
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of maculalactone A and derivatives for environmental fate tracking studies†
Samuel L.
Bader
,
Michael U.
Luescher
and
Karl
Gademann
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: karl.gademann@unibas.ch
First published on 20th October 2014
Abstract
Maculalactone A (1) constitutes a promising antifouling agent, inhibiting the formation of biofilms in marine and freshwater systems. In this study, we developed a new route, based on a late-stage formation of the butenolide core, leading to the total synthesis of maculalactone A (three steps, overall yield of 45%) and delivering material on a gram scale. In addition, analogues of the title compound were assayed concerning their biological activity, utilizing Artemia franciscana and Thamnocephalus platyurus. The most active analogue was functionalized with a rhodamine B fluorophore and was utilized in an in vivo staining experiment in Artemia salina. Two different tissues were found to accumulate this maculalactone A derivative.
Introduction
The accumulation of biological material on all surfaces immersed in biological fluids (referred to as biofouling) is a serious challenge for marine vessels, fresh water treatment and industrial plants.1–5 The ban of previously employed tributyltin coatings by international organizations further aggravated this issue.6 A bio-inspired approach to address this problem constitutes the use of natural products as antifouling agents.7,8 In recent years, marine butenolide natural products attracted interest, as they show good antifouling activity and quorum-sensing inhibition.9–12 In the context of our own research, we have developed several natural product based approaches to prevent biofouling, ranging from novel algicidal natural products,13–15 passivated metal oxide surfaces with antifouling properties16–18 to active surfaces, functionalized with natural products, for controlling or inhibiting bacterial growth.19,20The slowly growing cyanobacterium, Kyrtuthrix maculans, forms pure colonies on rocky shores, which, by the absence of other organisms in these colonies, suggests that some sort of chemical defence could be operating. Based on these observations, the Brown group isolated the maculalactones, a family of interesting secondary metabolites, from this cyanobacterium (Fig. 1). While maculalactone A (1), B, C and L consist of a benzylated γ-lactone, maculalactone D, E, F (2), G, H, I, J and K (3) include an additional phenylpropanoid unit forming a six membered carbocycle.21–24 Maculalactone A constitutes the most abundant congener in this series found in K. maculans. In addition, Brown et al. conducted preliminary antifouling experiments by submerging structures coated with a maculalactone A containing paint in the sea.25 After 12 weeks, a reduced area coverage with bivalves was observed. Although this reduction was partially compensated by an increased growth of algae, it demonstrated the potential of 1 as an antifouling agent. However, little is known about the structural requirements or the mode of action of maculalactone A (1). In addition, the isolation of larger amounts of 1 from the slow-growing producer is unpractical and the three published total syntheses offer overall yields below 10%.26–28 In this study, we have developed an efficient preparation of maculalactone A (1), performed a preliminary structure–activity relationship study and investigated the environmental fate of this compound in model crustaceans via labelled analogues.

Results and discussion
Improved total synthesis of maculalactone A
To access gram-scale amounts of maculalactone A (1) and its structural analogues, an improved synthesis had to be developed. The new route should not only result in a higher overall yield, but should also allow for the modification of the different benzyl groups. We first targeted an optimization of the published routes to maculalactone A.26–28 A survey of those revealed a general problem in the nucleophilic introduction of benzyl groups to the butenolide core. In a first phase, we tried to work around such a nucleophilic benzylation. Messorosh et al. recently described the radical benzylation of 2,3-dichloromaleic anhydride (4) in good yield.29 The resulting anhydride, 6, is a central intermediate in the synthesis shown by Brown et al. and Argade et al.26,27 However, the dichloride, 4, is expensive and difficult to prepare in the laboratory. The corresponding dibromide, 5, was synthesised30 and subjected to the same conditions, giving 6 in 28% yield (Scheme 1). Overall, this route led to no improvement when compared to the known syntheses of 1. | ||
| Scheme 1 Investigated shortcuts in the total syntheses shown by Brown et al. and Argade et al. The reaction from 4 to 6 was previously published by Messorosh et al.29 PTC = N-anthrylated quinine chloride, synthesised as described by Hofstetter et al.31 | ||
Alternatively, a shortcut in the route investigated by Argade27 used an electrophilic substitution at the γ-position of intermediate 7 (Scheme 1). An enantioselective phase-transfer catalyst mediated procedure was expected to give optically enriched maculalactone A. However, the benzyl group was directed to the α-position giving the achiral lactone 9.
Since an improvement of the previously published routes did not appear feasible, new strategies were considered. Therefore, we focused on strategies with a late stage butenolide formation, thereby, avoiding benzylic disconnections. Among the various known methods to synthesise butenolides, only few approaches allow substituents in all three positions.32–34
Alkene metathesis has been applied for butenolide syntheses by different groups.35–37 Diene 13 was therefore synthesised in a convergent fashion from the starting material 10, which was accessed as described by Pihko et al.38 (Scheme 2). The enal 10 was either converted to acid 11, applying a Pinnick oxidation, or benzylated in a 1,2-addition to obtain alcohol 12. The two parts were combined in a Steglich esterification, giving diene 13 in an overall yield of 42% and 38%, respectively. However, the subsequent metathesis reaction under optimized conditions and in the presence of Ti(OiPr)4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 37 resulted in a low conversion of 11%. Despite this disappointing result, this route allowed for the first time the modification of a single benzyl moiety.
