
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKelvin probe imaging of photo-injected electrons in metal oxide nanosheets from metal sulfide quantum dots under remote photochromic coloration†
A.
Kondo
a,
G.
Yin
a,
N.
Srinivasan
a,
D.
Atarashi
a,
E.
Sakai
a and
M.
Miyauchi
*ab
aDepartment of Metallurgy and Ceramics Science, Graduate School of Science and Engineering, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8552, Japan. E-mail: mmiyauchi@ceram.titech.ac.jp
bJapan Science and Technology Agency (JST), 4-1-8 Honcho Kawaguchi, Saitama 332-0012, Japan
First published on 15th June 2015
Abstract
Metal oxide and quantum dot (QD) heterostructures have attracted considerable recent attention as materials for developing efficient solar cells, photocatalysts, and display devices, thus nanoscale imaging of trapped electrons in these heterostructures provides important insight for developing efficient devices. In the present study, Kelvin probe force microscopy (KPFM) of CdS quantum dot (QD)-grafted Cs4W11O362− nanosheets was performed before and after visible-light irradiation. After visible-light excitation of the CdS QDs, the Cs4W11O362− nanosheet surface exhibited a decreased work function in the vicinity of the junction with CdS QDs, even though the Cs4W11O362− nanosheet did not absorb visible light. X-ray photoelectron spectroscopy revealed that W5+ species were formed in the nanosheet after visible-light irradiation. These results demonstrated that excited electrons in the CdS QDs were injected and trapped in the Cs4W11O362− nanosheet to form color centers. Further, the CdS QDs and Cs4W11O362− nanosheet composite films exhibited efficient remote photochromic coloration, which was attributed to the quantum nanostructure of the film. Notably, the responsive wavelength of the material is tunable by adjusting the size of QDs, and the decoloration rate is highly efficient, as the required length for trapped electrons to diffuse into the nanosheet surface is very short owing to its nanoscale thickness. The unique properties of this photochromic device make it suitable for display or memory applications. In addition, the methodology described in the present study for nanoscale imaging is expected to aid in the understanding of electron transport and trapping processes in metal oxide and metal chalcogenide heterostructure, which are crucial phenomena in QD-based solar cells and/or photocatalytic water-splitting systems.
1. Introduction
The interfacial heterostructure between metal oxides and metal chalcogenides is an essential factor affecting the efficiency of solar photovoltaic and photocatalytic water-splitting systems. Quantum dot (QD)-sensitized solar cells (QDSSCs) were developed in the 1990s as photoelectrodes with sunlight-harvesting capacity.1–7 QDSSCs do not require a solid layered p–n junction film, unlike silicon solar cells, as photo-generated electrons and holes are efficiently separated at the interface of the QDs and mesoporous metal oxide, such as TiO2. The solar-cell efficiencies of QDSSCs have recently exceeded 7%,6,7 and we had previously reported that the ratio of incident photons to current efficiency of QDSSCs exceeds 80% under short circuit conditions without the application of a bias potential.8–10 The high short circuit photocurrent of this material indicates the presence of a space charge layer or polar structure at the interface between the metal chalcogenide QDs and metal oxide particles. In addition to photovoltaic applications, metal chalcogenide QDs,11–14 such as cadmium telluride (CdTe),11 lead sulfide (PbS),12 cadmium sulfide (CdS),13 and cadmium selenide (CdSe)14 have been applied towards the sensitization of solar-driven water splitting systems. Such sensitized systems have the potential to generate hydrogen from water under visible-light irradiation owing to the narrow bandgap, high visible-light absorption coefficient, and efficient mobility of charge carriers with small effective mass.In addition to the use of metal chalcogenide QDs as light-harvesting materials in devices for solar energy production, the unique properties of the heterostructure formed by metal chalcogenide QDs and metal oxides have been applied towards the development of display and memory devices. For example, remote photochromic coloration has recently been reported in CdS QD-loaded tungsten oxide (WO3) particles.15 In this system, the photogenerated electrons in CdS are injected and trapped in WO3, where they act as blue color centers. Notably, this photochromic device has a responsive wavelength that is controllable by adjusting the size of the CdS QDs, and has a markedly higher decoloration rate than that of conventional metal–oxide photochromic devices using bandgap excitation of metal oxide. Injected electrons from QDs are trapped in the shallow state of the metal oxide and efficiently react with oxygen molecules in air.15 However, to better understand electron transport between QDs and metal oxides, nanoscale imaging of trapped electrons is essential and is expected to provide useful insight for developing efficient display and solar energy conversion devices.
