
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHistidine-rich stabilized polyplexes for cMet-directed tumor-targeted gene transfer†
Petra
Kos‡
a,
Ulrich
Lächelt‡
ab,
Annika
Herrmann
a,
Frauke Martina
Mickler
bc,
Markus
Döblinger
c,
Dongsheng
He
ab,
Ana
Krhač Levačić
a,
Stephan
Morys
a,
Christoph
Bräuchle
bc and
Ernst
Wagner
*ab
aPharmaceutical Biotechnology, Center for System-based Drug Research, Ludwig Maximilians University Munich, Butenandtstrasse 5-13, D-81377 Munich, Germany. E-mail: ernst.wagner@cup.uni-muenchen.de
bNanosystems Initiative Munich, Germany
cDepartment of Chemistry, Physical Chemistry, Ludwig Maximilians University Munich, Butenandtstrasse 5-13, D-81377 Munich, Germany
First published on 18th February 2015
Abstract
Overexpression of the hepatocyte growth factor receptor/c-Met proto oncogene on the surface of a variety of tumor cells gives an opportunity to specifically target cancerous tissues. Herein, we report the first use of c-Met as receptor for non-viral tumor-targeted gene delivery. Sequence-defined oligomers comprising the c-Met binding peptide ligand cMBP2 for targeting, a monodisperse polyethylene glycol (PEG) for polyplex surface shielding, and various cationic (oligoethanamino) amide cores containing terminal cysteines for redox-sensitive polyplex stabilization, were assembled by solid-phase supported syntheses. The resulting oligomers exhibited a greatly enhanced cellular uptake and gene transfer over non-targeted control sequences, confirming the efficacy and target-specificity of the formed polyplexes. Implementation of endosomal escape-promoting histidines in the cationic core was required for gene expression without additional endosomolytic agent. The histidine-enriched polyplexes demonstrated stability in serum as well as receptor-specific gene transfer in vivo upon intratumoral injection. The co-formulation with an analogous PEG-free cationic oligomer led to a further compaction of pDNA polyplexes with an obvious change of shape as demonstrated by transmission electron microscopy. Such compaction was critically required for efficient intravenous gene delivery which resulted in greatly enhanced, cMBP2 ligand-dependent gene expression in the distant tumor.
Introduction
As conventional cancer treatments often go hand in hand with severe side effects on normal tissues and organs as well as poor and intermittent therapeutic efficacy, targeted drug delivery represents an encouraging approach to enhance therapeutic efficiency while reducing the adverse effects. Gene therapy in particular offers an attractive opportunity to deal with cancer and many other diseases caused by genetic malfunction. Basic polymeric nucleic acid carriers as promising alternatives to viral vectors have been evolving for decades ranging from the “gold standard” polyethylenimine (PEI),1,2 dendrimers,3–6 chitosan,7,8 polylysine9–11 to various tailor-made synthetic carriers. The inherent heterogeneity of many polymeric carriers represents a troubling obstacle. Therefore a focus has been laid on sequence-defined oligomers of enhanced molecular precision with the possibility for structural tuning, implementation of multiple functional domains and development of structure–activity relationships.12–14Numerous potent carriers along with PEI comprise the 1,2-diaminoethane motif as a favorable structural element for nucleic acid binding, endosomal buffering and escape into the cytosol, enabling efficient gene transfer.15–18 Thus, we incorporated this 1,2-diaminoethane motif into artificial amino acids, such as succinoyl-tetraethylene pentamine (Stp) and succinoyl-pentaethylene hexamine (Sph). In combination with some natural amino acids for specific functionalities, they were applied in protected form in a recently established solid-phase synthesis platform for the design and synthesis of sequence-defined oligomers of different topologies showing encouraging transfection activity.19–22 Several studies describe promising delivery systems based on natural amino acids, containing e.g. lysine, histidine, arginine, proline.23–25 In a direct comparison to a similar only histidines and lysines containing carrier, branched oligoethanamino oligomer from our library was more potent. Apart from increased