
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH response and molecular recognition in a low molecular weight peptide hydrogel†
Stefanie C.
Lange
a,
Jan
Unsleber
a,
Patrick
Drücker
b,
Hans-Joachim
Galla
b,
Mark P.
Waller
a and
Bart Jan
Ravoo
*a
aOrganic Chemistry Institute, Westfälische Wilhelms-Universität Münster, Corrensstrasse 40, 48149 Münster, Germany. E-mail: b.j.ravoo@uni-muenster.de
bInstitute of Biochemistry, Westfälische Wilhelms-Universität Münster, Wilhelm-Klemm-Strasse 2, 48149 Münster, Germany
First published on 31st October 2014
Abstract
In this article we report the preparation and characterization of a peptide-based hydrogel, which possesses characteristic rheological properties, is pH responsive and can be functionalized at its thiol function. The tripeptide N-(fluorenyl-9-methoxycarbonyl)-L-Cys(acetamidomethyl)-L-His-L-Cys-OH 1 forms stable supramolecular aggregates in water leading to hydrogels above 1.5 wt%. Rheological analysis of the hydrogel revealed visco-elastic and shear thinning properties of samples containing 1.5 wt% of peptide 1. The hydrogel reversibly responds to pH changes. Below and above pH 6, electrostatic repulsion of the peptide results in a weakening of the three-dimensional gel network. Based on atomic force microscopy, small angle X-ray scattering and molecular dynamics simulations, it is proposed that the peptide assembles into nanostructures that tend to entangle at higher concentrations in water. The development of functional materials based on the peptide assemblies was possible via thiol–ene-click chemistry of the free thiol function at the C-terminal cysteine unit. As a proof of concept, the functionalization with adamantyl units to give 1-Ad was shown by molecular recognition of β-cyclodextrin vesicles. These vesicles were used as supramolecular cross-linkers of the assemblies of peptide 1 mixed with peptide 1-Ad leading to gel networks at a reduced peptide concentration.
Introduction
Self-assembly emerges as a powerful methodology for the preparation of nanostructured materials with dynamic and adaptive properties. Nowadays, the self-assembly in aqueous solution of macromolecular1 as well as low molecular weight building blocks2 into well-defined nanostructures such as fibers, networks and hydrogels is well established. However, the implementation of functionality into tailor made supramolecular architectures for application in biomedical technology is still a highly challenging area of research. In recent years, considerable progress has been made using macromolecular materials, such as coiled coil peptides that provide biomimetic fibrils,3 nanoparticles that sense proteins4 and co-polymers that act as chaperones.5In view of their biodegradability and biocompatibility, peptides are an excellent choice to construct bioactive functional materials in a bottom up approach. Peptides offer attractive and specific intermolecular interactions such as hydrogen bonds, electrostatic, hydrophobic, CH–π- and π–π-interactions and tend to self-assemble in aqueous media into fibers, vesicles and sheets, also giving rise to hydrogels.6 In some cases, the self-assembly of peptides can be directed by external stimuli, such as oxidation6e or pH.6j Stimulus-responsive self-assembly can also be observed for a variety of synthetic, non-peptide building blocks.7
It is well-known that the self-assembly of N-fluorenyl-9-methoxycarbonyl (Fmoc) protected dipeptides into π–π interlocked β-sheets leads to formation of hydrogels.6a–d,6i In the course of our work on dynamic libraries of peptides,8 we synthesized the tripeptide Fmoc-L-Cys(Acm)-L-His-L-Cys and by serendipity discovered that it has a pronounced tendency to form a hydrogel. In this article, we demonstrate the preparation and full characterization of hydrogels based on this peptide. Using a combination of rheology and microscopy as well as molecular dynamics simulations, we show that the peptide forms a hydrogel that possesses characteristic rheological properties, is pH responsive and can be functionalized at its thiol function. As a proof-of-concept, the peptide was functionalized with adamantane and shown to cross-link cyclodextrin vesicles as a result of selective host–guest inclusion. The formation of hydrogels from cyclodextrin vesicles (CDV) and adamantane-functionalized polymers was reported recently.9 Although the concept of a hydrogel containing supramolecular polymers and vesicles has been described before,6g the self-assembly of a hydrogel network based on molecular recognition of a supramolecular peptide polymer and vesicles is – to the best of our knowledge – unprecedented.
Results and discussion
The tripeptides Fmoc-L-Cys(Acm)-L-His-L-Cys 1 and Fmoc-L-Cys-L-His-L-Cys 2 (Fig. 1, left) were prepared with standard solid phase peptide synthesis using Fmoc-strategy and side chain protected amino acids in the natural L-form. Details of the synthesis and analysis of the peptides are provided in the Experimental section as well as in the ESI.† The analytical data obtained for peptides 1 and 2 are fully consistent with the molecular structures shown in Fig. 1. The peptide sequence features a number of important elements that are critical to its self-assembly in aqueous solution. The importance of the Fmoc group for the formation of hydrogels due to π–π-stacking of small peptides has been discussed in many recent publications.6a–d,10 Introduction of the polar imidazole side group equips the molecules with a second pH tunable moiety beside the carboxylic acids at the C-terminus. In addition, the free thiol functions of peptide 1 and 2 are useful for disulfide formation and disulfide exchange or functionalization in a thiol–ene-click reaction. Whereas peptide 2 does not show any tendency to form a gel in aqueous solution, the Acm protection of the N-terminal cysteine in peptide 1 changes the polarity of this unit completely. It was found to form a hydrogel above 1.5 wt% (Fig. 1, right), which is in agreement with previously reported Fmoc terminated small peptide hydrogels.6a–d,10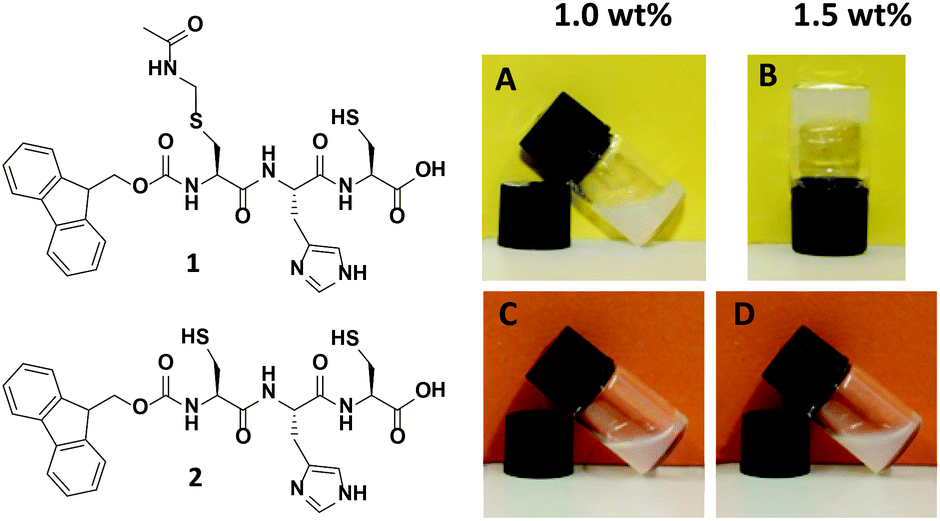 | ||
| Fig. 1 Structure of tripeptides Fmoc-L-Cys(Acm)-L-His-L-Cys-OH 1 and Fmoc-L-Cys-L-His-L-Cys-OH 2 (left) and visualization of gelation properties of 1 above 1.5 wt% compared to 2 (right). | ||
The mechanical properties of samples containing different amounts of peptide 1 were studied with rheology. For a detailed picture of the viscous and elastic contribution to the mechanical properties of the materials, oscillatory rheological measurements were performed (Fig. 2a). A strain amplitude sweep revealed constant extensions of the linear viscoelastic region although the plateau value of the storage modulus G′ drops drastically when less than 1.5 wt% 1 was used (G′ = 101–103 Pa). This can be explained by a decrease in rigidity of the hydrogel due to a diminishing number of entanglements and non-covalent cross-links. In Fig. 2b frequency sweep measurements of the storage and loss moduli (G′ and G′′) are shown. The curves are parallel, almost linear and the elastic component G′ dominates the viscous component G′′ for both concentrations, confirming the gel character of these samples.
