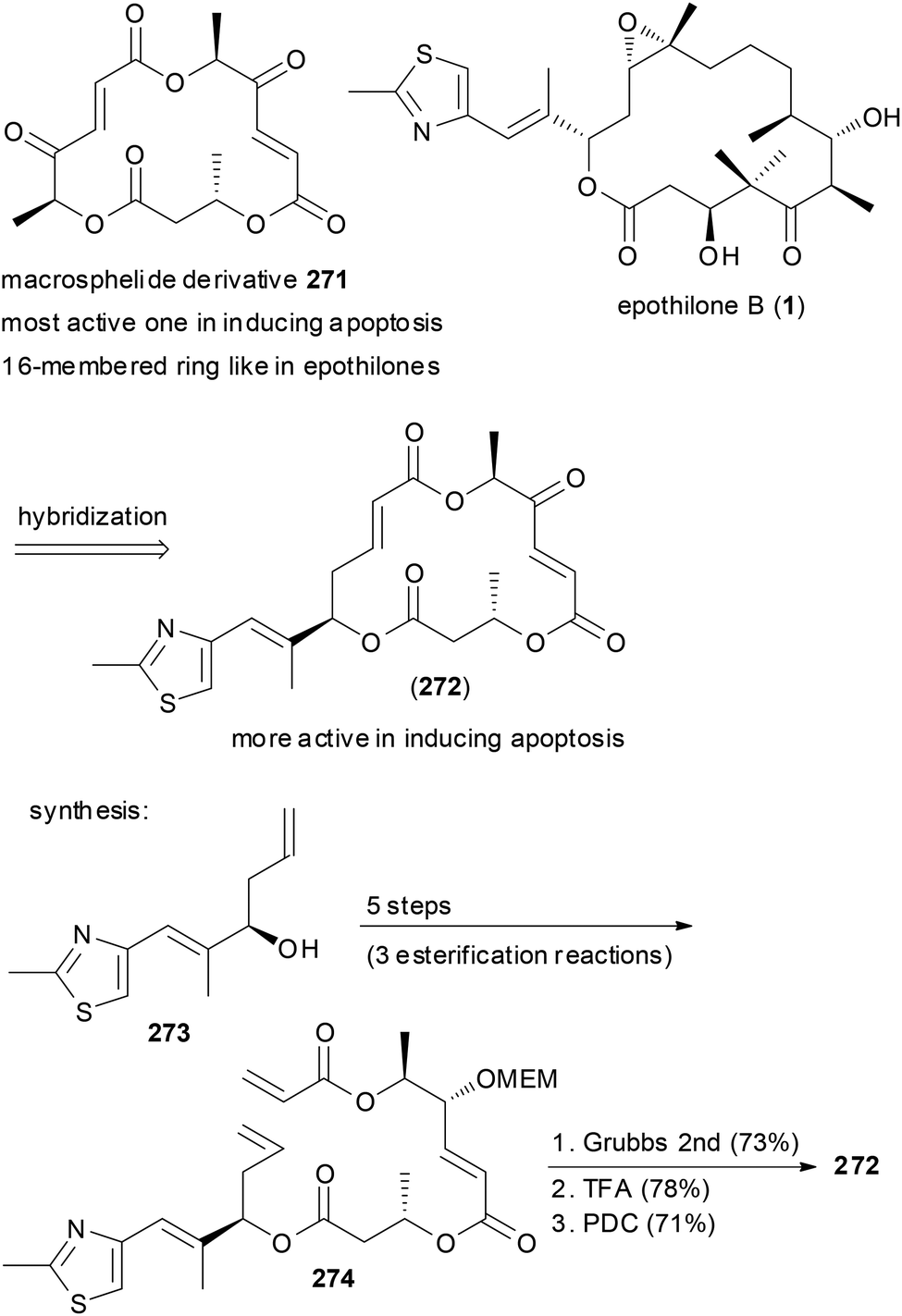
Macrocycles frequently have been turned into hybrid molecules. An option is to borrow a side chain from an active molecule and to attach it to a macrocyclic core. For example, some of the 16-membered macrosphelides are able to induce apoptosis in human lymphoma U937 cells. In an attempt to enhance their potency, hybrid macrosphelides were designed that contain the side chain of the epothilones (Scheme 26).135 The synthesis of hybrid 272 started from homoallylic alcohol 273, which was extended by three esterification reactions to triester 274. A ring-closing metathesis reaction and simple functional group manipulations led to NP analogue 272. This compound showed significantly higher apoptosis-inducing activity than the parent macrosphelides.
 | ||
| Scheme 26 Design and synthesis of macrosphelide–epothilone hybrids. | ||
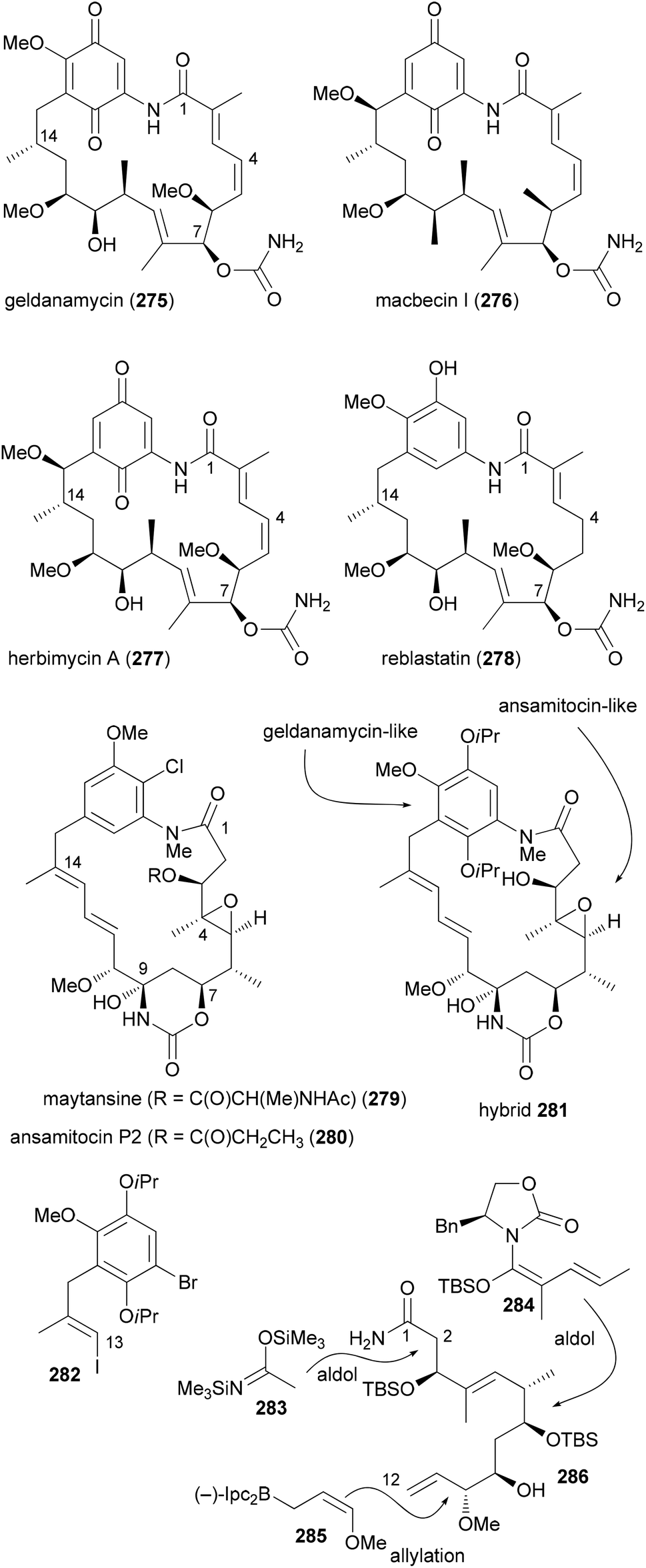
A more dramatic modification is the replacement of a region in a macrocycle with a section from another NP. This is best done by total synthesis. The Kirschning group designed hybrids that contain the aromatic core of geldanamycin (275) and the maytansine aliphatic part (Scheme 27).136 Geldanamycin (275) and maytansine (279) are representatives of the ansamycin antibiotics. The subfamily of the maytansinoids consists of several ansamitocins as well as maytansine. The geldanamycin137 subfamily includes macbecin I (276), herbimycin (277), and reblastin (278). While the maytansinoids share the same ring size for the macrolactam with the geldanamycins, they differ in their mode of action. Whereas the maytansinoids bind to and block the polymerization of tubulin, the cytotoxicity of the geldanamycins is due to their interaction with the heat shock protein Hsp90α. Therefore, the synthesis of hybrids like 281 seemed to be of interest and to make sense.
 | ||
| Scheme 27 Design and chemical synthesis of maytansine–geldanamycin hybrids. | ||
The aliphatic C1–C12 part 286 could be obtained from acrolein in a sequence of 12 steps utilizing asymmetric allylation and aldol reactions to create C–C bonds and the stereocenters (Scheme 27). A Heck coupling between vinyl iodide 282 and building block 286 was followed by an intramolecular copper-catalyzed amidation reaction. After the generation of the hydroxyketone functionality at C7–C9 a biotransformation attached the carbamoyl group at 7-OH.
Due to the isoproxy groups that could not be removed, hybrid 281 exists as a mixture of separable atropisomers. The minor isomer displayed significant activity against cancer cell lines (KB-3-1: 2.2 μg mL−1; SKOV-3: 4.5 μg mL−1). This activity might be connected to a mechanism that is different from the ansamycins. On the one hand 281 does not bind to Hsp90α. On the other hand its conformation is different from that of the maytansinoids. This example shows that it is difficult to predict reliably at the outset of a design the activity of a hybrid molecule.
The replacement of a larger section of a macrolactone with a fragment obtained from an amino acid is another strategy for obtaining hybrids of a NP. For example, latrunculin derived hybrid macrocyclic carbamates 287 were prepared using this rationale (Fig. 23).138 So far, however, biological data for these and other compounds are lacking.
 | ||
| Fig. 23 Hybrid structures designed based on latrunculin A. | ||
Molecules that bind to microtubules represent an important class of anticancer agents. Such molecules can stop cell cycle progression either by breaking up or stabilizing microtubules. Based on combretastatin A-4 (288), which causes disruption of microtubules through binding to β-tubulin, and pironetin (22), which inhibits tubulin assembly by targeting α-tubulin, the Marco group designed hybrids consisting of combretastatin A-4 (CoA4, 288) and simplified pironetin analogues linked together via a tether of variable length (Fig. 24).139 The simplified pironetins 291 and 292 contain at least the essential α-pyrone moiety. Compound 292 additionally features the 7-OH and the 9-OMe groups. The idea was to find improved compounds that can either bind α- or β-tubulin. Even though the exact distance between the two binding sites is not known, compounds might be found that address both sites simultaneously.
 | ||
| Fig. 24 Structures of pironetin analogue/combretastatin hybrids and the corresponding precursors. | ||
As described above (Scheme 2) the secondary alcohol functions were generated by Brown allylation reactions on the corresponding aldehydes. CoA4 derivatives equipped with a linker were prepared from CoA4 (288) by etherification with ω-bromo esters to give esters 289a–d followed by saponification to the acids 290a–d. A Yamaguchi esterification served to prepare the hybrids 293a–d and 294a–d (Fig. 24).
