
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of amorphous precursors in the crystallization of La and Nd carbonates†
Beatriz
Vallina
*a,
Juan Diego
Rodriguez-Blanco
ab,
Andrew P.
Brown
c,
Jesus A.
Blanco
d and
Liane G.
Benning
*ae
aSchool of Earth and Environment, University of Leeds, Leeds LS2 9JT, UK. E-mail: earbv@leeds.ac.uk; l.g.benning@leeds.ac.uk
bNano-Science Center, Department of Chemistry, University of Copenhagen H.C. Oersted Institute, C Bygn, 9 Universitetsparken 5, DK 2100 Copenhagen, Denmark
cInstitute for Materials Research, SCaPE, Faculty of Engineering, University of Leeds, LS2 9JT, UK
dDepartamento de Física, Universidad de Oviedo, Oviedo, E-33007, Spain
eResearch Center for Geosciences, GFZ, Interface Geochemistry Section, 14473, Potsdam, Germany
First published on 12th June 2015
Abstract
Crystalline La and Nd carbonates can be formed from poorly-ordered nanoparticulate precursors, termed amorphous lanthanum carbonate (ALC) and amorphous neodymium carbonate (ANC). When reacted in air or in aqueous solutions these precursors show highly variable lifetimes and crystallization pathways. We have characterized these precursors and the crystallization pathways and products with solid-state, spectroscopic and microscopic techniques to explain the differences in crystallization mechanisms between the La and Nd systems. ALC and ANC consist of highly hydrated, 10–20 nm spherical nanoparticles with a general formula of REE2(CO3)3·5H2O (REE = La, Nd). The stabilities differ by ∼2 orders of magnitude, with ANC being far more stable than ALC. This difference is due to the Nd3+ ion having a far higher hydration energy compared to the La3+ ion. This, together with temperature and reaction times, leads to clear differences not only in the kinetics and mechanisms of crystallization of the amorphous precursor La- and Nd-carbonate phases but also in the resulting crystallite sizes and morphologies of the end products. All crystalline La and Nd carbonates developed spherulitic morphologies when crystallization occurred from hydrous phases in solution at temperatures above 60 °C (La system) and 95 °C (Nd system). We suggest that spherulitic growth occurs due to a rapid breakdown of the amorphous precursors and a concurrent rapid increase in supersaturation levels in the aqueous solution. The kinetic data show that the crystallization pathway for both La and Nd carbonate systems is dependent on the reaction temperature and the ionic potential of the REE3+ ion.
1. Introduction
The formation of crystalline carbonates from supersaturated solutions often starts with the precipitation of a phase made of nanoparticles with diameters smaller than 100 nm, and which when characterized by conventional X-ray diffraction show an amorphous character.1–4 These amorphous precursors are usually unstable and sometimes transform to nanocrystalline, often metastable intermediate phases prior to the transformation into thermodynamically stable end products.5 However, such amorphous precursors can also remain stable and not crystallize for long time periods (days–years; e.g., phosphates or amorphous silica).6,7 When they are highly unstable (seconds to minutes) they are difficult to detect with conventional techniques because of their short lifetimes.8–10Most of the in-depth research on the formation and stability of amorphous precursors has focused on mineral systems that are key in biomineralization processes (e.g., amorphous Ca–Mg carbonates4,5,11–14 or amorphous Ca phosphates15,16). However, the search for potential materials for specialized industrial applications has recently also brought attention to the formation and stability of amorphous rare-earth element (REE) carbonate phases17–19 and the corresponding crystalline end products.
REEs are moderately abundant in the Earth's crust, but they are not concentrated enough to make them easily exploitable economically. The most important REE source in the world is the Bayan Obo deposit, China, where more than 90% of world's REE raw materials are extracted,20 and relatively high concentrations of REE have also been found in deep-sea ferromanganese nodules.21 Two of the most important REEs are lanthanum (La) and neodymium (Nd) and both belong to the Light Rare Earth Element (LREE) group, which tend to concentrate in carbonates and phosphates in 8- to 10-fold coordination.22,23 La and Nd have similar f-electron configurations and their ionic radii only differ by 0.07 Å (RLa = 1.15 Å; RNd = 1.08 Å; ionic potentials of 2.60 and 2.78 Å−1, respectively24–26). La is the most abundant trivalent REE with diverse industrial and technological applications including metallurgy,27 fluorescent biological labels,28 pollution control catalysts,18,20 gas sensors and CO2 absorbing ceramics,29 while Nd is an essential additive in neodymium–iron–boron (Nd2Fe14B) permanent magnets that are crucial in consumer electronics, energy materials, defence applications18,19 and is a key component of many lasers.30 Furthermore, Nd is an important component in glass technologies31 and geochronological rock dating.20
In the last decade or so the supply of La and Nd for all these applications has become constrained while the demand for a variety of new technologies has grown.20 According to the US Department of Energy,17 La has been classified as a near-critical REE, while Nd has been identified as one of the most critically needed REEs together with dysprosium, europium, terbium and yttrium. This classification is based on a balance between the importance of REEs for the development of clean energy and the supply of such REEs in the short (present–2020) and medium (2015–2025) terms. This increasing demand has driven the study of the origin and distribution of REEs in carbonate deposits. However, there is a lack of basic data regarding the mechanisms of crystallization of REE-bearing carbonates and, in particular, the first stages of formation.
Different natural or synthetic REE-bearing carbonates are known. These include the REE carbonates lanthanite [REE2(CO3)3·8H2O], tengerite [REE2(CO3)3·2–3H2O], hydroxylbastnasite [REECO3(OH)], and kozoite [REECO3(OH)], as well as several REE-bearing oxide carbonates (REE2O2CO3). The major REE carbonates occurring in natural REE deposits also contain F− as well as OH− in their structures (see references in Table SI-1†). Many studies have addressed the crystalline products of the synthesis of REE carbonates from aqueous solutions as a function of crystallization temperature, pressure, nature and concentrations of reagents or stirring.32–34 Furthermore, some studies have focused on the effect of the ionic radii of different REEs on the crystal structure of the crystalline product.33,35 However, information about the early stages of the precipitation reactions and the role that amorphous precursors or metastable intermediates may play in the crystallization pathways is surprisingly scarce. Recently, we have shown36 that dysprosium (Dy) carbonates form via the crystallization of a highly hydrated amorphous precursor (amorphous dysprosium carbonate, Dy2(CO3)3·4H2O). A few studies mention the formation of amorphous lanthanum37,38 and neodymium carbonates39–42 during the initial stages of precipitation, but the composition, basic characteristics or stability of these La or Nd amorphous phases as well as the subsequent crystallization pathways have so far not been addressed. Thus, we still lack information about the factors that affect the stability of these amorphous precursors and whether the degree of hydration or the kinetics of dehydration and crystallization of all amorphous REE carbonates is similar.
To address this, we have carried out a study on the formation and crystallization of La and Nd carbonates from aqueous solution. We have characterized the first formed phases and followed their transformation to crystalline REE carbonates both in air and in solution over a range of temperatures (25–220 °C) and reaction times (minutes to many months). The data reveal that the amorphous La and Nd carbonate precursors have temperature-dependent stability and that depending on reaction conditions the crystallization of REE-bearing carbonates proceeds via spherulitic growth and/or via dissolution–re-precipitation. Finally, the kinetic data revealed crystallization pathways that were primarily dependent on the ionic potential of the REE and the reaction temperature.
2. Experimental
Lanthanum and neodymium carbonates were obtained by mixing a solution of La(NO3)3·6H2O (10 mM, Alfa Aesar, 99.9% purity) or Nd(NO3)3·6H2O (10 mM, Alfa Aesar, 99.9% purity) with a Na2CO3 solution (10 mM, Fisher Scientific, 99.9% purity). Experiments were carried out at room temperature (21 °C) under constant and continuous stirring. Immediately after mixing a white (La-system) or pink (Nd-system) precipitate formed. Aliquots of these precipitates were treated using four different approaches. One batch was kept dry under ambient conditions (hereafter these experiments are termed dry-ambient) and analysed for up to 6 months by X-ray diffraction (see below) to assess whether or not they crystallized. A second dry batch was reacted in air at temperatures between 25 and 1000 °C. Changes in the properties of this dried, initial precipitate (i.e., its crystallization) were monitored using thermodiffraction (termed dry-heated). The final two batches were aged in the native solution either at 21 °C for up to two months (wet-ambient), or hydrothermally at 60, 95, 165 and 220 °C for up to 7 days (hydrothermal). The hydrothermal treatments were carried out in Teflon-lined stainless steel (40 mL) vessels at saturated water vapour pressures. Both the wet-ambient and hydrothermal suspensions were shaken regularly. All experiments were repeated a minimum of three and a maximum of six times.The initial precipitates, various intermediate products as well as the reaction products at the end of each treatment were quenched to room temperature and vacuum filtered through 0.2 μm polycarbonate membranes where required. The resulting solids were washed with water and isopropanol following the method described by Rodriguez-Blanco et al. (2008).4 All solid phases were characterized by powder X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), and scanning electron microscopy (SEM). The dried initial precipitates were also characterized by thermogravimetric analyses (TGA) and high-resolution transmission electron microscopy (HR-TEM), and crystallization was followed by time- and temperature-resolved powder X-ray thermodiffraction (XRTD).
Conventional powder XRD patterns were collected using a Bruker D8 powder X-ray diffractometer (CuKα1; 2θ range 10–75; 0.005° per step and 0.1 s per step), while powder XRTD was carried out in air using a Panalytical X'Pert Pro diffractometer equipped with an Anton Paar HTK 1200N High-Temperature Oven-Chamber (CuKα1,2; 2θ range 20–50 at 0.01° per step and 0.16 s per step; constant heating rate of 1 °C min−1 from 25 to 1000 °C). Crystallite sizes were calculated from the diffraction patterns using the Scherrer equation,43 with the assumption that the particles were stress-free and with pattern-matching refinement of the crystalline phases carried out using the Rietveld refinement software TOPAS.44 Furthermore, all crystallite sizes were calculated taking into account the instrumental line-broadening of the diffractometer by using an X-ray pattern of a silicon standard (2θ111 = 28.46° and FWHM = 0.049°). FTIR spectra were recorded on an A2-Technology Microlab Portable mid-IR spectrometer with a diamond internal reflection cell (DATR). The spectra were collected by adding 1024 scans in the 650–4000 cm−1 range at a resolution of 4 cm−1. Themogravimetric analyses (TGA) were carried out with a Mettler TA 4000 instrument, while heating the samples from 25 to 1000 °C at a rate of 10 °C min−1 under a N2 atmosphere. Images of the solids were acquired by field emission gun scanning electron microscopy (FEG-SEM, LEO 1530 Gemini, operated at 3 kV and with an in-lens detector, equipped with an Oxford Instruments energy-dispersive X-ray (EDX) analysis system; Isis) and by FEG-transmission electron microscopy (FEG-TEM: FEI CM200; operated at 197 kV and equipped with an Oxford Instruments energy-dispersive X-ray (EDX) analysis system (Isis) and a Gatan Imaging Filter (GIF-200)).