In the present study, Kelvin probe force microscopy (KPFM), which is a powerful tool to enable high-resolution surface potential and topography mapping of metal and semiconductor surfaces, was used to observe electrons injected into metal oxide from CdS QDs. For the nanoscale imaging analysis, CdS QDs were grafted onto the surface of atomically flat cesium tungstate (Cs4W11O362−) nanosheets. The bandgap of the Cs4W11O362− nanosheet is wider than that of CdS QDs and optically transparent in the visible light region. Further, its conduction band is more positive than that of CdS QDs,16 thus the Cs4W11O362− nanosheet is the suitable electron acceptor from QDs. KPFM imaging was used to map the work function of the CdS/Cs4W11O362− nanosheet heterostructure before and after visible-light irradiation. From the obtained KPFM images, charge transport and trapping processes between the two semiconductors were analyzed. In addition to nanoscale imaging, we investigated the macroscopic coloring phenomena of the remote photochromic reaction in CdS/Cs4W11O362− nanosheet thin films coated on glass substrates. An efficient photochromic reaction rate can be expected by using a single crystalline metal oxide nanosheet, owing to its large surface area, high carriers’ mobility, and efficient diffusion of oxygen molecules in the decoloration process. Based on the study of remote photochromism, the decoloration rate of the CdS/Cs4W11O362− nanosheet film was found to be markedly higher than that of the CdS-modified bulk WO3 particles due to the very short diffusion length of trapped electrons in the few nanometer thick nanosheets.
2. Experimental details
2.1 Synthesis of Cs4W11O362− nanosheets
Layered Cs4W11O362− nanosheets were exfoliated from bulk Cs6W11O36 by a chemical treatment. Powder forms of Cs2CO3 and WO3 were mixed at a molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11 and then ground to a fine powder with a mortar and pestle. The mixed powder was calcined at 1173 K for 6 h. After annealing, the resulting dark blue solid (Cs6W11O36) was ground into powder and 0.5 g of the powder was suspended in 50 ml of a 12 M hydrogen chloride (HCl) solution. After stirring the solution for 24 h at room temperature, the solution was centrifuged at 3000 rpm for 10 min and the acidic supernatant was discarded. Fifty milliliters of a 12 M HCl solution was then added and the suspension was again stirred for 24 h. The solution was centrifuged at 3000 rpm for 10 min and the formed yellow precipitate (protonated cesium tungstate; H2Cs4W11O36) was washed six times with pure water. The precipitate was dispersed in 50 mL of an aqueous tetra(n-butyl)ammonium hydroxide (TBAOH; Aldrich Ltd) solution to achieve neutral conditions. This suspension was shaken for 10 days at room temperature and yielded a colloidal suspension with a milky appearance.
:11 and then ground to a fine powder with a mortar and pestle. The mixed powder was calcined at 1173 K for 6 h. After annealing, the resulting dark blue solid (Cs6W11O36) was ground into powder and 0.5 g of the powder was suspended in 50 ml of a 12 M hydrogen chloride (HCl) solution. After stirring the solution for 24 h at room temperature, the solution was centrifuged at 3000 rpm for 10 min and the acidic supernatant was discarded. Fifty milliliters of a 12 M HCl solution was then added and the suspension was again stirred for 24 h. The solution was centrifuged at 3000 rpm for 10 min and the formed yellow precipitate (protonated cesium tungstate; H2Cs4W11O36) was washed six times with pure water. The precipitate was dispersed in 50 mL of an aqueous tetra(n-butyl)ammonium hydroxide (TBAOH; Aldrich Ltd) solution to achieve neutral conditions. This suspension was shaken for 10 days at room temperature and yielded a colloidal suspension with a milky appearance.