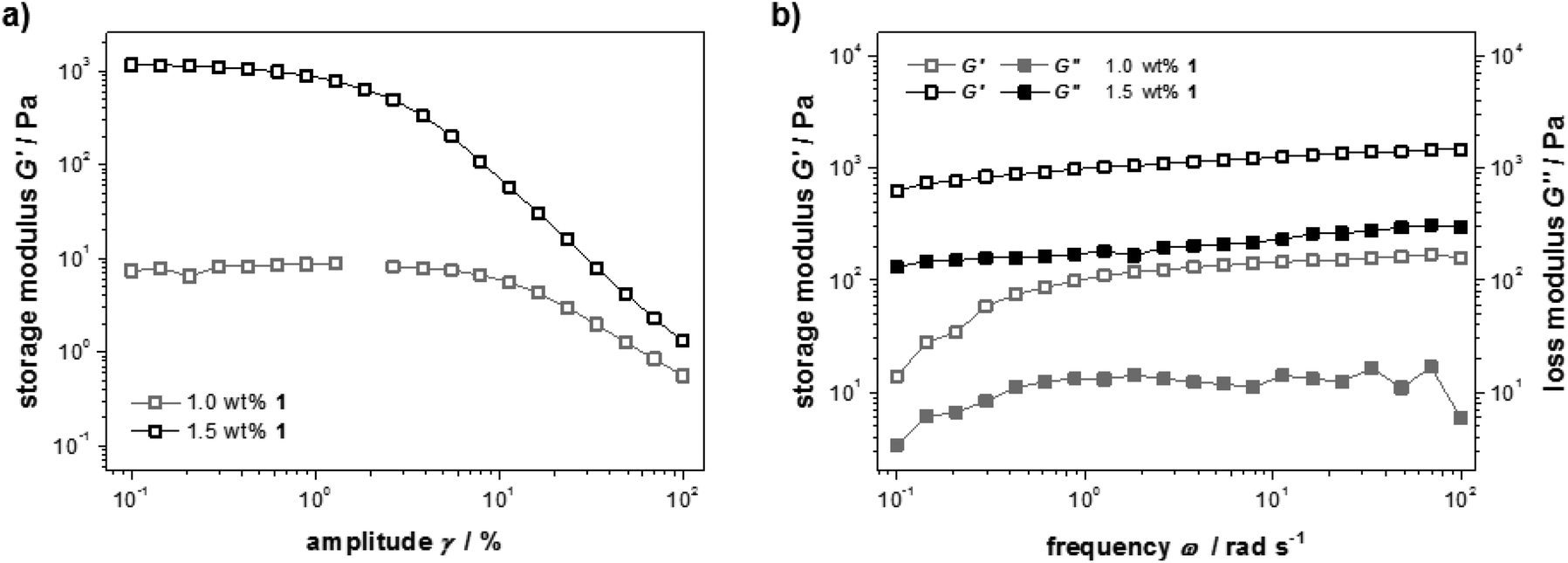 | ||
| Fig. 2 Oscillatory rheological measurements of solutions and hydrogels with 1.0 wt% and 1.5 wt% 1. (a) Storage modulus obtained from a strain-amplitude sweep performed at ω = 10 rad s−1. (b) Storage G′ and loss G′′ moduli obtained from a frequency sweep performed at γ = 0.5%. | ||
Steady shear flow experiments (Fig. 3a) showed flow curves with characteristic shear thinning behavior, despite classical Newtonian behavior up to a shear rate of ![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) = 0.01 s−1. The latter can be explained with overlapping flow curves of breaking and rebuilding junctions within the material at low rates, resulting in a constant viscosity and increasing shear stress. Here, the absolute values of the zero-shear viscosities (η0 = 103–104 Pa s) again indicate a dependency on the cross-link density. For increasing shear rates the materials show viscous flow, which likely indicates that the non-covalent interactions in the gel are easily broken under shear stress. Furthermore, the thixotropic behavior was examined in transient stress curves due to instantaneous changes in shear rate (Fig. 3b). Initially, the viscosity was measured at a shear rate of the linear regime ( = 0.5 s−1), next a breakdown of the bulk structure of the material was induced by applying a high-magnitude shear rate ( = 500 s−1) causing a steep decline in viscosity. To record the build-up curve subsequently the low shear rate of = 0.5 s−1 was used. The hydrogel with 1.5 wt% 1 show excellent thixotropic behavior and was found to recover within only a few seconds. The build-up curve of the less concentrated sample (1.0 wt%) increases much slower and not completely up to the initial value within the time range of the experiment, which is due to reduced entanglement of the supramolecular network at lower peptide concentration.
= 0.01 s−1. The latter can be explained with overlapping flow curves of breaking and rebuilding junctions within the material at low rates, resulting in a constant viscosity and increasing shear stress. Here, the absolute values of the zero-shear viscosities (η0 = 103–104 Pa s) again indicate a dependency on the cross-link density. For increasing shear rates the materials show viscous flow, which likely indicates that the non-covalent interactions in the gel are easily broken under shear stress. Furthermore, the thixotropic behavior was examined in transient stress curves due to instantaneous changes in shear rate (Fig. 3b). Initially, the viscosity was measured at a shear rate of the linear regime ( = 0.5 s−1), next a breakdown of the bulk structure of the material was induced by applying a high-magnitude shear rate ( = 500 s−1) causing a steep decline in viscosity. To record the build-up curve subsequently the low shear rate of = 0.5 s−1 was used. The hydrogel with 1.5 wt% 1 show excellent thixotropic behavior and was found to recover within only a few seconds. The build-up curve of the less concentrated sample (1.0 wt%) increases much slower and not completely up to the initial value within the time range of the experiment, which is due to reduced entanglement of the supramolecular network at lower peptide concentration.