In addition, we carried out turbidity experiments by mixing a solution of La(NO3)3·6H2O or Nd(NO3)3·6H2O (10 mM) with a solution of Na2CO3 (10 mM) in a cuvette inside a stirring module of a double beam UV-VIS spectrophotometer (Uvikon XL, SECOMAN Ltd). The development of turbidity was followed in situ and in a time-resolved manner by changes in UV-VIS spectra recorded at a wavelength of 450 nm every second for up to 17 hours to assess the formation and real time transformation of the initial precipitates.
Lastly, the saturation indexes (SI) of lanthanites (both La and Nd forms) in the initial solutions and prior to the formation of the first precipitates were calculated with the geochemical code PHREEQC45 using the only available solubility products for lanthanite-(La or Nd) in the literature.46 The saturation index is defined as
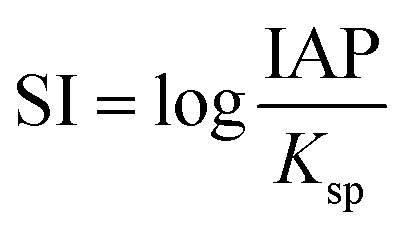 | (1) |
3. Results
3.1. The initial La and Nd precipitates and crystallisation under dry conditions
Upon mixing of the La(NO3)3·6H2O or Nd(NO3)3·6H2O and Na2CO3 starting solutions a white (La) or pink (Nd) precipitate was formed, respectively. The X-ray patterns of these precipitates were quite similar, showing only three humps centred at ∼15–20, 30 and 45° 2θ (marked with * in Fig. 1a and b, bottom patterns) indicating the presence of solely amorphous materials in both systems. Interestingly, both these amorphous phases remained stable when stored in a dry state in air (dry-ambient; Table 1), but the lifetimes were very different. The La-bearing amorphous phase stored in a dry state remained stable for ∼48 hours before crystallizing to lanthanite-(La) [La2(CO3)3·8H2O], ICSD-22224,47 while the dry Nd-bearing amorphous phase remained stable for up to 6 months. When dry-heated between 25 °C and 1000 °C with simultaneous recording of thermodiffraction patterns these amorphous phases behaved significantly different. The amorphous La-bearing precursor remained amorphous when heated up to 400 °C (lower patterns in Fig. 1a) and only at 450 °C did it start to transform into a crystalline compound identified as monoclinic La-dioxycarbonate (La2O2CO3; patterns at 450 ad 550 °C in Fig. 1a; ICDD PDF2 048-1113). Upon further heating to 700 °C this monoclinic La-dioxycarbonate transformed to hexagonal La-dioxycarbonate (La2O2CO3; ICDD PDF2 037-0804; pattern at 700 °C, Fig. 1a). Above 700 °C the hexagonal La-dioxycarbonate transformed to hexagonal La-oxide (La2O3; ICDD PDF2 005-0602), which remained the sole phase present between 800 and 1000 °C (Fig. 1a, upper pattern). In the case of the amorphous Nd-bearing phase, the thermodiffraction patterns revealed no Bragg peaks up to 450 °C (lower 3 patterns in Fig. 1b). By 550 °C the amorphous carbonate precursor transformed to monoclinic Nd-dioxycarbonate (Nd2O2CO3; Fig. 1b, pattern at 550 °C; ICDD PDF2 023-0421), at 700 °C (Fig. 1b, pattern at 700 °C) to cubic Nd-oxide (Nd2O3; ICDD PDF2 03-065-3184) and at 800 °C, the cubic Nd-oxide had transformed to hexagonal Nd-oxide (Nd2O3; Fig. 1b, upper pattern; ICDD PDF2 041-1089). This latter phase remained the only phase in all patterns up to 1000 °C.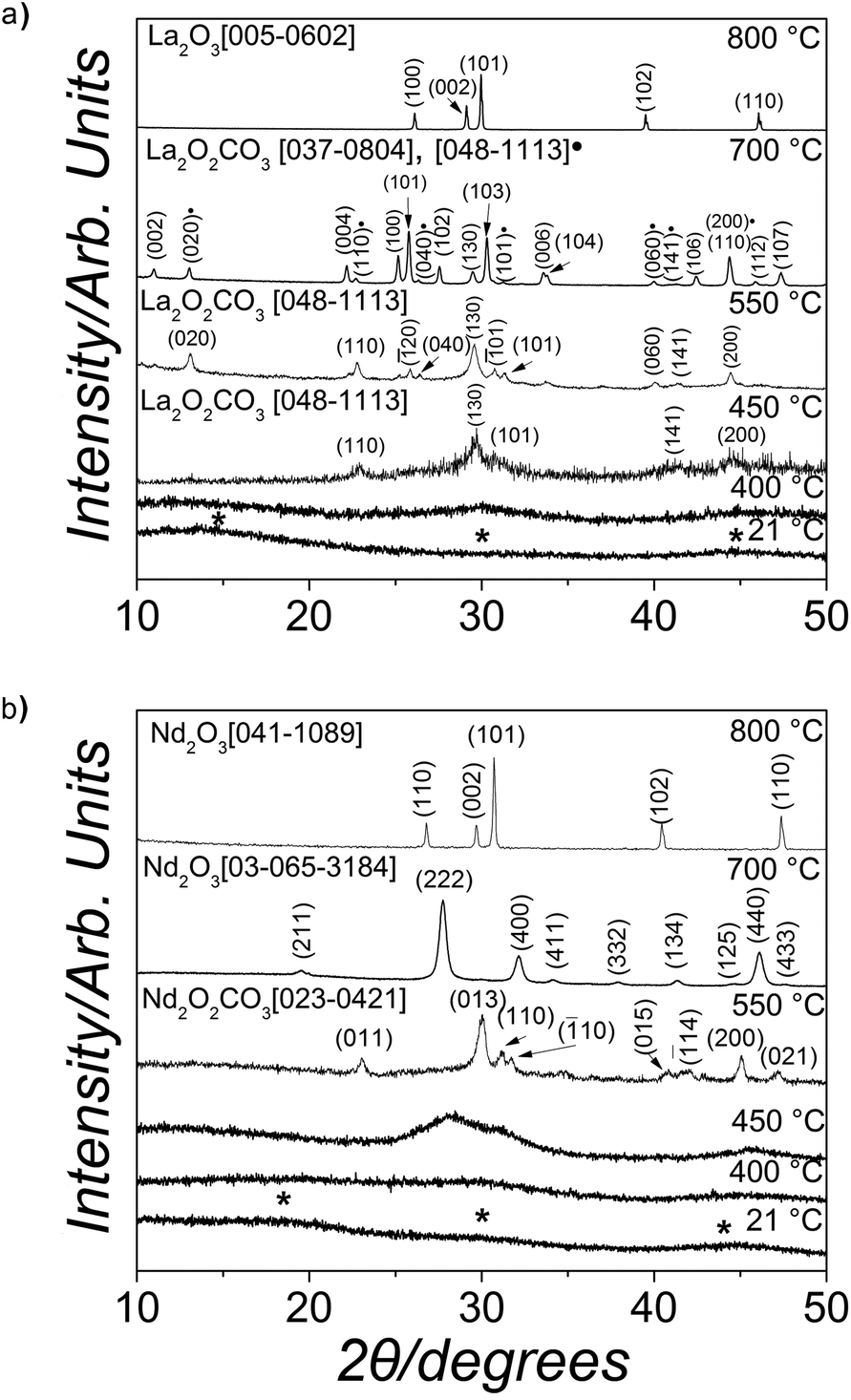 | ||
| Fig. 1 Selected powder X-ray diffraction patterns recorded from the lanthanum (a) and neodymium (b) solid precursor phases (bottom patterns in both cases) and their crystallization products when dry-heated from 25 °C to 1000 °C. The asterisks in the bottom patterns indicate the position of the humps in the amorphous precursors. | ||
| Exp. conditions | Temp. [°C] | Time [h] | Identity of the solid phase(s) (in the order of abundance) | Morphology of the most abundant phase (TEM/SEM) |
|---|---|---|---|---|
| Dry-ambient | 25 | 48 | ALC, (La2(CO3)3·5H2O), (x < 5), amorphous | Spheres |
| >48 | Lanthanite-(La) [La2(CO3)3·8H2O] (100%) | — | ||
| Dry-heated | 25–1000 | 0–13 (1 °C min−1 ramp) | ALC, (La2(CO3)3·5H2O), (x < 5), amorphous (≤400 °C) | Spheres |
| Monoclinic La-dioxycarbonate, La2O2CO3, a = 4.080 Å, b = 13.509 Å, c = 4.072 Å (450–700 °C) | (Not imaged) | |||
| Hexagonal La-dioxycarbonate, La2O2CO3, a = b = 4.078 Å, c = 15.950 Å (∼700–800 °C) | (Not imaged) | |||
| La-oxide, La2O3 (≥800 °C) | (Not imaged) | |||
| Wet-ambient | 21 | 4 | Lanthanite-(La) [La2(CO3)3·8H2O] (100%) | Rosettes |
| 24 | Lanthanite-(La) [La2(CO3)3·8H2O] (100%) | Rosettes | ||
| 1440 | Kozoite-(La) [LaCO3(OH)] (100%) | Rhombohedral prisms | ||
| Hydrothermal | 60 | 4 | Lanthanite-(La) [La2(CO3)3·8H2O] (100%) | Rosettes |
| 14 | Lanthanite-(La) [La2(CO3)3·8H2O] (100%) | Rosettes | ||
| 24 | Lanthanite-(La) [La2(CO3)3·8H2O] (∼60%) | Rosettes | ||
| Kozoite-(La) [LaCO3(OH)] (∼40%) | Ovoidal particles | |||
| 48 | Kozoite-(La) [LaCO3(OH)] (100%) | Dumbbell shapes | ||
| 95 | 1 | Lanthanite-(La) [La2(CO3)3·8H2O] (∼83%) | Rosettes | |
| Kozoite-(La) [LaCO3(OH)] (∼17%) | Oval particles | |||
| 3 | Kozoite-(La) [LaCO3(OH)] (∼99%) | Dumbbell shapes | ||
| Lanthanite-(La) [La2(CO3)3·8H2O] (∼1%) | Rosettes | |||
| 24 | Kozoite-(La) [LaCO3(OH)] (100%) | Dumbbell shapes | ||
| 165 | 4 | Kozoite-(La) [LaCO3(OH)] (∼88%) | Dumbbell shapes | |
| Hydroxylbastnasite-(La) [LaCO3(OH)] (∼12%) | Triangular pyramids | |||
| 24 | Kozoite-(La) [LaCO3(OH)] (∼70%) | Dumbbell shapes | ||
| Hydroxylbastnasite-(La) [LaCO3(OH)] (∼30%) | Triangular pyramids | |||
| 220 | 4 | Kozoite-(La) [LaCO3(OH)] (∼32%) | Dumbbell shapes | |
| Hydroxylbastnasite-(La) LaCO3(OH) (∼68%) | Triangular pyramids | |||
| 24 | Hydroxylbastnasite-(La) LaCO3(OH) (∼76%) | Triangular pyramids | ||
| Kozoite-(La) [LaCO3(OH)] (∼24%) | Dumbbell-like shapes | |||
| 168 | Hydroxylbastnasite-(La) LaCO3(OH) (100%) | Triangular pyramids |
The amorphous nature of both precursor materials identified by XRD was confirmed by high-resolution TEM. The images of these pristine phases revealed roughly spherical nanoparticles with diameters between 10 and 20 nm (Fig. 2a) that show no crystallinity by selected area electron diffraction (data not shown). Standardless quantification of EDX spectra (inset in Fig. 2a) from these amorphous nanoparticles revealed a La![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O atomic ratio of ∼2:8. The particles rapidly crystallized when exposed to the electron beam of the TEM for a few seconds, resulting in nanocrystals with an interplanar spacing of ∼2.9 Å (Fig. 2b). This value corresponds to the (101) d-spacing of hexagonal La-oxide (La2O3; ICDD PDF2 005-0602). The crystallization of the amorphous starting material to hexagonal La-oxide was also confirmed by quantification from EDX spectra, which showed a decrease in the amount of O from ∼2:8 La:O ratio in the amorphous precursor to ∼2:3 La:O ratio in La2O3. A similar behaviour was found for the Nd precursor material. High-resolution TEM images of the pristine Nd precursor showed no significant differences in morphology compared to the La bearing phase (Fig. 2c) and the electron beam also crystallized the initial nanoparticles after a few seconds of exposure. The newly formed nanocrystals showed an interplanar spacing of ∼3.2 Å (Fig. 2d), which matched with the (222) d-spacing of the cubic Nd-oxide (Nd2O3; ICDD PDF2 03-065-3184). EDX spectra from the neodymium amorphous precursor and cubic Nd-oxide showed the same Nd:O ratios as those obtained in the experiment with La (i.e. ∼2:8 and ∼2:3, before and after significant electron beam exposure respectively).