37 resulted in a low conversion of 11%. Despite this disappointing result, this route allowed for the first time the modification of a single benzyl moiety.
 | ||
| Scheme 2 Synthesis of maculalactone A by a late stage ring closing metathesis. | ||
Another potentially rewarding strategy constitutes the late stage preparation of the olefinic bond through an elimination approach. For this goal, we investigated a procedure originally reported by Avetisyan et al.39 in 1981 and later utilized in an industrial setting.40 Key to this strategy is the intramolecular cyclization of 3,6-dioxo-4-oxa-1-hexanoate derivative, 17, with a subsequent loss of ethanol and CO2, furnishing the desired butenolide (Scheme 3). In order to test this hypothesis, malonate 14 was saponified to the half-ester, 15, in a single step41 and converted to the corresponding anhydride with pivaloyl chloride. In the same pot, the anhydride intermediate was reacted with the known α-hydroxy ketone, 16, which itself is accessible via a Benzoin-type Umpolung reaction,42 to the corresponding keto-diester, 17.
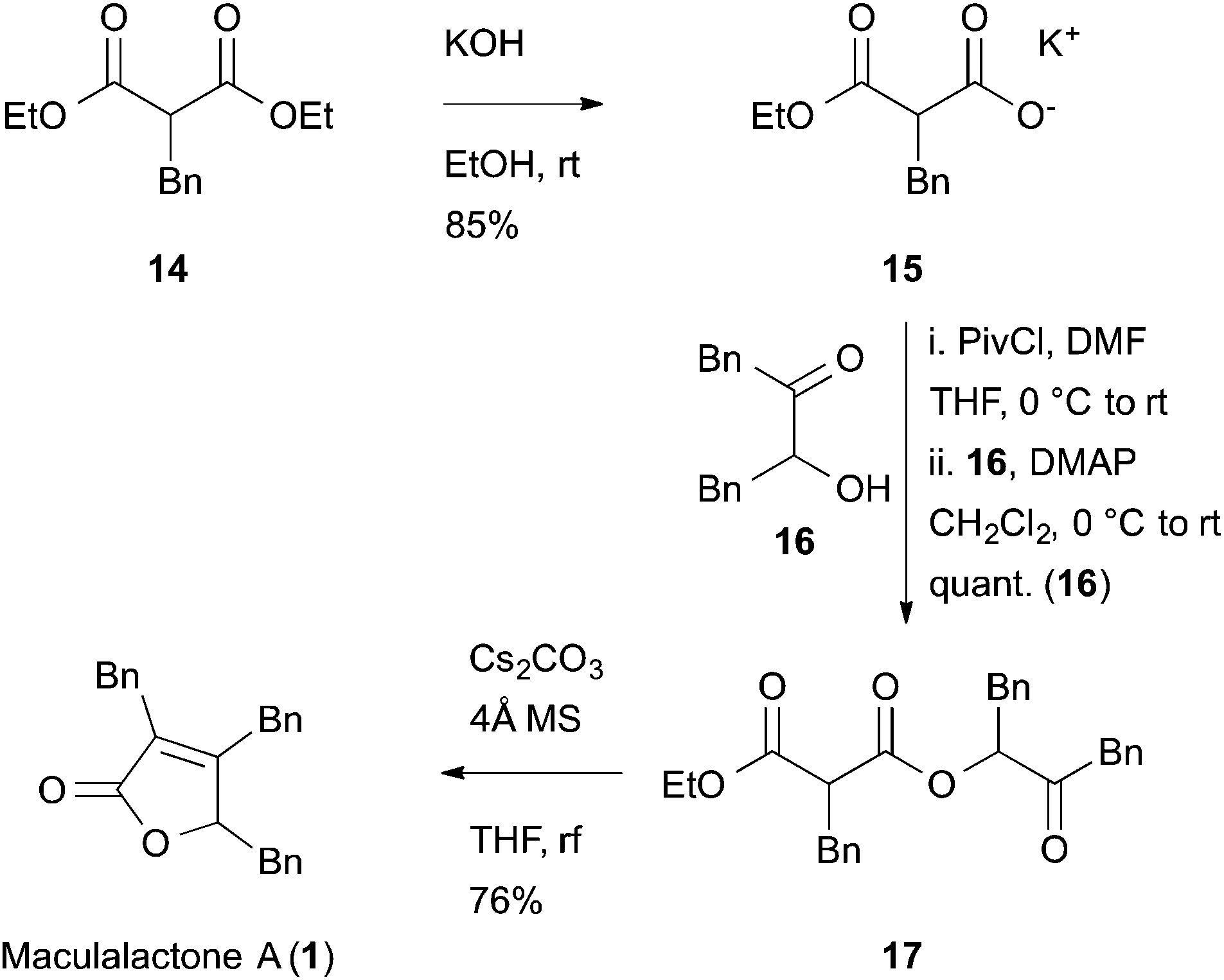 | ||
| Scheme 3 Synthesis of maculalactone A by a late stage butenolide formation. Synthesis of 15 from 14, as published by Goel et al.41 | ||
The key step, i.e. the late stage cyclization of 17 to butenolide 1, was addressed next. After optimizing the base, solvent and different additives, the cyclization was accomplished with caesium carbonate in refluxing THF, giving maculalactone A (1) in a yield of 76%. In order to drive the reaction to completion, and to prevent alcoholysis of 17 with the formed ethanol, the presence of 4 Å molecular sieves was found to be crucial. Interestingly, the intermolecular version of this reaction led to the formation of product 1, however, only little conversion could be observed.