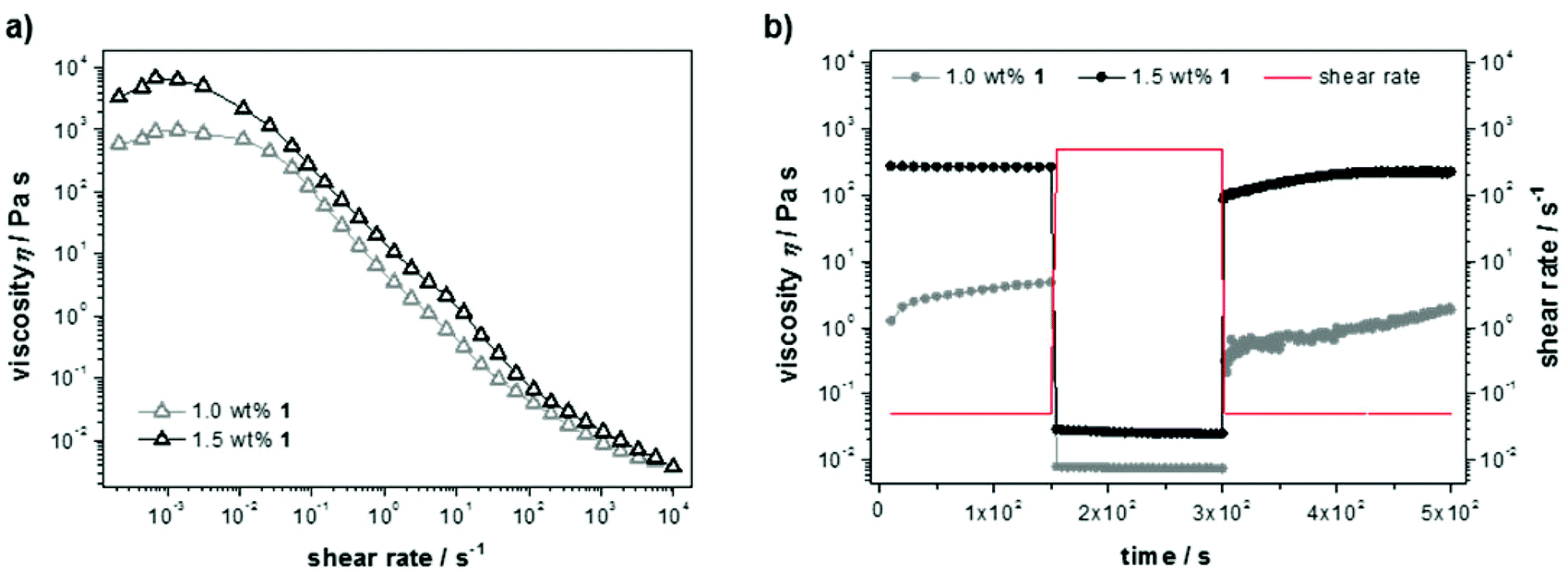 | ||
| Fig. 3 (a) Steady shear rheological measurements reveal the response to increasing shear stress of solutions and hydrogels with 1.0 wt% and 1.5 wt% 1. (b) Step-rate measurements of samples with 1.0 wt% and 1.5 wt% 1 display the different recovery of the hydrogel network immediately after the breakdown due to a high magnitude shear rate. | ||
In addition to its attractive mechanical properties, the obtained hydrogel exhibits a reversible pH response. In Fig. 4a two complete switching cycles are shown. The hydrogel was obtained every time physiological pH was reached, either from lower or higher pH values. The mechanical properties of different samples confirm an increased stability around pH 6, whereas acidic or basic conditions lead to lower viscosities (Fig. 4b). Images of the gels and solutions at different pH are provided in the ESI† (see ESI Fig. S5†). The viscosity values shown in Fig. 4b were determined with steady shear rheology measurements (see ESI Fig. S6†). Considering the pKa value for the imidazole unit of histidine (pKa = 6.0 for free histidine), it is reasonable to assume that the pH response of the gel originates primarily from the imidazole unit. We suggest that the peptide is zwitterionic at pH 6 (protonated imidazole and deprotonated C-terminus), whereas it becomes increasingly cationic at lower pH (pronated imidazole and C-terminus) and increasingly anionic at higher pH (deprotonated imidazole and C-terminus). It should be noted that above pH 8 the base labile Fmoc protection group is eliminated followed by a degradation of the hydrogel.11 As a consequence the peptide dissolves, while the Fmoc-group precipitates from solution (see ESI Fig. S5†).
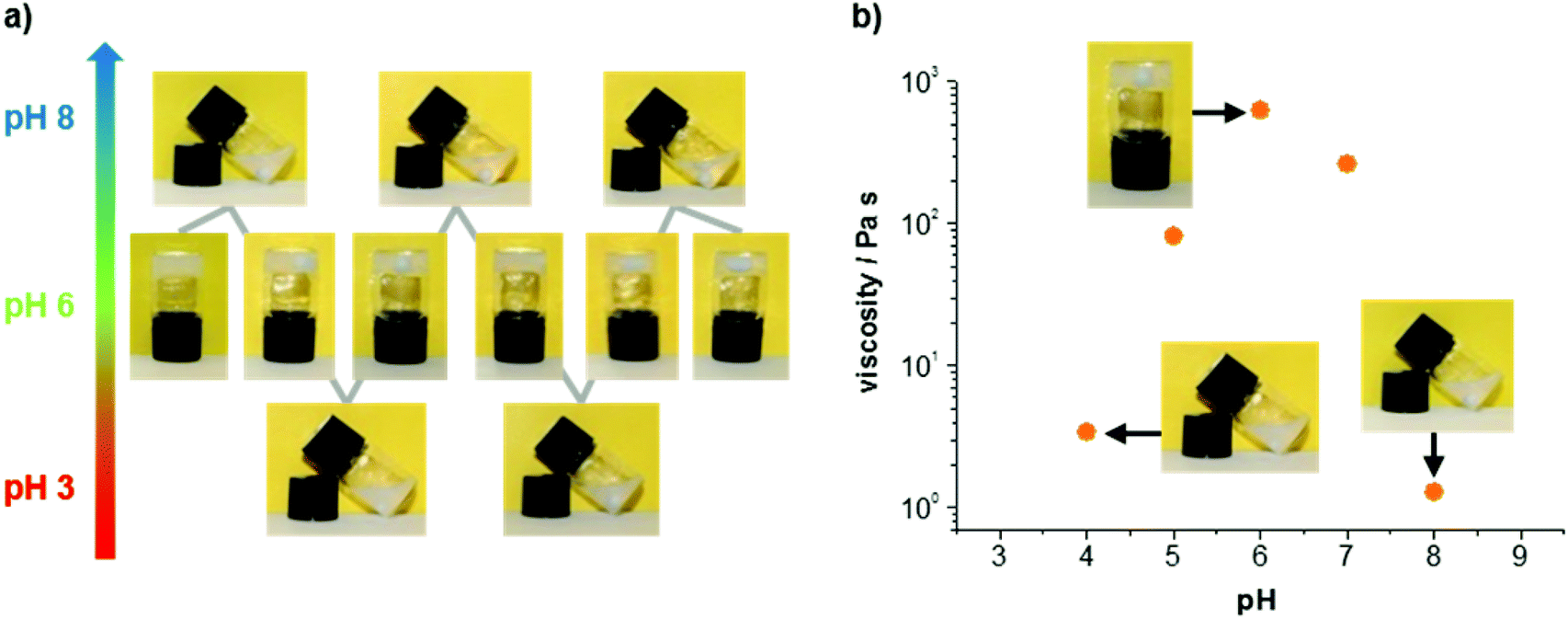 | ||
| Fig. 4 Visualization of the pH response of the hydrogel: (a) Cycles of gel formation due to pH stimuli; (b) pH response of the gel viscosity. The gel was prepared with 1.5 wt% 1 and the pH was adjusted with aqueous NaOH and trifluoroacetic acid. The viscosity was determined with steady shear rheology measurements. | ||
It should be emphasized that although hydrogels are only obtained for rather high concentrations of peptide 1 in water at pH 6, the peptide also forms supramolecular aggregates at much lower concentrations. In fact, the stability of the peptide assemblies under diluted conditions was demonstrated with circular dichroism spectroscopy. A pronounced circular dichroism characteristic for peptide self-assembly could be clearly observed even for concentrations down to 0.005 wt% (see ESI Fig. S7†). For morphological investigations atomic force microscopy (AFM) and small angle X-ray scattering (SAXS) measurements were performed. The scattering profiles of the SAXS experiments show a correlation peak at q = 0.23 Å−1, which is indicative for the presence of a molecular bilayer with a thickness of 2.8 nm (see ESI Fig. S8†). Based on results from Janmey12 and Ulijn,6c,d,10,13 Fmoc protection groups build up π–π-interactions in aqueous solutions leading to an overlapping of these units within the assemblies. For monomer 1 this implies a mean value of around 3 nm for the bilayer thickness, consistent with the SAXS data. Fig. 5 shows AFM images of diluted samples of the hydrogel with 0.01 wt% at pH 4 and pH 6 deposited on silicon wafers. In both cases, rod like assemblies were observed, which were up to 2 μm long. The height profiles of regions selected reveal rod widths of approximately 50 nm and multiple height steps, each 3 nm in height. The assembly into higher, stacked structures confirms the remarkable stability of the peptide assemblies even under dilute conditions. When comparing the samples at different pH values, it is observed that the assembly distribution varies drastically. While entangled assemblies are found at pH 6, only isolated assemblies are observed at pH 4, which is in good agreement with the macroscopic gelation behavior at pH 6. We propose that the entanglement of the assemblies is higher for the zwitterionic form of the peptide at pH 6 and decreases upon (partial) protonation at pH 4.