:O atomic ratio of ∼2:8. The particles rapidly crystallized when exposed to the electron beam of the TEM for a few seconds, resulting in nanocrystals with an interplanar spacing of ∼2.9 Å (Fig. 2b). This value corresponds to the (101) d-spacing of hexagonal La-oxide (La2O3; ICDD PDF2 005-0602). The crystallization of the amorphous starting material to hexagonal La-oxide was also confirmed by quantification from EDX spectra, which showed a decrease in the amount of O from ∼2:8 La:O ratio in the amorphous precursor to ∼2:3 La:O ratio in La2O3. A similar behaviour was found for the Nd precursor material. High-resolution TEM images of the pristine Nd precursor showed no significant differences in morphology compared to the La bearing phase (Fig. 2c) and the electron beam also crystallized the initial nanoparticles after a few seconds of exposure. The newly formed nanocrystals showed an interplanar spacing of ∼3.2 Å (Fig. 2d), which matched with the (222) d-spacing of the cubic Nd-oxide (Nd2O3; ICDD PDF2 03-065-3184). EDX spectra from the neodymium amorphous precursor and cubic Nd-oxide showed the same Nd:O ratios as those obtained in the experiment with La (i.e. ∼2:8 and ∼2:3, before and after significant electron beam exposure respectively).
 | ||
| Fig. 2 TEM images of the pristine amorphous lanthanum (a) and neodymium (c) carbonate precursor phases and the crystalline transformation products produced by irradiation with the high energy electron beam of the TEM for a few seconds (b and d respectively). The insets show the respective EDX spectra with the Cu peaks coming from the Cu support grid. Standardless quantification from these spectra gave a La:O and Nd:O atomic ratio of 2:8. | ||
The FTIR analysis of the amorphous La and Nd phases revealed identical ionic vibrations. Both spectra are characterized by typical carbonate and O–H vibrations (Fig. 3, lower and upper spectra correspond to amorphous La and Nd carbonate, respectively). The broad band between ∼2500 and 3700 cm−1 (marked as band 1; detailed assignments, see Table SI-2†) represents O–H stretching vibrations and corresponds to structural water. The most intense vibrations in both precursors were located between ∼1455 and 679 cm−1 (bands 2–8) and are characteristic of the main stretching vibrations of the carbonate ions. Combined with the X-ray thermodiffraction results (Fig. 1), this confirms that both La and Nd precursors are amorphous and strongly hydrated La and Nd carbonates. These phases will for simplicity hereafter be called ALC and ANC respectively, in line with other amorphous carbonates in the literature (e.g., amorphous calcium carbonate, ACC4,11,48).
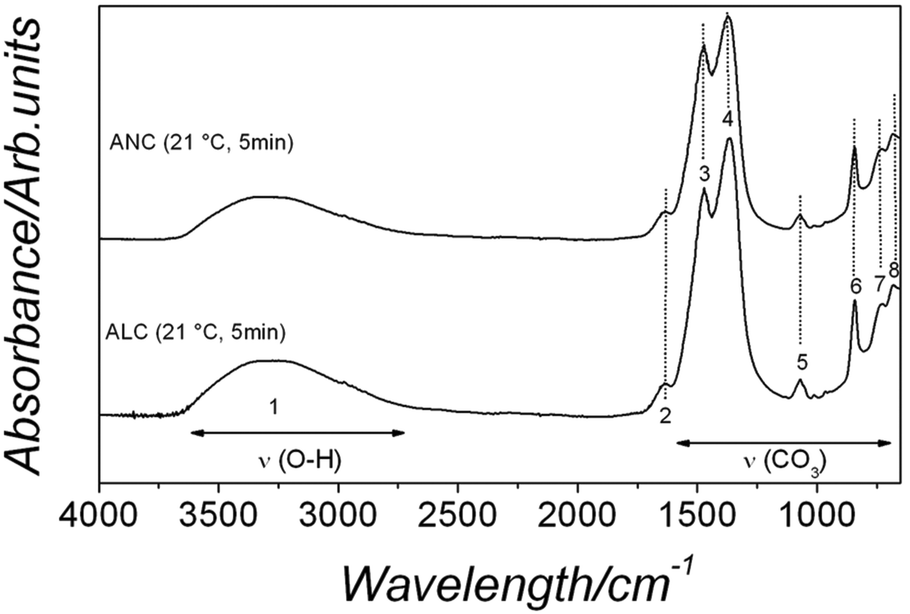 | ||
| Fig. 3 FTIR spectra of ALC and ANC showing the large OH band (1) and the principal carbonate bands (2–6). Details of the band assignments are presented in Table SI-1† and discussed in the text. | ||
Thermogravimetric analyses (Fig. 4a and b) show that upon gradual heating the total weight loss of ALC and ANC was ∼40%. For ALC, ∼16% of the total weight loss corresponds to the release of water at ∼100 °C (corresponding to the equivalent of five water molecules per formula unit [pfu]), while the remaining 23% mass loss was due to the decomposition of the carbonate at about 450 °C and its full transformation to hexagonal La-oxide above ∼700 °C. The dehydration and decarbonation processes of both amorphous precursors followed the same steps. However, in the case of ANC, the small weight loss above ∼700 °C (∼1%) was slower and more progressive compared to ALC (Fig. 4).