The screening for cytotoxicity was performed with two tumor cell lines (human colon adenocarcinoma HT-29 and breast adenocarcinoma MCF-7 cell lines) and, for comparison, with the normal human embryonic kidney cell line HEK-293. As it turned out all compounds were active in the low to medium micromolar range including the parent NPs (Table 4). Compounds 289b, 289c, 289d and 290d were active in both cell lines. Worth mentioning are compounds ent-293a, 294c, 289c, 289d and 290d that show pronounced selectivity for the tumor cell lines. Among the hybrids 293a–d and 294a–d that differ from each other in the length of the connecting spacer, the ones with a ten-carbon chain show maximum activity (293c and 294c). For all hybrids it seems that there is no simultaneous binding to both α- and β-tubulin. Comparing the hybrids 293a–c and their enantiomers to the pironetin analogue points to the fact that the cytotoxicity mainly originates from the CoA4 region. This example shows that a rational design of hybrids remains rather difficult.
| Compound | HT-29 | MCF-7 | HEK-293 |
|---|---|---|---|
| a Compounds with high toxicity towards both tumoral cell lines are marked in italics. | |||
| CoA4 (288) | 4.2 | 1 | 25 |
| Pironetin (22) | 8 | 18 | 46 |
| 293a | 12 | 52 | 39 |
| 293b | 32 | >300 | 80 |
| 293c | 10 | >300 | >300 |
| 293d | 107 | >300 | >300 |
| ent-293a | 3.5 | 17 | 114 |
| ent-293b | 19 | >300 | >300 |
| ent-293c | 38 | 69 | 59 |
| ent-293d | 64 | >300 | 72 |
| 294a | 15.4 | 31 | 22 |
| 294b | 44 | >300 | >300 |
| 294c | 2.9 | 5.6 | 22 |
| 294d | 50.8 | 21 | 81 |
| 289a | 60.1 | 10.4 | 121 |
| 290a | 50 | 2.9 | 67 |
| 289b | 3.5 | 9.6 | 9.6 |
| 290b | 16 | 17 | >300 |
| 289c | 8 | 3 | >300 |
| 290c | 103 | 5 | >300 |
| 289d | 5 | 9.6 | >300 |
| 290d | 1.9 | 4.4 | 39 |
| 291 | 109 | >300 | 35 |
| ent-291 | 81 | 109 | 119 |
| 292 | 25.4 | 2.5 | >300 |
Biosynthesis inspired synthesis of NP analogues
In many cases the biosynthesis of a NP is known. At least hypotheses regarding key transformations do exist in general. This is often provided whenever an alkaloid or terpene with a novel structure is published.140,141 For NPs from microorganisms a lot is known about the biosynthetic machinery and the genes that code for the structure of a NP. This is the case with polyketides from bacteria and partly from fungi.142 These genes are arranged in a sequence of modules. A loading module catches the starting carboxylic acid derivative, which is bound to the biosynthesis machinery as a thioester. Every module extends the chain by two carbon atoms via a Claisen condensation. The modules may contain additional domains that perform typical steps common to fatty acid biosynthesis, namely, reduction of the 3-ketoester to a 3-hydroxy ester (KR = keto reductase), elimination of water to give an enoate (DH = dehydratase) and conjugate reduction of the enoate (ER = enoyl reductase). Typical extender units are malonyl-CoA and methylmalonyl-CoA. Some other ones are known such as methoxymalonyl-CoA or allylmalonyl-CoA. Thus, by simply looking at the structures of the seco acid of a polyketide macrolactone, the domains, besides the ACP and KS that perform the Claisen condensations, can be predicted. This is shown in Fig. 25 for the immunosuppressive FK506 (295).143 Module 4 uses allylmalonyl-CoA, and modules 7 and 8 use methoxymalonyl-CoA as extender units. | ||
| Fig. 25 Biosynthesis of the polyketide FK506 (tacrolimus). Domains in modules might be inactive (red), non-functional (blue) or completely absent. KS, ketoacyl synthase (performs Claisen condensation; prior to the Claisen condensation the growing chain is attached to KS, the extender unit to ACP); AT, acyl transferase; DH, dehydratase; ER, enoyl reductase; KR, keto reductase; ACP, acyl carrier protein. | ||
Mutasynthesis
The detailed knowledge about the biosynthesis of polyketides can be exploited for the synthesis of NP analogues. In the case of mutasynthesis a mutant organism is generated where the synthesis of a natural building block has been blocked by deletion of the corresponding gene. Frequently, this refers to the starting unit of a polyketide which typically is very different from the extender units.144 Using mutasynthesis, the Kirschning group produced a range of geldanamycin derivatives that by total synthesis would not be available so efficiently.145 Thus, by feeding various 3-aminobenzoic acids to the mutant S. hygroscopicus K390-61-1, in which the synthesis of the natural starting unit was blocked, a range of novel geldanamycin analogues could be obtained (Scheme 28). The acids shown were accepted well and the process could be scaled-up. One limitation of mutasynthesis lies in the fact that only certain building blocks are accepted by the acyltransferase (AT) of the loading module. | ||
| Scheme 28 Mutasynthesis of nonbenzoquinone geldanamycin analogues by mutasynthesis. The analogues were obtained by feeding the acids to the blocked mutant of (K390-61-1) of S. hygroscopicus. | ||
Mutasynthesis can be combined with chemical synthesis (semisynthesis) if building blocks are incorporated that allow for subsequent selective transformations. This has been illustrated with the synthesis of ansamitocin analogues.146 For example, feeding of 3-amino-4-bromobenzoic acid (309) to the AHBA (−) blocked mutant of Actinosynnema pretiosum produced the ansamitocin P3 derivative 310. This compound turned out to be a suitable substrate for Pd-catalyzed cross-coupling reactions (Scheme 29).147
 | ||
| Scheme 29 Mutasynthesis followed by chemical modification. Both compounds were quite active [IC50 (pmol mL−1), KB-3-1 cell line, 310: 0.46, 311: 0.67]. | ||
In case an extender unit in a polyketide significantly differs from the standard acetate and propionate building blocks, mutasynthesis of analogues is possible by knocking out the synthesis of the unusual extender unit and adding substituted malonyl-CoA extender units. For example, the allyl group at C21 of the immunosuppressive FK506 is incorporated into the polyketide chain via the allyl malonyl-CoA extender unit 317 (Scheme 30). Investigations by Moore et al. showed that it is synthesized from 3-ethylacrylate 314. Reductive carboxylation is followed by introduction of the terminal double bond. For the final dehydrogenation the gene tcsD is responsible. By knocking out the tcsB gene that is involved in the synthesis of this building block (Claisen condensation to 313), a deletion mutant of Streptomyces sp. KCTC 11604BP was generated and used for the mutasynthesis of some FK506 analogues.148 By feeding trans-2-hexenoic acid (320), 4-methylpentanoic acid (322) and 4-fluorocrotonic acid (324) the known analogue 321 and the new analogues 323 and 325 were obtained. Biological testing showed 36-methyl-FK506 (323) to exhibit improved neurite outgrowth activity. However, the in vivo production levels turned out to be rather modest (0.06 mg mL−1 for 323).
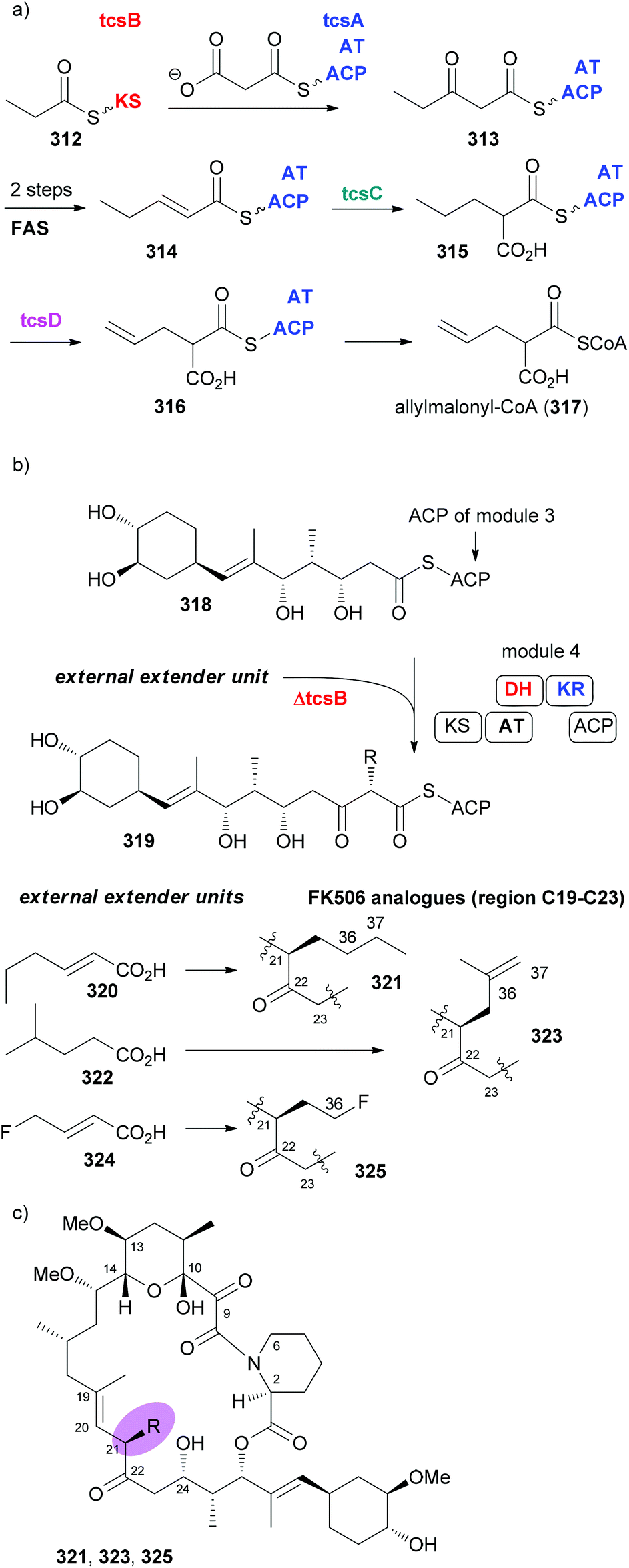 | ||
| Scheme 30 Mutasynthesis of FK506 analogues 321, 323, and 325 by feeding the acids 320, 322, and 324, respectively, to the ΔtcsB strain of Streptomyces sp. KCTC 11604BP. | ||
In order to replace parts of a NP that originate from common extender units, pathway engineering is necessary. For example, a range of geldanamycin analogues could be produced by replacing the original acyltransferase (AT) domains in various modules of the geldanamycin polyketide synthase (GdmPKS) with a malonyl AT domain from the rapamycin PKS. Thus, changing AT1 which incorporates normally methylmalonyl-CoA led to analogue 326 (Fig. 26).149 Other obtained analogues are also shown. In all cases production yields were sufficient for the characterization of the analogues and activity assays. Analogue 329 displayed a four-fold enhancement of the binding affinity for Hsp90.
