
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water-soluble aryl-extended calix[4]pyrroles with unperturbed aromatic cavities: synthesis and binding studies†
D.
Hernández-Alonso
a,
S.
Zankowski
a,
L.
Adriaenssens
a and
P.
Ballester
*ab
aInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans, 16. E-43007, Tarragona, Spain. E-mail: pballester@iciq.es
bCatalan Institution for Research and Advanced Studies (ICREA), Passeig Lluis Companys 23, E-08010 Barcelona, Spain
First published on 18th November 2014
Abstract
We report the synthesis of a novel, water-soluble aryl-extended calix[4]pyrrole receptor. The water-solubilising groups are placed at the lower rim of the receptor, leaving the binding pocket unperturbed and open for modification. Binding studies were performed with a series of pyridine N-oxides. These studies revealed the ability of the receptor to bind neutral and charged N-oxides in basified water with stability constants higher than 104 M−1.
Introduction
Calix[4]pyrroles are macrocyclic compounds, consisting of four pyrrole units and four connecting tetra substituted sp3 carbon atoms, the so-called meso carbons.1,2 Calix[4]pyrroles are well known receptors for anions,3 ion-pairs4 and electron rich molecules.5 Aryl-extended calix[4]pyrroles bear one aryl ring at each meso carbon.6,7 Depending on the relative orientation of these aryl substituents, aryl-extended calix[4]pyrroles exist as four configurational isomers. The α,α,α,α-isomer describes the aryl-extended calix[4]pyrrole with all aryl rings oriented in the same direction, i.e. all pointing above the calix[4]pyrrole core (Fig. 1). They are also conformationally flexible and adopt different conformations depending on the media in which they are dissolved.8 For example, in non-polar organic solvents the least polar 1,3-alternate conformation is the most energetically favourable. Upon the addition of an appropriate hydrogen-bond (H-bond) acceptor to the solution, the calix[4]pyrrole core undergoes a conformational change adopting the cone conformation. In this manner all four pyrrole N–H groups of the calix[4]pyrrole core can participate in hydrogen bonding interactions with the bound guest.9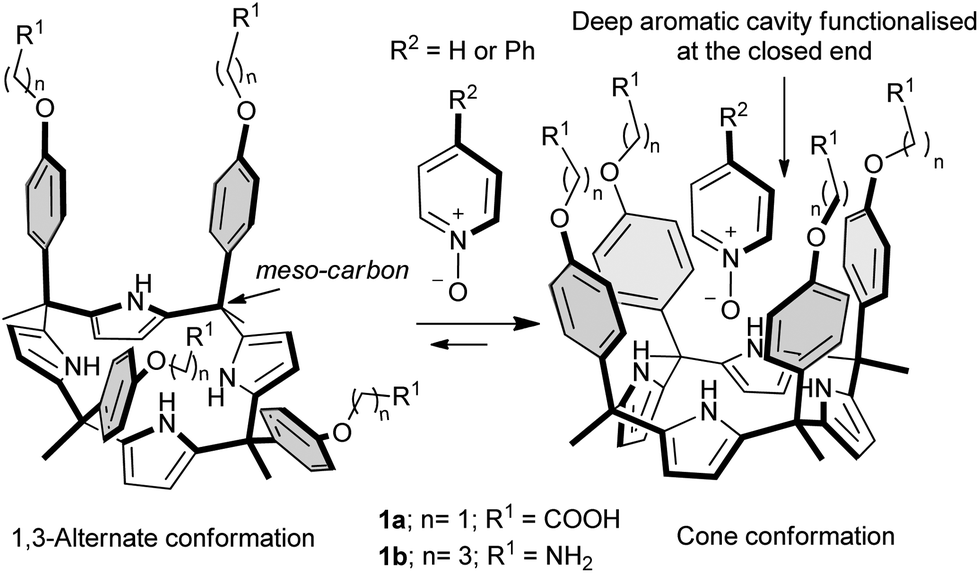 | ||
| Fig. 1 The α,α,α,α-isomers of aryl-extended calix[4]pyrroles undergo a conformational change from the 1,3-alternate conformation to the cone conformation when binding with guests capable of establishing H-bonds. | ||
In the cone conformation, the α,α,α,α-isomers of aryl-extended calix[4]pyrroles feature a deep aromatic cavity closed at one end by four converging pyrrole NHs. The dimensions of this cavity make it suitable for the inclusion of a sizeable number of electron-rich neutral guests and anions. The included guests interact in a directed manner with the NH groups resulting in a very specific relative orientation of the binding partners. Additional weak, non-covalent interactions such as CH–π and π–π stacking can also be established between the included guests and the aromatic walls of the receptor.
The fact that aryl-extended calix[4]pyrroles possess a deep hydrophobic cavity in which the pyrrole NHs converge and are sheltered from the bulk solvent makes them very attractive for molecular recognition studies of polar guests in aqueous media. Hydrogen bonding interactions occurring in polar solvents with high dielectric constants (especially water) are frequently much weaker than those in non-polar solvents.10,11 However, when the functional groups converge into an apolar cavity and are “sheltered” from the bulk water, the strength of their hydrogen bonding interactions becomes less solvent-dependent.12–15 Indeed, enzymes take advantage of this strategy to secure vital hydrogen bonding interactions that would otherwise be washed out by competition from water molecules. While such interactions are ubiquitous in Nature, the synthesis of water soluble receptors with functional groups converging into an apolar cavity is challenging.
Recently, the rich possibilities offered by the polar and deep aromatic binding pocket of “four wall” aryl-extended calix[4]pyrroles have led to their extensive use as synthetic receptors6,16 and binding units in supramolecular scaffolds.17,18 These studies have been almost exclusively undertaken in organic solvents. As supramolecular chemistry develops, the understanding of molecular recognition in water, that is to say the role and the magnitude of hydrophilic and hydrophobic interactions in controlling the binding strength and selectivity, will become more and more important.19,20
To date, and to the best of our knowledge, only two examples of water soluble aryl-extended calix[4]pyrroles have been reported in the literature.21,22 The example taken from the work of our group describes complexation studies in water with pyridyl N-oxides. The aryl extended calix[4]pyrroles 1 employed in the study were rendered water soluble by covalently introducing, in the para positions at their upper rims (meso-aryl groups), four ionisable groups, particularly amine and carboxylic groups. This synthetic strategy presented several design drawbacks. Firstly, the attachment of the solubilising groups to the meso-aryl substituents alters the electronic nature of the pristine aromatic cavity. We have previously shown that the electronic properties of the cavity are tuned by the electron-withdrawing or electron-donating character of the substituents present in the meso-aryl groups. Secondly and even more importantly, the presence of the solubilising groups at the upper rim impairs further elaboration of the aromatic cavity with other functional groups capable of interacting with the included guest or with other molecules providing higher order aggregates (i.e. dimeric capsules).