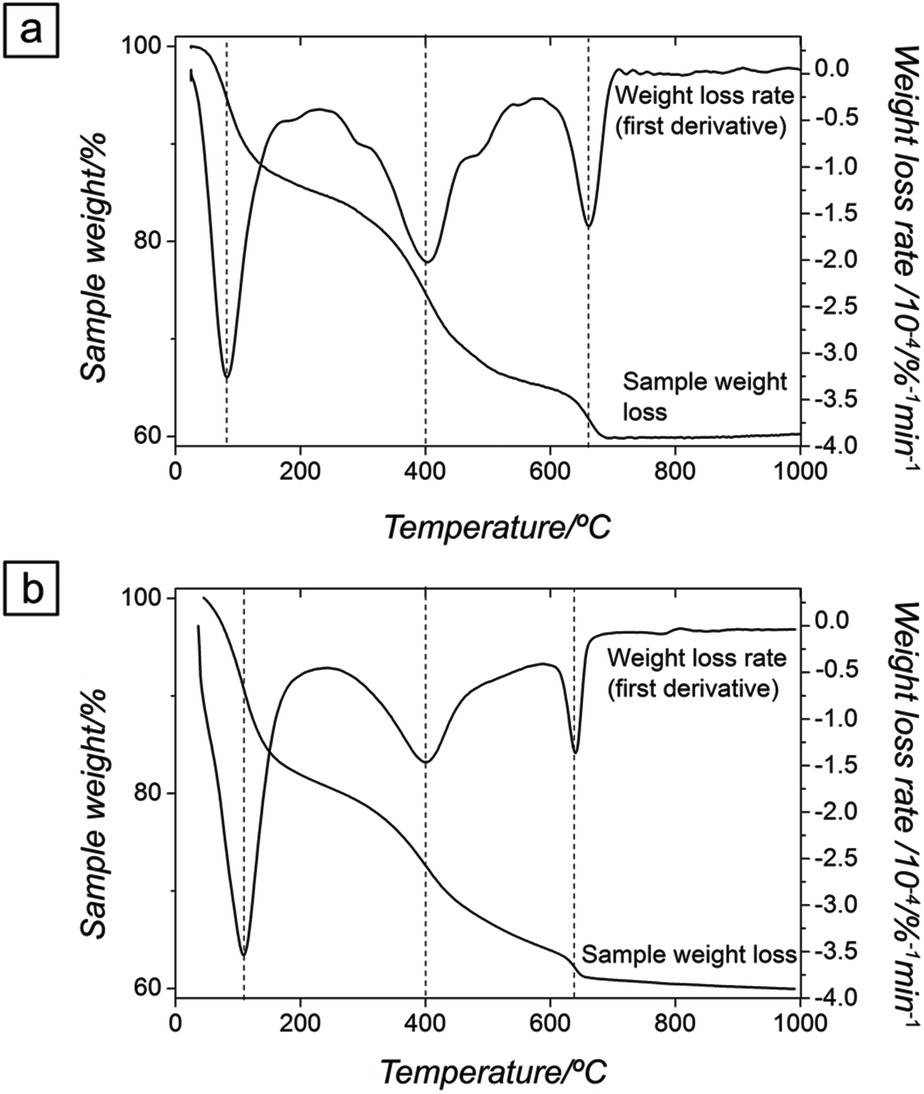 | ||
| Fig. 4 TGA (sample weight loss and weight loss rate curves) of the poorly-ordered precursor: ALC (a), ANC (b). The curves show a progressive loss of water and the final carbonate decomposition between 450 and ∼650 °C. | ||
3.2. Crystallization of ALC and ANC in aqueous solution
Compared to the dry conditions, the behaviour of ALC and ANC was completely different when reacted in the solutions in which they were initially formed (either under wet-ambient or hydrothermal conditions). Both ALC and ANC crystallized much faster compared to the dry conditions. Their crystallization also followed different pathways that were dependent on composition, temperature and reaction times (Tables 1 and 2).| Exp. conditions | Temp. [°C] | Time [h] | Identity of the solid phase(s) (in the order of abundance) | Morphology of the most abundant phase (TEM/SEM) |
|---|---|---|---|---|
| Dry-ambient | 25 | 4320 (6 months) | ANC, Nd2(CO3)3·xH2O (x < 5), amorphous | Spheres |
| Dry-heated | 25–1000 | 0–13 (1 °C per min ramp) | ANC, Nd2(CO3)3·xH2O (x < 5), amorphous (≤450 °C) | Spheres |
| Hexagonal Nd-dioxycarbonate Nd2O2CO3 (a = b = 3.988 Å, c = 13.120 Å) (∼550 °C) | (Not imaged) | |||
| Cubic Nd-oxide Nd2O3 (a = b = c = 11.072 Å) (∼700 °C) | (Not imaged) | |||
| Hexagonal Nd-oxide Nd2O3 (a = b = 3.830 Å, c = 5.999 Å) (≥800 °C) | (Not imaged) | |||
| Wet-ambient | 21 | 4 | ANC, Nd2(CO3)3·xH2O (x < 5), amorphous | Spheres |
| 24 | Lanthanite-(Nd) [Nd2(CO3)3·8H2O] | Crystal plates | ||
| 1440 | Tengerite-(Nd) [Nd2(CO3)3·2.5H2O)] (∼93%) | Acicular crystals | ||
| Lanthanite-(Nd) [Nd2(CO3)3·8H2O] (∼7%) | Crystal plates | |||
| Hydrothermal | 60 | 4 | ANC, Nd2(CO3)3·xH2O (x < 5), amorphous | Spheres |
| 14 | Tengerite-(Nd) [Nd2(CO3)3·2.5H2O)] (100%) | Acicular crystals | ||
| 24 | Tengerite-(Nd) [Nd2(CO3)3·2.5H2O)] (100%) | Acicular crystals | ||
| 48 | Tengerite-(Nd) [Nd2(CO3)3·2.5H2O)] (∼95%) | Acicular crystals | ||
| Kozoite-(Nd) [NdCO3(OH)] (∼4%) | Oval particles | |||
| 95 | 1 | ANC, Nd2(CO3)3·xH2O (x < 5), amorphous | Spheres | |
| 3 | Tengerite-(Nd) [Nd2(CO3)3·2.5H2O)] (∼60%) | Crystal plates (spherulitic) | ||
| Kozoite-(Nd) [NdCO3(OH)] (∼40%) | Dumbbells (spherulitic) | |||
| 24 | Kozoite-(Nd) [NdCO3(OH)] (100%) | Dumbbells (spherulitic) | ||
| 165 | 4 | Kozoite-(Nd) [NdCO3(OH)] (∼88%) | Elongated prisms | |
| Hydroxylbastnasite-(Nd) [NdCO3(OH)] (∼12%) | Triangular pyramids | |||
| 24 | Kozoite-(Nd) [NdCO3(OH)] (∼65%) | Elongated prisms | ||
| Hydroxylbastnasite-(Nd) [NdCO3(OH)] (∼35%) | Triangular pyramids | |||
| 220 | 4 | Kozoite-(Nd) [NdCO3(OH)] (∼41%); (*) | Elongated prisms | |
| Hydroxylbastnasite-(Nd) [NdCO3(OH)] (∼59%) | Triangular pyramids | |||
| 24 | Hydroxylbastnasite-(Nd) [NdCO3(OH) (∼99%) | Triangular pyramids | ||
| Kozoite-(Nd) [NdCO3(OH)] (∼1%) (*) | Elongated prims | |||
| 168 | Hydroxylbastnasite-(Nd) [NdCO3(OH)] (100%) | Triangular pyramids |
Under ambient conditions the development of turbidity in both systems when followed by in situ and time resolved UV-Vis spectroscopy (Fig. 5) revealed that in the ALC case, after ∼2 minutes the turbidity started to increase as a consequence of the progressive transformation of ALC to crystalline La carbonate phases. The absorbance reached a maximum value after 8 minutes, indicating the completion of the crystallization reaction. The subsequent decrease in turbidity is most probably a consequence of sedimentation of the crystalline particles. The end products of these experiments (Fig. 6a, bottom pattern; Table 1) were identified by XRD as the highly hydrated but crystalline lanthanite-(La) [La2(CO3)3·8H2O] (ICSD-2222447). Conversely, ANC, which also formed immediately after mixing, remained stable for more than ∼2 hours before it started to crystallize and its crystallization took ∼15 hours. The end product was also identified as a lanthanite-(Nd) ([Nd2(CO3)3·8H2O]; Fig. 6b, bottom pattern; Table 1). In both systems after 60 days at ambient temperature these lanthanite octahydrates recrystallized and transformed to kozoite-(La) [LaCO3(OH)] (ICSD 6180) in the La system, while in the Nd system, ∼90% of the initial lanthanite-(Nd) transformed to tengerite-(Nd) [Nd2(CO3)3·2–3H2O] (ICSD 1554; Fig. 6b and Table 2).
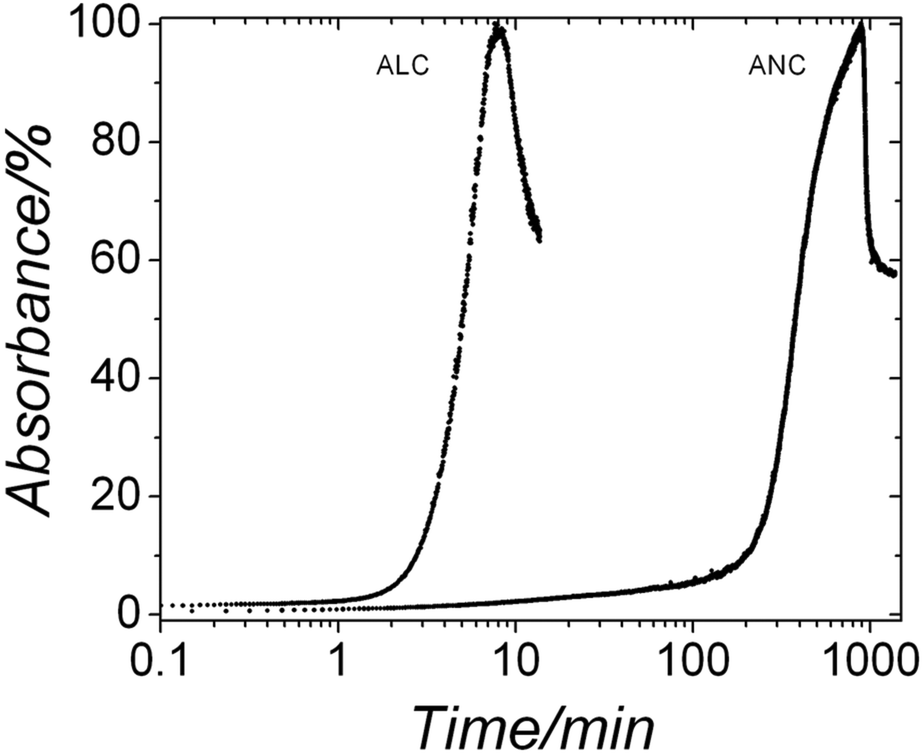 | ||
| Fig. 5 Turbidity curves from the in situ and time resolved UV-VIS experiments showing the evolution of the crystallization of ALC and ANC in aqueous solution at ambient temperature. | ||
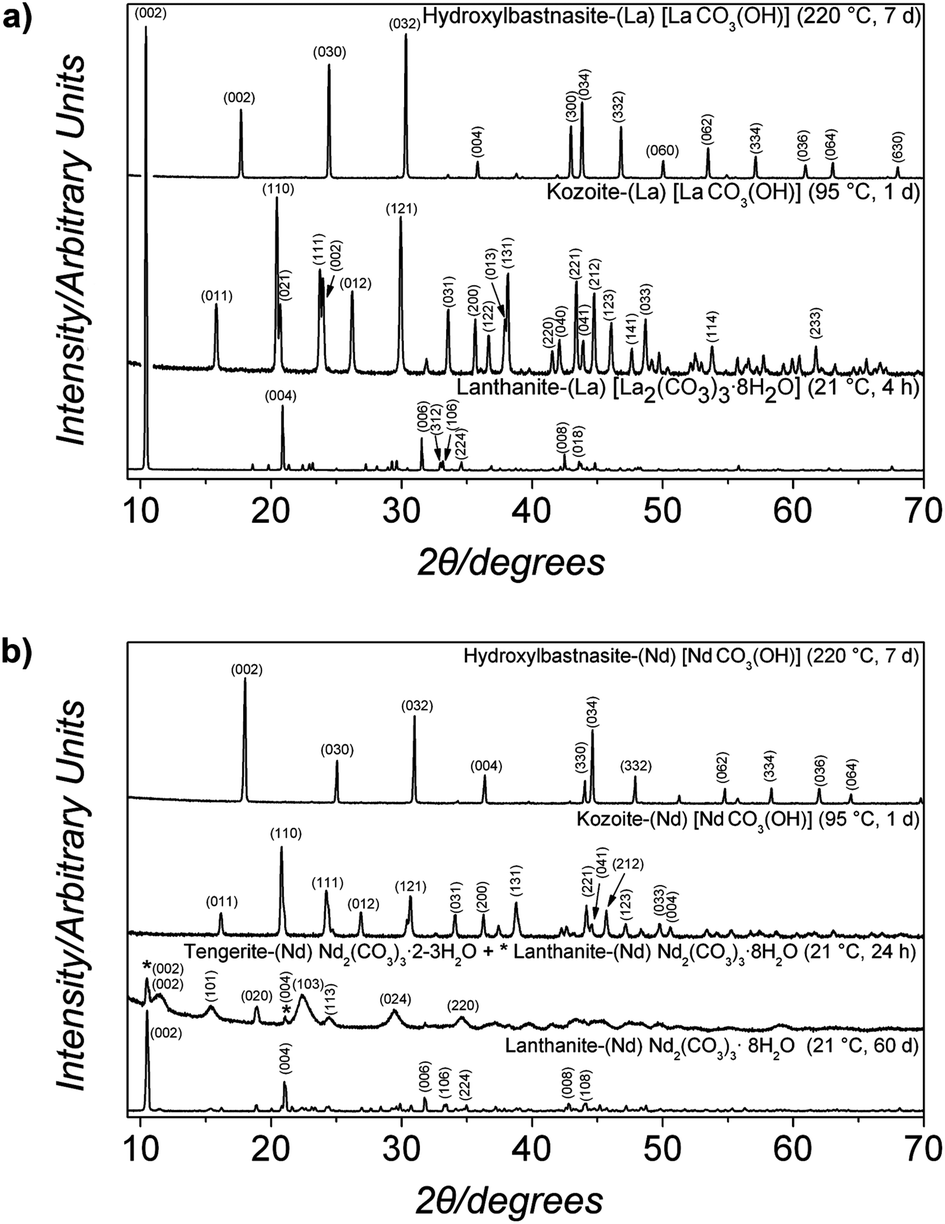 | ||
| Fig. 6 Powder X-ray diffraction patterns of the solids obtained in the (a) La and (b) Nd systems under wet-ambient conditions (21 °C) and under hydrothermal (60–220 °C) conditions. The patterns were indexed to (a) La2(CO3)3·8H2O, kozoite-(La) LaCO3(OH) and hydroxylbastnasite-(La) [LaCO3(OH)] and (b) Nd2(CO3)3·2–3H2O, Nd2(CO3)3·8H2O, kozoite-(Nd) [NdCO3(OH)], hydroxylbastnasite-(Nd) [NdCO3(OH)]. Full details about the indexing are presented in Fig. SI-1.† | ||
When the transformation was carried out also in aqueous media but at 60 °C (hydrothermal), ALC showed a similar behaviour to that at 21 °C. After 4 hours lanthanite-(La) was the only phase detected (Fig. 6a, lower pattern, and Table 1), after 24 hours ∼40% transformed to kozoite-(La), and the transformation was complete within 48 hours. Conversely, ANC transformed at 60 °C directly to tengerite-(Nd) within 14 hours with no lanthanite-(Nd) intermediate. After 48 hours only <5% of tengerite-(Nd) transformed to kozoite-(Nd) [NdCO3(OH)] (ICSD 6180). Not surprisingly, with increasing temperatures at 95 °C, the transformation was faster with the lanthanite-(La) being almost fully transformed to kozoite-(La) after only 3 hours (Fig. 6a, middle pattern; Table 1). However, it is worth noting that ANC remained stable for at least 1 hour before crystallizing to tengerite-(Nd) and then transforming completely to kozoite-(Nd) within 24 hours (middle pattern in Fig. 6b; Table 2).