2.2 Fabrication of the CdS QDs/Cs4W11O362− nanosheet film
Exfoliated Cs4W11O362− nanosheets were mono-dispersed in aqueous solution and these nanosheets were coated on a natural mica substrate (WAKO Ltd). The surface of mica was treated in poly(diallyldimethylammonium chloride) (PDDA) for effective adsorption of Cs4W11O362− nanosheets. Glass or mica substrates were alternately dipped into a colloidal solution of Cs4W11O362− nanosheets and a PDDA solution to fabricate multi-layered nanosheet films. After thin film fabrication, PDDA molecules were removed by UV-ozone treatment (UV253E, Filgen, Inc.). Next, CdS QDs were grafted onto Cs4W11O362− nanosheets by a successive ionic layer adsorption and reaction (SILAR) method. A thin film of Cs4W11O362− nanosheets was immersed into a 0.5 M Cd(NO3)2 aqueous solution for 5 s, washed with ethanol, and then immersed for another 5 s into a 0.25 M Na2S aqueous solution and washed with ethanol. This two-step immersion procedure is termed as one SILAR cycle, and the crystallite growth of CdS was controlled by repeating the SILAR cycles.2.3 Measurement and analysis
Cross-sectional images of the thin films were observed using scanning electron microscopy (SEM; model SU9000, Hitachi Co., Tokyo, Japan). Absorption spectra for thin films were recorded using a spectrophotometer (UV-vis, model V-660, JASCO Instruments Ltd) by a diffuse-reflectance method with BaSO4 as the background. Fluorescence spectra were recorded using a fluorescent spectrophotometer (F-7000, Hitachi Co., Tokyo, Japan). Surface morphologies and potential mapping of the mono-layered nanosheet film on the mica substrate were observed by scanning probe microscopy (model SPM-9700, Shimadzu Instruments, Japan). AFM and KPFM measurements were performed in the tapping mode under ambient conditions. For KPFM observation, the platinum and iridium coated cantilevers (PointProbe EFM, tip curvature: 20 nm, NanoWorld Ltd) were used, while a conventional silicon cantilever was used for AFM observation. X-ray photoelectron spectroscopy (XPS; model ESCA-5500MT, Perkin Elmer Instruments, Japan) analysis was conducted to determine the chemical states of cesium tungstate using standard Mg Kα X-rays. For visible light irradiation, a 150 W Xe lamp was used as the light source. The light was passed through a 430 nm optical UV cutoff filter (Y-43, Asahi Techno Glass Led), which allows wavelengths longer than 430 nm to pass. The spectrum of the light source was recorded by using a spectro-radiometer (USR-40D, Ushio Ltd).3. Results and discussion
The layered Cs4W11O362− nanosheets were exfoliated from bulk Cs6W11O36 by chemical treatment and were then monodispersed into single-crystalline structures. The Cs4W11O362− nanosheets were coated on glass or mica substrates in a high crystallographic c-plane orientation using a layer-by-layer alternate absorption method, and the thickness of the nanosheets was controlled by the number of dipping cycles (see the ESI, Fig. S1,† UV-Vis spectra).Fig. 1 shows atomic force microscopy (AFM) images of CdS QD-grafted Cs4W11O362− nanosheets coated onto a mica substrate. The line profile indicated that the thickness of the Cs4W11O362− nanosheet was about 2.3–2.6 nm, which is consistent with the reported thickness of a monolayer of a Cs4W11O362− nanosheet.16,17 AFM imaging revealed that the CdS QDs were mono-dispersed and grafted onto the surface of Cs4W11O362− nanosheets. The CdS QDs were a few nanometers in size when using one SILAR cycle. The size of the CdS QDs was controllable by the number of SILAR cycles, as shown in the UV-Vis spectra (ESI, Fig. S2†).