In this manuscript we report the synthesis and preliminary binding studies of a new tetraphenyl water-soluble aryl-extended calix[4]pyrrole α,α,α,α-2 (Scheme 1). The receptor features “innocent” water-solubilising groups located at the terminus of its meso-alkyl substituents. The solubilising groups are sufficiently distant to avoid any interference with the pristine characteristics of the deep aromatic binding site. We envision that the described synthetic strategy will permit further elaboration of the aromatic cavity of water soluble aryl extended calix[4]pyrroles through upper rim functionalization, eventually allowing access to higher order calix[4]pyrrole-based supramolecular structures in aqueous media.
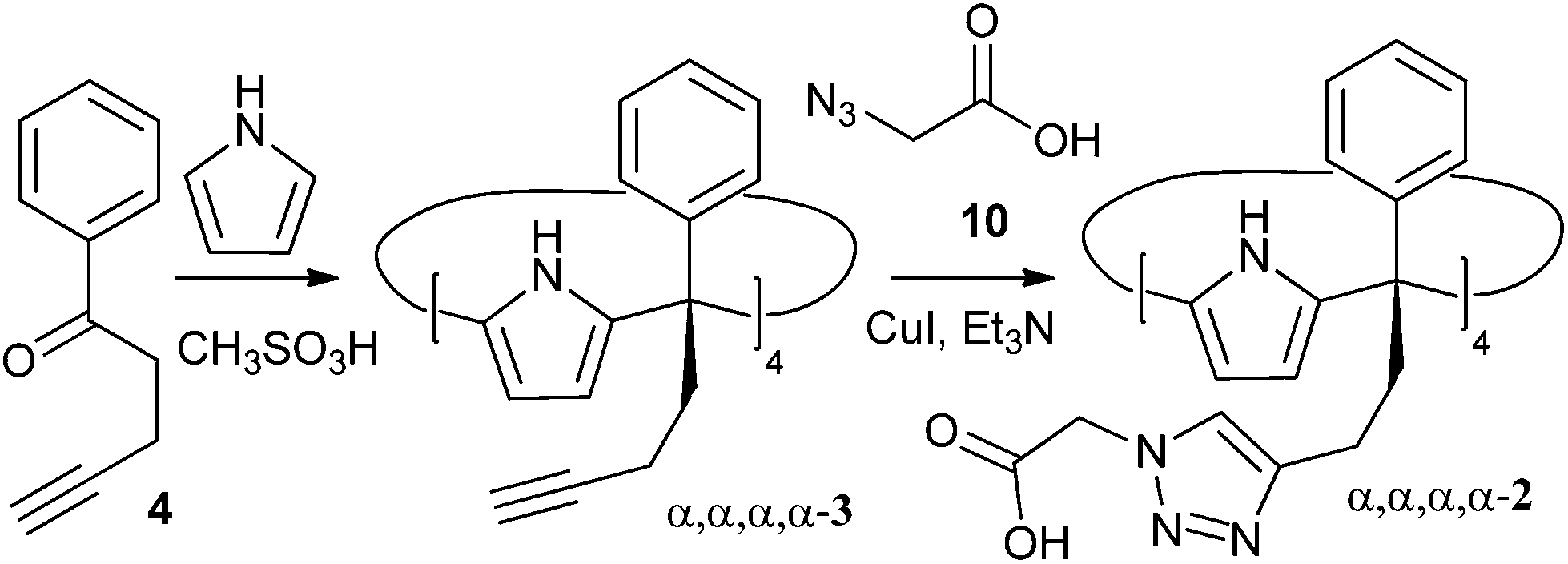 | ||
| Scheme 1 Synthetic scheme for the preparation of water soluble aryl extended calix[4]pyrrole α,α,α,α-2. | ||
Results and discussion
Synthesis and structural characterization
The novel water-soluble aryl-extended calix[4]pyrrole α,α,α,α-2 was synthesised in five synthetic steps. Conveniently, the ionisable water-solubilising groups are introduced in the final step. Thus, silica chromatography is an available purification technique in the synthesis of the calix[4]pyrrole precursor α,α,α,α-3 (Scheme 2). This is an essential aspect due to the inevitable need to remove the other configurational isomers and polymeric by-products from the crude reaction mixture obtained by the condensation of butynyl phenyl ketone 4 with pyrrole.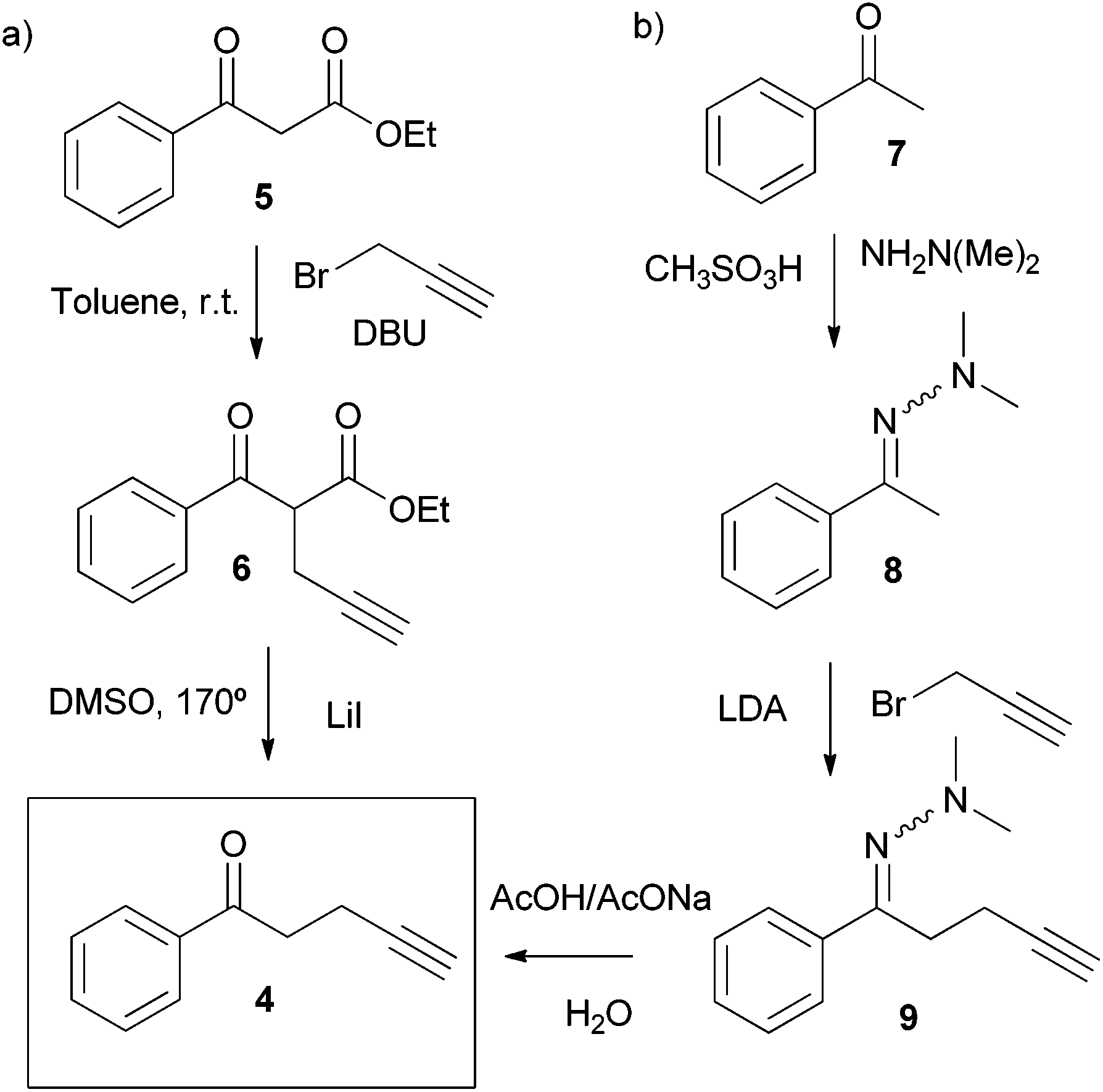 | ||
| Scheme 2 Synthetic routes for the preparation of butynyl phenyl ketone 4. | ||
The synthetic pathway to produce α,α,α,α-2 requires the preliminary preparation of the butynyl phenyl ketone 4 (Scheme 2). Initially, we proceeded by alkylation of the β-keto ester 5 with propargyl bromide in the presence of DBU followed by cleavage of the ester 6 and heat promoted decarboxylation of the corresponding β-keto acid (Scheme 2, pathway a). While the alkynyl ketone 4 could be obtained through this method, the decarboxylation step turned out to be capricious. A much better and reliable synthetic route for the preparation of the butynyl phenyl ketone 4 started from acetophenone 7 and employed the methodology developed by Zaman et al. (Scheme 2, pathway b).23