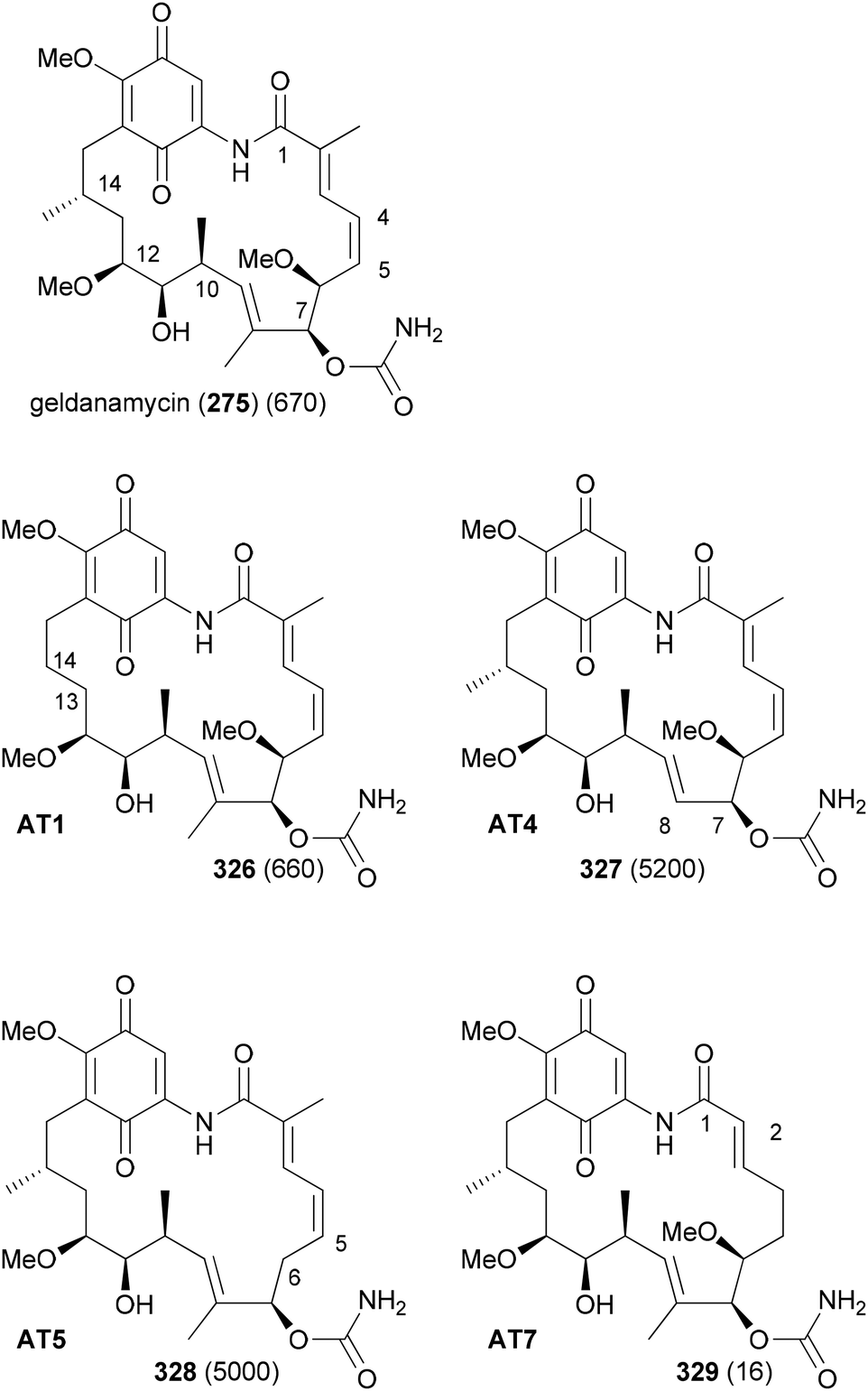 | ||
| Fig. 26 Geldanamycin analogues obtained by AT substitutions in the GdmPKS. AT1 incorporates chain atoms 13 and 14, AT4 atoms 7 and 8, AT5 atoms 5 and 6, and AT7 atoms 1 and 2. Values for Hsp90 Kd (nM) are given in brackets. | ||
If all the genes required for the synthesis of an unusual extender unit are engineered into a producer where the particular biosynthesis of the extender unit to be replaced is blocked, then novel NP analogues can be obtained as well. The group of Moore cloned the three genes anlF, anlG and anlE that produce the isobutyrylmalonyl-CoA building block in an ansalactam producer strain into the integrative vector pSET152. Gene expression was placed under the control of the constitutive ermE* promoter in vector pLD6. These plasmids were conjugated into the mutant Streptomyces sp. KCTC 11604BP ΔtcsB so that background production of FK506 was eliminated.150 With the gene tcsD, which is responsible for dehydrogenation, still intact, good production levels of 36-methyl-FK506 (323) could be realized (Scheme 31).
 | ||
| Scheme 31 Production of 36-methyl-FK506 via heterologous expression of the anIE-anIF-anIG gene cassette in a Streptomyces sp. KCTC 11640BP ΔtcsB mutant strain. The red enzymes are from an ansalactam A producer. | ||
Silent genes
The sequencing of bacterial genomes and the associated genetic analysis did provide some stunning facts. Thus, it is possible to predict the stereochemistry of secondary hydroxyl functions from the gene sequence of the corresponding keto reductases and the stereochemistry of methyl-bearing centers.151 Another finding was that in many polyketide synthases some genes are silent. That is, some enzymes are not operative. The exact reason for this is unknown. In order to shed some light on this phenomenon the total synthesis of NPs encoded by the complete polyketide synthases is a worthwhile task. This has recently been done for the macrolide soraphen A.152 For some time this NP was a promising antitumor and antifungal compound due to its ability to inhibit eukaryotic acetyl-coenzyme A carboxylase. According to the genetic analysis, modules 5 and 8 contain inactive domains (Fig. 27).153,154 Module 5 should produce a saturated section. Since the ER is not active an enoate double bond should remain (C8–C9). However soraphen A (335) has a double bond at C9–C10. This could either be the result of a deconjugation on the enoate or of a postketide transformation. Module 8 is lacking an ER and features inactive KH and DH domains. | ||
| Fig. 27 Structure of soraphen and arrangement of modules 1–8. The domains for modules 2, 5 and 8 are indicated together with the structure of the seco acid 338 corresponding to the full functional domains. KS = keto synthase, AT = acyl transferase, DH = dehydratase, KR = keto reductase, ER = enoyl reductase, TE = thioesterase. | ||
Based on the structure of soraphen A and the gene sequence, Kalesse et al. prepared the so-called paleo-soraphens A (339) and B (340) (Scheme 32). First, pentenal 341 was converted via an organocatalytic α-hydroxylation to building block 342. A cross-metathesis between 342 and 343 followed by hydrogenation delivered the C11–C17 part 345. The C3–C10 region 349 containing an array of five vicinal stereocenters could be constructed from Roche aldehyde 347 which was elongated on both sides using an Evans aldol reaction and a Marshall reaction, respectively. The vinyl iodide function resulted from hydrozirconation/iodination of the corresponding terminal alkyne. For the coupling of aldehyde 345 with vinyl iodide 349 a Nozaki–Hiyama–Kishi reaction came to use. After oxidation of the resulting alcohols (2:1 mixture), enone 350 was obtained. A chelation-controlled reduction [Zn(BH4)2, Et2O, −55 °C, 90%, d.r. > 25:1) delivered the anti-stereochemistry at C11/C12. Methylation and reductive ester cleavage furnished the advanced building block 351.
 | ||
| Scheme 32 Key transformations in the synthesis of the paleo-soraphens A and B. | ||
From there, six routine steps including chain extension by Wittig reaction and Shiina macrolactonization led to paleo-soraphen A (339). One additional step, namely the hydrogenation of the double bond of 351 in the presence of the Crabtree catalyst, was required to reach paleo-soraphen B (340).
Biological tests showed both compounds to have much lower antifungal activity (Table 5). The cytotoxic activity was also almost gone.
| Compound | Pythium debaryanum | Botrytis cinerea | Mucor hiemalis |
|---|---|---|---|
| Paleo-soraphen A (339) | 0.4 | >40 | 21 |
| Paleo-soraphen B (340) | >40 | >40 | 16 |
| Soraphen A (335) | 0.045 | 0.0035 | 0.0006 |
The mode of action of the two paleo-soraphens was subsequently probed by impedance profiling. The principle of this method is that molecules with a similar mode of action will show similar time-dependent impedance curves. A hierarchical cluster analysis indicated paleo-soraphen A to be in close proximity to soraphen A. In contrast, paleo-soraphen B turned up in a cluster with camptothecin, a well-known topoisomerase inhibitor.
While in the case at hand this concept did not lead to highly potent compounds it appears promising for the selection of targets for synthesis programmes. If this observation should be general, it might also reveal regions of NPs that are important for the biological activity. Thus, the parts which originate from modules with silent domains would then be important for activity.
Propionate-scanning
It is obvious that the molecular scaffold and substituents on a NP determine its affinity and selectivity to a cellular target. The scaffold together with operating conformational effect secures directionality and distance of functional groups that are involved in direct interactions with the target protein. Many NPs are characterized by a molecular built. This means that they are assembled from small building blocks as in the case of peptides, polyketides or terpenes. In a way they can be considered natural oligomers. However, in general it is not known what determines the selection of a building block for a certain position. To what extent this is random or driven by evolution remains largely unclear. In peptides an alanin scan is often used to address the importance of amino acids at certain positions.155,156In polyketides two main building blocks, acetate and propionates, are common. A propionate results in a methyl substituent on the backbone. There are polyketides that consist of only one type of building block. For example, zearalenone (353) and curvularin (355) are solely acetate based NPs (Fig. 28). On the other hand erythronolide B (356) only contains propionate building blocks.
 | ||
| Fig. 28 Structures of natural products that are only made up of one type of building block. Key polyketide intermediates for zearalenone and curvularin are also shown. Red bold bonds indicate the acetate carbons. The 6-OH in 356 originates from an oxidation. | ||
Typically polyketide NPs are made up of both types of building blocks. Thus, the antitumor compound dictyostatin (357) is constructed from seven acetates and six propionates (Fig. 29). It is also worth mentioning that plants and microorganisms themselves produce NP analogues in large numbers. Thus, with all natural product classes rather similar compounds have been isolated over the years. In fact, it is nowadays not that easy to discover really novel natural products. With regard to polyketides, epothilone A (358) and B (1) represent a pair of macrolactones that only differ in one methyl group. The myxobacterium strain Sporangium cellulosum produces 22 mg mL−l epothilone A (358) and 11 mg mL−l epothilone B (1).157 The additional methyl group (propionate instead of acetate) in epothilone B results in about ten times stronger antitumor activity. In the case of the statins also several similar compounds are known that only differ in a few substituents. While compactin (359), isolated from the fungus Penicillium citrinum, has too many side effects, lovastatin (mevacor) (360) from Aspergillus terreus and pravastatin (mevalotin) (361) from the bacterium Nocardia autotrophica are in clinical use. Due to their inhibitory effect on HMG reductase, the statins are used as cholesterin lowering drugs. However, one should mention that designed NP mimetics like atorvastatin have a higher market share. These examples underscore the importance of substituents for biological activity.
 | ||
| Fig. 29 Structure of dictyostatin (357), a mixed polyketide, and examples of NP analogues produced by nature. | ||
Our goal in this project was to investigate the role of an additional methyl group in the simple lactones zearalenone (353) and curvularin (355). The plan was not to use genetic engineering but rather organic synthesis. We wanted to consider biosynthetic aspects, meaning that the methyl group would be positioned on the scaffold where it would make sense from a biosynthesis point of view. Thus, replacing the second acetate with propionate led to propionate analogues like 362 and 363 (Fig. 30).158 In tests for cytotoxicity several analogues showed activity in the low micromolar range, comparable with or better than zearalenone itself. Thermal shift assays performed on Hsp90 revealed a rough correlation of the Tm-shifts159 with the IC50 values. A higher Tm-shift means stronger binding to Hsp90.
 | ||
| Fig. 30 Zearalenone analogues with a propionate as a second building block. The IC50 values against L929 mouse fibroblasts and the Tm-shift values against Hsp90 are given in brackets. | ||
Shifting the methyl group to the third acetate produced rather weak analogues (Fig. 31). Thus, it seems that a methyl group at this position interferes with binding to target proteins relevant for the cytotoxic effects. The corresponding aryl methyl ethers showed even weaker activity.
 | ||
| Fig. 31 Zearalenone analogues with a propionate as the third building block. The IC50 values against L929 mouse fibroblasts are given in brackets. Here Tm-shift values were not determined. | ||
A propionate at position four (5′-Me) led to a rather interesting analogue 367 (Fig. 32). As evaluated by a thermal shift assay it binds to fewer kinase targets than zearalenone.160 Binding to Hsp90 was completely blocked by the additional methyl group. Instead, analogue 367 was found to bind to human carbonyl reductase 1 (CBR 1) in the presence of the oxidized cofactor NADP+ (IC50 = 210 nM).