We postulate the intramolecular attack of the deprotonated malonate to the ketone, followed by an intramolecular transesterification (liberation of EtOH) and formation of a β-lactone as a working hypothesis for this interesting reaction. Subsequent CO2 release would then result in maculalactone A (1). The mechanism was studied by a series of experiments; however, direct mass spectrometric detection of the intermediates was not possible. Interestingly, running the reaction in sealed tubes did not lead to product formation. Additional experiments will be necessary to study the mechanism of this transformation.
Based on this short approach, maculalactone A (1) was synthesised in an overall yield of 45% in three linear steps from the inexpensive, commercially available starting material, 14. In addition, this strategy allows for the differentiation of the benzyl groups via their sequential regioselective introduction. We took advantage of this approach to access analogues as described below.
Syntheses of maculalactone A derivatives and SAR studies
For further investigations of the mode of action, environmental fate studies and potential applications in antifouling surface coatings, we were interested in modified maculalactone A derivatives. First, however, the appropriate position for the modifications had to be identified in a structure–activity relationship study (SAR). We initially proposed the para-positions of the respective benzyl groups to have a minimal influence on the biological activity and, therefore, the three structural isomers, 25, 26 and 27, each bearing a methoxy group in the para-position, were synthesised (Scheme 4). Malonate, 18, was synthesised based on a reported procedure43 and converted to its potassium salt, 19, at reduced temperature to prevent decarboxylation. The differently substituted α-hydroxy ketones Bn–CO–CH(OH)–PMB (20) and PMB–CO–CH(OH)–Bn (21) were prepared from the corresponding aldehydes via formation of the TMS protected cyanohydrins, followed by the addition of the respective Grignard reagent, as described in the literature.44,45 The formation of ester, 22–24, proceeded smoothly, as did the ring closure (yields of 40 to 69% over two steps). | ||
| Scheme 4 Synthesis of first generation maculalactone A analogues, 25–27. PMB = para-methoxybenzyl. | ||
We were then interested in profiling the biological activity of maculalactone A and its analogues. In the work of Brown et al., the LC50 values of extracted maculalactone A were determined in ecotoxicity assays utilizing different stages of Tetraclita japonica, Balanus amphitrite, Ibla cumingii and Artemia salina,25 of which the latter is often used for reference toxicity experiments. For comparison, we evaluated the compounds in the related Artemia franciscana as well as the sensitive freshwater crustacean, Thamnocephalus platyurus, which is well-established in our group.46,47
The assays show a comparable (same order of magnitude) but slightly lower toxicity of the synthetic maculalactone A (1) in A. franciscana when compared to the report of Brown et al.25 This difference is either caused by racemic 1 or by differences in the assay set-up, and should not be of concern. Interestingly, compound 25, featuring the PMB group in the α-position, exhibits an increased toxicity in A. franciscana, whereas isomeric lactones 26 and 27 display lower or no toxic effects on the test organism (Fig. 2). In addition to the toxicity, the assays exposed a strong effect on the swimming ability of the test organisms. While fast movement is observed in both the blank and in a 1 μM maculalactone A solution, upon increased concentration (10 μM), the ability of the animals to swim in a controlled manner is strongly impaired (for a video, see the ESI†).
 | ||
| Fig. 2 Biological evaluation of the first generation maculalactone A analogues. Grey: A. franciscana, black: T. platyurus. Values are given as mean of three independent experiments with ten animals; error bars denote the standard error of the mean (SEM). | ||
As analogue 25 showed a similar or higher toxicity as the parent maculalactone A, it was used for further derivatisation studies. The methoxy group was quantitatively cleaved in the presence of boron tribromide to give phenol 28, which was coupled with different residues (Scheme 5). First, etherification with a triethylene glycol unit48,49 gave the short PEGylated lactone 29. After mesitylation of phenol 28, it was coupled with an octylborane derivative in a Suzuki reaction, affording compound 30. Furthermore, phenol 28 was added to the activated PEG azide 31. By a Click reaction, this intermediate was connected to the rhodamine B derivative 3250 giving the fluorescently labelled maculalactone A analogue, 33. Additionally, compound 34, with a free hydroxyl group instead of the maculalactone A moiety, was synthesised as a negative control. However, this compound proved to be unstable (amide hydrolysis), and, after repeated purification, could only be obtained in very low yield.