This robust synthetic pathway has the advantage of starting from simple and readily available acetophenones, meaning that water-soluble calix[4]pyrroles displaying a wide variety of meso aryl groups should be accessible. The acid mediated cyclocondensation of alkynyl ketone 4 and pyrrole to give α,α,α,α-3 was very sluggish (Scheme 1). After four days, the crude reaction mixture consisted of a rich array of products, including the desired α,α,α,α-isomer of the calix[4]pyrrole 3 (Scheme 2), other configurational isomers, and higher order condensation products. Column chromatography purification of the crude mixture, followed by recrystallization with acetonitrile eventually afforded the α,α,α,α-isomer of calix[4]pyrrole 3 in low yield but with excellent purity.
The configuration of the α,α,α,α-3 isomer was confirmed by X-ray diffraction of a single crystal that grew from the acetonitrile solution.‡ In the solid state, α,α,α,α-3 adopts the cone conformation with a tilted molecule of acetonitrile included in its deep aromatic cavity. The nitrogen atom of the included acetonitrile is bound to the four pyrrolic NH groups, with an average N⋯N distance of 3.26 Å for the hydrogen bonds (Fig. 2).
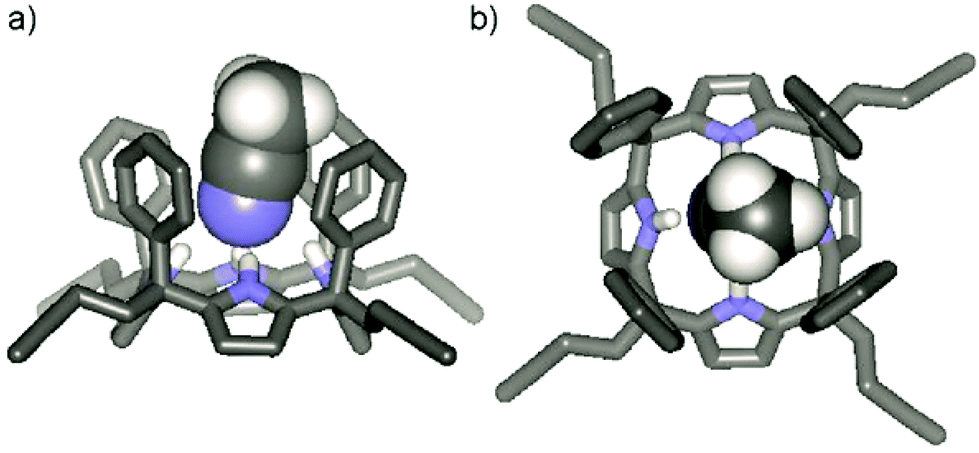 | ||
| Fig. 2 (a) Side view and (b) top view of the X-ray structure of the aryl extended calix[4]pyrrole CH3CN ⊂ α,α,α,α-3. For clarity, non-polar hydrogen atoms have been omitted. The included acetonitrile molecule is shown in CPK representation. | ||
Finally, the water-soluble tetracarboxylic-functionalised receptor α,α,α,α-2 was obtained in good yield via 1,3 Hüisgen cycloaddition with azide 10 under standard copper(I) catalysed conditions (Scheme 1). Gratifyingly, calix[4]pyrrole α,α,α,α-2 is readily soluble in a variety of aqueous solutions such as diluted NaOH and, most notably, phosphate buffer saline (PBS) at pH 7.4, a solution often used to mimic biological media.