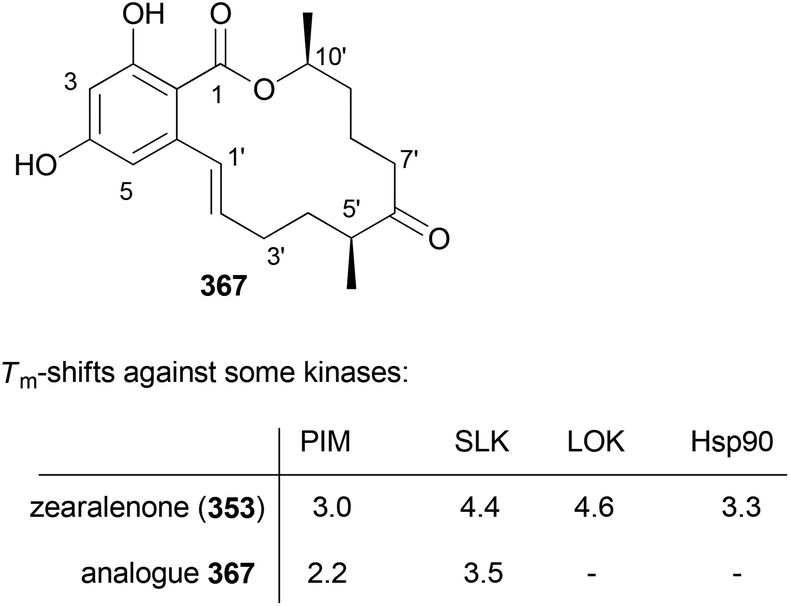 | ||
| Fig. 32 Zearalenone analogue 367 does not bind to Hsp90 and is less strong than other kinases according to DSF. DSF = differential scanning fluorometry. | ||
Shifting the methyl group to the next acetate (3′-Me) also produced an analogue 368 that was more active in the cell proliferation assay than zearalenone (353) (Fig. 33).161 The dissociation constant of 368 for Hsp90 was determined with 130 nM.
 | ||
| Fig. 33 Zearalenone analogues with a propionate as the fifth building block. The IC50 values against L929 mouse fibroblasts are given in brackets. | ||
While we could make these analogues, each substitution pattern required a separate synthetic strategy. Key steps for each group of analogues are summarized in the following schemes. Thus, the synthesis of compounds of type 362 started with the Frater alkylation product 372 which via alkynoate 374 was converted to Weinreb amide 375 (Scheme 33). Grignard addition eventually led to alkenol 376. Its esterification with acid 377 under Mitsunobu conditions furnished ester 378. Ring-closing metathesis [Grubbs 2nd, toluene, 80 °C, 5 h (85%)] and deprotection steps led to analogue 362.
 | ||
| Scheme 33 Synthesis strategy towards zearalenone analogue 362. | ||
For benzolactones of type 364 an rcm-strategy could be used as well (Scheme 34). In the assembly of the aliphatic building block 381 an Evans aldol reaction between 5-hexenal and carboxylic acid derivative 379 set the stereochemistry at C7′. In some of these analogues, the C7′ center was prone to epimerization.
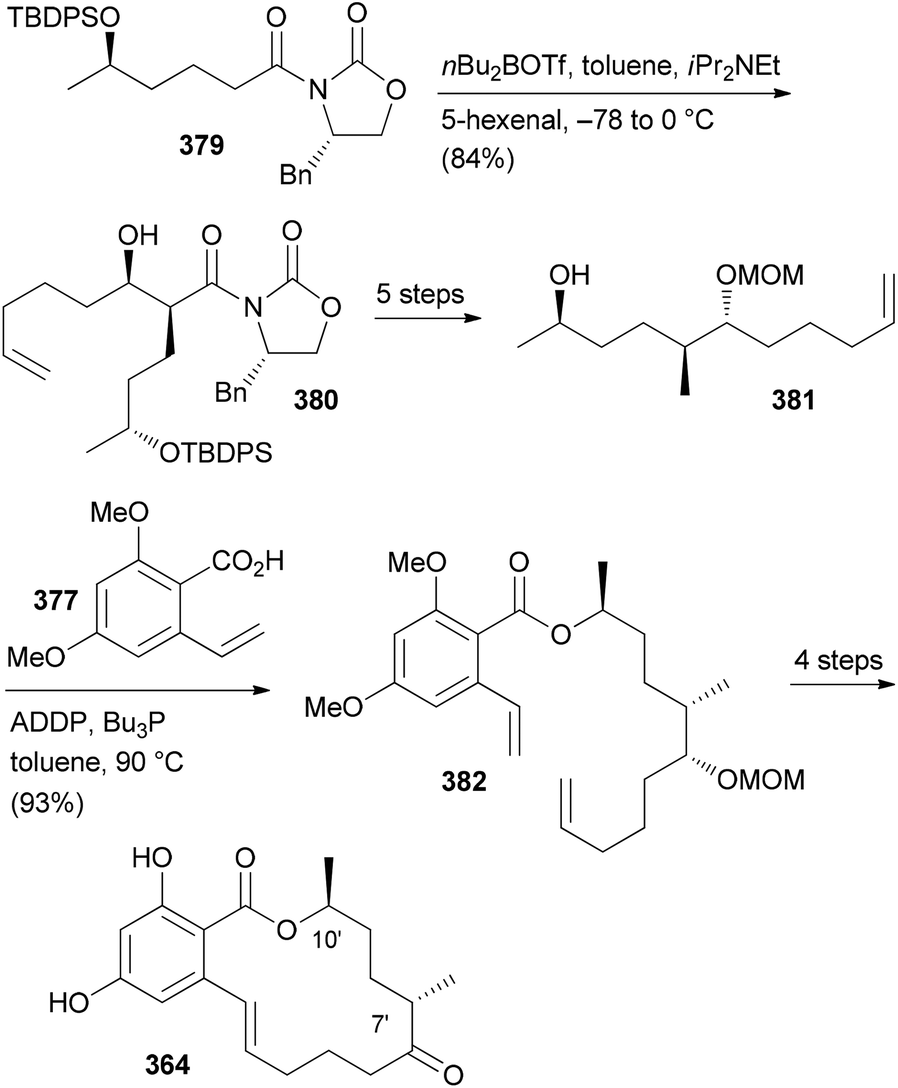 | ||
| Scheme 34 Synthesis strategy towards zearalenone analogue 358. ADDP = azodicarboxylic acid dipiperidide. | ||
A different strategy was employed for the synthesis of analogue 367. Thus, alkynol 383 resulting from a Marshall–Tamaru reaction served as the precursor for alkenol 384 (Scheme 35).160 Its hydroboration and coupling with vinyl iodide 385 led to hydroxy acid 387. Macrolactone formation under Mitsunobu conditions followed by routine steps delivered zearalenone analogue 367.
 | ||
| Scheme 35 Synthesis strategy towards zearalenone analogue 367. | ||
Yet another approach was chosen for analogue 368 (Scheme 36). Here aldehyde 390 was converted to propargylic alcohol 391. Protection and hydrogenation gave the building block 392. The derived sulfone 393 was combined with aldehyde 394via Julia–Kocienski olefination. Carboxylation followed by protecting group manipulation and macrolactonization furnished benzolactone 368. However, even for simple compounds like the polyketide zearalenone the synthesis effort is substantial. Even though we just studied zearalenone analogues it can be concluded that methyl groups modulate biological activity. This is also known from classical drug discovery programmes.162 But with analogue 367 we found one case where the additional methyl group redirects the molecule to another target protein. Such cases seem quite interesting. A similar scanning could be performed with hydroxyl functions or combinations of methyl and hydroxyl groups.