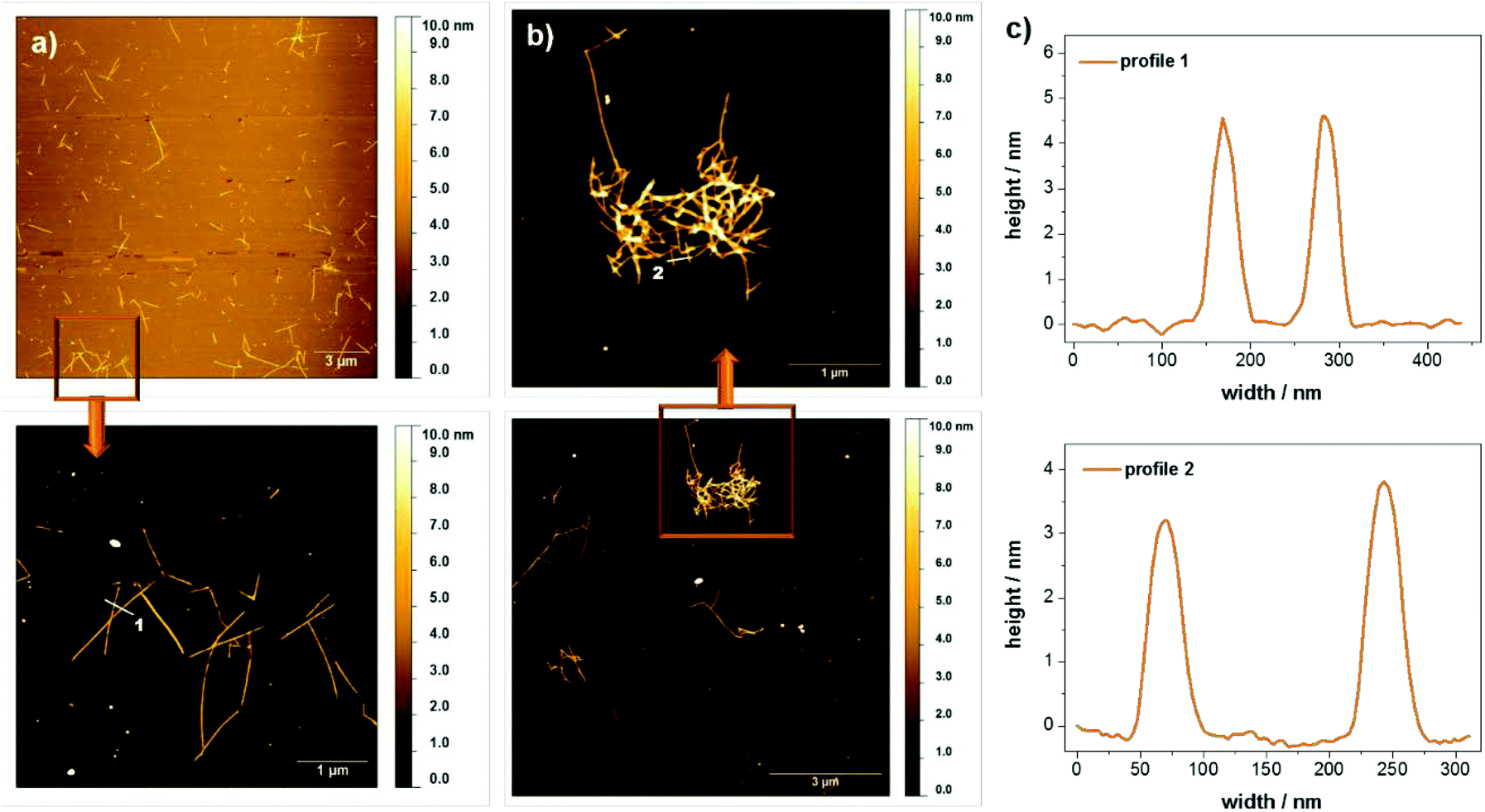 | ||
| Fig. 5 AFM images of 100-fold diluted hydrogel with 1.5 wt% 1 at (a) pH 4 and (b) pH 6 show rod like structures which assemble in a different manner. (c) The height profiles 1 and 2 were marked within the zoom images. Silicon wafers were used as substrate. | ||
The free C-terminal thiol function of peptide 1 is prone to oxidation under air exposure. In that case, it would be expected that disulfide formation between two monomers of 1 occurs at pH 8, resulting in an ultrastable, densely cross-linked hydrogel since not only two monomers in the same assembly may form an intra-assembly disulfide bond, but also two different assemblies can form inter-assembly disulfide bonds. AFM images of a sample of oxidized material confirm a highly cross-linked network of peptide assemblies (Fig. 6).
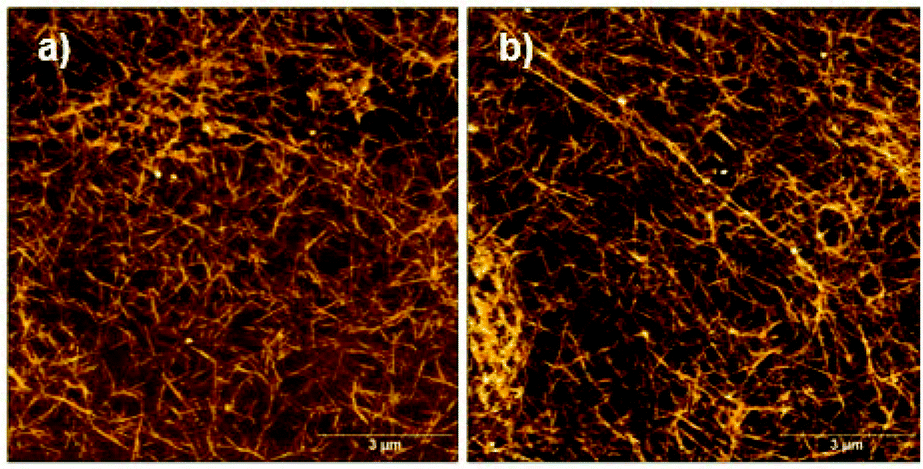 | ||
| Fig. 6 AFM images of samples with 0.01 wt% 1 at pH 8 after prolonged exposure to air. | ||
Molecular dynamics (MD) simulations of peptides 1 and 2 within the proposed assembly were performed to verify the importance of the Acm protection group concerning the gelation behavior of peptide 1. The calculated average bilayer thicknesses of 3.144 nm for 1 and 2.968 nm for 2 (Fig. 7) are in good agreement with the previously discussed morphologies of the assemblies of 1. Considering that the outer monomers lack neighbors, because the simulation only represents a fraction of a larger system, the assembly of 1 holds its shape as a bilayer during the time scale of the calculation (Fig. 8a), whereas the assembly of 2 falls apart from the initial structure to form a disordered conglomerate (Fig. 8b). However, the peptides behave like amphiphiles and arrange with the hydrophilic part facing water, while the hydrophobic Fmoc-protection groups overlap at the inner part of the bilayer.