When the amorphous precursors were reacted hydrothermally at temperatures above 95 °C, only anhydrous crystalline compounds were observed in both systems (Table 2). At 165 °C, kozoite-(Nd) was the main phase formed within the first 24 hours and with time its crystallization to a different polymorph, hydroxylbastnasite-(Nd) [NdCO3(OH)] (ICSD 2880), was observed. The isomorph, hydroxylbastnasite-(La) [LaCO3(OH)],35,49 was the product of the transformation of kozoite-(La) [LaCO3(OH)] in the La system (Table 1). The phase evolution of the experiments carried out at 220 °C is similar to those at 165 °C in that at both temperatures, the final product was a hydroxylbastnasite-(REE) (Fig. 6a and b, upper patterns). In the La system, kozoite-(La) was always observed at the beginning of the experiment and its transformation to hydroxylbastnasite-(La) [LaCO3(OH)] was completed within 7 days. This evolution was similar in the Nd system, but in some cases kozoite-(Nd) was not observed and hydroxylbastnasite-(Nd) formed directly from ANC within the first three hours of reaction.
The comparison between the XRD patterns of REE-bearing carbonate isomorphs obtained in identical experiments in the La and Nd systems showed larger unit cell parameters and Scherrer crystallite sizes for the La-bearing carbonates (lanthanite, kozoite, hydroxylbastnasite) than for the Nd-bearing isomorphs (Table 3).
| Mineral | Cell parameters | La | Nd | Unit cell parameters as a function of the ionic radius of the RE3+ ion | Crystallite size, La [nm] | Crystallite size, Nd [nm] |
|---|---|---|---|---|---|---|
| Lanthanite (21 °C) | a [Å] | 8.9894 | 8.8889 | a = 1.4357 r + 7.3383 | >200 | 31 |
| b [Å] | 9.5734 | 9.4299 | b = 2.05 r + 7.2159 | |||
| c [Å] | 17.0265 | 16.8996 | c = 1.8129 r + 14.942 | |||
| Vol [Å3] | 1465 | 1416 | — | |||
| Tengerite (60 °C) | a [Å] | — | 6.2288 | — | — | 14 |
| b [Å] | — | 9.4316 | — | |||
| c [Å] | — | 15.5623 | — | |||
| Vol [Å3] | — | 914 | — | |||
| Kozoite (165 °C) | a [Å] | 5.0357 | 4.9662 | a = 0.9929 r + 3.8939 | 113 | 54 |
| b [Å] | 8.5835 | 8.4982 | b = 1.2186 r + 7.1821 | |||
| c [Å] | 7.4023 | 7.2325 | c = 2.4257 r + 4.6127 | |||
| Vol [Å3] | 320 | 305 | — | |||
| Hydroxylbastnasite (220 °C) | a [Å] | 12.6294(95) | 12.3579 | a = 3.8786 r + 8.169 | 121 | 109 |
| c [Å] | 10.0321(78) | 9.9025 | c = 1.8514 r + 7.903 | |||
| Vol [Å3] | 1386 | 1309 | — |
The FEG-SEM images (Fig. 7 and 8) produced in the wet-ambient (21 °C) experiments showed that the first crystalline La-carbonate phase formed from the ALC, lanthanite-(La), consisted of small (2–4 μm) crystal plates in rosette-like aggregates with a maximum diameter of ∼10 μm (Fig. 7a). After a long reaction time (60 days) these transformed into 1–2 μm sized kozoite-La prisms (Fig. 7b). In the hydrothermal experiments at 60 and 95 °C, small oval particles (<3 μm in size) were observed (Fig. 7c), corresponding to the neoformed crystals of kozoite-(La) identified by XRD at 24 h (60 °C) and 1 h (95 °C). After further aging or above 165 °C, kozoite-(La) was present as well-developed elongated sub-micron-sized prisms grouped in dumbbell shaped aggregates (up to 8 μm in size; Fig. 7d). Finally, these elongated prisms transformed to triangular pyramids of hydroxylbastnasite-(La) (<3 μm in size, Fig. 7e), which were the crystalline end products at 165 and 220 °C (Fig. 7f).
 | ||
| Fig. 7 FEG-SEM images of the crystalline lanthanum end product phases of the wet-ambient and hydrothermal treatments: (a) La2(CO3)3·8H2O rosettes obtained at 21 °C. (b) Well-faceted prisms of kozoite-(La) obtained at 21 °C. (c) La2(CO3)3·8H2O crystals and ellipsoidal crystals of kozoite-(La) at the beginning of the transformation and (d) dumbbell-shaped kozoite-(La) obtained at 165 °C. (e) Transformation of the dumbbell-shaped kozoite-(La) into triangular hydroxylbastnasite-(La) pyramids and (f) hydroxylbastnasite-(La) pyramids obtained as the final product of the hydrothermal treatments at 165 °C; the same final products with the same shapes were also obtained at 220 °C. | ||
 | ||
| Fig. 8 FEG-SEM images of the crystalline neodymium end product phases of the wet-ambient and hydrothermal treatments: (a) crystal plates of Nd2(CO3)3·8H2O obtained at 21 °C. (b) Acicular needle-like crystals of tengerite-(Nd) obtained at 60 °C. (c) Spherulitic, plate-like tengerite-(Nd) obtained at 95 °C. (d) Dumbbell-shaped kozoite-(Nd) obtained at 95 °C. (e) Triangular hydroxylbastnasite-(Nd) pyramids obtained at 165 °C. (f) Spherulitic hydroxylbastnasite-(Nd) [Nd(CO3)OH] obtained as a direct product of the transformation of the amorphous Nd carbonate precursor, ANC, at 220 °C. | ||
The morphologies observed in the Nd system (Fig. 8) at 21 °C and after 4 hours were individual, non-aggregated crystal plates of lanthanite-(Nd) (up to 12 μm in size, Fig. 8a) that transformed within 2 months to acicular tengerite-(Nd) crystals with a maximum length of 3 μm (Fig. 8b). At 95 °C these tengerite-(Nd) crystals clustered into sheets with dumbbell shapes (Fig. 8c). After several hours these in turn transformed to kozoite-(Nd) (Fig. 8d), whose morphology was similar to that described above for kozoite-(La). Kozoite-(Nd) was also observed at 165 °C and at 220 °C, forming directly from ANC (Fig. SI-4†). Finally, the morphology of hydroxylbastnasite-(Nd) formed from kozoite-(Nd) at 165 and 220 °C was similar to its La counterpart (Fig. 8e). However, in some experiments at 220 °C hydroxylbastnasite-(Nd) formed directly from ANC and lead to the formation of 10–20 μm cauliflower-like structures composed of micrometer-sized ellipsoidal crystals (Fig. 8f).