Binding studies
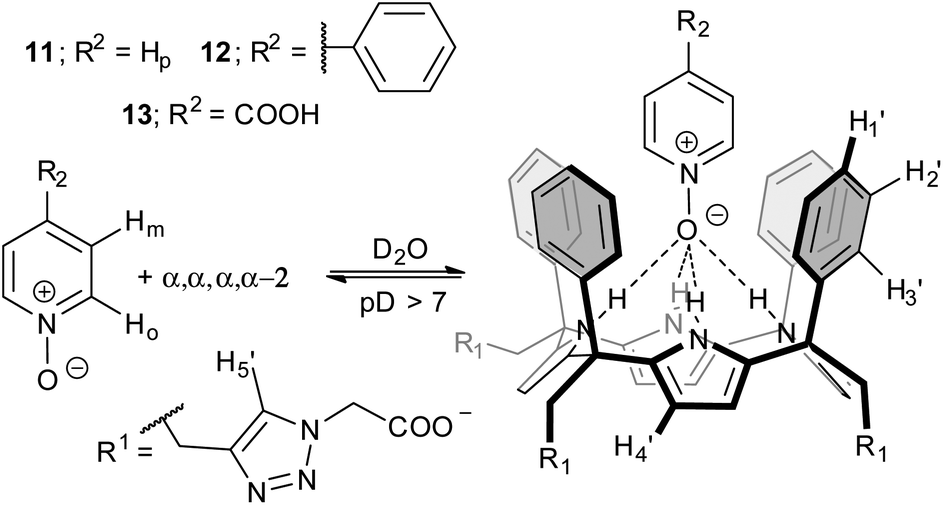 | ||
| Fig. 3 Equilibrium involved in the formation of the inclusion complexes PNO ⊂ α,α,α,α-2 and the proposed structure for the binding geometry. | ||
Initially, we probed the interaction of calix[4]pyrrole α,α,α,α-2 with different PNO derivatives using 1H NMR spectroscopy in a basic D2O solution (pD between 7.2 and 7.6) adjusted with NaOD. The 1H NMR spectrum of α,α,α,α-2 shows sharp signals and the characteristics of an averaged C4v symmetry (Fig. 4a). The β-pyrrolic protons resonate as a singlet at δ = 6.1 ppm, suggesting that the free host might adopt the cone conformation in solution.21
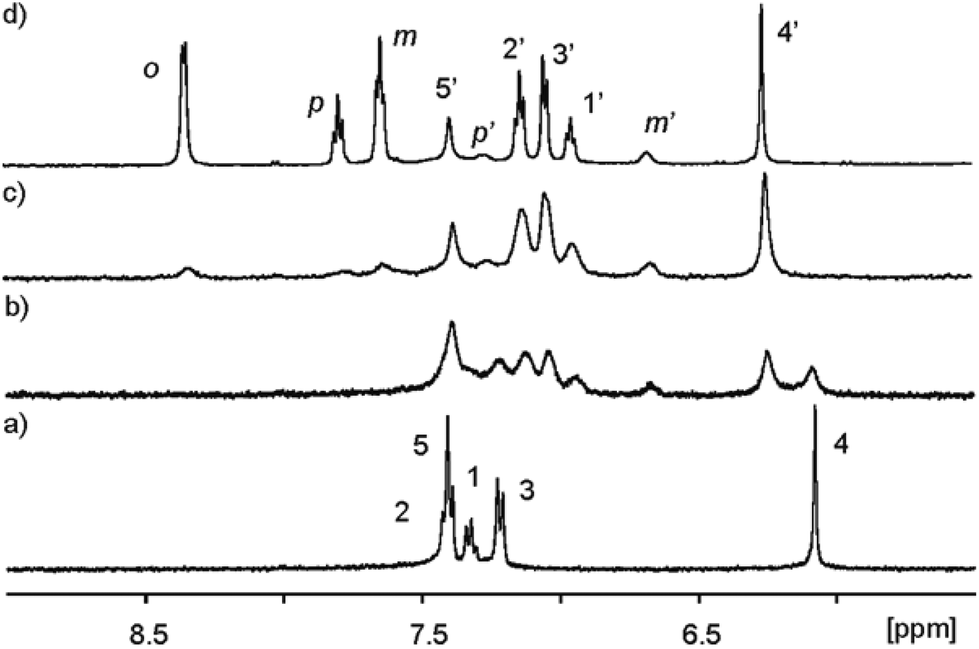 | ||
| Fig. 4 Selected downfield regions of the 1H NMR spectra (400 MHz, D2O adjusted to pD ≈ 7.6 with NaOD, 298 K) acquired during the titration of calix[4]pyrrole α,α,α,α-2 (1 mM) with incremental amounts of 11. (a) α,α,α,α-2; (b) α,α,α,α-2 + 0.6 eq. 11; (c) α,α,α,α-2 + 1.1 eq. 11; (d) α,α,α,α-2 + 3.4 eq. 11. Primed letters and numbers correspond to proton signals in the encapsulation complex 11 ⊂ α,α,α,α-2. See Fig. 3 for proton assignment and ESI† for the analogous titration with 13. | ||
As increasing amounts of PNO 11 were added to ∼1 mM aqueous solutions of α,α,α,α-2, the 1H NMR spectra of the resulting mixtures showed that the signals corresponding to the free receptor, α,α,α,α-2, broadened and decreased in intensity. Concomitantly, a new set of broad signals appeared and grew at the expense of the signals of free α,α,α,α-2 (Fig. 4b). This new set of signals was assigned to the protons of the calix[4]pyrrole receptor in the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inclusion complex 11 ⊂ α,α,α,α-2. The new proton signals corresponding to the meta and para pyridyl protons of the included 11 resonating at δ ≈ 6.5 (Δδ = −0.95 ppm) and 7.1 ppm (Δδ = −0.52 ppm), respectively, also became evident. The pyridyl proton ortho to the nitrogen atom of 11 (not shown in Fig. 4 owing to the overlap with the broad water signal in this experiment, see Fig. 5) is the most affected by the magnetic anisotropy of the four meso-phenyl substituents and experiences a large upfield shift (Δδ = −4.0 ppm). This observation shows that the N-oxide group of bound 11 must be located at the bottom of the aromatic cavity of α,α,α,α-2 supporting the proposed binding geometry in which the N-oxide forms four hydrogen bonds with the pyrrole NH.21 When slightly more than one equivalent of 11 was added, the proton signals corresponding to the free calix disappeared (Fig. 4c). Only the proton signals of the host in the inclusion complex 11 ⊂ α,α,α,α-2 were detected. The addition of an excess of 11 did not provoke changes in the chemical shifts of the inclusion complex but sharpened the proton signals. The proton signals corresponding to the free 11 in excess resonate as separate signals in the downfield region of the spectrum. Taken together, these results indicate that: (a) the free species and the 1:1 host–guest complex are involved in a chemical exchange process that is slow on the 1H NMR chemical shift time scale; (b) the stability constant for the 11 ⊂ α,α,α,α-2 can be estimated to be larger than 104 M−1; and (c) the hydrogen bonded N-oxide 11 is rotating on its vertical axis when included in the deep aromatic binding pocket of α,α,α,α-2.