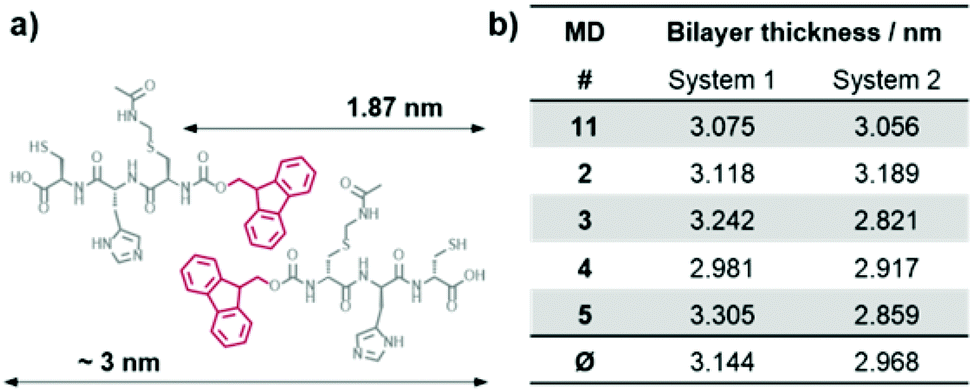 | ||
| Fig. 7 (a) Schematic representation of the estimated membrane thickness from the calculated length of peptide 1 taking into account overlapping Fmoc-protection groups in the core of the bilayer. (b) Average thickness of the bilayer models of peptide 1 and 2, calculated from 100 evenly distributed snapshots for each independent 5 ns MD run. | ||
 | ||
| Fig. 8 Selected snapshots from the MD simulations of the three decameric structures of 1 and 2. (a) Assembly of peptide 1 at around 3.6 ns with an average bilayer thickness of 3.144 nm. (b) Assembly of peptide 2 at around 3.6 ns. Water is omitted from this figure for clarity. | ||
Furthermore, the tendency for the Acm protected peptide 1 to build up supramolecular assemblies, in comparison to peptide 2 lacking the Acm protection group, was confirmed by a higher aggregation propensity (see ESI Fig. S9†). The aggregation propensity is calculated as the sum of the solvent accessible surface areas of all individual monomers divided by the solvent accessible surface area of the assembly. Therefore an increased aggregation propensity indicates aggregation while a value of 1 indicates the diffusion of free monomers. The aggregation propensity has previously been used by Tuttle and co-workers to evaluate the aggregation of dipeptides.14 Because our simulations started from a tight assembly, peptide 2 reveals an aggregation propensity not much lower than that of peptide 1, but the increasing trajectory of peptide 1 shows a trend to an even higher aggregation, while peptide 2 does not.
In the final part of this paper, we demonstrate that it is possible to use the hydrogel as a supramolecular scaffold of a low molecular weight peptide that offers addressable functions for molecular recognition. Functionalization of peptide 1 with adamantane to give 1-Ad was easily achieved via thiol–ene click chemistry using an alkene-terminated adamantane Ad-en (Scheme 1). The inclusion of adamantane into the cavity of β-cyclodextrin (CD) is well studied and has previously been applied in soft supramolecular nanomaterials.9,15 The affinity and selectivity (binding constant Ka ∼ 104 M−1) of this particular host–guest system are suitable to mediate the incorporation of β-cyclodextrin vesicles (CDV) in the peptide scaffold.
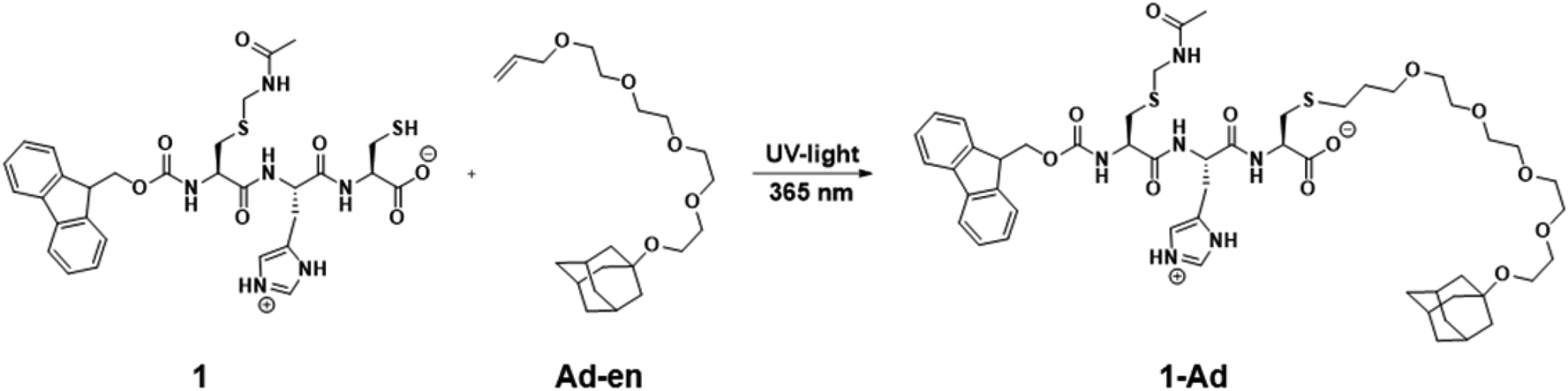 | ||
| Scheme 1 Functionalization of peptide 1 with adamantane via thiol–ene click reaction. Conditions: DMPA, acetonitrile, UV-light irradiation λ = 365 nm, 6 h, RT. | ||
The functionalization of the scaffold with adamantane side groups for host–guest inclusion complexation with CDV is illustrated in Fig. 9. A functionalized hydrogel was prepared simply by mixing peptide 1 with adamantane functionalized peptide 1-Ad in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. When a diluted, viscous solution of the adamantane functionalized gel with 1.0 wt% total peptide is mixed with a dispersion of CDV (10 mM, 120 μL) a hydrogel is obtained, whereas no gel is formed with control samples prepared from unfunctionalized 1 and CDV at 1.0 wt% (see ESI Fig. S10†). Since the adamantane on the peptide and the CD at the surface of the CDV form strong host–guest inclusion complexes, it can be assumed that the CDV act as supramolecular gel stabilizer due to the formation of multiple additional non-covalent cross-links of the peptide aggregates. This is in agreement with our previously reported polymer-based system, where CDV were incorporated into a gel network of guest polymers and host vesicles.9 To the best of our knowledge, this is the first example of a supramolecular small peptide scaffold in aqueous solution that bears reactive groups that are accessible for one step functionalization resulting in tunable properties of the assembly.
:1 molar ratio. When a diluted, viscous solution of the adamantane functionalized gel with 1.0 wt% total peptide is mixed with a dispersion of CDV (10 mM, 120 μL) a hydrogel is obtained, whereas no gel is formed with control samples prepared from unfunctionalized 1 and CDV at 1.0 wt% (see ESI Fig. S10†). Since the adamantane on the peptide and the CD at the surface of the CDV form strong host–guest inclusion complexes, it can be assumed that the CDV act as supramolecular gel stabilizer due to the formation of multiple additional non-covalent cross-links of the peptide aggregates. This is in agreement with our previously reported polymer-based system, where CDV were incorporated into a gel network of guest polymers and host vesicles.9 To the best of our knowledge, this is the first example of a supramolecular small peptide scaffold in aqueous solution that bears reactive groups that are accessible for one step functionalization resulting in tunable properties of the assembly.
