
6-Substituted 1,2-benzoxathiine-2,2-dioxides are isoform-selective inhibitors of human carbonic anhydrases IX, XII and VA†
Muhammet
Tanc
ab,
Fabrizio
Carta
*b,
Andrea
Scozzafava
b and
Claudiu T.
Supuran
*ab
aUniversità degli Studi di Firenze, NEUROFARBA Department, Section of Pharmaceutical Chemistry, Via Ugo Schiff 6, 50019 Sesto Fiorentino (Florence), Italy. E-mail: claudiu.supuran@unifi.it
bUniversità degli Studi di Firenze, Polo Scientifico, Laboratorio di Chimica Bioinorganica, Rm. 188, Via della Lastruccia 3, 50019 Sesto Fiorentino (Florence), Italy. E-mail: fabrizio.carta@unifi.it
First published on 27th October 2014
Abstract
A series of 6-substituted 2-benzoxathiine-2,2-dioxides were synthesized starting from 2,5-dihydroxybenzaldehyde, and then screened in vitro for their inhibition properties against five human carbonic anhydrase (hCA, EC 4.2.1.1) isoforms. All the compounds showed excellent selectivity against the mitochondrial (hCA VA) and the tumor associated (hCA IX and XII) enzymes.
The 1,2-benzoxathiine 2,2-dioxides, also called sulfocoumarins, are bioisosters of the coumarins and were recently reported as potent and isoform selective carbonic anhydrase inhibitors (CAIs).1 In analogy to the coumarin class, their inhibition mechanism starts with the CA-mediated hydrolysis of the intramolecular sulfonic acid ester to give, upon geometrical isomerisation, the trans vinyl sulfonic acid, which in turn binds to the zinc-coordinated water molecule (Fig. 1).2
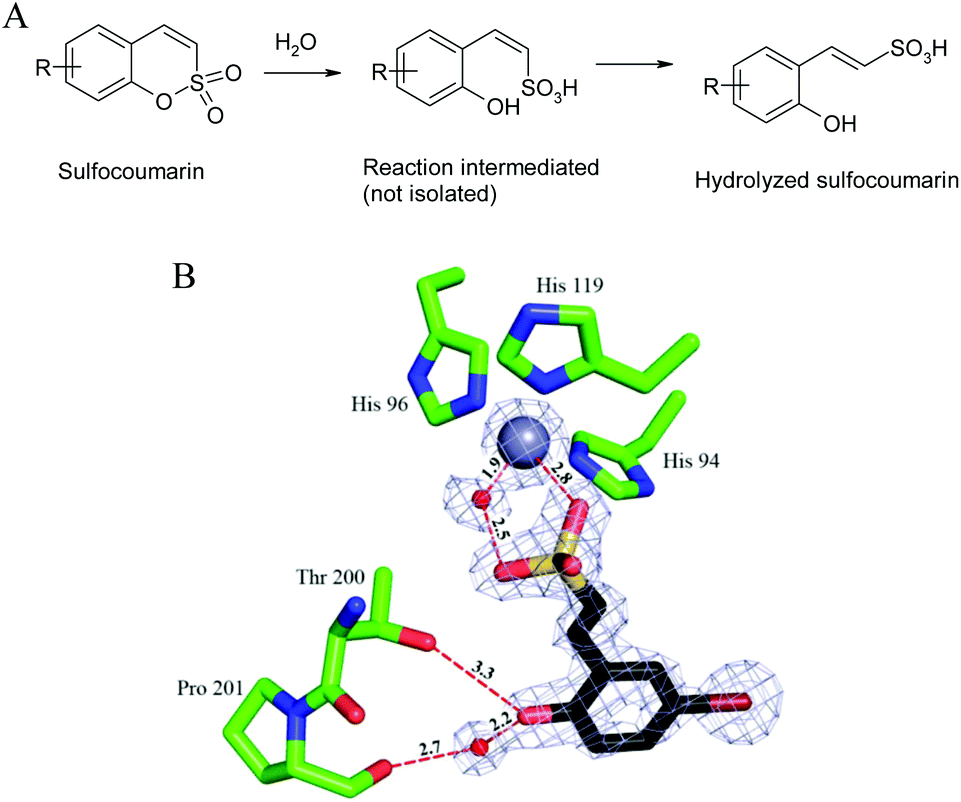 | ||
| Fig. 1 CA inhibition mechanism of sulfocoumarins. (A) The sulfocoumarin undergoes an enzyme-mediated hydrolysis with formation of the trans-2-hydroxy-phenyl-ω-ethenylsulfonic acid. (B) The sulfonic acid binds to the CA II active site, by anchoring of the sulfonic acid group to the zinc-coordinated water molecule. The Zn(II) ion (central larger sphere), its three His ligands (His94, 96 and 119), the water molecule coordinated to the zinc (small sphere) as well as active site residues Thr200 and Pro201 involved in the binding of the hydrolyzed sulfocoumarin are shown, as determined by X-ray crystallography (PDB file 4BCW).1 | ||
CAIs of the sulfonamide type have been in clinical use for the treatment of glaucoma, obesity, epilepsy and as diuretics for almost 60 years.3–5 The use of CAIs for pharmaceutical applications relies on the diverse distribution of the CAs under physio/pathological conditions within the tissues. For instance the antiglaucoma drugs mainly target CA II, IV and XII, diuretics CA II, IV, XII and XIV, and antiepileptics CA VII and XIV, whereas the isoforms IX and XII are strictly correlated with the tumors.6–10 The main drawback associated with their use is the lack of selectivity in inhibiting the various enzymatic isoforms, which results in a plethora of side effects.3–10