The FTIR spectra of the crystalline phases are dominated by much sharper carbonate vibrations compared with the respective amorphous precursors (Fig. SI-2 and SI-3, Table SI-2†). Furthermore, in both systems the OH vibrations of the lanthanites are much bigger confirming the higher water content (8 water molecules pfu) compared to 5 water molecules pfu in the amorphous precursor, consistent with the TGA data (Fig. 4). There is also a clear decrease of the structural water content at higher temperatures, again confirming the transition from highly hydrous crystalline lanthanites to the hydroxylated La and Nd carbonate phases. Furthermore, multiple band splittings were observed in the spectra of the crystalline carbonates. For example, the ν3 asym. CO3 peak at 1409 cm−1 (band 16 in Fig. SI-2a, SI-3a, and Table SI-2†) in both the La and Nd kozoite (La/NdCO3OH) appear as a split peak in the La and Nd- hydroxylbastnasite (La/NdCO3OH) with two peaks at 1425 and 1392 (bands 21 and 22 in Fig. SI-2a and SI-3a; Table SI-2†).
4. Discussion
4.1. Stability of amorphous La and Nd carbonates
The crystallization of La and Nd carbonates takes place via poorly-ordered hydrated metastable precursors. Taking into account the La:O or Nd:O ratios of 2:8 from the EDX analyses (Fig. 2), the presence of water and carbonate confirmed by FTIR (Fig. 3) and the weight loss shown by thermogravimetric analyses (Fig. 4), we deduced an idealized chemical formula for ALC and ANC as La2(CO3)3·5H2O and Nd2(CO3)3·5H2O respectively. The decomposition reactions upon dry-heating proceeded via the reactions:| La2(CO3)3·5H2O → La2O3 + 5H2O + 3CO2 |
| Nd2(CO3)3·5H2O → Nd2O3 + 5H2O + 3CO2. |
Our UV-Vis results (Fig. 5) revealed that the lifetimes of ALC and ANC were ∼9 and ∼900 min respectively; ∼1 and ∼2 orders of magnitude longer than those measured for the pure amorphous calcium carbonate (ACC), which is stable for <2 minutes at 21 °C.5,13 Finally, our hydrothermal data (Tables 1 and 2) show that ANC was stable for at least 1 hour even when reacted at 95 °C. Interestingly, two other amorphous REE carbonates are known to be even more stable than ANC. These are amorphous Dy carbonate (∼3 days36) and amorphous Yb carbonate (AYbC), which do not crystallize after one week of reaction at 120 °C.32 Taken together, all these data show that there is a direct proportionality between the ionic potential of the REE3+ (2.60 Å−1 for La3+, 2.78 Å−1 for Nd3+, 3.03 Å−1 for Dy3+ and 3.22 Å−1 for Yb3+) and the lifetimes of the REE carbonate precursors (9 min for ALC, 900 min for ANC, 2880 min for ADC and more than 10000 min for AYbC). Fig. 9 shows this proportionality: the larger the ionic potential of the REE3+, the longer the stability of the amorphous REE carbonate. A similar trend is known to occur with amorphous Ca and Mg carbonates: amorphous Mg carbonate is stable for several days at ambient temperature (ionic potential of Mg2+ = 3.07 Å−1,50) compared to ACC (<2 min; ionic potential of Ca2+ = 2.02 Å−1,5). The breakdown of ACC prior to its crystallization is known to occur by the progressive loss of structural water within the disordered framework of the precursor.11,12,50–52 Di Tommaso and de Leeuw (2010)53 have attributed the higher stability of amorphous Mg carbonate compared to ACC to the stronger hydration shell of the Mg2+ ion: because of its larger ionic potential, the energy needed to dehydrate the Mg2+ ion is larger than Ca2+. In the case of amorphous REE-bearing carbonates, the ionic potential of La3+ (2.60 Å−1) is smaller than Nd3+ (2.78 Å−1), Dy3+ (3.03 Å−1) or Yb3+ (3.19 Å−1) and therefore the energy required to dehydrate the La3+ ion in ALC will be lower compared to the Nd3+ ion in ANC. Similarly, the exceptional stabilities of amorphous Dy carbonate36 and amorphous Yb carbonate32 are in agreement with the larger ionic potential of these heavier lanthanides.
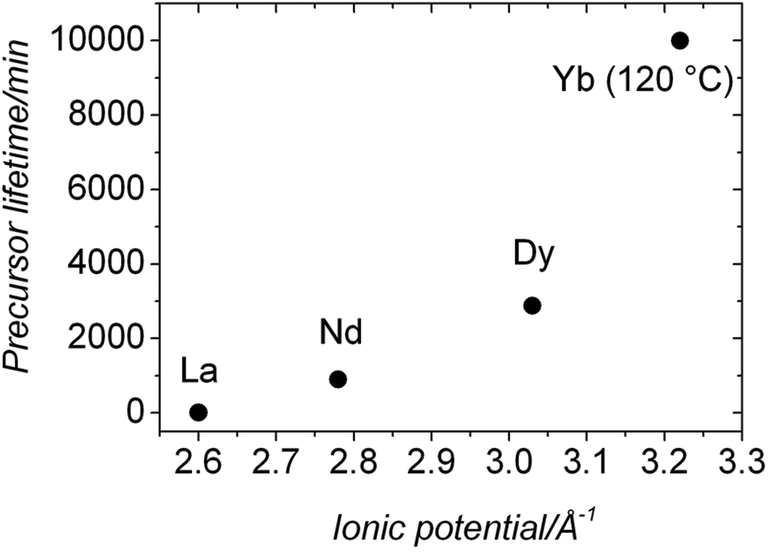 | ||
| Fig. 9 Plot showing the proportionality between the stability of the La, Nd, Dy and Yb amorphous carbonate precursors and the ionic potential of the REE3+ involved in the precursor. | ||
The different crystallization pathways of ALC and ANC during the thermodiffraction experiments (dry heated) (Fig. 1) suggest that a higher dehydration energy is needed for the Nd3+ ion in ANC compared to La3+ in ALC. Thermodiffraction data (Fig. 1) showed that both ALC and ANC crystallized to REE2O2CO3 and then transformed to hexagonal REE2O3 above 400–450 °C, that is after the decomposition of the CO3 ion.54 However, the transformation of cubic Nd-dioxycarbonate to hexagonal Nd-oxide occurred via a cubic Nd-oxide (Fig. 1b, pattern at 700 °C). Fedorov et al. (2002)55 observed that this transformation from cubic to hexagonal Nd-oxide was completed at ∼760 °C and was irreversible. They explained that this may be a consequence of crystallographic shear triggered by the release of strongly bonded residual water. Conversely, in our La-system, hexagonal La-dioxycarbonate transformed directly to hexagonal La-oxide above 700 °C because the La3+ ion requires less energy than Nd3+ to dehydrate and thus all structural water had been already released prior to reaching 700 °C; the sample weight remained constant above 700 °C in the La system (Fig. 4a). In the Nd system a small but progressive mass loss is still observed above 700 °C (Fig. 4b), which is consistent with the release of structural water during the transformation from cubic to hexagonal Nd-oxide.
4.2. The crystallization of La and Nd carbonates in solution: morphologies, mechanisms and pathways
The breakdown of ALC and ANC in solution was translated into the crystallization of a variety of La and Nd hydrated carbonates and hydroxycarbonates with different compositions, structures and stabilities (Tables 2S and 3S†). Our hydrothermal experiments revealed an evolution consistent with a progressive dehydration, from the octahydrate lanthanite-(REE) [REE2(CO3)3·8H2O] to the di-trihydrate tengerite-(REE) [REE2(CO3)3·2–3H2O], to kozoite-(REE) [REECO3OH] and finally to hydroxylbastnasite-(REE) [REECO3OH]. This evolution is not unexpected, as the anhydrous phases are usually more insoluble, denser and more stable than the hydrated polymorphs. There are also some mineralogical differences between both systems: in the La system, tengerite was completely absent because the structure of lanthanite-type carbonates only forms when the ionic radii of the REE is equal or larger than Nd, and tengerite-type carbonate structures are more typical for heavier REE with ionic radii smaller than Nd.32,49,56 Our data also show that the unit cell parameters are larger in the La-bearing compounds compared to the Nd counterparts (Table 3), again a consequence of the larger ionic radius of La than Nd.33,35The morphologies of the transformed crystalline La and Nd carbonates (Fig. 7 and 8) were only in some cases (Fig. 8c) similar to those reported in the literature.42 In many experiments the resulting morphologies were consistent with ‘Category 2’ spherulites described by Gránásy et al. (2005)57 and Andreassen et al. (2010).58 The best examples are kozoite-(La) (Fig. 7c–e) and kozoite-(Nd) (Fig. 8e), but also tengerite-(Nd) (Fig. 8c) and hydroxylbastnasite-(Nd) (Fig. 8f), the latter being very similar to spherulitic vaterite morphologies (CaCO3).8,59 It is worth noting however that in the La and Nd systems, these spherulitic morphologies were only found at temperatures >60 °C (La system) and >95 °C (Nd system) and only when the crystallization occurred directly from the amorphous precursors. The only exception was the crystallization of spherulitic kozoite >60 °C as this formed either directly from the amorphous precursor or through hydrated lanthanites (Tables 1 and 2). A summary of the crystallization products for the La and Nd carbonate systems at different temperatures is shown in Fig. 10.