efficacy, toxicity of artificial amino acid–based oligomers remains low.26 Still, the efficient in vivo targeting to cancerous tissues presents a considerable challenge. The upregulation of surface receptors in cancer tissues enables selective targeting to tumor cells using various targeting ligands. Antibodies and antibody fragments,27–29 aptamers,30 glycoproteins,31,32 small molecules33–35 and peptides36–40 are just a few of targeting ligand classes that can recognize receptors over-expressed in tumors. Especially peptides have gained increasing attention based on the straight-forward identification of high-affinity and high-selectivity binding sequences by phage display, their low molecular weight, and efficient tumor penetration. In this regard, for example peptides containing the arginine-glycine-aspartic acid sequence (RGD) binding to integrin receptors37,39,41 and or directed towards growth factor receptors (EGFR, VEGFR)42–44 have shown vast promise. In the current work, we focused on the receptor tyrosine kinase HGFR/c-Met, which is overexpressed in epithelial-derived tumors as well as in stromal and interstitial cell-derived tumors such as sarcomas.45 Binding of the natural ligand hepatocyte growth factor (HGF) to c-Met stimulates cell motility and migration, triggers mitogenesis and morphogenesis and thereby promotes oncogenesis and tumor progression. Therefore, different approaches based on c-Met signaling have been refined in cancer treatment: development of (1) antagonists preventing HGF receptor binding to cell surface c-Met, (2) cytosolic active tyrosine kinase inhibitors and (3) antagonists of the interactions between activated receptors and downstream effectors.46 To date, targeting c-Met overexpressed in cancer tissues has been mostly limited to a variety of receptor binding antibodies intended primarily for in vivo imaging.47–49 Conjugation of an anti-c-Met antibody fragment to the chemotherapeutic drug doxorubicin led to more effective antitumor activity.50 The anti-Met nanobodies attached to the cross-linked albumin nanoparticles have shown potential as a system for lysosomal delivery of drugs.51 Nguyen et al. demonstrated enhanced gene transfer selectivity to hepatocarcinoma cells using retrovirus displaying single-chain variable-fragment (scFv) directed against the c-Met receptor.52 Surprisingly, the c-Met proto oncogene, despite its known oncological relevance, up to now has not been utilized as target receptor for non-viral gene delivery. In the present work, for the first time a potent c-Met binding peptide53,54 herein called cMBP2, initially developed by phage display library screening as diagnostic agent for tumor imaging, was applied as a targeting ligand for receptor-mediated gene transfer. The cMBP2 ligand was conjugated to monodisperse sequence-defined oligomers, comprising polyethylene glycol (PEG) units for shielding and 1,2-diaminoethane motif containing artificial amino acids for alleviation of crucial steps in gene delivery. Furthermore, additional histidines were implemented in the oligomer core for improved endosomal escape. Terminal cysteines were provided for disulfide-based increased pDNA polyplex stability and redox-sensitive cargo release within the cells.55–57 The novel cMBP2-decorated polyplexes resulted in remarkable target-specific gene transfer efficiency in vitro and, upon proper pDNA compaction, also in vivo.
Experimental section
Materials
Fmoc-Ala-Wang resin was obtained from Novabiochem (Darmstadt, Germany). Protected Fmoc-α-amino acids, 2-chlorotrityl chloride resin, N,N-dimethylformamide (DMF), N,N-diisopropylethylamine (DIPEA) and trifluoroacetic acid (TFA) were purchased from Iris Biotech (Marktredewitz, Germany). Triisopropylsilane (TIS), 1-hydroxybenzotriazole (HOBt), tris(2-carboxyethyl)phosphine (TCEP), and Triton X-100 were purchased from Sigma-Aldrich (Munich, Germany). (Benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP) and microreactors were obtained from MultiSynTech (Witten, Germany). Fmoc-N-amido-dPEG24-acid was purchased from Quanta Biodesign (Powell, Ohio, USA). Cell culture media, antibiotics and fetal calf serum (FCS) were purchased from Invitrogen (Karlsruhe, Germany), HEPES from Biomol GmbH (Hamburg, Germany) and glucose from