 | ||
| Scheme 5 Deprotection and modification of the most active maculalactone A analogue, 25. EtO(CH2CH2O)3Ts was prepared as described in the literature.48,49 Compound 32 was synthesised as described by Yan et al.50 | ||
Consequently, the PEGylated compound, 29, and the fluorescently labelled, rhodamine B derivative, 33, were subjected to the toxicity assay with A. franciscana (Fig. 3). Surprisingly, compound 29, with the short PEG residue, did not display any toxicity in this assay. However, the rhodamine B bearing analogue, 33, exhibited toxicity comparable to the one observed for 1. Therefore, this fluorescent analogue, 33, was used as a model to examine the distribution of maculalactone A in crustaceans.
 | ||
| Fig. 3 Toxicological tests of modified maculalactone A analogues using A. franciscana as test organism. Values are given as mean of three independent experiments with ten animals; error bars denote the standard error of the mean (SEM). | ||
Visualization of the maculalactone A distribution in Artemia salina
The media of A. franciscana was found to be incompatible with the subsequent visualization steps (fixation with formalin). Therefore, a more robust species of A. salina was employed for the in vivo experiments with the rhodamine B labelled maculalactone A derivative, 33.A marker concentration of 1 μM was found to be optimal, as no toxic effects were determined at this level. The two day old animals were incubated for 24 hours in 1 μM solutions of the marked maculalactone A, 33, the negative control, 34, rhodamine B and a blank solution. This was followed by a washing procedure were the crustaceans were twice transferred into fresh media and incubated for three hours. Finally, the larvae were fixed in a formalin solution in PBS buffer. The stained and fixed organisms were examined by fluorescence microscopy under identical lighting conditions (see the ESI† for the micrographs). While rhodamine B led to a general staining of the entire body, the negative control, 34, did not show any fluorescence at all. In contrast, the maculalactone A marker, 33, led to a well defined distribution in the animal with a selective staining of certain organs (Fig. 4). A highly stained rod-shaped tissue in the centre of the body is observed, which can be attributed to the intestine,51 and is sharply terminated towards the tail region.
 | ||
| Fig. 4 Overlay of four microscope pictures of A. salina nauplia, stained with compound 33 (visual and fluorescence images). Scale bar = 100 μm. | ||
Further support for this observation stems from the examination using a confocal microscope, which disclosed more details of this region. Slices through the animal (Fig. 5) reveal a tube-like structure, which is in agreement with the hypothesis that the intestine, but not its content, is stained by the marker, 33. Further evidence is presented by the well-defined localization of the fluorescently labelled natural product, 33, which can be rationalized by a selective staining and little or no diffusion. A similar, well-defined staining is observed towards the head region (see the ESI† for details). In summary, the evidence presented supports the notion that the labelled maculalactone A derivative is accumulated in the animal in well-defined tissue. However, as little is known about the anatomy of A. salina in larvae stage, a more detailed assignment of the involved tissues will require further experiments based on the preliminary successful use of the fluorescently labelled probe, 33.
 | ||
| Fig. 5 The tail region of Artemia salina nauplia stained with compound 33. Visual and fluorescence images; 10 μm slides in the z-axis; scale bar = 50 μm. | ||
Conclusions
In conclusion, we have developed a new and efficient route to maculalactone A (1) that delivered gram-amounts of material in three steps and 45% overall yield. The key step of the synthesis employed a late stage butenolide formation, involving an intramolecular malonate addition with subsequent extrusion of CO2. This route enabled the preparation of maculalactone analogues, 25, 26 and 27, of which their biological activity was compared in a toxicity assay using A. franciscana and T. platyurus. Significant activity was observed with compound 25, which was further modified. The rhodamine B labelled derivative, 33 proved to be the most interesting for tissue distribution and environmental fate studies in A. salina. Staining of specific tissues of the organisms supported the hypothesis that selective targets can be reached by maculalactone in the animal. However, further investigations are required to understand the distribution of maculalactone A (1) in living organisms, which should be enabled by the probe reported herein.Experimental