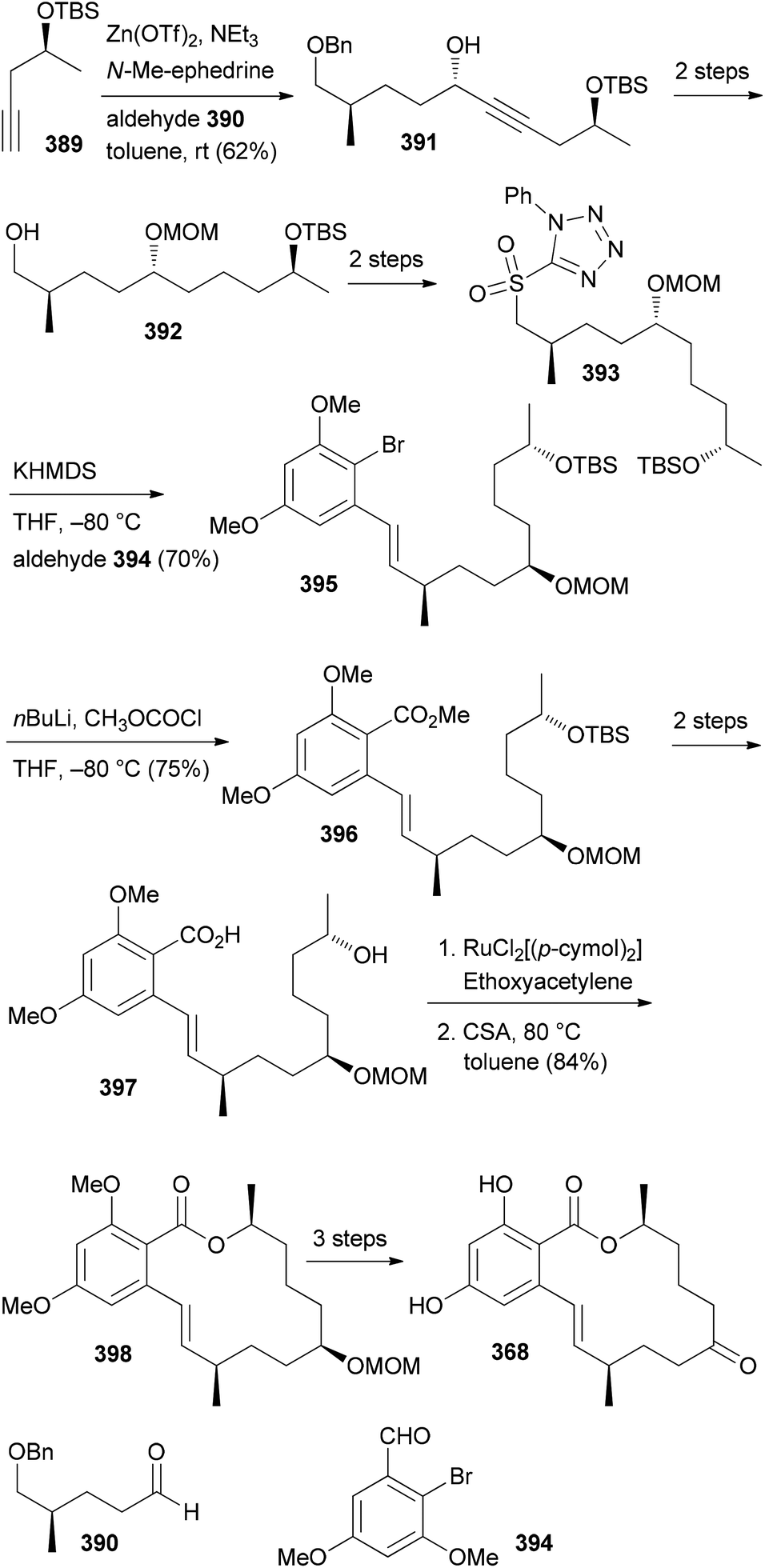 | ||
| Scheme 36 Synthesis strategy towards zearalenone analogue 368. | ||
Conclusions
This review shows that natural product analogues can be prepared by organic synthesis. However, even for simple compounds like, for example, the polyketide zearalenone, the synthesis effort required is substantial. But it is less a problem how we can prepare a certain molecule, but rather a problem concerning which molecules chemists should prepare. After the era of combinatorial chemistry the goal should be to prepare as few as possible molecules to achieve a molecule with the desired properties. Thus, some kind of preselection seems highly desirable in order to avoid the bottleneck organic synthesis. However, even in case the target is known, reliable and accurate docking of flexible NPs or similar compounds has so far appeared impossible. Thus, for many years to come, organic chemists will work like hunters and gatherers. While prediction of the biological activity of a designed molecule will remain difficult, strategies based on genomics appear promising.163 This would require chemists to acquire the relevant skills. Another option would be to use a brute force approach and to put some emphasis on the development of docking software. Thus, with a suitable structure generator that would generate virtual NP analogues based on biosynthesis rules, subsequent docking might be able to suggest promising compounds for synthesis. Until then the design and synthesis of NP analogues will rely largely on trial and error, supplemented with hunches and experience of scientists. In addition, one should not forget the role of serendipity for discovery and the advancement of science.Acknowledgements
Financial support by the Federate State of Baden-Württemberg, the Fonds der Chemischen Industrie, and in particular the DFG is gratefully acknowledged. Part of this work was carried out within the framework of EU COST Action CM0804 (Chemical Biology with Natural Products). I also thank Dr Florenz Sasse (HZI Braunschweig) and his team for performing cellular assays with our compounds for many years.Notes and references
- (a) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311 CrossRef CAS PubMedG. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 3670 CrossRef CAS PubMed; (b) B. B. Mishra and V. K. Tiwari, Eur. J. Med. Chem., 2011, 46, 4769 CrossRef CAS PubMed.
- (a) A. L. Harvey, Drug Discovery Today, 2008, 13, 894 CrossRef CAS PubMed; (b) C. Cordier, D. Morton, S. Murrison, A. Nelson and C. O'Leary-Steele, Nat. Prod. Rep., 2008, 25, 719 RSC; (c) K. Grabowski, K.-H. Baringhaus and G. Schneider, Nat. Prod. Rep., 2008, 25, 892 RSC; (d) N. I. Vasilevich, R. V. Kombarov, D. V. Genis and M. A. Kirpichenok, J. Med. Chem., 2012, 55, 7003 CrossRef CAS PubMed.
- A. Ganesan, Curr. Opin. Chem. Biol., 2008, 12, 306 CrossRef CAS PubMed.
- E. Kellenberger, A. Hofmann and R. J. Quinn, Nat. Prod. Rep., 2011, 28, 1483 RSC.
- L. Ruddigkeit, L. C. Blum and J.-L. Reymond, J. Chem. Inf. Model., 2012, 53, 56 CrossRef PubMed.
- M. Awale, R. van Deursen and J.-L. Reymond, J. Chem. Inf. Model., 2013, 53, 509 CrossRef CAS PubMed.
- M. Gütlein, A. Karwath and S. Kramer, J. Cheminf., 2012, 4, 7, DOI:10.1186/1758-2946-4-7.
- A. M. Virshup, J. Contreras-Garcia, P. Wipf, W. Yang and D. N. Beratan, J. Am. Chem. Soc., 2013, 135, 7296 CrossRef CAS PubMed.
- R. M. Wilson and S. J. Danishefsky, J. Org. Chem., 2006, 71, 8329 CrossRef CAS PubMed.
- For a review, see: A. M. Szpilman and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9592 CrossRef CAS PubMed.
- J. P. Nandy, M. Prakesch, S. Khadem, P. T. Reddy, U. Sharma and P. Arya, Chem. Rev., 2009, 109, 1999 CrossRef CAS PubMed.
- For recent reviews, see: (a) K.-H. Altmann, F. Z. Gaugaz and R. Schiess, Mol. Diversity, 2011, 15, 383 CrossRef CAS PubMed; (b) M. J. Edelman and M. Shvartsbeyn, Clin. Lung Cancer, 2012, 13, 171 CrossRef CAS PubMed; (c) S. Kaiser, J. J. Muller, P. E. Froehlich, S. Cristina Baggio Gnoatto and A. M. Bergold, Anti-Cancer Agents Med. Chem., 2013, 13, 1057 CrossRef CAS.
- K. B. Kim and C. M. Crews, Nat. Prod. Rep., 2013, 30, 600 RSC.
- P. M. Wright, I. B. Seiple and A. G. Myers, Angew. Chem., Int. Ed., 2014, 53, 8840–8869 CrossRef CAS PubMed.
- One can find countless cases for DTS in the literature, for example on dictyostatin: (a) W. Zhu, M. Jiménez, W.-H. Jung, D. P. Camarco, R. Balachandran, A. Vogt, B. W. Day and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 9175 CrossRef CAS PubMed; (b) I. Paterson, G. J. Naylor, N. M. Gardner, E. Guzmán and A. E. Wright, Chem. – Asian J., 2011, 6, 459 CrossRef CAS PubMed; (c) L. L. Vollmer, M. Jiménez, D. P. Camarco, W. Zhu, H. N. Daghestani, R. Balachandran, C. E. Reese, J. S. Lazo, N. A. Hukriede, D. P. Curran, B. W. Day and A. Vogt, Mol. Cancer Ther., 2011, 10, 994 CrossRef CAS PubMed; (d) J. Gallon, J. Esteban, S. Bouzbouz, M. Campbell, S. Reymond and J. Cossy, Chem. – Eur. J., 2012, 18, 11788 CrossRef CAS PubMed.
- For some reviews, see: (a) M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46 CrossRef PubMed; (b) T. E. Nielsen and S. L. Schreiber, Angew. Chem., Int. Ed., 2008, 47, 48 CrossRef CAS PubMed; (c) R. J. Spandl, A. Bender and D. R. Spring, Org. Biomol. Chem., 2008, 6, 1149 RSC; (d) C. Cordier, D. Morton, S. Murrison, A. Nelson and C. O'Leary-Steele, Nat. Prod. Rep., 2008, 25, 719 RSC; (e) W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 1 CrossRef PubMed; (f) C. Serba and N. Winssinger, Eur. J. Org. Chem., 2013, 4195 CrossRef CAS PubMed.
- For example: (a) C. Chen, X. Li, C. S. Neumann, M. M. C. Lo and S. L. Schreiber, Angew. Chem., Int. Ed., 2005, 44, 2249 CrossRef CAS PubMed; (b) S. Su, D. E. Acquilano, J. Arumugasamy, A. B. Beeler, E. L. Eastwood, J. R. Giguere, P. Lan, X. Lei, G. K. Min, A. R. Yeager, Y. Zhou, J. S. Panek, J. K. Snyder, S. E. Schaus and J. A. Porco Jr., Org. Lett., 2005, 7, 2751 CrossRef CAS PubMed; (c) V. Duplan, C. Serba, J. Garcia, G. Valot, S. Barluenga, M. Hoerlé, M. Cuendet and N. Winssinger, Org. Biomol. Chem., 2014, 12, 370 RSC.
- A. Fürstner, D. Kirk, M. D. B. Fenster, C. Aiessa, D. De Souza, C. Nevado, T. Tuttle, W. Thiel and O. Müller, Chem. – Eur. J., 2007, 13, 135 CrossRef PubMed.
- Y. Kashman, A. Groweiss and U. Shmueli, Tetrahedron Lett., 1980, 21, 3629 CrossRef CAS.
- S. Kudrimoti, S. A. Ahmed, P. R. Daga, A. E. Wahba, S. I. Khalifa, R. J. Doerksen and M. T. Hamann, Bioorg. Med. Chem., 2009, 17, 7517 CrossRef CAS PubMed.
- K. A. El Sayed, M. A. Khanfar, H. M. Shallal, A. Muralidharan, B. Awate, D. T. A. Youssef, Y. Liu, Y.-D. Zhou, D. G. Nagle and G. Shah, J. Nat. Prod., 2008, 71, 396 CrossRef CAS PubMed.
- A. Fürstner, D. De Souza, L. Turet, M. D. B. Fenster, L. Parra-Rapado, C. Wirtz, R. Myott and C. W. Lehmann, Chem. – Eur. J., 2007, 13, 115 CrossRef PubMed.
- I. Paterson, D. Y. K. Chen, M. J. Coster, J. L. Aceña, J. Bach and D. J. Wallace, Org. Biomol. Chem., 2005, 3, 2431 CAS.
- For a review, see: J. Pietruszka, Angew. Chem., Int. Ed., 1998, 37, 2629 CrossRef CAS.
- A. B. Smith III, C. A. Risatti, O. Atasoylu, C. S. Bennett, J. Liu, H. Cheng, K. TenDyke and Q. Xu, J. Am. Chem. Soc., 2011, 133, 14042 CrossRef PubMed.
- M. Carda, J. Murga, S. Díaz-Oltra, J. García-Pla, J. Paños, E. Falomir, C. Trigili, J. F. Díaz, I. Barasoain and J. A. Marco, Eur. J. Org. Chem., 2013, 1116 CrossRef CAS PubMed.
- A. F. Petri, F. Sasse and M. E. Maier, Eur. J. Org. Chem., 2005, 1865 CrossRef CAS PubMed.
- Among the benzolactones, radicicol served as a prominent lead structure for analogue synthesis because of its binding to Hsp90: (a) E. Moulin, V. Zoete, S. Barluenga, M. Karplus and N. Winssinger, J. Am. Chem. Soc., 2005, 127, 6999 CrossRef CAS PubMed; (b) X. Lei and S. J. Danishefsky, Adv. Synth. Catal., 2008, 350, 1677 CrossRef CAS PubMed; (c) R. R. A. Kitson and C. J. Moody, J. Org. Chem., 2013, 78, 5117 CrossRef CAS PubMed.
- The benzolactone enamides might be considered a subclass of the resorcylic acid lactones. These are covered in the following review: N. Winssinger and S. Barluenga, Chem. Commun., 2007, 22 RSC.