Since the identification of the sulfonamides and their bioisosters, the sulfamates and the sulfonates, as CAIs many efforts have been made for the development of specific inhibitors. In this context the “tail approach” was the preferred one. It consisted of the chemical manipulation of a compound at its tail-end, which resulted in a modification of its physical/chemical properties.3,4 In addition several novel CAI scaffolds have been identified such as the polyamines,11 phenols,12 dithiocarbamates,13 xanthates,14 the coumarins2 and their structural derivatives such as the thiocoumarins, 2-thioxo-coumarins and coumarin oximes.6a,15 The sulfocoumarins represent the latest class of CAIs identified and show high selectivity profiles.1,7 The inhibition mechanisms of many of these compounds were determined by X-ray crystallography of the enzyme-inhibitor adducts.2,11–14
In our previous reports we explored the CA kinetic profiles of a small series of 6-tetrazolyl and 6-triazolyl-substituted sulfocoumarins as well as the effects derived by the introduction of different substituents at the 7 position.1,7 As extension herein we report the synthesis, characterization and in vitro CA inhibition of a small series of 6-substituted sulfocoumarins.
All the compounds were prepared according to the general strategy of Zalubovskis's group16 by reacting the commercially available 2,5-dihydroxybenzaldehyde with benzyl bromide to afford 1 which was treated with magnesium bromide for selective de-benzylation.17 Then mesylation of the phenol 2 was followed by intramolecular cyclisation in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to afford the racemic 4-hydroxy-6-benzyloxy-3,4-dihydrosulfocoumarin 4, which was deprotected under Pd-C catalyzed hydrogenation and then dehydrated by means of its triflate derivative 6. As expected such a compound proved to be unstable and was directly subjected to treatment with tetraethylammonium hydroxide to afford the 6-hydroxy-substituted sulfocoumarin 7. At this point the hydroxyl group in 7 was alkylated with 2-chloroethanol through an in situ Filkenstein reaction in the presence of potassium carbonate as a base to afford 8, which in turn was treated with tosylchloride in pyridine to give 9. Finally compound 11 was obtained via the Mitsunobu coupling reaction of 7 with the freshly prepared Boc-protected 2-aminoethanol 10 (Scheme 1).