 | ||
| Fig. 1 AFM image (left) of the CdS/Cs4W11O362− nanofilm produced using 1 SILAR cycle, and a line profile (right) of the area in the nanosheet indicated by the dotted line in the AFM image. | ||
UV-Vis spectra were used to estimate the band-gap values based on the fact that the optical excitation of Cs4W11O362− and CdS proceeds as indirect and direct transitions, respectively. Fig. 2 shows the relationship between the absorption coefficients and photon energies of the bare Cs4W11O362− nanosheet and CdS/Cs4W11O362− nanosheet films. The bare Cs4W11O362− nanosheet was optically transparent in the visible-light region and had an estimated bandgap of 3.6 eV. This bandgap was wider than that of bulk tungsten oxide (2.5–2.8 eV)18–21 due to the quantum confinement effect. In contrast, the CdS/Cs4W11O362− nanosheet films exhibited visible-light absorption, which was attributable to the excitation of CdS QDs. In addition, the bandgap of CdS/Cs4W11O362− was controllable by adjusting the number of SILAR cycles. Fluorescence spectroscopy also revealed that the emission wavelength from band-to-band recombination in CdS was red-shifted with increased numbers of SILAR cycles (see ESI, Fig. S3†).
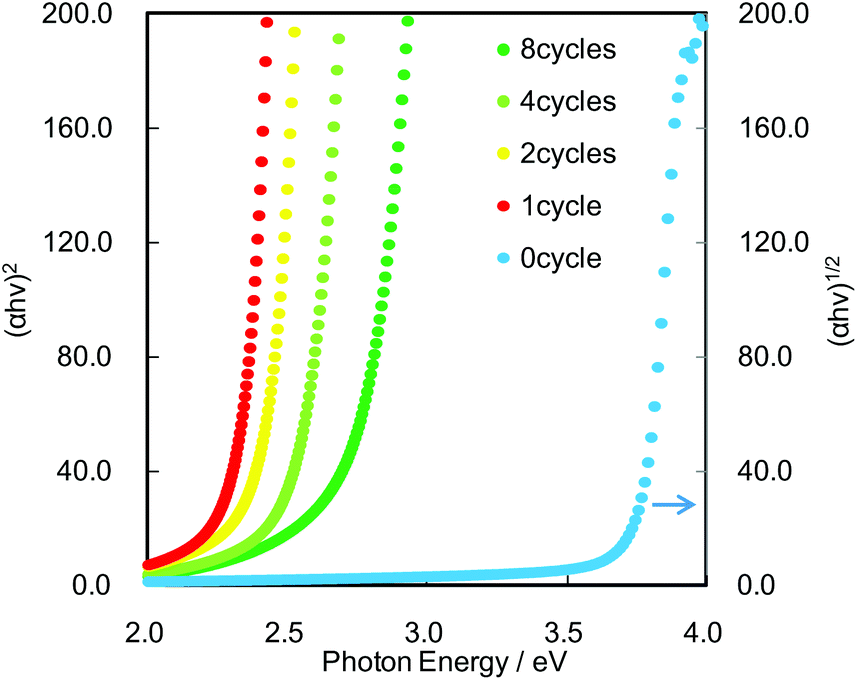 | ||
| Fig. 2 Optical absorption coefficient versus photon energy. Bandgap values were estimated based on the fact that the optical excitation of Cs4W11O362− and CdS proceeds as indirect and direct transitions, respectively. | ||
We next performed AFM and KPFM imaging of the CdS QD-grafted monolayered Cs4W11O362− nanosheet film before and after visible-light irradiation. In the AFM image (Fig. 3(a)), CdS QDs and Cs4W11O362− nanosheets were clearly observed on the mica substrate. Fig. 3(b) shows the KPFM image before visible light irradiation, which reveals the mapping of work function on the same area with Fig. 3(a). Work function is the energy of the Fermi level versus the vacuum level, thus the work function becomes smaller when electrons are located at negative potential sites. According to the result in Fig. 3(b), the work function of CdS QDs was determined to be smaller than that of Cs4W11O362−, as the Fermi level of the exposed CdS surface is more negative than that of Cs4W11O362−. These results are reasonable, because both CdS and Cs4W11O362− are n-type semiconductors, and the conduction band of the former is higher than that of the latter. The work function of the mica substrate is higher than those of CdS and Cs4W11O362−, because mica is an insulator and has positive Fermi level as compared to CdS and Cs4W11O362−. Fig. 3(c) shows a KPFM image after visible-light irradiation of the same area of the CdS QDs and Cs4W11O362− nanosheets shown in Fig. 3(a) and (b). A Xe lamp with a UV-cutoff filter was used as the light source to excite only CdS QDs (ESI, Fig. S4† shows the spectrum of the Xe light source). Under visible-light irradiation, the Fermi level of the Cs4W11O362− nanosheet became more negative in the near regions of CdS QDs (point (i) in Fig. 3(c)), but did not markedly change in regions where CdS QDs were not deposited (point (ii) in Fig. 3(c)). These trends were also observed on a different area, where many QDs were deposited (point (iii)), and the work function of this area became smaller after visible light irradiation. These results indicate that visible light irradiation influences the potential of nanosheets near the heterojunction region. For comparison, we performed KPFM imaging of the bare Cs4W11O362− nanosheet film, but the work function of the bare nanosheet did not change after light irradiation, as the Cs4W11O362− nanosheet did not absorb visible light (see ESI, Fig. S5†).