All chemicals have been purchased from Acros, Alfa Aesar, Fluka or Sigma-Aldrich and were used without further purification if not otherwise reported. Solvents applied for chemical transformations were either puriss quality or HPLC grade solvents. Dry solvents were purchased from Sigma-Aldrich. Technical grade solvents for extractions and chromatography were distilled before usage. Air or moisture sensitive reactions were set up in dry glassware under nitrogen atmosphere. All synthetic transformations have been monitored by either thin layer chromatography (TLC) or 1H-NMR spectroscopy. Yields refer to purified, dried and spectroscopically pure compounds. TLC was performed on Merck silica gel 60 F254 plates. The developed plates were examined under UV light at 254 nm and stained with an aqueous KMnO4–Na2CO3 solution. Concentration was performed by rotary evaporation at 40 °C under reduced pressure (unless otherwise stated). Flash chromatography was performed using silica gel 60 (230–400 mesh) from Merck or Silicycle with a forced flow eluent at 0.1–0.3 bar pressure. All 1H- and 13C-NMR spectra were recorded using a Bruker DPX-NMR 400 MHz (1H) and 100 MHz (13C) spectrometer or a Bruker Avance DRX 500 MHz (1H) and 125 MHz (13C) spectrometer at room temperature. Chemical shifts (δ-values) are reported in ppm, spectra were calibrated related to the solvents’ residual proton chemical shifts, multiplicity is reported as follows: s = singlet, d = doublet, t = triplet or m = multiplet, and the coupling constant, J, in Hz. IR spectra were recorded on a Varian 800 FT-IR ATR spectrophotometer. The absorptions are reported in cm−1. Mass spectra were recorded either on a high-resolution Bruker maXis 4G spectrometer using electrospray ionization (HRMS) or a Finnigan MAT 95Q spectrometer using electronic ionization (EI-MS). Elementary analysis (EA) was measured on a Perkin-Elmer Analysator 240. HPLC analysis was performed with a Dionex-Chromatography system using a P680 pump and a PDA-100 detector. Preparative reversed-phase HPLC was performed using Varian Prep Star pumps and a Dionex UltiMate 3000 RS detector or a Dionex P680 pump and a Dionex PDA-100 detector. Melting points (Mp) were determined using a Büchi B545 apparatus in open capillaries and are uncorrected. Absorptions spectra (UV/VIS) were recorded on a Shimadzu UV-1650 PC instrument. Emission spectra were recorded on a Shimadzu RF-5301 PC instrument. Additional procedures and characterization data can be found in the ESI.†Potassium 2-benzyl-3-ethoxy-3-oxopropanoate (15)
A published procedure41 was modified as follows: to a solution of diethyl benzylmalonate (14, 5.11 g, 20.0 mmol, 1.00 eq.) in EtOH (20 ml) a freshly filtered solution of potassium hydroxide (1.39 g, 21.0 mmol, 1.05 eq.) in EtOH (20 ml) was added at room temperature. The reaction was stirred for six hours at room temperature. The precipitate was filtered off and the filtrate was concentrated to a yellowish foam. The residue was suspended twice in Et2O and again concentrated. After drying at high vacuum, the foam was triturated overnight in Et2O (50 ml). The precipitate was isolated by filtration to give the title compound, 15, (4.44 g, 17 mmol, 85%) as a white solid. 1H-NMR (400 MHz, D2O): 7.30–7.22 (m, 2H), 7.22–7.15 (m, 3H), 4.06–3.96 (m, 2H), 3.51 (dd, J = 8.8, 7.4 Hz, 1H), 3.05–3.00 (m, 2H), 1.07 (t, J = 7.1 Hz, 3H). 13C-NMR (101 MHz, D2O): 176.0, 173.3, 139.1, 128.7, 128.6, 126.6, 62.0, 57.3, 35.0, 13.2. IR (neat): 3396, 2980, 1710, 1598, 1496, 1454, 1359, 1321, 1234, 1150, 1095, 1061, 1028, 950, 914, 858, 830, 745, 698. HRMS: Calculated for C12H13O4 [M–K]−: 221.0819, found: 221.0822. Mp: 120 °C (decomposition). The analytical data match those reported in the literature.413-Hydroxy-1,4-diphenylbutan-2-one (16)
A published procedure42 was modified as follows: to a solution of sodium acetate (328 mg, 4.00 mmol, 0.200 eq.) and freshly distilled phenyl acetaldehyde (4.77 ml, 40.0 mmol, 2.00 eq.) in EtOH (20 ml), 3-benzyl-5-(2-hydroxyethyl)-4-methylthiazolium chloride (540 mg, 2.00 mmol, 0.100 eq.) was added. The mixture was heated to reflux for three hours, cooled down and mixed with ice water (20 ml). After stirring for another hour, the mixture was saturated with NaCl and extracted three times with EtOAc. The org. layers were washed with brine, combined, dried with Na2SO4 and concentrated. The crude product was purified by column chromatography (SiO2, pentane–EtOAc (7:1 to 5:1)) to give the title compound, 16, (2.9 g, 12 mmol, 61%) as a yellow liquid, which crystallized after several days. 1H-NMR (500 MHz, CDCl3): 7.39–7.26 (m, 6H), 7.25–7.12 (m, 4H), 4.51 (ddd, J = 7.4, 5.5, 4.9 Hz, 1H), 3.81 (d, J = 15.9 Hz, 1H), 3.75 (d, J = 15.9 Hz, 1H), 3.22 (d, J = 5.6 Hz, 1H), 3.15 (dd, J = 14.1, 4.7 Hz, 1H), 2.90 (dd, J = 14.1, 7.4 Hz, 1H). 13C-NMR (126 MHz, CDCl3): 209.2, 136.5, 133.1, 129.6, 129.5, 128.9, 128.8, 127.5, 127.2, 76.8, 45.9, 40.3. The analytical data match those reported in the literature.42,44