- For some reviews regarding the biology of V-ATPases, see: (a) E. J. Bowman and B. J. Bowman, J. Bioenerg. Biomembr., 2005, 37, 431 CrossRef CAS PubMed; (b) K. W. Beyenbach and H. Wieczorek, J. Exp. Biol., 2006, 209, 577 CrossRef CAS PubMed; (c) M. Huss and H. Wieczorek, J. Exp. Biol., 2009, 212, 341 CrossRef CAS PubMed; (d) M. Perez-Sayans, J. M. Somoza-Martin, F. Barros-Angueira, J. M. G. Rey and A. Garcia-Garcia, Cancer Treat. Rev., 2009, 35, 707 CrossRef CAS PubMed.
- X.-S. Xie, D. Padron, X. Liao, J. Wang, M. G. Roth and J. K. De Brabander, J. Biol. Chem., 2004, 279, 19755 CrossRef CAS PubMed.
- For total syntheses, see: (a) A. Bhattacharjee, O. R. Seguil and J. K. De Brabander, Tetrahedron Lett., 2001, 42, 1217 CrossRef CAS; (b) K. C. Nicolaou, D. W. Kim and R. Baati, Angew. Chem., Int. Ed., 2002, 41, 3701 CrossRef CAS; (c) Q. Su and J. S. Panek, J. Am. Chem. Soc., 2004, 126, 2425 CrossRef CAS PubMed; (d) S. S. Palimkar and J. Uenishi, Org. Lett., 2010, 12, 4160 CrossRef CAS PubMed.
- A. F. Petri, A. Bayer and M. E. Maier, Angew. Chem., Int. Ed., 2004, 43, 5821 CrossRef CAS PubMed.
- Other groups also used transannular pyran formation: (a) F. Hilli, J. M. White and M. A. Rizzacasa, Tetrahedron, 2011, 67, 5054 CrossRef CAS PubMed; (b) Y.-H. Jung, Y.-J. Kim, J. Lee and J. Tae, Chem. – Asian J., 2007, 2, 656 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, D. W. Kim, R. Baati, A. O'Brate and P. Giannakakou, Chem. – Eur. J., 2003, 9, 6177 CrossRef CAS PubMed; (b) S. S. Palimkar, J. Uenishi and H. Ii, J. Org. Chem., 2011, 77, 388 CrossRef PubMed.
- M. Bauer and M. E. Maier, Org. Lett., 2002, 4, 2205 CrossRef CAS PubMed.
- C. Herb and M. E. Maier, J. Org. Chem., 2003, 68, 8129 CrossRef CAS PubMed.
- C. Herb, A. Bayer and M. E. Maier, Chem. – Eur. J., 2004, 10, 5649 CrossRef CAS PubMed.
- C. Herb, F. Dettner and M. E. Maier, Eur. J. Org. Chem., 2005, 728 CrossRef CAS PubMed.
- J. Ritschel, F. Sasse and M. E. Maier, Eur. J. Org. Chem., 2007, 78 CrossRef CAS PubMed.
- S. Lebreton, X.-S. Xie, D. Ferguson and J. K. De Brabander, Tetrahedron, 2004, 60, 9635 CrossRef CAS PubMed.
- For the synthesis of some other salicylihalamide analogues, see: (a) Y. Sugimoto, K. Konoki, M. Murata, M. Matsushita, H. Kanazawa and T. Oishi, J. Med. Chem., 2009, 52, 798 CrossRef CAS PubMed; (b) S. Tang and K. L. Erickson, J. Nat. Prod., 2008, 71, 898 CrossRef CAS PubMed.
- S. Lebreton, J. Jaunbergs, M. G. Roth, D. A. Ferguson and J. K. De Brabander, Bioorg. Med. Chem. Lett., 2008, 18, 5879 CrossRef CAS PubMed.
- K. H. Müller, G. A. Spoden, K. D. Scheffer, R. Brunnhöfer, J. K. De Brabander, M. E. Maier, L. Florin and C. P. Muller, Antimicrob. Agents Chemother., 2014, 58, 2905 CrossRef PubMed.
- J. A. Hall, B. R. Kusuma, G. E. L. Brandt and B. S. J. Blagg, ACS Chem. Biol., 2014, 9, 976 CrossRef CAS PubMed.
- For a recent review, see: J. Franke, S. Eichner, C. Zeilinger and A. Kirschning, Nat. Prod. Rep., 2013, 30, 1299 RSC.
- L. Jundt, H. Steinmetz, P. Luger, M. Weber, B. Kunze, H. Reichenbach and G. Höfle, Eur. J. Org. Chem., 2006, 5036 CrossRef CAS PubMed.
- V. V. Vintonyak and M. E. Maier, Angew. Chem., Int. Ed., 2007, 46, 5209 CrossRef CAS PubMed.
- A. Fürstner, M. Bindl and L. Jean, Angew. Chem., Int. Ed., 2007, 46, 9275–9278 CrossRef PubMed.
- M. Fouché, L. Rooney and A. G. M. Barrett, J. Org. Chem., 2012, 77, 3060 CrossRef PubMed.
- B. R. Kusuma, G. E. L. Brandt and B. S. J. Blagg, Org. Lett., 2012, 14, 6242 CrossRef CAS PubMed.
- For a recent review, see: A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 2794 CrossRef PubMed.
- (a) V. V. Vintonyak and M. E. Maier, Org. Lett., 2008, 10, 1239 CrossRef CAS PubMed; (b) V. V. Vintonyak, M. Calà, F. Lay, B. Kunze, F. Sasse and M. E. Maier, Chem. – Eur. J., 2008, 14, 3709 CrossRef CAS PubMed.
- M. Bindl, L. Jean, J. Herrmann, R. Müller and A. Fürstner, Chem. – Eur. J., 2009, 15, 12310 CrossRef CAS PubMed.
- V. V. Vintonyak, B. Kunze, F. Sasse and M. E. Maier, Chem. – Eur. J., 2008, 14, 11132 CrossRef CAS PubMed.
- A. E. Wright, J. C. Botelho, E. Guzmán, D. Harmody, P. Linley, P. J. McCarthy, T. P. Pitts, S. A. Pomponi and J. K. Reed, J. Nat. Prod., 2007, 70, 412 CrossRef CAS PubMed.
- (a) W. Youngsaye, J. T. Lowe, F. Pohlki, P. Ralifo and J. S. Panek, Angew. Chem., Int. Ed., 2007, 46, 9211 CrossRef CAS PubMed; (b) S. K. Woo, M. S. Kwon and E. Lee, Angew. Chem., Int. Ed., 2008, 47, 3242–3244 CrossRef CAS PubMed; (c) D. W. Custar, T. P. Zabawa and K. A. Scheidt, J. Am. Chem. Soc., 2008, 130, 804 CrossRef CAS PubMed; (d) H. Fuwa, S. Naito, T. Goto and M. Sasaki, Angew. Chem., Int. Ed., 2008, 47, 4737–4739 CrossRef CAS PubMed; (e) O. A. Ulanovskaya, J. Janjic, M. Suzuki, S. S. Sabharwal, P. T. Schumacker, S. J. Kron and S. A. Kozmin, Nat. Chem. Biol., 2008, 4, 418 CrossRef CAS PubMed; (f) I. Paterson and N. A. Miller, Chem. Commun., 2008, 4708 RSC; (g) X. Guinchard and E. Roulland, Org. Lett., 2009, 11, 4700 CrossRef CAS PubMed; (h) Y. Cui, W. Tu and P. E. Floreancig, Tetrahedron, 2010, 66, 4867 CrossRef CAS PubMed; (i) H. Fuwa, A. Saito and M. Sasaki, Angew. Chem., Int. Ed., 2010, 49, 3041 CrossRef CAS PubMed; (j) A. K. Ghosh, K. A. Shurrush and Z. L. Dawson, Org. Biomol. Chem., 2013, 11, 7768 RSC.
- For the most recent one, see: G. V. M. Sharma, S. V. Reddy and K. V. S. Ramakrishna, Org. Biomol. Chem., 2012, 10, 3689 CAS.
- (a) H. Fuwa, A. Saito, S. Naito, K. Konoki, M. Yotsu-Yamashita and M. Sasaki, Chem. – Eur. J., 2009, 15, 12807 CrossRef CAS PubMed; (b) D. W. Custar, T. P. Zabawa, J. Hines, C. M. Crews and K. A. Scheidt, J. Am. Chem. Soc., 2009, 131, 12406 CrossRef CAS PubMed; (c) Y. Cui, R. Balachandran, B. W. Day and P. E. Floreancig, J. Org. Chem., 2012, 77, 2225 CrossRef CAS PubMed; (d) H. Fuwa, M. Kawakami, K. Noto, T. Muto, Y. Suga, K. Konoki, M. Yotsu-Yamashita and M. Sasaki, Chem. – Eur. J., 2013, 19, 8100 CrossRef CAS PubMed; (e) H. Fuwa, T. Noguchi, M. Kawakami and M. Sasaki, Bioorg. Med. Chem. Lett., 2014, 24, 2415 CrossRef CAS PubMed.
- For a review, see: G. S. Bagavananthem Andavan and R. Lemmens-Gruber, Mar. Drugs, 2010, 8, 810 CrossRef CAS PubMed.
- Y.-Y. Xu, C. Liu and Z.-P. Liu, Curr. Org. Synth., 2013, 10, 67 CAS.
- (a) B. Kunze, R. Jansen, F. Sasse, G. Höfle and H. Reichenbach, J. Antibiot., 1995, 48, 1262 CrossRef CAS; (b) R. Jansen, B. Kunze, H. Reichenbach and G. Höfle, Liebigs Ann., 1996, 285 CAS.
- (a) T. M. Zabriskie, J. A. Klocke, C. M. Ireland, A. H. Marcus, T. F. Molinski, D. J. Faulkner, C. Xu and J. C. Clardy, J. Am. Chem. Soc., 1986, 108, 3123 CrossRef CAS; (b) P. Crews, L. V. Manes and M. Boehler, Tetrahedron Lett., 1986, 27, 2797 CrossRef CAS.
- A. Tripathi, J. Puddick, M. l. R. Prinsep, M. Rottmann and L. T. Tan, J. Nat. Prod., 2010, 73, 1810 CrossRef CAS PubMed.
- (a) Isolation: P. D. Boudreau, T. Byrum, W.-T. Liu, P. C. Dorrestein and W. H. Gerwick, J. Nat. Prod., 2012, 75, 1560 CrossRef CAS PubMed; (b) Total synthesis: D. Wang, S. Song, Y. Tian, Y. Xu, Z. Miao and A. Zhang, J. Nat. Prod., 2013, 76, 974 CrossRef CAS PubMed.
- See, for example: A. K. Ghosh and D. K. Moon, Org. Lett., 2007, 9, 2425 CrossRef CAS PubMed and references cited therein.
- T.-S. Hu, R. Tannert, H.-D. Arndt and H. Waldmann, Chem. Commun., 2007, 3942 RSC.
- H. Waldmann, T.-S. Hu, S. Renner, S. Menninger, R. Tannert, T. Oda and H.-D. Arndt, Angew. Chem., Int. Ed., 2008, 47, 6473–6477 CrossRef CAS PubMed.
- U. Eggert, R. Diestel, F. Sasse, R. Jansen, B. Kunze and M. Kalesse, Angew. Chem., Int. Ed., 2008, 47, 6478–6482 CrossRef CAS PubMed.
- (a) F. Sasse, B. Kunze, T. M. A. Gronewold and H. Reichenbach, J. Natl. Cancer Inst., 1998, 90, 1559 CrossRef CAS PubMed; (b) L.-G. Milroy, S. Rizzo, A. Calderon, B. Ellinger, S. Erdmann, J. Mondry, P. Verveer, P. Bastiaens, H. Waldmann, L. Dehmelt and H.-D. Arndt, J. Am. Chem. Soc., 2012, 134, 8480 CrossRef CAS PubMed.