 | ||
| Fig. 3 AFM and KPFM images for the CdS/Cs4W11O362− nanofilm. (a) AFM image, (b) KPFM image before visible-light irradiation, and (c) KPFM image after visible-light irradiation for 5 min. Points (i) and (iii) indicate the sites of CdS QD deposition, and point (ii) is the exposed surface of the Cs4W11O362− nanosheet. (d) XPS spectra for the W-4f orbital of CdS/Cs4W11O362− after visible-light irradiation. The black line shows the measured XPS spectrum, and the red and blue lines are fitted curves for W6+ and W5+ species, respectively. | ||
X-ray photoelectron spectroscopy (XPS) spectra for the tungsten (W)-4f orbital of the CdS/Cs4W11O362− film prepared on a glass substrate were also recorded after visible-light irradiation (Fig. 3(d)). The shape of the XPS curve was deconvoluted into two doublets: a main doublet at 36.1 eV that could be assigned to W-4f7/2 orbital, and a second doublet at 38.2 eV that was attributable to W-4f5/2, which corresponds to the W6+ oxidation state. In addition to the signals for W6+ species, doublet signals with the same gap (2.1 eV) were overlapped at the lower binding energy than W6+ signals after visible light irradiation. These additional signals appeared at 35.1 and 37.2 eV, and were assigned to W5+ oxidation states, which form small polarons. In XPS measurements performed before visible-light irradiation, no W5+ species were detected. Moreover, the valence states of other elements, including sulfur (S), cesium (Cs), and cadmium (Cd), did not change after visible-light irradiation of the CdS/Cs4W11O362− film (see ESI, Fig. S6†). These results indicate that excited electrons in CdS QDs are injected into the Cs4W11O362− nanosheet, resulting in a reduction of the Fermi level of the Cs4W11O362− nanosheet as electrons are trapped as W5+ species. Electron injection from QDs to metal oxide in the present study proceeds under the same process as that in a quantum dot sensitized solar cell.8,9 The potential of excited electrons in the conduction band of CdS QD is higher than that of the Cs4W11O362− nanosheet,16,22 thus the excited electrons in QDs can be injected into nanosheets. Injected electrons in the conduction band of the Cs4W11O362− nanosheet are still in the excited states, thus these electrons are trapped at more stable sites as color centers, which have been observed as W5+ species in the nanosheet crystal by XPS analysis. According to the KPFM image after light irradiation (Fig. 3(c)), electrons were injected into an area extending several tens of nanometers from the junction point with QDs, indicating that the electrons were hopping and diffused into the Cs4W11O362− nanosheet. The electron diffusion length estimated here is reasonable, as a previous study reported that the space charge layer of Cs4W11O362− without a dopant was several micrometers.16 When the Cs4W11O362− nanosheet was stored under dark conditions in ambient air for several minutes, the Fermi level of the nanosheet returned to its initial state, as the trapped electrons rapidly reacted with the oxygen molecules in air.