:1 inclusion complex 11 ⊂ α,α,α,α-2. The new proton signals corresponding to the meta and para pyridyl protons of the included 11 resonating at δ ≈ 6.5 (Δδ = −0.95 ppm) and 7.1 ppm (Δδ = −0.52 ppm), respectively, also became evident. The pyridyl proton ortho to the nitrogen atom of 11 (not shown in Fig. 4 owing to the overlap with the broad water signal in this experiment, see Fig. 5) is the most affected by the magnetic anisotropy of the four meso-phenyl substituents and experiences a large upfield shift (Δδ = −4.0 ppm). This observation shows that the N-oxide group of bound 11 must be located at the bottom of the aromatic cavity of α,α,α,α-2 supporting the proposed binding geometry in which the N-oxide forms four hydrogen bonds with the pyrrole NH.21 When slightly more than one equivalent of 11 was added, the proton signals corresponding to the free calix disappeared (Fig. 4c). Only the proton signals of the host in the inclusion complex 11 ⊂ α,α,α,α-2 were detected. The addition of an excess of 11 did not provoke changes in the chemical shifts of the inclusion complex but sharpened the proton signals. The proton signals corresponding to the free 11 in excess resonate as separate signals in the downfield region of the spectrum. Taken together, these results indicate that: (a) the free species and the 1:1 host–guest complex are involved in a chemical exchange process that is slow on the 1H NMR chemical shift time scale; (b) the stability constant for the 11 ⊂ α,α,α,α-2 can be estimated to be larger than 104 M−1; and (c) the hydrogen bonded N-oxide 11 is rotating on its vertical axis when included in the deep aromatic binding pocket of α,α,α,α-2.
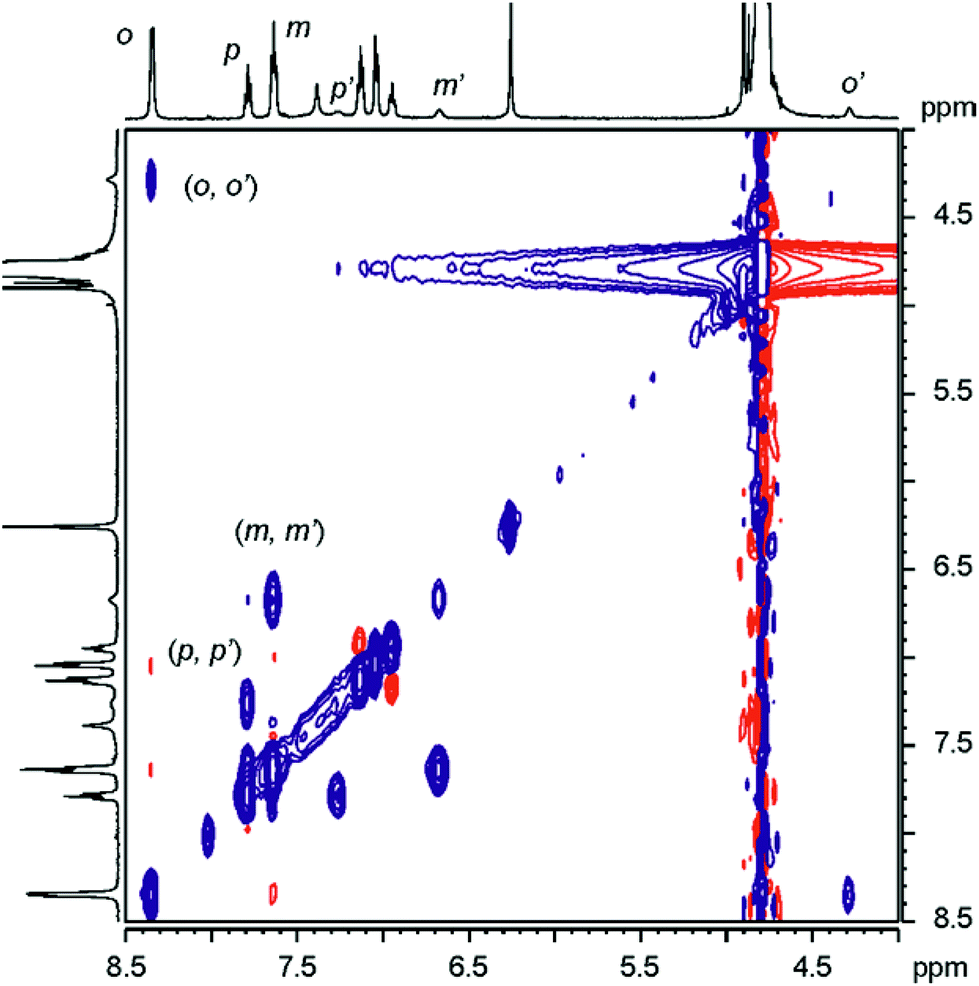 | ||
| Fig. 5 Selected region of ROESY 1H NMR spectrum of complex 11 ⊂ α,α,α,α-2 in the presence of free 11 in excess. The cross-peaks resulting from chemical exchange between protons of free and included 11 are indicated. Primed letters correspond to signals of the protons in the complex. | ||
The binding geometry of the 11 ⊂ α,α,α,α-2 complex and the kinetics of the binding process were also investigated by performing a ROESY experiment on the sample containing an excess of N-oxide 11.
The ROESY spectrum (Fig. 5) revealed the existence of positive cross peaks between the signals assigned to the protons of 11 in the inclusion complex and those of free 11. This finding demonstrates that although the chemical exchange between the free and bound guest is slow on the chemical shift scale it is fast on the relaxation time scale (complex lifetime <0.1 s). The observation of a cross peak between the signal of the proton alpha to the nitrogen atom of free 11 (ortho, Ho) and the highest upfield shifted signal belonging to the protons of bound 11 confirms the location of the N-oxide group near the closed end of the cavity. Completely analogous results were obtained for the titration with N-oxide of the phenyl pyridine 12.
The obtained results for the titration of α,α,α,α-2 with isonicotinic acid 13 deserve a small comment (Fig. S1 and S2†). Isonicotinic acid 13, in basic media, presents two functional groups known to bind strongly to calix[4]pyrrole units, the N-oxide and the carboxylate.26,27 The inclusion of 13, as the carboxylate, in the deep functionalised cavity of α,α,α,α-2 produced a single set of separated proton signals for the complex 13− ⊂ α,α,α,α-2. The complex was formed quantitatively when 1 equiv. of 13 was added to the basic solution. The ROESY experiment performed with a solution containing the inclusion complex 13− ⊂ α,α,α,α-2 and free 13− in excess showed that the highest upfield shifted proton of the included N-oxide is involved in a chemical exchange exclusively with the proton ortho to the nitrogen atom in the free guest. In short, the carboxylate derivative of isonicotinic 13 binds selectively through the N-oxide group to the α,α,α,α-2 receptor. The carboxylate function of bound 13− is exposed to the bulk solution and its negative charge seems to have a reduced effect in the binding strength of the complex (estimated Ka(13− ⊂ α,α,α,α-2) > 104 M−1) when compared with those estimated for the neutral N-oxides.