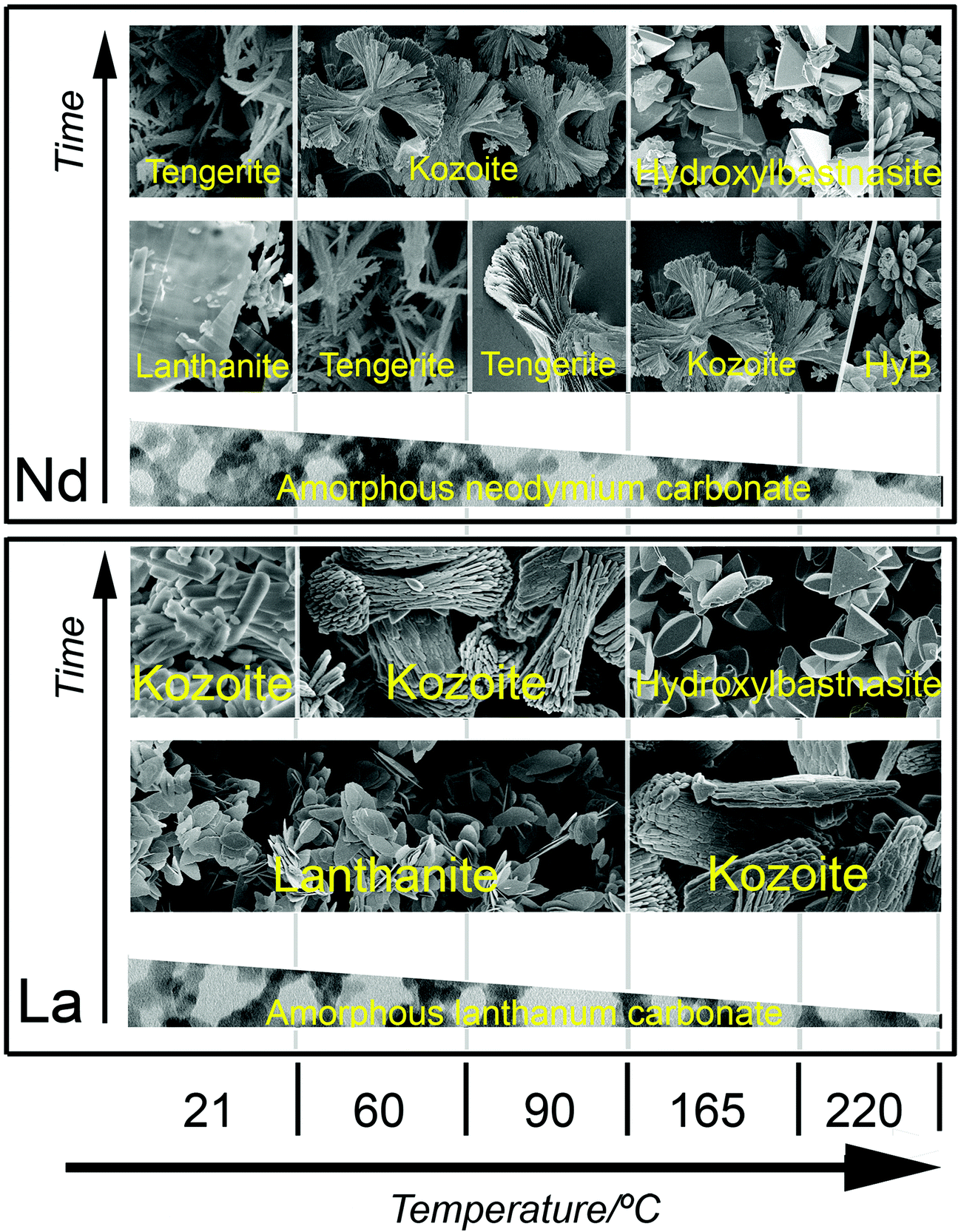 | ||
| Fig. 10 Summary of the crystallization products for the La and Nd systems at different temperatures in the wet-ambient and hydrothermal treatments. | ||
The spherulitic growth mechanism57,60 results in the crystallization of aggregates of particles with very distinctive spheroidal morphologies. It is considered to occur via ‘secondary’ nucleation: after the formation of a single nucleus, the growth takes place as a consequence of the continuous nucleation of new particles with random orientations on the surfaces of the growing spherulite.57 This ‘secondary’ nucleation is usually referred to as ‘growth front nucleation’ and often leads to the formation of sheaf-like morphologies.57 In the carbonate system, spherulitic growth has been described for the crystallization of vaterite,8,58,61 calcite62 and aragonite.59 This mechanism requires a strong crystallization driving force in order to maintain a continuous growth front nucleation during spherulitic growth. In aqueous systems this driving force is provided by a high level of supersaturation with respect to the solid phase.
For carbonates, Andreassen et al. (2010)58 and Beck and Andreassen (2010)62 suggested that a saturation index greater than 2–3 is necessary for spherulitic growth to occur. To determine the saturation index (SI) the activities of the ions in the aqueous solution and the solubility product of the crystallizing solid phases need to be known. Solubility data for La and Nd carbonates are limited to lanthanite-(La) and lanthanite-(Nd).46 Using these as analogues for all crystalline La and Nd carbonates we calculated the SI values of lanthanite-(La) and lanthanite-(Nd) prior to solution mixing (i.e. before the formation of any amorphous precursor) as 12.91 and 11.01. These are larger than the critical values suggested by Andreassen et al. (2010)58 and Beck and Andreassen (2010)62 thus, it is not surprising that we observed spherulitic growth. The high SI values suggest that the supersaturation condition for spherulitic growth would have been fulfilled if the crystals were formed directly from solution. Nevertheless in our experiments La- and Nd-bearing crystalline carbonates formed via amorphous precursors, meaning that the aqueous solution had to be in equilibrium with ALC or ANC prior to the formation of the crystalline phases. There is however, no data about the solubility products of these precursors available and therefore calculating SI values for crystalline La and Nd carbonates when the aqueous solution was in equilibrium with ALC or ANC is impossible. Yet, it is well known that amorphous precursors are usually more soluble than the crystalline phases they transform into. The best-known example is ACC, which is one order of magnitude more soluble than vaterite or calcite.48
Furthermore, as discussed above, the breakdown of amorphous precursors involves temperature-dependent dissolution and dehydration processes.5,11–13,50–52 This means that the faster an amorphous precursor breaks down, the bigger the supersaturation levels are during the crystallization, favouring spherulitic growth. In the La and Nd system, the spherulitic morphologies provide unambiguous evidence of large supersaturation levels during crystallization. Therefore by taking all these data as a whole, we hypothesize that >60 °C, high supersaturation levels were reached and this promoted the rapid breakdown of the La and Nd amorphous carbonate precursors, leading to the crystallization of La and Nd carbonates via a spherulitic growth process (e.g., Fig. SI-4†).
In addition to supersaturation, two other factors need to be taken into account in order to understand the drive to grow spherulitic morphologies in the La and Nd systems: (a) ionic potentials and (b) heating process. We have discussed above that the dehydration energies needed for the breakdown of ALC and ANC differ and that this is a consequence of the difference in ionic potentials between the La3+ and the Nd3+ ions. This was evidenced in both the dry-heated (Fig. 2) and UV-Vis (wet-ambient) (Fig. 5) experiments. Note however, that if the desired thermal regime in the reactor is reached slowly due to thermal buffering effects, the amorphous precursor may break down before the target hydrothermal temperature is reached. This may lead to a slower dissolution of the amorphous precursor and hence a slower achievement of the high supersaturation levels needed for the next stage. Therefore, the combination of high supersaturation levels, differences in ionic potentials and lifetime of the precursor phase (also dependent on ionic potential of the REE3+ ion) all affect the crystallization of La and Nd carbonates.
Spherulitic growth occurred at 95 °C in the Nd system and 60 °C in the La system because the Nd3+ ion in ANC required higher dehydration energy compared to the La3+ ion in ALC. This also explains the fact that at temperatures above 95 °C it was possible to obtain spherulitic tengerite-(Nd) (at 95 °C), kozoite-(Nd) (at 165 °C) and hydroxylbastnasite-(Nd) (at 220 °C) directly from the precursor. However, when these minerals were obtained as a product of recrystallization from a different crystalline Nd carbonate (e.g. hydroxylbastnasite from kozoite, Fig. 8e), the morphologies were not spherulitic. We suggest that in these cases the difference in solubility of the minerals was too small to promote spherulitic growth, similar to what happens in the CaCO3 system (e.g. vaterite–calcite transformation5,13).
In the La system, once lanthanite-(La) formed, it recrystallized to kozoite-(La), a transformation that also involves a dehydration process. At ambient temperature, this recrystallization was too slow to promote spherulitic growth, but lanthanite rapidly transformed to spherulitic kozoite-(La) at higher (60 °C) temperatures. It is possible to explain this spherulitic growth of kozoite-(La) from lanthanite-(La) at greater than 60 °C because of the highly hydrated and layered structure of lanthanite49 and the lower dehydration energy of La3+ compared to Nd3+, which would allow a rapid dissolution of lanthanite at these temperatures (similarly, in the Nd system the dissolution of tengerite-(Nd) also seems to be fast enough to promote the spherulitic growth of kozoite-(Nd)). At 165 and 220 °C no lanthanite-(La) was observed and kozoite-(La) most probably formed directly from ALC. However, due to the short lifetime of ALC, the formation of spherulitic hydroxylbastnasite-(La) at 220 °C did not occur in our experiments because the primary crystallization of spherulitic kozoite-(La) took place before the final experimental temperature was reached. Once kozoite-(La) crystallized, its transformation to hydroxylbastnasite-(La) via spherulitic growth was not favoured because of the small differences in solubility between kozoite-(La) and hydroxylbastnasite-(La). Our experiments suggest that the kozoite–hydroxylbastnasite transformation is likely to occur via a dissolution–reprecipitation mechanism (SEM images and XRD data, Fig. 7e and Table 1).
The stronger hydration shell of the Nd3+ ion compared to the La3+ also affects the Scherrer crystallite size in the hydrothermal experiments. The Scherrer crystallite sizes were always smaller in the Nd-bearing polymorphs than in their La-bearing isomorphs (Table 3). Similarly to the dry-heated experiments, we consider that this effect is produced by the higher energy needed for release of water molecules from the Nd3+ ion compared to La3+ before incorporation to the lattice of the growing crystals. A similar effect has also been observed for the Mg2+ ion in the CaCO3 carbonate system.63 In the Nd system, the release of water from the hydration shell of Nd3+ would be incomplete,55 especially at low temperatures, affecting the crystallinity of the forming solid.
5. Conclusions
The crystallization of La or Nd bearing-carbonates is always initiated by the nucleation and growth of amorphous La or Nd carbonate precursors. Both of the precursors are highly hydrated nanoparticulate compounds with similar spherical morphologies and sizes. However, the most striking difference between the amorphous La and Nd precursors is related to their stabilities. These depend directly on the ionic potential of the REEs: the higher ionic potential the longer the lifetime of the amorphous phase because the energy needed to dehydrate the Nd3+ ion is higher compared to the La3+. This is translated into a two order of magnitude longer lifetime of ANC than ALC. After the breakdown of the precursors under different dry heated, ambient or hydrothermal conditions and over a range of temperatures, they transform to a variety of crystalline REE carbonates with different structures, compositions and stabilities. Hydrated carbonates like lanthanite-(REE) and tengerite (only stable for the Nd system) were obtained at lower temperatures (21, 60 and 95 °C), whereas at higher temperatures (165 and 220 °C) anhydrous carbonates like kozoite-(REE) and hydroxylbastnasite-(REE) were dominant. The morphology of these minerals depends on the temperature and the kinetics of crystallization. The general behaviour is that above 60 °C (La system) and 95 °C (Nd system) the minerals develop spherulitic morphologies when they crystallize directly form amorphous precursors. This development of the spherulitic growth requires high supersaturation levels that can only be obtained by the rapid dissolution of the amorphous precursors or the hydrated crystalline carbonates in some specific cases.The amorphous La and Nd carbonates have enormous potential in the materials science industry if they can be exploited for the controlled development of REE-bearing crystalline carbonates with specific properties and morphologies. Our results show transformation products of varying morphologies resulting from different crystallization mechanisms that operate even with a simple transformation method, opening new perspectives on the synthesis of REE-bearing carbonates for advanced technological applications. This study describes for the first time the crystallization sequences of these REE-bearing carbonates. This knowledge will be essential in the design of new separation methods of La and Nd during processing of REE ores.