In addition to the nanoscale imaging, we investigated the macroscopic color change that occurs in the CdS/Cs4W11O362− nanosheet composite film upon light irradiation (SEM images are shown in Fig. S7†). Fig. 4(a) shows optical absorption spectra of the film before and after visible-light irradiation. Photos of the CdS/Cs4W11O362− composite film before and after visible-light irradiation are also shown in the inset of Fig. 4(a). The CdS/Cs4W11O362− composite film turned blue after the visible-light irradiation, indicating that remote photochromic coloration occurred in the QDs/nanosheet heterostructure (Fig. 4(a), inset). Specifically, photogenerated electrons in the CdS QDs were injected into the Cs4W11O362− nanosheet, where they become trapped and generate W5+ species, which form the dark blue color centers in the nanosheet. The observed color change was reversible and was clearly detectable by the naked eye, as shown in the inset photos of Fig. 4(a).
 | ||
| Fig. 4 Remote photochromic properties of the CdS/Cs4W11O362− film. (a) UV-Vis spectra for the film in the decoloration state (before light irradiation) and coloration state (after light irradiation). Inset figures show photos of the coloration and decoloration states of the CdS/Cs4W11O362− film. (b) Decay in color (at 850 nm) for thin films under dark conditions in ambient air. Visible light was irradiated for first 5 hours, and then the samples were stored under the dark conditions. Data for CdS/WO3 were previously reported by Zhao et al.15 | ||
One of the most unique properties of the CdS QDs/Cs4W11O362− nanosheet film is the efficiency of the visible-light induced color change. We therefore compared the switching properties of the present CdS/Cs4W11O362− nanosheet with those previously reported for CdS/WO3 particles.15Fig. 4(b) shows changes in the absorbance values of the CdS QDs/Cs4W11O362− nanosheet film at a wavelength of 850 nm in ambient air under dark conditions after visible light irradiation. The absorbance of CdS QDs/Cs4W11O362− after visible-light irradiation was larger than that of CdS QDs/bulk WO3. Furthermore, the decoloration rate of the CdS/Cs4W11O362− nanosheet under dark conditions was markedly higher than that of CdS/WO3. Examination of the decoloration process under various atmospheres, including air and nitrogen with different temperatures is shown in Fig. 5. Blue color was retained under a nitrogen atmosphere, while it was converted into the initial color under the existence of oxygen. Further, the heat treatment accelerated the decoloration rate, indicating that decoloration was attributable to the oxidation of trapped electrons in reduced tungsten species. Our previous spectroscopic analysis demonstrated that the trapped state of Cs4W11O362− was shallower than that of bulk WO3.16 In addition, the diffusion length of trapped electrons in the Cs4W11O362− nanosheet is much shorter than that of bulk WO3 particles, as the thickness of the former (2.3 nm) is markedly smaller than that of the latter. Thus, the high decoloration rate observed for the Cs4W11O362− nanosheet is due to efficient electron transfer within the nanoscale sheet-like structure.