In general, the 1H NMR titration results reveal that the aryl-extended calix[4]pyrrole's hydrophobic pocket of α,α,α,α-2 is adequate to protect the pyrrole NHs from bulk water allowing them to effectively form hydrogen bonds with pyridyl N-oxide guests. Although the establishment of hydrogen bonding interactions is the main factor responsible for the selectivity towards the N-oxide group (hydrophilic binding), the partial desolvation of the host and the guest required for the formation of the tight inclusion complexes (π–π and CH–π interactions) plays an important role in determining the binding strength (hydrophobic binding).
All the ITC titrations performed with the PNO series 11–13 and receptor α,α,α,α-2 showed excellent fits to the theoretical binding isotherm for a 1:1 complex formation (see ESI† for titrations). The fit of the titration data to the theoretical isotherms allowed the accurate calculation of stability constant (Ka) and enthalpy (ΔH) values for the formation of the complexes. From these values, we derived the free energy of binding (ΔG) and the entropic (TΔS) component for each process. The obtained results are summarized in Table 1.
| N-oxide | Medium/pH |
K
a × 104a |
ΔG | ΔHa | TΔS |
|---|---|---|---|---|---|
| a The values presented are the mean of those obtained from at least two ITC titration experiments. Errors (standard deviation) are determined to be less than 3%. b pH of water solutions was adjusted with NaOH. | |||||
| 11 | Water/7.2b | 4.3 | −6.2 | −4.5 | 1.7 |
| 12 | Water/7.2b | 20.0 | −7.2 | −7.4 | −0.2 |
| 13 | Water/7.2b | 4.5 | −6.3 | −6.4 | −0.1 |
In complete agreement with the 1H NMR titrations, the ITC results indicate the formation of thermodynamically highly stable complexes (Ka > 104 M−1). In all cases, the binding process is mainly enthalpically driven. The entropic term is comparatively small and in the case of N-oxide 11 it also favours binding. These thermodynamic characteristics are in agreement with a tight fit of the interacting molecules in the complex and support the selective binding observed for the N-oxide group.28
Association constant values for the inclusion complexes of PNOs 11 and 12 with calix[4]pyrrole receptor 1a, having four carboxy groups at the upper rim, were previously reported.21 The reported values were measured using UV-vis titrations in H2O solutions (pH = 7.2) at concentrations that were similar to those used for the ITC experiments described here. Thus, the comparison of the measured magnitudes in the two studies is reasonable. The binding constant value determined for the 11 ⊂ 1a complex was Ka = 1.6 ± 0.2 × 104 M−1. This value is very close to the stability constant determined in this study for the 11 ⊂ α,α,α,α-2 complex. This indicates that moving the water solubilising anionic groups from the upper rim to the lower rim has a minimum impact on the binding of a small neutral molecule like pyridine-N-oxide 11 with the calix[4]pyrrole core. We foresee that the change of location of the solubilizing groups should allow the functionalization of the upper rim of the calix[4]pyrrole receptor with groups that provide higher affinity and selectivity.
In contrast with the result abovementioned, the reported binding constant for the 12 ⊂ 1a (Ka = 2.4 ± 1.3 × 103 M−1) was two orders of magnitude smaller than the one measured in this study for the related 12 ⊂ α,α,α,α-2 complex. This result indicated that the location of the water solubilising groups at the lower rim of a calix[4]pyrrole scaffold had a positive effect in the binding of larger N-oxides like 12. In this case, the use of the alternative synthetic methodology for the placement of the solubilising groups at the lower rim without further modification of the upper rim was rewarding.
Interestingly, the binding constant values determined for PNOs 11 and [13]− with receptor α,α,α,α-2 are almost identical. This surprised us, as we expected the negatively charged [13]−N-oxide to form a thermodynamically weaker complex with the also negatively charged, in basified water, calix[4]pyrrole α,α,α,α-2.§ In water, the strong repulsive electrostatic interactions established between negative charges are highly attenuated, allowing the two molecules to form a tight complex.
Not surprisingly, however, the thermodynamic characteristics of the two binding processes are indeed significantly different. Meanwhile, the stability constant for the complex formed between phenyl-substituted N-oxide 12 and receptor α,α,α,α-2 is almost five-fold larger than that of N-oxide 11. At first sight, this result could be attributed to the higher hydrophobicity of phenyl pyridyl N-oxide 12 with respect to pyridyl N-oxide 11. Larger apolar aromatic surfaces translate into higher hydrophobicity and a stronger hydrophobic effect because of the favourable increase in desolvation entropy. However, in the case at hand, the enhanced binding of 12 with respect to 11, results from an enthalpic gain on complex formation. In addition, the contacts in the surface area for complexes 12 ⊂ α,α,α,α-2 and 11 ⊂ α,α,α,α-2 are predicted to be similar, only the end of the pyridyl N-oxide residue is included in the binding site (Fig. 6). The thermodynamic characteristics suggest a tighter geometry for the 12 ⊂ α,α,α,α-2 complex that, as expected, is accompanied by a reduction in entropy gain, when compared to the looser complex formed with 11.