1-Ethyl 3-(3-oxo-1,4-diphenylbutan-2-yl) 2-benzylmalonate (17) as a mixture of diastereomers
To a suspension of potassium 2-benzyl-3-ethoxy-3-oxopropanoate (15, 19.0 g, 72.8 mmol, 1.40 eq.) in dry THF (250 ml) at 0 °C, DMF (201 μl, 5.20 mmol, 0.050 eq.) was added followed by the dropwise addition of pivaloyl chloride (11.0 ml, 88.4 mmol, 1.70 eq.) over five minutes. During the addition of pivaloyl chloride, the reaction mixture turned to a solution and after another 10 minutes to a gel. This gel was then kept for one hour at 0 °C and for 2.5 hours at room temperature, during which the gel turned back into a solution. The resulting solution was concentrated, dried in high vacuum, again dissolved in dry CH2Cl2 (250 ml), concentrated and dried again in high vacuum without exceeding a temperature of 25 °C. The residue was dissolved in dry CH2Cl2 (250 ml) and cooled to 0 °C. The addition of 4-dimethylaminopyridine (635 mg, 5.20 mmol, 0.100 eq.) to the reaction mixture was followed by the addition of a solution of 3-hydroxy-1,4-diphenylbutan-2-one (16, 12.5 g, 52.0 mmol, 1.00 eq.) in dry CH2Cl2 (50 ml) over 10 minutes. The reaction was allowed to reach room temperature overnight before it was quenched at 0 °C with a mixture of water and a sat. aq. NaHCO3 solution (1:1 v/v). The layers were separated and the aq. layer was extracted three times with CH2Cl2. The org. layers were washed with a sat. aq. NH4Cl solution, combined, dried with Na2SO4 and concentrated. The crude product was purified by column chromatography (SiO2, pentane–EtOAc (6:1)) to give the title compound, 17, (23.1 g, 52 mmol, quantitative) as a yellowish liquid. Rf (SiO2, pentane–Et2O (2:1)): 0.7. 1H-NMR (500 MHz, CDCl3): 7.32–7.20 (m, 8H), 7.20–7.15 (m, 2H), 7.14–7.09 (m, 2H), 7.06–6.99 (m, 3H), 5.35–5.25 (m, 1H), 4.21–4.04 (m, 2H), 3.76–3.69 (m, 1H), 3.58 (s, 1H), 3.50 (d, J = 16.9 Hz, 0.5H), 3.38 (d, J = 16.9 Hz, 0.5H), 3.26–3.11 (m, 2H), 3.06–2.93 (m, 2H), 1.19–1.11 (m, 3H). 13C-NMR (126 MHz, CDCl3): 204.4, 204.3, 168.5, 168.4, 168.4, 168.3, 137.7, 137.6, 135.7, 135.5, 132.9, 132.9, 129.9, 129.9, 129.6, 129.6, 129.0, 129.0, 128.7, 128.7, 128.7, 127.3, 127.3, 127.2, 127.1, 127.1, 79.6, 79.4, 61.9, 61.9, 53.8, 53.7, 46.5, 46.4, 37.1, 34.7, 34.7, 14.1, 14.1. IR (neat): 3031, 2982, 2936, 1732, 1497, 1454, 1369, 1276, 1229, 1149, 701, 633. HRMS: Calculated for C28H32NO5 [M + NH4]+: 462.2275, found: 462.2274. EI-MS (70 eV) m/z (%): 444 (3, [M]+), 353 (9), 205 (43), 160 (10), 159 (100), 131 (68), 91 (94).
Maculalactone A (1)
A two neck round bottom flask was equipped with a condenser and a device to run the condensed solvent over a bed of 4 Å molecular sieves. In the flask, a mixture of cesium carbonate (1.23 g, 3.78 mmol, 0.200 eq.) and THF (400 ml) was heated until the suspension was strongly refluxing. After refluxing for 30 minutes, a solution of 1-ethyl 3-(3-oxo-1,4-diphenylbutan-2-yl) 2-benzylmalonate (17, 8.40 g, 18.9 mmol, 1.00 eq.) in THF (20 ml) was added over 25 minutes. The reaction was further stirred under strong reflux. After 3.5 hours, the finished reaction was cooled to room temperature. The mixture was filtrated over a bed of SiO2 and the solids were washed with TBME. The filtrate was concentrated. The crude product was purified by recrystallization from water and methanol to give the title compound, 1, (5.1 g, 14 mmol, 76%) as off-white crystals. 1H-NMR (400 MHz, CDCl3): 7.34–7.23 (m, 6H), 7.22–7.10 (m, 5H), 7.03 (d, J = 6.5 Hz, 2H), 6.92–6.85 (m, 2H), 4.99–4.89 (m, 1H), 3.92 (d, J = 15.6 Hz, 1H), 3.63 (d, J = 15.3 Hz, 1H), 3.57 (d, J = 15.3 Hz, 1H), 3.48 (d, J = 15.6 Hz, 1H), 3.23 (dd, J = 14.5, 3.9 Hz, 1H), 2.82 (dd, J = 14.6, 6.1 Hz, 1H). 13C-NMR (101 MHz, CDCl3): 173.7, 161.8, 137.8, 136.1, 135.0, 129.6, 129.2, 128.8, 128.7, 128.3, 127.5, 127.3, 126.5, 81.7, 77.4, 38.1, 33.4, 29.5. The analytical data match those reported in the literature.26Acknowledgements
The support by the Swiss National Science Foundation (200020_130475) is gratefully acknowledged. We thank Dr Heinz Nadig for MS, Werner Kirsch for EA, Verena Grundler for HPLC and Nik Hostettler for emission spectroscopic measurements.Notes and references
- D. M. Yebra, S. Kiil and K. Dam-Johansen, Prog. Org. Coat., 2004, 50, 75–104 CrossRef CAS PubMed.