 | ||
| Fig. 5 Changes in absorbance at 850 nm under various dark conditions (ambient air at 298 K, ambient air at 373 K, nitrogen at 298 K). | ||
We also investigated the repeatability of the reversible color change in the CdS/Cs4W11O362− nanosheet film and the result is shown in Fig. 6. We confirmed that the coloration and decoloration of the film could be repeated several times; however, the absorbance values in the colored state slightly decreased with each cycle, indicating that the CdS QDs undergo photo-corrosion. We speculate that most of the photogenerated holes in CdS QDs react with water molecules or contaminants in air to produce oxygen or carbon dioxide through a photocatalytic oxidation process,23,24 since the absorbed photon number was 8000 times larger than the number of created W5+ species in the Cs4W11O362− nanosheet film. Notably, when we introduced a gaseous sacrificial agent, such as methanol, into the atmosphere during light irradiation, the stability of the CdS/Cs4W11O362− nanosheet was improved (see ESI, Fig. S8†). These results suggest that the long-term stability of QDSSC systems is improved by the introduction of a sacrificial agent. In addition, the previous findings from studies on core–shell QD structures suggest that the use of a surface passivation coating may lead to higher stabilities.25,26
 | ||
| Fig. 6 Repeatability of the reversible color change in the CdS/Cs4W11O362− nanosheet film by visible light irradiation and stored under the dark conditions in air. Initial absorbance of the coloration state was defined as 100% at a wavelength of 850 nm. | ||
4. Conclusions
We performed nanoscale imaging of trapped electrons in metal oxide nanosheet and quantum dot heterostructure by KPFM and revealed that excited electrons in CdS QDs are injected into Cs4W11O362− nanosheets and cause a negative shift in the work function of the nanosheet near the junction with CdS QDs. These findings provide insight into the electron transport and trapping processes that occur in metal oxide and metal chalcogenide heterostructure, such as that found in quantum dot-based solar cells and photocatalytic water-splitting systems. Further, the present research provides the unique photochromic device, termed remote photochromism with multi-color tunability and a rapid color-change response. As the photochromic device is driven by the visible-light excitation of QDs, the color change is tunable by the quantum size effect of QDs. In addition, because the diffusion length of trapped electrons in the nanosheet is very short, the electrons easily react with oxygen molecules in the decoloration process. While the present KPFM imaging has been performed on the interface between metal sulfide quantum dots and metal oxide nanosheet, this technique can be useful to discuss the charge transport phenomena at various interfaces, such as p–n junction semiconductors, cocatalyst promoter modified metal oxide photocatalysts, and organic–inorganic hetero-junctions. The methodology described in the present study for imaging nanoscale charge carriers is expected to aid in the development of solar cells and photocatalysts, as well as unique display and memory devices.Acknowledgements
This work was supported by a grant from the JST, PRESTO and ACT-C programmes. We also thank Mr A. Genseki at the Center for Advanced Materials Analysis for help with the SEM characterization, and also acknowledge Mr Greg Newton for the critical reading of the manuscript.References
- D. Liu and P. V. Kamat, J. Phys. Chem., 1993, 97, 10769–10773 CrossRef CAS.
- R. Vogel, P. Hoyer and H. Weller, J. Phys. Chem., 1994, 98, 3183–3188 CrossRef CAS.
- L. Kronik, N. Ashkenasy, M. Leibovitch, E. Fefer, Y. Shapira, S. Gorer and G. Hodes, J. Electrochem. Soc., 1998, 145, 1748–1755 CrossRef CAS PubMed.
- A. J. Nozik, Physica E, 2002, 14, 115–120 CrossRef CAS.
- Q. Shen, D. Arae and T. Toyoda, J. Photochem. Photobiol., A, 2004, 164, 75–80 CrossRef CAS PubMed.
- A. H. Ip, S. M. Thon, S. Hoogland, O. Voznyy, D. Zhitomirsky, R. Debnath, L. Levina, L. R. Rollny, G. H. Carey, A. Fischer, K. W. Kemp, I. J. Kramer, Z. Ning, A. J. Labelle, K. W. Chou, A. Amassian and E. H. Sargent, Nat. Nanotechnol., 2012, 7, 577–582 CrossRef CAS PubMed.