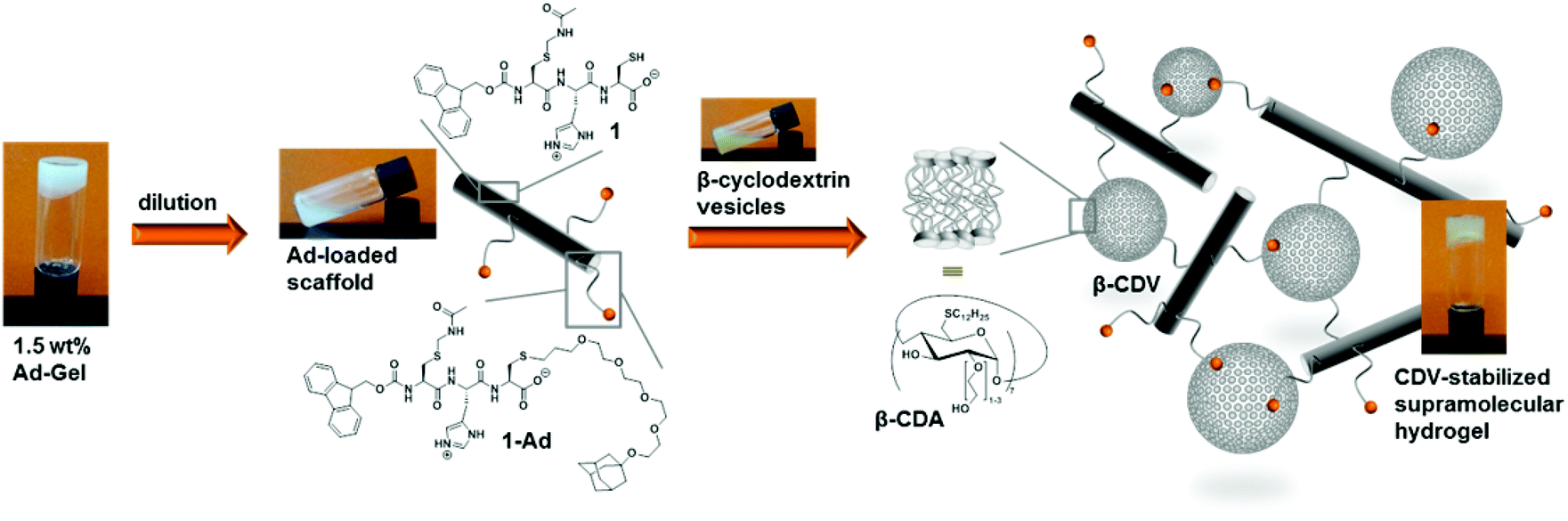 | ||
| Fig. 9 Formation of a stabilized supramolecular hydrogel of adamantane functionalized peptide scaffolds (1:1-Ad mixed 9:1 and cyclodextrin vesicles (CDV) of amphiphilic β-cyclodextrin host molecules (β-CDA) via multivalent host–guest inclusion complexes at pH 6 (final concentrations are 0.75 wt% of 1 and 2 mM (0.6 wt%) CDV). | ||
Response of the hydrogels towards competitive host and guest clearly demonstrates that the gel formation is due to specific interaction of the adamantane groups of the peptide scaffold and the CD cavities exposed on the CDV: Addition of an excess of competitive host (β-CD) or guest (1-adamantylamine hydrochloride) to the gel caused the disruption of the gel network as the interactions of the scaffold with the vesicles were inhibited (see ESI Fig. S11†). In either case, solutions of lower viscosity were obtained. These results clearly confirm the highly specific functionalization of the scaffold with adamantane as model for molecular recognition processes within the hydrogel.
Furthermore, the stability of the self-assembled peptide scaffold against dilution offers an alternative opportunity to use the peptide as a readily functionalized template in even more dilute samples. The interaction between the peptide scaffold (1:1-Ad mixed 9:1) and CDV under dilute conditions was monitored with optical density measurements. To this end, a solution of the adamantane functionalized peptide scaffold (10 μL, 1.5 wt%) was added to a solution of CDV (1 mL, [CDA] = 50 μM). Molecular recognition of the adamantanes on the peptide and the CD cavities on the vesicles induce a strong increase in the optical density (ca. 0.45 in 60 min) due to the formation of host–guest complexes resulting in vesicle aggregation (Fig. 10a). The minor increase of the optical density of the control sample (0.1 directly upon mixing) of non-functionalized peptide is attributed to the scattering of the scaffold itself. The size of the obtained aggregates was measured with dynamic light scattering (Fig. 10b). A 50 μM solution of CDV shows an average vesicle size of around 100 nm. The average size of the vesicles remains unchanged when the non-functionalized peptide scaffold and the CDV are mixed. In contrast, the apparent size the CDV after addition of the adamantane functionalized peptide scaffold shows is much larger, i.e. around 6000 nm. Thus, both the optical density and the light scattering measurements confirm molecular recognition of the adamantane-functionalized peptide scaffold and the CDV.
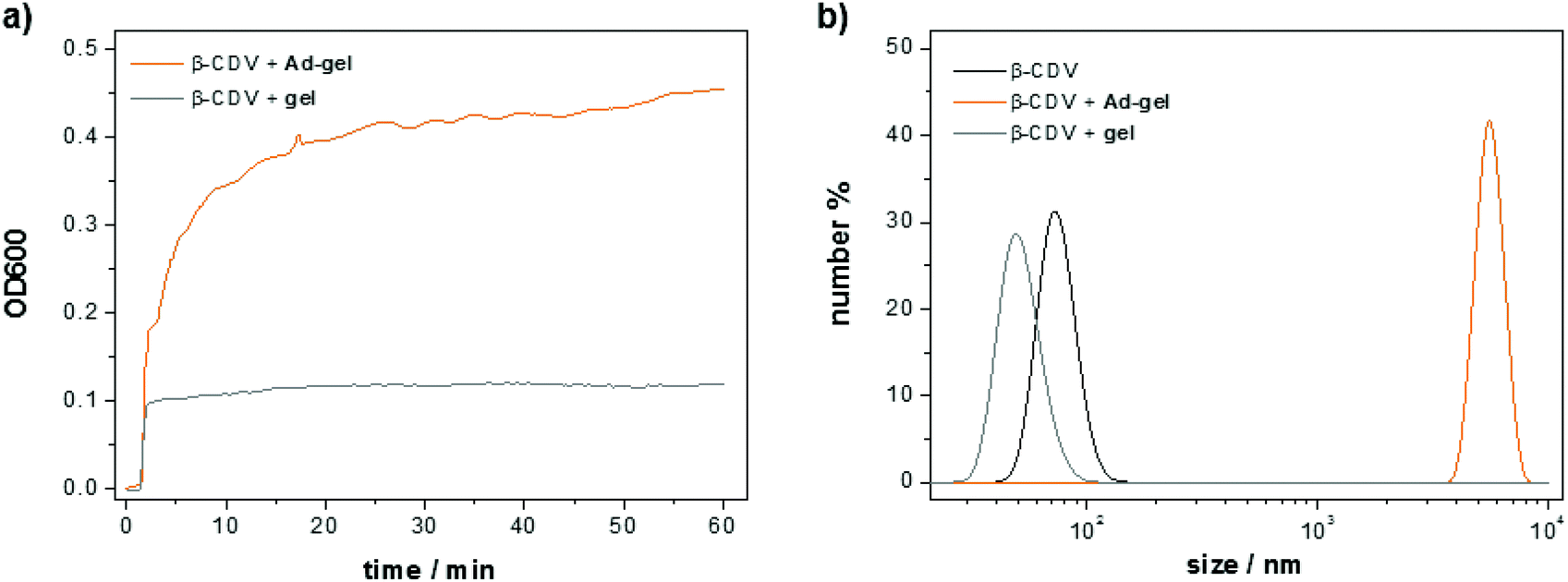 | ||
| Fig. 10 Peptide scaffold mediated vesicle aggregation: (a) optical density at 600 nm of two samples of CDV (50 μM or 0.0015 wt%) after addition of 10 μL gel of peptide 1 and gel of peptide 1:peptide 1-Ad (mixed 9:1), respectively, each at 1.5 wt% (final concentration of peptide: 0.015 wt%). (b) Dynamic light scattering of bare CDV and both samples of the OD600 measurement. | ||
Conclusion
In summary we demonstrated that the tripeptide Fmoc-L-(Acm)Cys-L-His-L-Cys 1 self-assembles into supramolecular bilayer nanostructures, which entangle into a pH responsive gel network above 1.5 wt% in water with characteristic rheomechanical properties. Furthermore, peptide 1 was successfully functionalized with adamantane guest units via thiol–ene-click chemistry for molecular recognition with cyclodextrin vesicles. To the best of our knowledge, this is the first example of a supramolecular tripeptide scaffold in aqueous solution that bears reactive groups that are accessible for one step functionalization resulting in tunable properties of the entire assembly.Experimental section
Materials and methods
All side chain and Fmoc protected amino acids used for SPPS were purchased from Novabiochem (Merck Biosciences AG, Läufelfingen, Switzerland), all other chemicals from Sigma Aldrich (Taufkirchen, Germany) or Acros Organics (Schwerte, Germany) were used without further purification. All solvents were dried according to the conventional methods before use. DMF for SPPS had peptide synthesis grade. All reactions were carried out in oven-dried flasks and were stirred magnetically. Purification of final compounds was achieved with a preparative HPLC system using a semipreparative HPLC column. NMR spectra were recorded on a 400 MHz spectrometer. Mass spectra were recorded in the ESI mode. IR spectra were recorded on a 3100 FT-IR equipped with a MKII Golden Gate Single Reflection ATR unit. Ultrapure water was used in all analytical measurements and for gel preparations. AFM samples were prepared from a 100-fold diluted sample of the hydrogel with 1.5 wt%. A drop of the solution was air-dried on oxidized silicon surfaces and analyzed. For rheology and SAXS experiments standard hydrogel samples with 1.5 wt% 1 were used. OD600 measurements were performed on a double beam UV-spectrometer using a constant wavelength of λ = 600 nm and PMMA cuvettes. To a solution of CDV [50 μM], 10 μL of the hydrogel with 1.5 wt% of peptide 1 or the mixture 1 with 10% 1-Ad was added.Standard operation procedures for SPPS