- T. S. Wood and T. G. Marsh, Water Res., 1999, 33, 609–614 CrossRef CAS.
- H. A. Jenner, J. W. Whitehouse, C. J. L. Taylor and M. Khalanski, Hydroécologie Appliquée, 1998, 10, I–225 CrossRef.
- S. E. Coester and T. E. Cloete, Crit. Rev. Microbiol., 2005, 31, 213–232 CrossRef PubMed.
- T. Vladkova, in Marine and Industrial Biofouling, ed. H.-C. Flemming, P. S. Murthy, R. Venkatesan and K. Cooksey, Springer, Berlin Heidelberg, 2008, vol. 4, pp. 135–163 Search PubMed.
- B. Antizar-Ladislao, Environ. Int., 2008, 34, 292–308 CrossRef CAS PubMed.
- L. D. Chambers, K. R. Stokes, F. C. Walsh and R. J. K. Wood, Surf. Coat. Technol., 2006, 201, 3642–3652 CrossRef CAS PubMed.
- M. Salta, J. A. Wharton, P. Stoodley, S. P. Dennington, L. R. Goodes, S. Werwinski, U. Mart, R. J. K. Wood and K. R. Stokes, Philos. Trans. R. Soc. London, Ser. A, 2010, 368, 4729–4754 CrossRef CAS PubMed.
- N. Fusetani, Nat. Prod. Rep., 2011, 28, 400–410 RSC.
- Y.-F. Zhang, K. Xiao, K. H. Chandramouli, Y. Xu, K. Pan, W.-X. Wang and P.-Y. Qian, PLoS One, 2011, 6, e23803 CAS.
- Y. Li, F. Zhang, Y. Xu, K. Matsumura, Z. Han, L. Liu, W. Lin, Y. Jia and P.-Y. Qian, Biofouling, 2012, 28, 857–864 CrossRef CAS PubMed.
- P.-Y. Qian, Y. H. Wong and Y. Zhang, Proteomics, 2010, 10, 3435–3446 CrossRef CAS PubMed.
- J. F. Blom, T. Brütsch, D. Barbaras, Y. Bethuel, H. H. Locher, C. Hubschwerlen and K. Gademann, Org. Lett., 2006, 8, 737–740 CrossRef CAS PubMed.
- C. Portmann, C. Prestinari, T. Myers, J. Scharte and K. Gademann, ChemBioChem, 2009, 10, 889–895 CrossRef CAS PubMed.
- S. Bonazzi, D. Barbaras, L. Patiny, R. Scopelliti, P. Schneider, S. T. Cole, M. Kaiser, R. Brun and K. Gademann, Bioorg. Med. Chem., 2010, 18, 1464–1476 CrossRef CAS PubMed.
- S. Zürcher, D. Wäckerlin, Y. Bethuel, B. Malisova, M. Textor, S. Tosatti and K. Gademann, J. Am. Chem. Soc., 2006, 128, 1064–1065 CrossRef PubMed.
- J.-Y. Wach, B. Malisova, S. Bonazzi, S. Tosatti, M. Textor, S. Zürcher and K. Gademann, Chem. – Eur. J., 2008, 14, 10579–10584 CrossRef CAS PubMed.
- S. Saxer, C. Portmann, S. Tosatti, K. Gademann, S. Zürcher and M. Textor, Macromolecules, 2010, 43, 1050–1060 CrossRef CAS.
- J.-Y. Wach, S. Bonazzi and K. Gademann, Angew. Chem., Int. Ed., 2008, 47, 7123–7126 CrossRef CAS PubMed.
- J. Gomes, A. Grunau, A. K. Lawrence, L. Eberl and K. Gademann, Chem. Commun., 2013, 49, 155–157 RSC.
- W.-Y. Tsui, G. A. Williams and G.D. Brown, Phytochemistry, 1996, 43, 1083–1085 CrossRef CAS.
- S.-C. Lee and G. D. Brown, J. Nat. Prod., 1998, 61, 29–33 CrossRef CAS PubMed.