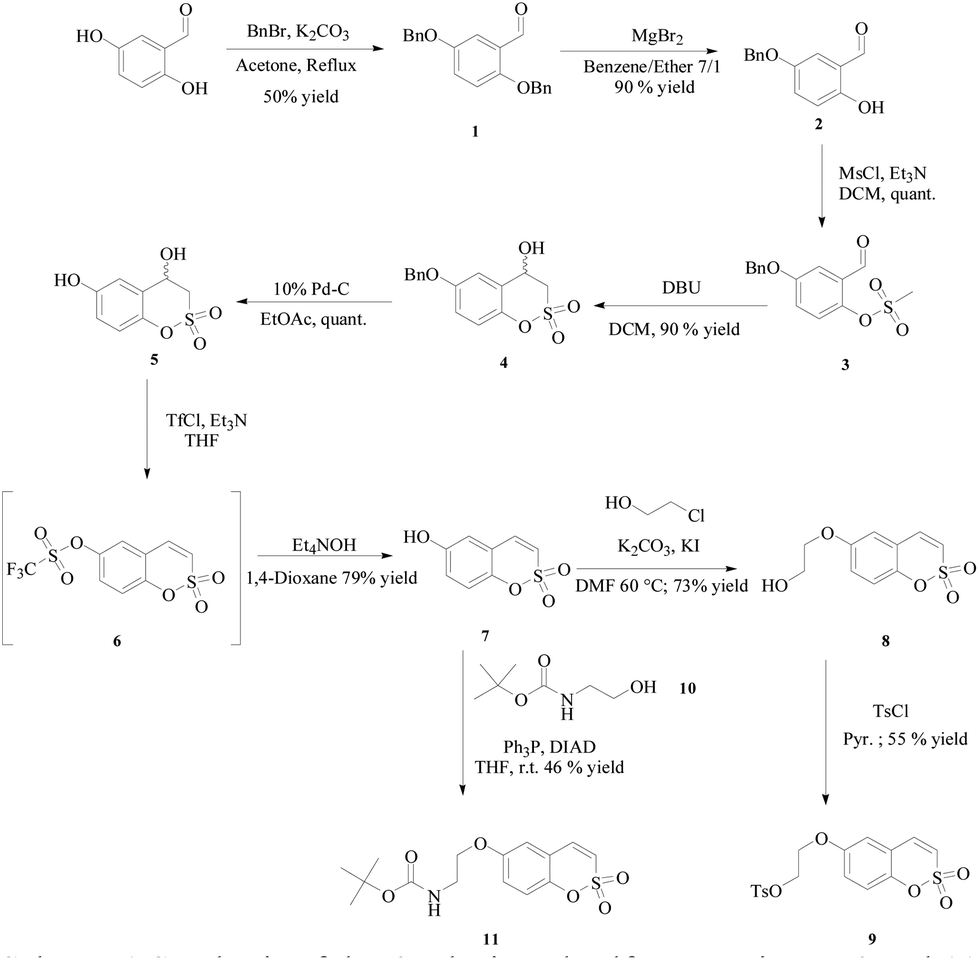 | ||
| Scheme 1 Synthesis of the 6-substituted sulfocoumarins 7–9 and 11. | ||
The obtained compounds 4, 5, 7–9 and 11 were screened in vitro as inhibitors against the most abundant and cytosolic CAs (hCAI and II), the mitochondrial (hCA VA) and the transmembrane tumor-associated hCA IX and XII in comparison with acetazolamide (AAZ) as a standard. In consideration of the previous reports1,7 we can summarize that the main influencing factor for isozyme selectivity is the substitution at the phenyl ring. In particular all the compounds showed to be ineffective inhibitors of the cytosolic hCAs I and II (Table 1).
| Entry | K i | ||||
|---|---|---|---|---|---|
| hCAI (μM) | hCA II (μM) | hCAVA (μM) | hCAIX (nM) | hCAXII (nM) | |
| a Errors in the range of ±5% of the reported values, from three different assays. b Data retrieved from ref. 1. | |||||
| 4 | >100 | >100 | 89 | 83.7 | 36.2 |
| 5 | >100 | >100 | 42 | 36.2 | 16.5 |
| 7 | 91b | >100 | 96 | 300b | 204b |
| 8 | >100 | >100 | 0.06 | 26.8 | 10.4 |
| 9 | >100 | >100 | 42 | 50.3 | 684 |
| 11 | >100 | >100 | 87 | 11.5 | 9.8 |
| AAZ | 0.25 | 0.012 | 0.06 | 25 | 5.7 |
Herein for the first time we report the kinetic profiles of the 6-substituted sulfocoumarins 7–9, 11 and their precursors 4 and 5 against the mitochondrial hCA VA, with Kis in the range of 0.06–96 μM (Table 1). Such an isoform is receiving much attention as it was proved to be a suitable target for the development of new and effective antiobesity agents.5b,18 As a preliminary structure–activity-relationship (SAR) study here we report that the introduction at the 7-position in the sulfocoumarin scaffold of the ethanolic moiety (as for the compound 8) resulted in a significant reduction of the Ki for the hCA VA (0.06 μM). Further manipulation at the alcoholic terminal such as in compounds 9 and 11, or elimination of the alkyl chain as for 7, suppressed any isoform selectivity (Table 1). Currently an exhaustive SAR investigation on this kind of compound is ongoing. All the compounds of the series are potent inhibitors of the transmembrane and tumor-associated isoform hCAs IX and XII with Kis spanning between 9.8 and 684 nM (Table 1). As for the hCA VA isoform a defined SAR is not feasible at the moment. However some key elements are evident: (i) the introduction of an alkyl substituent, as for compound 8, drastically reduced the Ki values (26.8 and 10.4 nM for the hCAs IX and XII respectively). Such a behaviour is analogous to that previously reported for the mitochondrial isoform. (ii) The introduction of the lipophilic and bulky substituent Boc-moiety at the end of the chain, as for the compound 11, accounted for selectivity against the tumor associated CAs. Such an effect was not observed when the tosyl group was introduced instead (compound 9). Probably hydrophobic interactions occurring between the Boc-group of 11 and specific amino acids present at the rim of the enzymatic cavities particularly contribute to the stabilization of the inhibitor-enzyme complex.