- A. Schmauder, S. Müller and M. E. Maier, Tetrahedron, 2008, 64, 6263 CrossRef CAS PubMed.
- A. Schmauder, L. D. Sibley and M. E. Maier, Chem. – Eur. J., 2010, 16, 4328 CrossRef CAS PubMed.
- A. Zhdanko, A. Schmauder, C. I. Ma, L. D. Sibley, D. Sept, F. Sasse and M. E. Maier, Chem. – Eur. J., 2011, 17, 13349 CrossRef CAS PubMed.
- For the synthesis of jasplakinolide analogues, see: (a) A. K. Ghosh, Z. L. Dawson, D. K. Moon, R. Bai and E. Hamel, Bioorg. Med. Chem. Lett., 2010, 20, 5104 CrossRef CAS PubMed; (b) L.-G. Milroy, S. Rizzo, A. Calderon, B. Ellinger, S. Erdmann, J. Mondry, P. Verveer, P. Bastiaens, H. Waldmann, L. Dehmelt and H.-D. Arndt, J. Am. Chem. Soc., 2012, 134, 8480 CrossRef CAS PubMed.
- R. Tannert, L.-G. Milroy, B. Ellinger, T.-S. Hu, H.-D. Arndt and H. Waldmann, J. Am. Chem. Soc., 2010, 132, 3063 CrossRef CAS PubMed.
- C. I. Ma, K. Diraviyam, M. E. Maier, D. Sept and L. D. Sibley, J. Nat. Prod., 2013, 76, 1565 CrossRef CAS PubMed.
- For some reviews, see: (a) S. Bonnal, L. Vigevani and J. Valcárcel, Nat. Rev. Drug Discovery, 2012, 11, 847 CrossRef CAS PubMed; (b) S. M. Dehm, Cancer Res., 2013, 73, 5309 CrossRef CAS PubMed; (c) J. Zhang and J. L. Manley, Cancer Discovery, 2013, 3, 1228 CrossRef CAS PubMed; (d) A. G. Matera and Z. Wang, Nat. Rev. Mol. Cell Biol., 2014, 15, 108 CrossRef CAS PubMed.
- A. Yokoi, Y. Kotake, K. Takahashi, T. Kadowaki, Y. Matsumoto, Y. Minoshima, N. H. Sugi, K. Sagane, M. Hamaguchi, M. Iwata and Y. Mizui, FEBS J., 2011, 278, 4870 CrossRef CAS PubMed.
- (a) T. Sakai, T. Sameshima, M. Matsufuji, N. Kawamura, K. Dobashi and Y. Mizui, J. Antibiot., 2004, 57, 173 CrossRef CAS; (b) T. Sakai, N. Asai, A. Okuda, N. Kawamura and Y. Mizui, J. Antibiot., 2004, 57, 180 CrossRef CAS.
- M. Seki-Asano, T. Okazaki, M. Yamagishi, N. Sakai, Y. Takayama, K. Hanada, S. Morimoto, A. Takatsuki and K. Mizoue, J. Antibiot., 1994, 47, 1395 CrossRef CAS.
- B. G. Isaac, S. W. Ayer, R. C. Elliott and R. J. Stonard, J. Org. Chem., 1992, 57, 7220 CrossRef CAS.
- (a) H. Nakajima, B. Sato, T. Fujita, S. Takase, H. Terano and M. Okuhara, J. Antibiot., 1996, 49, 1196 CrossRef CAS; (b) H. Nakajima, S. Takase, H. Terano and H. Tanaka, J. Antibiot., 1997, 50, 96 CrossRef CAS.
- (a) X. Liu, S. Biswas, M. G. Berg, C. M. Antapli, F. Xie, Q. Wang, M.-C. Tang, G.-L. Tang, L. Zhang, G. Dreyfuss and Y.-Q. Cheng, J. Nat. Prod., 2013, 76, 685 CrossRef CAS PubMed; (b) H. He, A. S. Ratnayake, J. E. Janso, M. He, H. Y. Yang, F. Loganzo, B. Shor, C. J. O'Donnell and F. E. Koehn, J. Nat. Prod., 2014, 77, 1864 CrossRef CAS PubMed.
- M. Sato, N. Muguruma, T. Nakagawa, K. Okamoto, T. Kimura, S. Kitamura, H. Yano, K. Sannomiya, T. Goji, H. Miyamoto, T. Okahisa, H. Mikasa, S. Wada, M. Iwata and T. Takayama, Cancer Sci., 2014, 105, 110 CrossRef CAS PubMed.
- S. M. Dehm, Clin. Cancer Res., 2013, 19, 6064 CrossRef CAS PubMed.
- S. Osman, B. J. Albert, Y. Wang, M. Li, N. L. Czaicki and K. Koide, Chem. – Eur. J., 2011, 17, 895 CrossRef CAS PubMed.
- L. Fan, C. Lagisetti, C. C. Edwards, T. R. Webb and P. M. Potter, ACS Chem. Biol., 2011, 6, 582 CrossRef CAS PubMed.
- C. Lagisetti, A. Pourpak, T. Goronga, Q. Jiang, X. Cui, J. Hyle, J. M. Lahti, S. W. Morris and T. R. Webb, J. Med. Chem., 2009, 52, 6979 CrossRef CAS PubMed.
- M. K. Gundluru, A. Pourpak, X. Cui, S. W. Morris and T. R. Webb, Med. Chem. Commun., 2011, 2, 904 RSC.
- V. P. Kumar and S. Chandrasekhar, Org. Lett., 2013, 15, 3610 CrossRef CAS PubMed.
- S. Müller, T. Mayer, F. Sasse and M. E. Maier, Org. Lett., 2011, 13, 3940 CrossRef PubMed.
- K. Arai, S. Buonamici, B. Chan, L. Corson, A. Endo, B. Gerard, M.-H. Hao, C. Karr, K. Kira, L. Lee, X. Liu, J. T. Lowe, T. Luo, L. A. Marcaurelle, Y. Mizui, M. Nevalainen, M. W. O'Shea, E. S. Park, S. A. Perino, S. Prajapati, M. Shan, P. G. Smith, P. Tivitmahaisoon, J. Y. Wang, M. Warmuth, K.-M. Wu, L. Yu, H. Zhang, G.-Z. Zheng and G. F. Keaney, Org. Lett., 2014, 16, 5560 CrossRef CAS PubMed.
- R. Villa, M. K. Kashyap, D. Kumar, T. J. Kipps, J. E. Castro, J. J. La Clair and M. D. Burkart, J. Med. Chem., 2013, 56, 6576 CrossRef CAS PubMed.
- C. Lagisetti, M. V. Yermolina, L. K. Sharma, G. Palacios, B. J. Prigaro and T. R. Webb, ACS Chem. Biol., 2013, 9, 643 CrossRef PubMed.
- R. M. Kanada, D. Itoh, M. Nagai, J. Niijima, N. Asai, Y. Mizui, S. Abe and Y. Kotake, Angew. Chem., Int. Ed., 2007, 46, 4350 CrossRef CAS PubMed.
- A. K. Ghosh and D. D. Anderson, Org. Lett., 2012, 14, 4730 CrossRef CAS PubMed.
- I. Shiina, H. Fukui and A. Sasaki, Nat. Protocols, 2007, 2, 2312 CAS.
- S. Müller, F. Sasse and M. E. Maier, Eur. J. Org. Chem., 2014, 1025 CrossRef PubMed.
- For the concept and some reviews, see: (a) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40 CrossRef CAS PubMed; (b) P. A. Wender, Tetrahedron, 2013, 69, 7529 CrossRef CAS PubMed; (c) P. A. Wender, Nat. Prod. Rep., 2014, 31, 433 RSC.
- G. R. Pettit, C. L. Herald, D. L. Doubek, D. L. Herald, E. Arnold and J. Clardy, J. Am. Chem. Soc., 1982, 104, 6846 CrossRef CAS.
- For a review about the synthesis of bryostatins, see: K. J. Hale and S. Manaviazar, Chem. – Asian J., 2010, 5, 704 CrossRef CAS PubMed.
- Y. Lu, S. K. Woo and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 13876 CrossRef CAS PubMed.
- P. A. Wender, J. C. Horan and V. A. Verma, Org. Lett., 2006, 8, 5299 CrossRef CAS PubMed.
- P. A. Wender, J. Baryza, C. E. Bennett, C. Bi, S. E. Brenner, M. O. Clarke, J. C. Horan, C. Kan, E. Lacôte, B. Lippa, P. G. Nell and T. M. Turner, J. Am. Chem. Soc., 2002, 124, 13648 CrossRef CAS PubMed.
- P. A. Wender, J. L. Baryza, S. E. Brenner, B. A. DeChristopher, B. A. Loy, A. J. Schrier and V. A. Verma, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6721 CrossRef CAS PubMed.
- M. J. Yu, W. Zheng, B. M. Seletsky, B. A. Littlefield and Y. Kishi, in Annual Reports in Medicinal Chemistry, ed. J. E. Macor, Academic Press, New York, 2011, ch. 14, vol. 46, pp. 227–241 Search PubMed.
- M. J. Yu, W. Zheng and B. M. Seletsky, Nat. Prod. Rep., 2013, 30, 1158 RSC.
- C. Zambaldo, K. K. Sadhu, G. Karthikeyan, S. Barluenga, J.-P. Daguer and N. Winssinger, Chem. Sci., 2013, 4, 2088 RSC.
- For some reviews, see: (a) M. Kaiser, S. Wetzel, K. Kumar and H. Waldmann, Cell. Mol. Life Sci., 2008, 65, 1186 CrossRef CAS PubMed; (b) S. Wetzel, R. S. Bon, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 10800 CrossRef CAS PubMed; (c) S. Rizzo and H. Waldmann, Chem. Rev., 2014, 114, 4621 CrossRef CAS PubMed; (d) H. van Hattum and H. Waldmann, J. Am. Chem. Soc., 2014, 136, 11853 CrossRef CAS PubMed.
- B.-W. Chen, C.-H. Chao, J.-H. Su, C.-W. Tsai, W.-H. Wang, Z.-H. Wen, C.-Y. Huang, P.-J. Sung, Y.-C. Wu and J.-H. Sheu, Org. Biomol. Chem., 2011, 9, 834 CAS.
- S. Díaz, J. Cuesta, A. González and J. Bonjoch, J. Org. Chem., 2003, 68, 7400 CrossRef PubMed.
- For a review, see: D. H. Mac, S. Chandrasekhar and R. Grée, Eur. J. Org. Chem., 2012, 5881 CrossRef CAS PubMed.
- (a) S. Wetzel, K. Klein, S. Renner, D. Rauh, T. I. Oprea, P. Mutzel and H. Waldmann, Nat. Chem. Biol., 2009, 5, 581 CrossRef CAS PubMed; (b) K. Klein, O. Koch, N. Kriege, P. Mutzel and T. Schäfer, Mol. Inf., 2013, 32, 964 CrossRef CAS PubMed.
- (a) B. M. McArdle, M. R. Campitelli and R. J. Quinn, J. Nat. Prod., 2006, 69, 14 CrossRef CAS PubMed; (b) D. Camp, R. A. Davis, M. Campitelli, J. Ebdon and R. J. Quinn, J. Nat. Prod., 2011, 75, 72 CrossRef PubMed.
- S. Rizzo and H. Waldmann, Chem. Rev., 2014, 114, 4621 CrossRef CAS PubMed.