- J. Jean, S. Chang, P. R. Brown, J. J. Cheng, P. H. Rekemeyer, M. G. Bawendi, S. Gradečak and V. Bulović, Adv. Mater., 2013, 25, 2790–2796 CrossRef CAS PubMed.
- Z. Liu, M. Miyauchi, Y. Uemura, Y. Cui, K. Hara, Z. Zhao, K. Sunahara and A. Furube, Appl. Phys. Lett., 2010, 96, 233107-1-3 Search PubMed.
- H. Wang, M. Miyauchi, Y. Ishikawa, A. Pyatenko, N. Koshizaki, Y. Li, L. Li, X. Li, Y. Bando and D. Golberg, J. Am. Chem. Soc., 2011, 133, 19102–19109 CrossRef CAS PubMed.
- Z. Liu, Y. Li, Z. Zhao, Y. Cui, K. Hara and M. Miyauchi, J. Mater. Chem., 2010, 20, 492–497 RSC.
- H. M. Chen, C. K. Chen, Y.-C. Chang, C.-W. Tsai, R.-S. Liu, S.-F. Hu, W.-S. Chang and K.-H. Chen, Angew. Chem., Int. Ed., 2010, 49, 5966–5969 CrossRef CAS PubMed.
- Y. Jin-nouchi, T. Hattori, Y. Sumida, M. Fujishima and H. Tada, ChemPhysChem, 2010, 11, 3592–3595 CrossRef CAS PubMed.
- Y.-L. Lee, C.-F. Chi and S.-Y. Liau, Chem. Mater., 2010, 22, 922–927 CrossRef CAS.
- K. Zhang and L. Guo, Catal. Sci. Technol., 2013, 3, 1672–1690 CAS.
- Z.-G. Zhao, Z.-F. Liu and M. Miyauchi, Adv. Funct. Mater., 2010, 20, 4162–4167 CrossRef CAS PubMed.
- M. Miyauchi, A. Kondo, D. Atarashi and E. Sakai, J. Mater. Chem. C, 2014, 2, 3732–3737 RSC.
- K. Fukuda, K. Akatsuka, Y. Ebina, R. Ma, K. Takada, I. Nakai and T. Sasaki, ACS Nano, 2008, 2, 1689–1695 CrossRef CAS PubMed.
- A. Sclafani, L. Palmisano, G. Marcí and A. M. Venezia, Sol. Energy Mater. Sol. Cells, 1998, 51, 203–219 CrossRef CAS.
- G. R. Bamwenda and H. Arakawa, Appl. Catal., A, 2001, 210, 181–191 CrossRef CAS.
- J. R. Darwent and A. Mills, J. Chem. Soc., Faraday Trans. 2, 1982, 78, 359–367 RSC.
- M. Miyauchi, Y. Nukui, D. Atarashi and E. Sakai, ACS Appl. Mater. Interfaces, 2013, 5, 9770–9776 CAS.
- N. Srinivasan, Y. Shiga, D. Atarashi, E. Sakai and M. Miyauchi, Appl. Catal., B, 2015, 179, 113–121 CrossRef CAS PubMed.
- N. M. Dimitrijevic, S. Li and M. Graetzel, J. Am. Chem. Soc., 1984, 106, 6565–6569 CrossRef CAS.
- Z. Khan, T. R. Chetia, A. K. Vardhaman, D. Barpuzary, C. V. Sastri and M. Qureshi, RSC Adv., 2012, 2, 12122–12128 RSC.
- D. V. Talapin, I. Mekis, S. Götzinger, A. Kornowski, O. Benson and H. Weller, J. Phys. Chem. B, 2004, 108, 18826–18831 CrossRef CAS.
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321–1324 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr02405f |
| This journal is © The Royal Society of Chemistry 2015 |