Loading of the resin (SOP 1) Loading of the resin was performed according to literature under an atmosphere of argon.16 The amino acid (2 eq. relative to resin loading) is dissolved in dry DCM containing a small amount of DMF (solvent grade) and added to 2-chlorotrityl resin (loading 1.5 mmol g−1 resin). After addition of DIPEA (2 eq. relative to resin loading) the mixture is agitated by a slow stream of argon for 5 min. Following another addition of DIPEA (3 eq. relative to resin loading), agitation is continued for 1 h. The remaining reactive groups of the resin are quenched with MeOH p. a. (1 mL g−1 resin) for 15 min. After filtration of the resin it was washed consecutively with DCM p. a. (2 × 20 mL), DMF SPPS s. g. (3 × 20 mL), DCM p. a. (3 × 20 mL) and MeOH p. a. (3 × 20 mL) and dried in high vacuum until a constant weight is reached. Step-wise chain elongation via SPPS (SOP 2) SPPS was performed according to literature.17 Dry beads prepared as described in SOP 1 were swelled in DMF p.a. for 45 min while shaking the reaction vessel. After sucking of the DMF p.a., the Fmoc-group is cleaved with 20% piperidine in DMF for 20 min. The resin is washed with DMF p.a. (4 times) and alternately with DCM p.a. (3 times) and isopropanol p.a. (3 times) to fully remove the piperidine. Success of the cleavage was controlled by the Kaiser test. Equal amounts of a 5% ninhydrin solution in EtOH, 80% phenole in EtOH and 0.001 M KCN in pyridine were mixed with a few washed beads and heated for 1 min to 100 °C. A blue color of the beads indicated free amine functions on the resin.18 The coupling step was performed by suspending the resin in a solution of the according Fmoc-protected amino acid (3 eq.) and Oxyma pure® (4.0 eq.) in DMF (SPPS grade). After addition of DIPCDI (3.0 eq.), the suspension is shaken for 2 h and then the solvent is filtered off. Washing of the beads with DMF (SPPS grade, 4 times) was followed by the Kaiser test, which should not result in a color change if the coupling was successful. The deprotection and coupling steps were continued until the desired peptide sequence was obtained. Cleavage of resin and removal of permanent protection groups (SOP 3) Cleavage of the resin and removal of permanent protection groups was performed according to literature.[see SOP1/2] Prior to splitting of the peptide from the resin, the N terminal Fmoc-group has to be removed by treatment with 20% piperidine in DMF for 20 min. After washing with DMF p.a. (4 times) and alternately with DCM (2 times) and isopropanol p.a. (2 times), the beads were suspended in a solution of TFA:H2O:EDT:TIS = 94:2.5:2.5:1 and stirred for 8 h at r.t.. After filtration, the beads were washed with TFA (5 times) and the filtrate was concentrated until the peptide started to precipitate. Addition of cold Et2O resulted in complete precipitation and the suspension was kept overnight in the freezer. The peptide was collected by filtration, washed with Et2O and dried in high vacuum to yield the peptides as white, hygroscopic crude products, which were stored under argon. Purification by preparative HPLC (SOP 4) Purification of the crude peptides were achieved by reversed phase preparative HPLC using a gradient of 95% to 20% A within 45 min. MALDI analysis of collected fractions determines the product and analytical HPLC confirmed the purity of each fraction before they were combined. Freeze drying provided the desired products as fluffy foams.
Synthesis of 1-Ad
The synthesis of adamantine-functionalized gelator 1-Ad was achieved in three steps. Alkene functionalized adamantane Ad-TEG-en (obtained in two steps) was coupled in a thiol–ene-click reaction in situ to the free thiol of peptide 1.Preparation of hydrogels
Hydrogel samples were prepared by stirring appropriate amounts of freshly grounded 1 in distilled water at 40 °C over night in a closed container. Cooling of the samples to room temperature resulted in homogeneous hydrogels. The samples were used for a maximum of one week to avoid cross-linking due to disulfide formation. For the adamantane functionalized gels 10 mol% of 1-Ad and 90 mol% of 1 were used instead of only 1. The Ad-gel was prepared according to the described procedure for the unfunctionalized hydrogel. For the incorporation of CDV in the peptide hydrogel, a 10 mM solution of CDV was prepared according to literature13 and 120 μL of this solution was added to a 1.0 wt% viscous Ad-gel sample. The solutions were mixed and left for at least 2 h to provide a homogeneous hydrogel.Molecular dynamics simulations
The initial monomer geometries of 1 and 2 were generated by geometry optimization with MOPAC201220 at the PM721 level of theory. The systems were setup as three neighboring decameric stacks with each alternating peptide monomer being twisted by 180 degrees, only the central decameric stack was used in the collection of statistics. Each of the systems was solvated in a 4 nm sphere of TIP3 water. The solvent was relaxed (solute was constrained) with a 20 ps MD simulation and the solvation procedure was repeated several times to fill in any intermediate voids that had appeared. The solvated systems were then calculated for 250 ps equilibration, followed by 5 independent MD simulations of 5 ns each. All molecular dynamics simulations were carried out using CHARMM Version c35b3.22Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft DFG (SFB858) is gratefully acknowledged. We thank the European Synchrotron Radiation Facility in Grenoble (France) for the beam time at the high-brilliance beamline ID02. The authors are grateful for the assistance from Dr G. Lotze and Dr T. Narayanan in Grenoble as well as for the help with measurements and data analysis by Dr P. Besenius, Dr Sabine Himmelein and Ms S. Kumbhar. This work was supported by COST CM1005 “Supramolecular Chemistry in Water”.Notes and references
- (a) B. Balakrishnan and R. Banerjee, Chem. Rev., 2011, 111, 4453–4474 CrossRef CAS PubMed; (b) S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 122, 7623–7626 CrossRef; (c) E. A. Appel, X. J. Loh, S. T. Jones, F. Biedermann, C. A. Dreiss and O. A. Scherman, J. Am. Chem. Soc., 2012, 134, 11767–11773 CrossRef CAS PubMed; (d) Y. Yan, A. de Keizer, A. A. Martens, C. Luis Pinto Oliveira, J. Skov Pedersen, F. A. de Wolf, M. Drechsler, M. A. Cohen Stuart and N. A. M. Besseling, Langmuir, 2009, 25, 12899–12908 CrossRef CAS PubMed; (e) E. A. Appel, J. del Barrio, X. Jun Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- (a) J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed; (b) J. H. Jung, J. A. Rim, W. S. Han, S. J. Lee, Y. J. Lee, E. J. Cho, J. S. Kim, Q. Ji and T. Shimizu, Org. Biomol. Chem., 2006, 4, 2033–1038 RSC; (c) A. Saha, B. Roy, A. Esterrani and A. K. Nandi, Org. Biomol. Chem., 2011, 9, 770–776 RSC; (d) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed; (e) M. Ikeda, Bull. Chem. Soc. Jpn., 2013, 86(1), 10–24 CrossRef CAS.