- H.-F. Wong, G. A. Williams and G. D. Brown, Phytochemistry, 2002, 60, 425–429 CrossRef CAS.
- S.-C. Lee, G. A. Williams and G. D. Brown, Phytochemistry, 1999, 52, 537–540 CrossRef CAS.
- G. D. Brown, H.-F. Wong, N. Hutchinson, S.-C. Lee, B. K. K. Chan and G. A. Williams, Phytochem. Rev., 2004, 3, 381–400 CrossRef CAS PubMed.
- G. D. Brown and H.-F. Wong, Tetrahedron, 2004, 60, 5439–5451 CrossRef CAS PubMed.
- A. Kar, S. Gogoi and N. P. Argade, Tetrahedron, 2005, 61, 5297–5302 CrossRef CAS PubMed.
- R. J. Duffy, K. A. Morris, R. Vallakati, W. Zhang and D. Romo, J. Org. Chem., 2009, 74, 4772–4781 CrossRef CAS PubMed.
- A. V. Messorosh, A. V. Trukhin and E. V. Eliseenkov, Tetrahedron, 2008, 64, 10849–10852 CrossRef CAS PubMed.
- M. Dubernet, V. Caubert, J. Guillard and M.-C. Viaud-Massuard, Tetrahedron, 2005, 61, 4585–4593 CrossRef CAS PubMed.
- C. Hofstetter, P. S. Wilkinson and T. C. Pochapsky, J. Org. Chem., 1999, 64, 8794–8800 CrossRef CAS PubMed.
- T. M. Ugurchieva and V. V. Veselovsky, Russ. Chem. Rev., 2009, 78, 337–373 CrossRef CAS PubMed.
- Y. S. Rao, Chem. Rev., 1976, 76, 625–694 CrossRef CAS.
- A. A. Avetisyan and M. T. Dangyan, Russ. Chem. Rev., 1977, 46, 643–656 CrossRef PubMed.
- S. Krehl, D. Geißler, S. Hauke, O. Kunz, L. Staude and B. Schmidt, Beilstein J. Org. Chem., 2010, 6, 1188–1198 CrossRef CAS PubMed.
- G. Hughes, M. Kimura and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 11253–11258 CrossRef CAS PubMed.
- A. Fürstner, O. R. Thiel, L. Ackermann, H.-J. Schanz and S. P. Nolan, J. Org. Chem., 2000, 65, 2204–2207 CrossRef.
- A. Erkkilä and P. M. Pihko, J. Org. Chem., 2006, 71, 2538–2541 CrossRef PubMed.
- A. A. Avetisyan, R. G. Nazaryan, A. N. Dzhandzhaoanyan, V. I. Votyakov, S. V. Khlyustov, G. V. Vladyko, V. Y. Klimovich, L. V. Korobchenko, M. N. Shashikhina and S. V. Zhavrid, Khim.-Farm. Zh., 1982, 16, 783–786 CAS.
- S. Furrer, P. Schieberle and A. Burdack-Freitag, Flavorant and Fragrance Compounds, WO2007107023 (A1), 2007 Search PubMed.
- O. P. Goel and U. Krolls, Tetrahedron Lett., 1983, 24, 163–166 CrossRef CAS.
- H. Stetter and R. Y. Rämsch, Synthesis, 1981, 477–478 CrossRef CAS.
- S. Shiotani, H. Okada, T. Yamamoto, K. Nakamata, J. Adachi and H. Nakamoto, Heterocycles, 1996, 43, 113–126 CrossRef CAS PubMed.
- G. Pattenden, N. A. Pegg and R. W. Kenyon, J. Chem. Soc., Perkin Trans. 1, 1991, 2363–2372 RSC.
- M. Gill, M. J. Kiefel and D. A. Lally, Tetrahedron Lett., 1986, 27, 1933–1934 CrossRef CAS.
- K. Gademann, C. Portmann, J. F. Blom, M. Zeder and F. Jüttner, J. Nat. Prod., 2010, 73, 980–984 CrossRef CAS PubMed.
- C. Portmann, J. F. Blom, K. Gademann and F. Jüttner, J. Nat. Prod., 2008, 71, 1193–1196 CrossRef CAS PubMed.
- M. J. Baena, J. Buey, P. Espinet and C. E. García-Prieto, J. Organomet. Chem., 2005, 690, 998–1010 CrossRef CAS PubMed.
- T. Tamura, K. Yoshida, T. Hachida, M. Tsuchiya, M. Nakamura, Y. Kazue, N. Tachikawa, K. Dokko and M. Watanabe, Chem. Lett., 2010, 39, 753–755 CrossRef CAS.
- R. Yan, E. El-Emir, V. Rajkumar, M. Robson, A. P. Jathoul, R. B. Pedley and E. Årstad, Angew. Chem., Int. Ed., 2011, 50, 6793–6795 CrossRef CAS PubMed.
- T. Bartolomaeus, B. Quast and M. Koch, Zoomorphology, 2009, 128, 247–262 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob02042a |
| This journal is © The Royal Society of Chemistry 2015 |