Conclusions
Herein we reported a series of 6-substituted sulfocoumarins obtained by intramolecular cyclization of the corresponding methanesulfonic acid esters. We investigated compounds 4, 5, 7–9 and 11 for their inhibition activities against five hCA isoforms.As expected for the 6-substituted sulfocoumarins, the compounds showed to be ineffective in inhibiting the cytosolic hCAs I and II, whereas they showed interesting profiles against the tumor associated hCAs IX and XII with Kis in the range of 9.8–684 nM.
For the first time we report the 6-substituted sulfocoumarins as effective inhibitors of the mitochondrial hCA VA isoform, with compound 8 as the strongest in the series (Ki 0.06 μM).
These findings are of particular importance for the future development of new and effective inhibitors against the mitochondrial CA isoforms having antiobesity pharmacological applications. In particular the lack of safe and effective drugs for the treatment of obesity and/or obesity-related pathologies makes these compounds particularly interesting for developing new therapeutics.
Acknowledgements
This work was financed by two FP7 EU grants (METOXIA and DYNANO).Notes and references
- K. Tars, D. Vullo, A. Kazaks, J. Leitans, A. Lends, A. Grandane, R. Zalubovskis, A. Scozzafava and C. T. Supuran, J. Med. Chem., 2013, 56, 293 CrossRef CAS PubMed.
- (a) A. Maresca, C. Temperini, H. Vu, N. B. Pham, S. A. Poulsen, A. Scozzafava, R. J. Quinn and C. T. Supuran, J. Am. Chem. Soc., 2009, 131, 3057 CrossRef CAS PubMed; (b) A. Maresca, C. Temperini, L. Pochet, B. Masereel, A. Scozzafava and C. T. Supuran, J. Med. Chem., 2010, 53, 335 CrossRef CAS PubMed.
- F. Carta, C. T. Supuran and A. Scozzafava, Future Med. Chem., 2014, 10, 1149 CrossRef PubMed.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168 CrossRef CAS PubMed.
- (a) E. Masini, F. Carta, A. Scozzafava and C. T. Supuran, Expert Opin. Ther. Pat., 2013, 23, 705 CrossRef CAS PubMed; (b) C. T. Supuran and F. Carta, Expert Opin. Ther. Pat., 2013, 23, 725 CrossRef PubMed; (c) M. Aggarwal and R. McKenna, Expert Opin. Ther. Pat., 2012, 22, 903 CrossRef CAS PubMed; (d) F. Carta and C. T. Supuran, Expert Opin. Ther. Pat., 2013, 23, 681 CrossRef CAS PubMed.
- (a) F. Carta, A. Maresca, A. Scozzafava and C. T. Supuran, Bioorg. Med. Chem., 2012, 20, 2266 CrossRef CAS PubMed; (b) R. A. Davis, D. Vullo, A. Maresca, C. T. Supuran and S. A. Poulsen, Bioorg. Med. Chem., 2013, 21, 1539 CrossRef CAS PubMed.
- (a) M. Tanc, F. Carta, M. Bozdag, A. Scozzafava and C. T. Supuran, Bioorg. Med. Chem., 2013, 15, 4502 CrossRef PubMed; (b) A. Grandane, M. Tanc, R. Zalubovskis and C. T. Supuran, Bioorg. Med. Chem., 2014, 5, 1256 CrossRef PubMed; (c) A. Grandane, M. Tanc, R. Zalubovskis and C. T. Supuran, Bioorg. Med. Chem., 2014, 5, 1522 CrossRef PubMed.