- M. G. Ryadnov and D. N. Woolfson, J. Am. Chem. Soc., 2004, 126, 7454–7455 CrossRef CAS PubMed.
- A. Laromaine, L. Koh, M. Murugesan, R. V. Ulijn and M. M. Stevens, J. Am. Chem. Soc., 2007, 129, 4156–4157 CrossRef CAS PubMed.
- H. Jones, J. Dalmaris, M. Wright, J. H. G. Steinke and A. D. Miller, Org. Biomol. Chem., 2006, 4, 2568–22574 CAS.
- (a) Y. Zhang, H. Gu, Z. Yang and B. Xu, J. Am. Chem. Soc., 2003, 125, 13680–13681 CrossRef CAS PubMed; (b) Z. Yang, H. Gu, Y. Zhang, L. Wang and B. Xu, Chem. Commun., 2004, 208–209 RSC; (c) V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS; (d) A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS; (e) J. W. Sadownik and R. V. Ulijn, Chem. Commun., 2010, 46, 3481–3483 RSC; (f) X.-D. Xu, C.-S. Chen, B. Lu, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, J. Phys. Chem. B, 2010, 114, 2365–2372 CrossRef CAS PubMed; (g) A. Brizard, M. Stuart, K. van Bommel, A. Friggeri, M. de Jong and J. van Esch, Angew. Chem., Int. Ed., 2008, 47, 2063–2066 CrossRef CAS PubMed; (h) G. Cheng, V. Castelletto, C. M. Moulton, G. E. Newby and I. W. Hamley, Langmuir, 2010, 26, 4990–4998 CrossRef CAS PubMed; (i) D. M. Ryan and B. L. Nilsson, Polym. Chem., 2012, 3, 18–33 RSC; (j) H. Frisch, J. P. Unsleber, D. Lüdeker, M. Peterlechner, G. Brunklaus, M. Waller and P. Besenius, Angew. Chem., Int. Ed., 2013, 52, 10097–10101 CrossRef CAS PubMed; (k) S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- (a) B. Rybtchinski, ACS Nano, 2011, 5, 6791–6818 CrossRef CAS PubMed; (b) T. Fenske, H. G. Korth, A. Mohr and C. Schmuck, Chem. – Eur. J., 2012, 18, 738–755 CrossRef CAS PubMed.
- (a) M. Rauschenberg, S. Bandaru, M. P. Waller and B. J. Ravoo, Chem. – Eur. J., 2014, 20, 2770–2782 CrossRef CAS PubMed; (b) M. Rauschenberg, S. Bomke, U. Karst and B. J. Ravoo, Angew. Chem., Int. Ed., 2010, 49, 7340–7345 CrossRef CAS PubMed; (c) M. Rauschenberg, E. C. Fritz, C. Schulz, T. Kaufmann and B. J. Ravoo, Beilstein J. Org. Chem., 2014, 10, 1354–1364 CrossRef CAS PubMed.
- S. Himmelein, V. Lewe, M. C. A. Stuart and B. J. Ravoo, Chem. Sci., 2014, 5, 1054–1058 RSC.
- (a) S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS PubMed; (b) R. Orbach, I. Mironi-Harpaz, L. Adler-Abramovich, E. Mossou, E. P. Mitchell, V. T. Forsyth, E. Gazit and D. Seliktar, Langmuir, 2012, 28, 2015–2022 CrossRef CAS PubMed; (c) I. H. Lin, L. S. Birchall, N. Hodson, R. V. Ulijn and S. J. Webb, Soft Matter, 2013, 9, 1188–1193 RSC.
- G. B. Fields, Methods in Molecular Biology, Peptide Synthesis Protocols, Chapter 2: Methods for removing the Fmoc group, ed. M. W. Pennington and B. M. Dunn, Humana Press Inc., Totowa, NJ, 1994, vol. 35, p. 19 Search PubMed.
- R. Vegners, I. Shestakova, I. Kalvinsh, R. M. Ezzell and P. A. Janmey, J. Pept. Sci., 1995, 1, 371–378 CrossRef CAS PubMed.
- (a) A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS; (b) H. Xu, A. K. Das, M. Horie, M. S. Shaik, A. M. Smith, Y. Luo, R. Collins, S. Y. Liem, A. Song, P. L. A. Popelier, M. L. Turner, P. Xiao, I. A. Kinloch and R. V. Ulijn, Nanoscale, 2010, 2, 960–966 RSC.
- P. W. J. M. Frederix, R. V. Ulijn, N. T. Hunt and T. Tuttle, J. Phys. Chem. Lett., 2011, 2, 2380–2384 CrossRef CAS PubMed.
- (a) J. Voskuhl, M. C. A. Stuart and B. J. Ravoo, Chem. – Eur. J., 2010, 16, 2790–2796 CrossRef CAS PubMed; (b) U. Kauscher and B. J. Ravoo, Beilstein J. Org. Chem., 2012, 8, 1543–1551 CrossRef CAS PubMed; (c) F. Versluis, J. Voskuhl, M. C. A. Stuart, J. B. Bultema, S. Kehr, B. J. Ravoo and A. Kros, Soft Matter, 2012, 8, 8770–8777 RSC; (d) J. Voskuhl, T. Fenske, M. C. A. Stuart, B. Wibbeling, C. Schmuck and B. J. Ravoo, Chem. – Eur. J., 2010, 16, 8300–8306 CrossRef CAS PubMed.
- (a) K. Barlos, D. Gatos, J. Kallitsis, G. Papaphotiu, P. Sotiriu, Y. Wenqing and W. Schafer, Tetrahedron Lett., 1989, 30, 3943 CrossRef CAS; (b) K. Barlos, O. Chatzi, D. Gatos and G. Stavropoulos, Int. J. Pept. Protein Res., 1991, 37, 513 CAS; (c) R. Bollhagen, M. Schmiedberger, K. Barlos and E. Grell, J. Chem. Soc., Chem. Commun., 1994, 2559 RSC.
- L. A. Carpino and A. El-Faham, Tetrahedron, 1999, 55, 6813 CrossRef CAS.
- E. Kaiser, R. L. Colescott, C. D. Bossinger and P. I. Cook, Anal. Biochem., 1970, 34, 595 CrossRef CAS.
- A. Mulder, T. Auletta, A. Sartori, S. Del Ciotto, A. Casnati, R. Ungaro, J. Huskens and D. N. Reinhoudt, J. Am. Chem. Soc., 2004, 126, 6627–6636 CrossRef CAS PubMed.
- J. J. P. Stewart, MOPAC2012. Stewart Computational Chemistry Colorado Springs CO, 2012 Search PubMed.
- J. Stewart, J. Mol. Model, 2013, 19, 1–32 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, D. J. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob02069c |
| This journal is © The Royal Society of Chemistry 2015 |