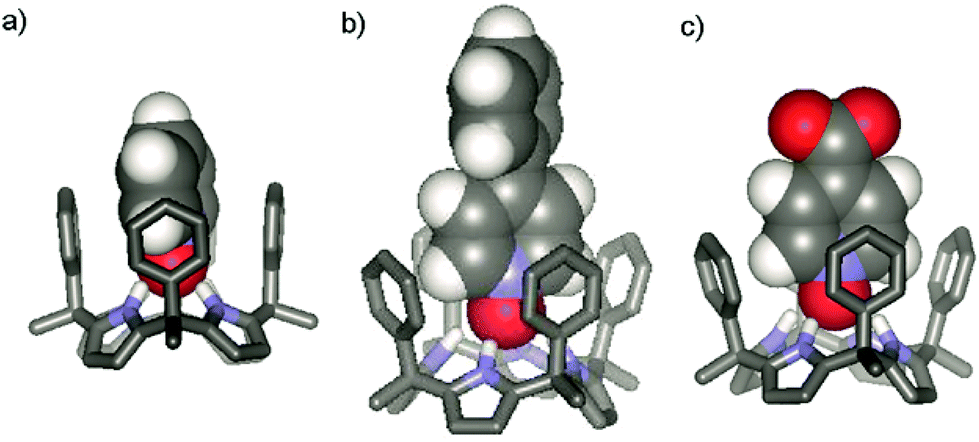 | ||
| Fig. 6 SCIGRESS (v 3.0.0, Fujitsu Ltd.) energy minimized (MO-G PM6 in water) structures of the inclusion complexes of the N-oxides with α,α,α,α-2: (a) 11 ⊂ α,α,α,α-2; (b) 12 ⊂ α,α,α,α-2; and (c) 13 ⊂ α,α,α,α-2. The meso-alkyl chains of the receptor were modelled as methyl groups. For clarity, only the polar hydrogen atoms of the receptor are shown and the included guest is represented as the CPK model. | ||
Conclusion
In conclusion, we have described the synthesis of a novel water-soluble aryl-extended calix[4]pyrrole α,α,α,α-2. In contrast to previous examples, the water-solubilising groups are distal to the binding site. This leaves the binding site and the upper rim of the calix[4]pyrrole pristine, making possible the future functionalization of the meso-aryl rings, thus, permitting the construction of more complicated calix[4]pyrroles. The receptor α,α,α,α-2 forms thermodynamically and kinetically stable complexes with a series of pyridyl N-oxide guests in aqueous solution. The binding geometry of these complexes was probed using 1H NMR titrations and their thermodynamic characteristics were determined using ITC experiments. The obtained results also demonstrated that calix[4]pyrrole α,α,α,α-2 preferentially engages in hydrogen bonding interactions of the four pyrrole NHs with a N-oxide moiety than with a carboxylate group. Nevertheless, the anionic α,α,α,α-2 receptor binds negatively charged pyridyl N-oxides with high affinity. Our results showcase the ability of the receptor's aromatic pocket to protect the pyrrole N–H groups and ensure selective hydrogen-bonding interactions even in water.Experimental
General
Reagents were obtained from commercial suppliers and used without further purification. All solvents were commercially obtained and used without further purification except for pyrrole which was distilled (50 mbar, 58–59 °C) and then stored in the freezer for further use. Where indicated, solvents were degassed by 3 freeze–pump–thaw cycles. Routine 1H NMR and 13C NMR spectra were recorded on a Bruker Avance 300 (300 MHz for 1H NMR), Bruker Avance 400 (400 MHz for 1H NMR), Bruker Avance 500 (500 MHz for 1H NMR) ultrashield spectrometer, or on a Bruker Avance III 500 (500 MHz for 1H NMR) with a QNP cryoprobe. The deuterated solvents (Aldrich) used are indicated in the Experimental section; chemical shifts are given in ppm. For CDCl3 the peaks were referenced relative to the solvent residual peak δH = 7.26 ppm and δC = 77.0 ppm. For D2O the peaks were referenced relative to the solvent residual peak δH = 4.79 ppm. All NMR J values are given in Hz. High resolution mass spectra were obtained on a MicroTOF II (Bruker Daltonics) ESI as ionization mode Mass Spectrometer or on a Bruker Ultraflex MALDI-TOF-TOF Mass spectrometer. Column chromatography was performed with silica gel, technical grade, pore size 60 Å, and 230–400 mesh particle size. Crystal structure determination was carried out using a Bruker-Nonius diffractometer equipped with an APEX 2 4K CCD area detector, a FR591 rotating anode with MoKα radiation, Montel mirrors as the monochromator and a Kryoflex low temperature device (T = 100 K). Full sphere data collection was carried out by omega and phi scans. Programs used: data collection Apex2 V. 1.0–22 (Bruker-Nonius 2004), data reduction Saint + Version 6.22 (Bruker-Nonius 2001) and absorption correction SADABS V. 2.10 (2003). Crystal structure solution was achieved using direct methods as implemented in SHELXTL Version 6.10 (Sheldrick, Universität Göttingen (Germany), 2000) and visualized using the XP program. Missing atoms were subsequently located from difference Fourier synthesis and added to the atom list. Least-squares refinement on F2 using all measured intensities was carried out using the program SHELXTL Version 6.10 (Sheldrick, Universität Göttingen (Germany), 2000).Procedures and characterisation data
Step 2 alkylation and hydrolysis of the hydrazone.23 Lithium diisopropylamide (6.29 mL, 1.96 M, 12.33 mmol, 2 equiv.), freshly titrated using diphenylacetic acid as an indicator, was added to a solution at −74 °C (achieved using a dry ice/acetone bath) of hydrazine 8 (1 g, 6.16 mmol, 1 equiv.) in THF (15 mL, freshly distilled from sodium benzophenone) under an argon atmosphere. After 1 hour, 3-bromoprop-1-yne (80% solution in toluene, 0.69 mL, 6.16 mmol, 1 equiv.) was added dropwise via a syringe. The reaction was stirred overnight while coming to RT. The next day, the reaction was quenched by adding 40 mL of saturated NH4Cl(aq). The reaction mixture was extracted with diethylether (3 × 20 mL) and the organic extracts were combined, dried over Na2SO4, filtered and concentrated under vacuum to give a brown residue. Acetic acid 3 mL, sodium acetate 1.45 g, water 0.5 mL and THF 3 mL were added to the resulting residue and the reaction was stirred under air until complete hydrolysis of the hydrazone was observed by 1H NMR (typically 7 hours). The reaction mixture was quenched by adding NaOH(aq) (2 M) at 0 °C until the pH of the mixture was basic as judged by litmus paper. The mixture was stirred for 20 min and then 20 mL of diethyl ether were added. The two phases were separated and the aqueous layer was extracted with diethyl ether (2 × 20 mL). The organic extracts were combined, washed with 50 mL of water, and then with 50 mL of brine. Finally the organic extract was dried over Na2SO4, filtered and concentrated under vacuum, affording 0.82 g of the crude product as a black solid. The solid was purified by column chromatography (40 g SiO2 column, 1:1 DCM–hexane eluent, product RF = 0.5). The ketone 4 was obtained as light yellow crystals which were then sublimed in a bulb to bulb apparatus (0.1 mmHg, 80 °C), affording a white microcrystalline powder (0.63 g, 65%).The 1H NMR spectrum is in agreement with that previously reported in the literature.31
:35, product RF = 0.4). Two separate fractions containing the product were isolated: the first fraction (11.6 mg) contained the desired product and other configurational isomers. The second fraction (78 mg) contained the α,α,α,α-3 isomer and an unidentified impurity with similar RF = 0.4. Both fractions were separately dissolved in a minimum of hot acetonitrile. The solutions were cooled at 0 °C and the α,α,α,α-3 isomer crystalized as small colorless cubes, 21.9 mg, >1% yield. 1H NMR (300 MHz, CDCl3, 25 °C): δ (ppm) 7.25 (bs, 4H), 7.18–7.07 (m, 20H), 5.92 (d, J = 2.7 Hz, 8H), 2.56 (m, 8H), 2.09 (m, 8H), 1.95 (t, J = 2.6 Hz, 4H); 13C{1H}NMR (125 MHz, CDCl3 25 °C): δ (ppm) 135.1, 128.2, 128.1, 127.0, 106.4, 100.2, 84.5, 68.6, 48.6, 39.3, 15.0; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C60H53N4 829.4265; Found 829.4282; IR ṽ (cm−1) 2116, 2961, 3301, 3370, 3404; m.p. 214–224 °C decomposition.