Acknowledgements
This research was supported by the Marie Curie EU-FP7 CO2-REACT Research and Training Network under contract RG.EVE.10.1025-004. The authors would like to thank the Cohen Laboratories in the School of Earth and Environment, the Leeds Electron Microscopy and Spectroscopy Centre (LEMAS) at the Faculty of Engineering (University of Leeds), and the Spanish Ministry of Science and Innovation (MICINN-12-MAT2011-27573-C04-02). We also would like to thank the reviewer for his suggestions and constructive comments.References
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332 CrossRef CAS PubMed.
- F. C. Meldrum and R. P. Sear, Science, 2008, 322, 1802–1803 CrossRef CAS PubMed.
- L. B. Gower, Chem. Rev., 2008, 108, 4551 CrossRef CAS PubMed.
- J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, Mineral. Mag., 2008, 72, 283 CrossRef CAS PubMed.
- J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, Nanoscale, 2011, 3, 265 RSC.
- T. Roncal-Herrero, J. D. Rodriguez-Blanco, L. G. Benning and E. H. Oelkers, Cryst. Growth Des., 2009, 9, 5197 CAS.
- D. J. Tobler, S. Shaw and L. G. Benning, Geochim. Cosmochim. Acta, 2009, 73, 5377 CrossRef CAS PubMed.
- P. Bots, J. D. Rodriguez-Blanco, T. Roncal-Herrero, S. Shaw and L. G. Benning, Cryst. Growth Des., 2012, 12, 3806 CAS.
- A. E. S. Van Driessche, L. G. Benning, J. D. Rodriguez-Blanco, M. Ossorio, P. Bots and J. M. García-Ruiz, Science, 2012, 336, 69 CrossRef CAS PubMed.
- T. Roncal-Herrero, J. D. Rodriguez-Blanco, E. H. Oelkers and L. G. Benning, J. Nanopart. Res., 2011, 13, 4049 CrossRef CAS.
- A. V. Radha, T. Z. Forbes, C. E. Killian, P. U. P. A. Gilbert and A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16348 CrossRef PubMed.
- A. L. Goodwin, F. M. Michel, B. L. Phillips, D. A. Keen, M. T. Dove and R. J. Reeder, Chem. Mater., 2010, 22, 3197 CrossRef CAS.
- J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, Am. Mineral., 2015, 100, 1172 CrossRef.
- J. D. Rodriguez-Blanco, S. Shaw, P. Bots, T. Roncal-Herrero and L. G. Benning, Geochim. Cosmochim. Acta, 2014, 127, 204 CrossRef CAS PubMed.
- Z. Z. Zyman, D. V. Rokhmistrov and V. I. Glushko, J. Mater. Sci. Mater. Med., 2010, 21, 123 CrossRef CAS PubMed.
- C. Combes and C. Rey, Acta Biomater., 2010, 6, 3362 CrossRef CAS PubMed.
- D. Bauer, D. Diamond, J. Li, M. McKittrick, D. Sandalow and P. Telleen, Critical Materials Strategy, US Department of Energy, Washington, DC, 2011, http://energy.gov/node/349057 Search PubMed.
- G. P. Hatch, Elements, 2012, 8, 341 CrossRef.
- A. R. Chakhmouradian and F. Wall, Elements, 2012, 8, 333 CrossRef CAS.
- S. B. Castor and J. B. Hedrick, in Industrial Minerals and Rocks, ed. J. E. Kogel, N. D. Trivedi and J. M. Barker, Society for Mining, Metallugy and Exploration, 2006, pp. 769–792 Search PubMed.
- Y. C. Cui, J. H. Liu, X. W. Ren and X. F. Shi, J. Rare Earths, 2009, 27, 169 CrossRef.
- A. J. Ragavan and D. V. Adams, J. Nucl. Mater., 2009, 389, 394 CrossRef CAS PubMed.
- A. P. Gysi and A. E. William-Jones, Chem. Geol., 2015, 392, 87 CrossRef CAS PubMed.
- L. B. Railsback, Geology, 2003, 31, 737 CrossRef CAS PubMed.
- G. H. Cartledge, J. Am. Chem. Soc., 1928a, 50, 2855 Search PubMed.
- G. H. Cartledge, J. Am. Chem. Soc., 1928b, 50, 2863 Search PubMed.
- H. Gamsjäger, Pure Appl. Chem., 1995, 67, 535 CrossRef.
- H. Niu, Q. Min, Z. Tao, J. Song, C. Mao, S. Zhang and Q. Chen, J. Alloys Compd., 2011, 509, 744 CrossRef CAS PubMed.
- B. Bakiz, F. Guinneton, M. Arab, A. Benlhachemi and J. R. Gavarria, M. J. Condensed Matter., 2010, 12, 60 Search PubMed.
- M. J. Norman, J. E. Andrew, T. H. Bett, R. K. Clifford, J. E. England, N. W. Hopps, K. W. Parker, K. Porter and M. Stevenson, Appl. Opt., 2002, 41, 3497 CrossRef CAS.
- K. V. Kumar and A. S. Kumar, Opt. Mater., 2012, 35, 12 CrossRef PubMed.
- K. Nagashima, H. Wakita and A. Mochizuki, Bull. Chem. Soc. Jpn., 1973, 46, 152 CrossRef CAS.
- Z. Han, P. Xu, K. R. Ratinac and G. Q. Lu, J. Cryst. Growth, 2004, 273, 248 CrossRef CAS PubMed.
- S. Liu, R. Ma, R. Jiang and F. Luo, J. Cryst. Growth, 1999, 206, 88 CrossRef CAS.
- K. Michiba, T. Tahara, I. Nakai, R. Miyawaki and S. Matsubara, Z. Kristallogr., 2011, 226, 518 CrossRef CAS.
- B. Vallina, J. D. Rodriguez-Blanco, A. P. Brown, J. A. Blanco and L. G. Benning, J. Nanopart. Res., 2013, 15, 1438 CrossRef.
- J. M. Haschke, J. Solid State Chem., 1975, 12, 115 CrossRef CAS.
- M. L. Panchula and M. Akinc, J. Eur. Ceram. Soc., 1996, 16, 833 CrossRef CAS.
- J. W. Ding, Y. X. Li, H. Ting, X. M. Luo, Y. Yang and X. B. He, Chin. J. Inorg. Chem., 2005, 21, 1213 CAS.
- Y. L. Wu, W. L. Sun, X. P. Feng, X. Xu and Y. X. Li, Chin. J. Inorg. Chem., 2007, 23, 550 CAS.
- S. Liu, R. Ma, R. Jiang and F. Luo, J. Cryst. Growth, 1999, 203, 454 CrossRef CAS.
- Y. X. Li, T. Huang, J. M. Luo and X. Z. Zhou, Chin. J. Inorg. Chem., 2005, 21, 1561 CAS.
- A. L. Patterson, Phys. Rev., 1939, 56, 978 CrossRef CAS.
- A. A. Coelho, TOPAS: General Profile and Structure Analysis Software for Powder Diffraction Data, 2003 Search PubMed.
- D. Parkhurst , Water Res. Invest. Report. 1998. PHREEQC (Version 2)-A Computer Program for Speciation, Batch-Reaction, One Dimensional Transport, and Inverse Geochemical Calculations. USGS 99.
- M. E. Essington and S. V. Mattigod, Soil Sci. Soc. Am. J., 1985, 49, 1387 CrossRef CAS.
- P. Bayliss and A. A. Levinson, Am. Mineral., 1988, 73, 422 CAS.
- L. Brečević and A. E. Nielsen, J. Cryst. Growth, 1989, 98, 504 CrossRef.
- M. Leskelä and L. Niinistö, in Handbook of the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner, Jr. and L. Eyring, Elsevier B.V., 1986, pp. 203–334 Search PubMed.
- A. Navrotsky, Mineral. Mag., 2012, 76, 2159 Search PubMed.
- M. Saharay, A. O. Yazaydin and R. J. Kirkpatrick, J. Phys. Chem. B, 2013, 117, 3328 CrossRef CAS PubMed.
- P. Raiteri and J. D. Gale, J. Am. Chem. Soc., 2010, 132, 17623 CrossRef CAS PubMed.
- D. Di Tommaso and N. H. de Leeuw, Phys. Chem. Chem. Phys., 2010, 12, 894 RSC.
- A. K. Galwey and M. E. Brown, in Thermal decomposition of ionic solids, Elsevier B.V., 1999, pp. 345–364 Search PubMed.
- P. P. Fedorov, M. V. Nazarkin and R. M. Zakalyukin, Crystallogr. Rep., 2002, 47, 281 CrossRef CAS.
- V. Philippini, T. Vercouter, A. Chaussé and P. Vitorge, J. Solid State Chem., 2008, 181, 2143 CrossRef CAS PubMed.
- L. Gránásy, T. Pusztai, G. Tegze, J. A. Warren and J. F. Douglas, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2005, 72, 011605 CrossRef.
- J. P. Andreassen, E. M. Flaten, R. Beck and A. E. Lewis, Chem. Eng. Res. Des., 2010, 88, 1163 CrossRef CAS PubMed.
- K. K. Sand, J. D. Rodriguez-Blanco, E. Makovicky, L. G. Benning and S. S. L. Stipp, Cryst. Growth Des., 2012, 12(2), 842 CAS.
- A. G. Shtukenberg, Y. O. Punin, E. Gunn and B. Kahr, Chem. Rev., 2012, 112, 1805 CrossRef CAS PubMed.
- J. P. Andreassen, J. Cryst. Growth, 2005, 274, 256 CrossRef CAS PubMed.
- R. Beck and J. Andreassen, Cryst. Growth Des., 2010, 10, 2934 CAS.
- A. Mucci and J. W. Morse, Geochim. Cosmochim. Acta, 1983, 47, 217 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr01497b |
| This journal is © The Royal Society of Chemistry 2015 |