- See also: I. D. Jenkins, F. Lacrampe, J. Ripper, L. Alcaraz, P. Van Le, G. Nikolakopoulos, P. de Almeida Leone, R. H. White and R. J. Quinn, J. Org. Chem., 2008, 74, 1304 CrossRef PubMed.
- B. Dinda, S. Debnath and R. Banik, Chem. Pharm. Bull., 2011, 59, 803 CrossRef CAS.
- H. Takayama, Z.-J. Jia, L. Kremer, J. O. Bauer, C. Strohmann, S. Ziegler, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2013, 52, 12404–12408 CrossRef CAS PubMed.
- K. C. Morrison and P. J. Hergenrother, Nat. Prod. Rep., 2014, 31, 6 RSC.
- P. K. Chowdhury, A. Prelle, D. Schomburg, M. Thielmann and E. Winterfeldt, Liebigs Ann. Chem., 1987, 1095–1099 CrossRef CAS PubMed.
- For a recent application, see: J.-P. Krieger, G. Ricci, D. Lesuisse, C. Meyer and J. Cossy, Angew. Chem., Int. Ed., 2014, 53, 8705–8708 CrossRef CAS PubMed.
- S. Bäurle, T. Blume, A. Mengel, C. Parchmann, W. Skuballa, S. Bäsler, M. Schäfer, D. Sülzle and H.-P. Wrona-Metzinger, Angew. Chem., Int. Ed., 2003, 42, 3961–3964 CrossRef PubMed.
- R. J. Rafferty, R. W. Hicklin, K. A. Maloof and P. J. Hergenrother, Angew. Chem., Int. Ed., 2014, 53, 220–224 CrossRef CAS PubMed.
- R. W. Huigens III, K. C. Morrison, R. W. Hicklin, T. A. Flood Jr., M. F. Richter and P. J. Hergenrother, Nat. Chem., 2013, 5, 195 CrossRef PubMed.
- A. J. Grenning, J. K. Snyder and J. A. Porco, Org. Lett., 2014, 16, 792 CrossRef CAS PubMed.
- For reviews, see: (a) G. Mehta and V. Singh, Chem. Soc. Rev., 2002, 31, 324 RSC; (b) L. F. Tietze and H. P. Bell, Angew. Chem., Int. Ed., 2003, 42, 3996 CrossRef CAS PubMed; (c) K. Suzuki, Chem. Rec., 2010, 10, 291 CrossRef CAS PubMed.
- For a review, see: K. C. Nicolaou, J. Org. Chem., 2009, 74, 951 CrossRef CAS PubMed.
- Y.-M. Yan, J. Ai, Y.-N. Shi, Z.-L. Zuo, B. Hou, J. Luo and Y.-X. Cheng, Org. Lett., 2014, 16, 532 CrossRef CAS PubMed.
- For a review, see: S. Ziegler, V. Pries, C. Hedberg and H. Waldmann, Angew. Chem., Int. Ed., 2013, 52, 2744 CrossRef CAS PubMed.
- B. G. Isaac, S. W. Ayer, R. C. Elliott and R. J. Stonard, J. Org. Chem., 1992, 57, 7220 CrossRef CAS.
- M. Hasegawa, T. Miura, K. Kuzuya, A. Inoue, S. Won Ki, S. Horinouchi, T. Yoshida, T. Kunoh, K. Koseki, K. Mino, R. Sasaki, M. Yoshida and T. Mizukami, ACS Chem. Biol., 2011, 6, 229 CrossRef CAS PubMed.
- B. Meunier, Acc. Chem. Res., 2007, 41, 69 CrossRef PubMed.
- F. Coslédan, L. Fraisse, A. Pellet, F. o. Guillou, B. Mordmüller, P. G. Kremsner, A. Moreno, D. Mazier, J.-P. Maffrand and B. Meunier, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17579 CrossRef PubMed.
- For some reviews, see: (a) P. J. Hajduk and J. Greer, Nat. Rev. Drug Discovery, 2007, 6, 211 CrossRef CAS PubMed; (b) C. W. Murray and D. C. Rees, Nat. Chem., 2009, 1, 187 CrossRef CAS PubMed; (c) D. E. Scott, A. G. Coyne, S. A. Hudson and C. Abell, Biochemistry, 2012, 51, 4990 CrossRef CAS PubMed; (d) M. Baker, Nat. Rev. Drug Discovery, 2013, 12, 5 CrossRef CAS PubMed.
- Y. Matsuya, T. Kawaguchi, K. Ishihara, K. Ahmed, Q.-L. Zhao, T. Kondo and H. Nemoto, Org. Lett., 2006, 8, 4609 CrossRef CAS PubMed.
- G. Jürjens and A. Kirschning, Org. Lett., 2014, 16, 3000 CrossRef PubMed.
- For a review, see: J. Franke, S. Eichner, C. Zeilinger and A. Kirschning, Nat. Prod. Rep., 2013, 30, 1299 RSC.
- M. Aeluri, B. Dasari and P. Arya, Org. Lett., 2015, 17, 472 CrossRef CAS PubMed ; See also: S. K. R. Guduru, R. Jimmidi, G. S. Deora and P. Arya, Org. Lett., 2015, 17, 480 CrossRef PubMed.
- S. Torijano-Gutiérrez, C. Vilanova, S. Díaz-Oltra, J. Murga, E. Falomir, M. Carda and J. A. Marco, Eur. J. Org. Chem., 2014, 2284 CrossRef PubMed.
- For some recent examples, see: (a) H. Lou, S. Zheng, T. Li, J. Zhang, Y. Fei, X. Hao, G. Liang and W. Pan, Org. Lett., 2014, 16, 2696 CrossRef CAS PubMed; (b) L. Song, H. Yao and R. Tong, Org. Lett., 2014, 16, 3740 CrossRef CAS PubMed.
- (a) Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2014, 136, 2450 CrossRef CAS PubMed; (b) M. Isegawa, S. Maeda, D. J. Tantillo and K. Morokuma, Chem. Sci., 2014, 5, 1555 RSC.
- (a) M. A. Fischbach, C. T. Walsh and J. Clardy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4601 CrossRef CAS PubMed; (b) J. Young and R. E. Taylor, Nat. Prod. Rep., 2008, 25, 651 RSC; (c) C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688 CrossRef CAS PubMed.
- H. Motamedi and A. Shafiee, Eur. J. Biochem., 1998, 256, 528 CAS.
- For some recent reviews, see: (a) A. Kirschning, F. Taft and T. Knobloch, Org. Biomol. Chem., 2007, 5, 3245 RSC; (b) J. Kennedy, Nat. Prod. Rep., 2008, 25, 25 RSC; (c) A. Kirschning and F. Hahn, Angew. Chem., Int. Ed., 2012, 51, 4012 CrossRef CAS PubMed.
- S. Eichner, H. G. Floss, F. Sasse and A. Kirschning, ChemBioChem, 2009, 10, 1801 CrossRef CAS PubMed.
- For a review, see: F. Taft, S. Eichner, T. Knobloch, K. Harmrolfs, J. Hermane and A. Kirschning, Synlett, 2012, 1416 CAS.
- F. Taft, M. Brünjes, H. G. Floss, N. Czempinski, S. Grond, F. Sasse and A. Kirschning, ChemBioChem, 2008, 9, 1057 CrossRef CAS PubMed.
- S. Mo, D. H. Kim, J. H. Lee, J. W. Park, D. B. Basnet, Y. H. Ban, Y. J. Yoo, S.-w. Chen, S. R. Park, E. A. Choi, E. Kim, Y.-Y. Jin, S.-K. Lee, J. Y. Park, Y. Liu, M. O. Lee, K. S. Lee, S. J. Kim, D. Kim, B. C. Park, S.-g. Lee, H. J. Kwon, J.-W. Suh, B. S. Moore, S.-K. Lim and Y. J. Yoon, J. Am. Chem. Soc., 2011, 133, 976 CrossRef CAS PubMed.
- K. Patel, M. Piagentini, A. Rascher, Z.-Q. Tian, G. O. Buchanan, R. Regentin, Z. Hu, C. R. Hutchinson and R. McDaniel, Chem. Biol., 2004, 11, 1625 CrossRef CAS PubMed.
- A. Lechner, M. C. Wilson, Y. H. Ban, J.-y. Hwang, Y. J. Yoon and B. S. Moore, ACS Synth. Biol., 2012, 2, 379 CrossRef PubMed.
- (a) A. Kitsche and M. Kalesse, ChemBioChem, 2013, 14, 851 CrossRef CAS PubMed; (b) O. Hartmann and M. Kalesse, Angew. Chem., Int. Ed., 2014, 53, 7335 CrossRef CAS PubMed.
- H.-H. Lu, A. Raja, R. Franke, D. Landsberg, F. Sasse and M. Kalesse, Angew. Chem., Int. Ed., 2013, 52, 13549 CrossRef CAS PubMed.
- J. Ligon, S. Hill, J. Beck, R. Zirkle, I. Molnár, J. Zawodny, S. Money and T. Schupp, Gene, 2002, 285, 257 CrossRef CAS.
- S. C. Wenzel, R. M. Williamson, C. Grünanger, J. Xu, K. Gerth, R. A. Martinez, S. J. Moss, B. J. Carroll, S. Grond, C. J. Unkefer, R. Müller and H. G. Floss, J. Am. Chem. Soc., 2006, 128, 14325 CrossRef CAS PubMed.
- Representative examples: (a) Y. Chen, M. Bilban, C. A. Foster and D. L. Boger, J. Am. Chem. Soc., 2002, 124, 5431 CrossRef CAS PubMed; (b) J. Nam, D. Shin, Y. Rew and D. L. Boger, J. Am. Chem. Soc., 2007, 129, 8747 CrossRef CAS PubMed.
- For a review, see: V. J. Hruby, Nat. Rev. Drug Discovery, 2002, 1, 847 CrossRef CAS PubMed.
- G. Höfle, N. Bedorf, H. Steinmetz, D. Schomburg, K. Gerth and H. Reichenbach, Angew. Chem., Int. Ed. Engl., 1996, 35, 1567 CrossRef PubMed.
- M. Ugele, F. Sasse, S. Knapp, O. Fedorov, A. Zubriene, D. Matulis and M. E. Maier, ChemBioChem, 2009, 10, 2203 CrossRef CAS PubMed.
- F. H. Niesen, H. Berglund and M. Vedadi, Nat. Protocols, 2007, 2, 2212 CAS.
- T. J. Zimmermann, F. H. Niesen, E. S. Pilka, S. Knapp, U. Oppermann and M. E. Maier, Bioorg. Med. Chem., 2009, 17, 530 CrossRef CAS PubMed.
- C. Rink, F. Sasse, D. Matulis, A. Zubriene and M. E. Maier, Chem. – Eur. J., 2010, 16, 14469 CrossRef CAS PubMed.
- For reviews, see: (a) C. S. Leung, S. S. F. Leung, J. Tirado-Rives and W. L. Jorgensen, J. Med. Chem., 2012, 55, 4489 CrossRef CAS PubMed; (b) H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed.
- J. R. Doroghazi, J. C. Albright, A. W. Goering, K.-S. Ju, R. R. Haines, K. A. Tchalukov, D. P. Labeda, N. L. Kelleher and W. W. Metcalf, Nat. Chem. Biol., 2014, 10, 963 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Prof. Richard R. Schmidt, Univ. of Konstanz, on the occasion of his 80th birthday. |
| This journal is © The Royal Society of Chemistry 2015 |