Acknowledgements
The authors thank Gobierno de España MINECO (project CTQ2011-23014), Severo Ochoa Excellence Accreditation 2014-2018 (SEV-2013-0319), COST Action CM1005 and the ICIQ Foundation for funding. We also thank Eduardo C. Escudero-Adán for X-ray crystallographic data.Notes and references
- P. A. Gale, J. L. Sessler and V. Kral, Chem. Commun., 1998, 1–8 RSC.
- P. A. Gale, P. Anzenbacher and J. L. Sessler, Coord. Chem. Rev., 2001, 222, 57–102 CrossRef CAS.
- J. L. Sessler, D. E. Gross, W. S. Cho, V. M. Lynch, F. P. Schmidtchen, G. W. Bates, M. E. Light and P. A. Gale, J. Am. Chem. Soc., 2006, 128, 12281–12288 CrossRef CAS PubMed.
- S. K. Kim and J. L. Sessler, Acc. Chem. Res., 2014, 47, 2525–2536 CrossRef CAS PubMed.
- W. E. Allen, P. A. Gale, C. T. Brown, V. M. Lynch and J. L. Sessler, J. Am. Chem. Soc., 1996, 118, 12471–12472 CrossRef CAS.
- P. Anzenbacher, K. Jursíková, V. M. Lynch, P. A. Gale and J. L. Sessler, J. Am. Chem. Soc., 1999, 121, 11020–11021 CrossRef CAS.
- L. Bonomo, E. Solari, G. Toraman, R. Scopelliti, M. Latronico and C. Floriani, Chem. Commun., 1999, 2413–2414 RSC.
- J. R. Blas, J. M. Lopez-Bes, M. Marquez, J. L. Sessler, F. J. Luque and M. Orozco, Chem. – Eur. J., 2007, 13, 1108–1116 CrossRef PubMed.
- Y. D. Wu, D. F. Wang and J. L. Sessler, J. Org. Chem., 2001, 66, 3739–3746 CrossRef CAS PubMed.
- C. Allott, H. Adams, P. L. Bernad, C. A. Hunter, C. Rotger and J. A. Thomas, Chem. Commun., 1998, 2449–2450 RSC.
- V. M. Rotello, E. A. Viani, G. Deslongchamps, B. A. Murray and J. Rebek, J. Am. Chem. Soc., 1993, 115, 797–798 CrossRef CAS.
- B. Sookcharoenpinyo, E. Klein, Y. Ferrand, D. B. Walker, P. R. Brotherhood, C. F. Ke, M. P. Crump and A. P. Davis, Angew. Chem., Int. Ed., 2012, 51, 4586–4590 CrossRef CAS PubMed.
- M. Torneiro and W. C. Still, J. Am. Chem. Soc., 1995, 117, 5887–5888 CrossRef CAS.
- J. D. Howgego, C. P. Butts, M. P. Crump and A. P. Davis, Chem. Commun., 2013, 49, 3110–3112 RSC.
- G. Joshi and A. P. Davis, Org. Biomol. Chem., 2012, 10, 5760–5763 CAS.
- G. Gil-Ramírez, E. C. Escudero-Adán, J. Benet-Buchholz and P. Ballester, Angew. Chem., Int. Ed., 2008, 47, 4114–4118 CrossRef PubMed.
- M. Chas and P. Ballester, Chem. Sci., 2012, 3, 186–191 RSC.
- M. Espelt and P. Ballester, Org. Lett., 2012, 14, 5708–5711 CrossRef CAS PubMed.
- E. A. Kataev and C. Muller, Tetrahedron, 2014, 70, 137–167 CrossRef CAS PubMed.
- G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS PubMed.
- B. Verdejo, G. Gil-Ramírez and P. Ballester, J. Am. Chem. Soc., 2009, 131, 3178–3179 CrossRef CAS PubMed.
- K. D. Bhatt, D. J. Vyas, B. A. Makwana, S. M. Darjee and V. K. Jain, Spectrochim. Acta, Part A, 2014, 121, 94–100 CrossRef CAS PubMed.
- S. Zaman, M. Kitamura and A. D. Abell, Aust. J. Chem., 2007, 60, 624–626 CrossRef CAS.
- L. Adriaenssens and P. Ballester, Chem. Soc. Rev., 2013, 42, 3261–3277 RSC.
- L. Adriaenssens, J. L. Acero Sanchez, X. Barril, C. O'Sullivan and P. Ballester, Chem. Sci., 2014, 5, 4210–4215 RSC.
- D. E. Gross, D. W. Yoon, V. M. Lynch, C. H. Lee and J. L. Sessler, J. Inclusion Phenom. Macrocyclic Chem., 2010, 66, 81–85 CrossRef CAS PubMed.
- J. Kříž, J. Dybal, E. Makrlík, Z. Sedláková and V. Kašička, Chem. Phys. Lett., 2013, 561–562, 42–45 CrossRef PubMed.
- Highly selective substrate binding should be enthalpically driven see: F. Diederich, Cyclophanes, The Royal Society of Chemistry, Cambridge, 1991 Search PubMed.
- H. F. Motiwala, B. Gülgeze and J. Aubé, J. Org. Chem., 2012, 77, 7005–7022 CrossRef CAS PubMed.
- S. D. Sharma and S. B. Pandhi, J. Org. Chem., 1990, 55, 2196–2200 CrossRef CAS.
- H. Imagawa, T. Kurisaki and M. Nishizawa, Org. Lett., 2004, 6, 3679–3681 CrossRef CAS PubMed.
- C. K. Y. Chun and R. J. Payne, Aust. J. Chem., 2009, 62, 1339–1343 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for novel compounds, additional 1H NMR titrations and 2D ROESY experiments mentioned in the text, details describing determination of the thermodynamic binding properties in solution using ITC and the fits of the obtained thermograms to a 1:1 binding model. CCDC 1027046. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob02108h |
‡ Crystal data. C62H55N5, M = 870.11, triclinic, a = 8.9089(3); b = 14.1642(4); c = 19.4386(6) Å, U = 2372.83(13) Å3, T = 100(2) K, space group: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, 18604 reflections were measured, 10409 unique (Rint = 0.0242) which were used in all calculations. The final wR(F2) was 0.1357 (all data). CCDC 1027046 contains the supplementary crystallographic data for this paper. , Z = 2, 18604 reflections were measured, 10409 unique (Rint = 0.0242) which were used in all calculations. The final wR(F2) was 0.1357 (all data). CCDC 1027046 contains the supplementary crystallographic data for this paper. |
| § The protonation constants for the receptor α,α,α,α-2 were not determined, but in analogy to compound 1a, it is expected that in water solution at pH = 7.2 the former will be exclusively presented as a tetraanionic species. |
| This journal is © The Royal Society of Chemistry 2015 |