- (a) V. Alterio, A. Di Fiore, K. D'Ambrosio, C. T. Supuran and G. De Simone, Chem. Rev., 2012, 112, 4421 CrossRef CAS PubMed; (b) D. Neri and C. T. Supuran, Nat. Rev. Drug Discovery, 2011, 10, 767 CrossRef CAS PubMed; (c) M. Aggarwal, B. Kondeti and R. McKenna, Bioorg. Med. Chem., 2013, 21, 1526 CrossRef CAS PubMed.
- (a) C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2012, 27, 759 CrossRef CAS PubMed; (b) C. T. Supuran, A. Scozzafava and A. Casini, Med. Res. Rev., 2003, 23, 146 CrossRef CAS PubMed; (c) S. Pastorekova, S. Parkkila, J. Pastorek and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2004, 19, 199 CrossRef CAS PubMed.
- (a) C. T. Supuran, Bioorg. Med. Chem. Lett., 2010, 20, 3467 CrossRef CAS PubMed; (b) C. T. Supuran, Future Med. Chem., 2011, 3, 1165 CrossRef CAS PubMed; (c) M. Aggarwal, C. D. Boone, B. Kondeti and R. McKenna, J. Enzyme Inhib. Med. Chem., 2013, 28, 267 CrossRef PubMed; (d) A. Jain, G. M. Whitesides, R. S. Alexander and D. W. Christianson, J. Med. Chem., 1994, 37, 2100 CrossRef CAS; (e) L. Baranauskiene, M. Hilvo, J. Matuliene, D. Golovenko, E. Manakova, V. Dudutiene, V. Michailoviene, J. Torresan, J. Jachno, S. Parkkila, A. Maresca, C. T. Supuran, S. Grazulis and D. Matulis, J. Enzyme Inhib. Med. Chem., 2010, 25, 863 CrossRef CAS PubMed.
- F. Carta, C. Temperini, A. Innocenti, A. Scozzafava, K. Kaila and C. T. Supuran, J. Med. Chem., 2010, 53, 5511 CrossRef CAS PubMed.
- S. K. Nair, P. A. Ludwig and D. W. Christianson, J. Am. Chem. Soc., 1994, 116, 3659 CrossRef CAS.
- F. Carta, M. Aggarwal, A. Maresca, A. Scozzafava, R. McKenna, E. Masini and C. T. Supuran, J. Med. Chem., 2012, 55, 1721 CrossRef CAS PubMed.
- F. Carta, A. Akdemir, A. Scozzafava, E. Masini and C. T. Supuran, J. Med. Chem., 2013, 56, 4691 CrossRef CAS PubMed.
- F. Carta, D. Vullo, A. Maresca, A. Scozzafava and C. T. Supuran, Bioorg. Med. Chem. Lett., 2012, 6, 2182 CrossRef PubMed.
- A. Grandane, S. Belyakov, P. Trapencieris and R. Zalubovskis, Tetrahedron, 2012, 68, 5541 CrossRef CAS PubMed.
- H. Ken-Ichiro, A. Yamazoe, Y. Ishibashi, N. Kusaka, Y. Oono and H. Nozaki, Bioorg. Med. Chem., 2008, 16, 5331 CrossRef PubMed.
- R. L. Arechederra, A. Waheed, W. S. Sly, C. T. Supuran and S. D. Minteer, Bioorg. Med. Chem., 2013, 21, 1544 CrossRef CAS PubMed.
- An Applied Photophysics stopped-flow instrument was used for assaying the CA catalysed CO2 hydration activity. Phenol red (at a concentration of 0.2 mM) was used as an indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining the constant ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction were used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of the inhibitor (0.1 mM) were prepared in distilled–deionized water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min–6 h at room temperature (15 min) or 4 °C (6 h) prior to the assay, in order to allow for the formation of the E–I complex. Data from Table 1 were obtained after 6 h incubation of the enzyme and the inhibitor, as for the sulfocoumarins and coumarins reported earlier.1,7 The inhibition constants were obtained by non-linear least squares methods using PRISM 3, as reported earlier,8a and represent the mean from at least three different measurements. All CA isoforms were recombinant ones obtained in-house as reported earlier.1,7.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical data and spectra (1H, 13C NMR) for all products. See DOI: 10.1039/c4ob02155j |
| This journal is © The Royal Society of Chemistry 2015 |