Merck (Darmstadt, Germany). For in vitro use, plasmid pCMVLuc (encoding Photinus pyralis luciferase under control of the CMV promoter)58 was produced with the Qiagen Plasmid Giga Kit (Qiagen, Hilden, Germany) according to the manufacturer specifications. For in vivo experiments, pCMVLuc produced and purified by Plasmid Factory GmbH (Bielefeld, Germany) was applied. pDNA Cy5-labeling kit and the kit for direct covalent attachment of amine functional groups to pDNA were obtained from Mirus Bio (Madison, WI, USA). Luciferase cell culture lysis buffer and D-luciferin sodium salt were obtained from Promega (Mannheim, Germany).Targeting peptide synthesis
Both c-Met targeting peptides and the four scrambled sequences were synthesized using a 2-chlorotrityl resin preloaded with Fmoc-Lys(ivDde)-OH as solid support. The peptides were sequentially assembled at the α-amino function of the preloaded lysine using standard Fmoc chemistry conditions and L-amino acids. cMBP1 (sequence N- to C-terminal: YLFSVHWPPLKA) and cMBP2 (KSLSRHDHIHHH) were synthesized using an Applied Biosystems ABI 431A automated peptide synthesizer. For the random creation of scrambled sequences computer generated permutations of cMBP2 were obtained from an online sequence generator (RANDOM.ORG). The resulting four scrambled sequences cMBP2sc1 (N- to C-terminal: LHHHDRKSSIHH), cMBP2sc2 (KSHHRDHIHLHS), cMBP2sc3 (HHSIHRLHHKSD) and cMBP2sc4 (RKIHHHLHSHSD) were synthesized in parallel using a Syro Wave (Biotage, Uppsala, Sweden) parallel peptide synthesizer. After the final Fmoc deprotection step, the N-termini of the peptides were protected by reaction with 10 equivalents of di-tert-butyl dicarbonate (Boc anhydride) and N,N-diisopropylethylamine (DIPEA) in dichloromethane (DCM) for one hour. Subsequently the ivDde protecting group at the ε-amino function of the C-terminal lysine was removed by repeated incubation with 2% hydrazine monohydrate in N,N-dimethylformamide (DMF) with regular exchange of the deprotection solution every 5 minutes. The deprotection progress was monitored photometrically by detection of the cleavage product at 290 nm. To investigate the purity and identity of the targeting ligands cMBP1 and cMBP2, small portions of the resin bound peptides were cleaved and isolated for subsequent analysis by RP-HPLC and ESI-MS. The analytical data can be found in the ESI.† The remaining resin bound solid-phase ligands were used for subsequent oligomer synthesis.Oligomer synthesis
The synthesis of oligomers containing targeting peptides was continued by sequential assembly at the deprotected ε-amino function of the C-terminal L-lysine. Artificial Fmoc-oligoamino acids Fmoc-Stp(Boc3)-OH and Fmoc-Sph(Boc4)-OH were synthesized as described before.20,21 Oligomers of artificial oligoamino acids were synthesized manually under standard Fmoc solid phase peptide synthesis conditions using syringe microreactors. Coupling steps were carried out using 4 eq. Fmoc-amino acid, 4 eq. HOBT, 4 eq. PyBop and 8 eq. DIPEA in DCM–DMF 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (10 mL g−1 resin) and 1 h incubation time. Fmoc deprotection was accomplished by 4 × 10 min incubation with 20% piperidine in DMF (10 mL g−1 resin). After each coupling and deprotection step a washing procedure comprising 3 × 1 min DMF, 3 × 1 min DCM incubation (10 mL g−1 resin) and a Kaiser59 test were performed. In case of a positive result of the Kaiser test after coupling, the last coupling step was repeated. In case of a negative result after deprotection, the last deprotection step was redone. Symmetrical branching points were introduced using Fmoc-Lys(Fmoc)-OH. Finally, all peptides were cleaved off the resin by incubation with TFA–TIS–H2O 95:2.5:2.5 (10 mL g−1 resin) for 90 min. The cleavage solution was concentrated under reduced pressure and peptides were precipitated in 50 mL pre-cooled MTBE–n-hexane 1:1. All oligomers were purified by size exclusion chromatography using an Äkta purifier system (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) based on a P-900 solvent pump module, a UV-900 spectrophotometrical detector, a pH/C-900 conductivity module, a Frac-950 automated fractionator, a Sephadex G-10 column and 10 mM hydrochloric acid solution–acetonitrile 7:3 as solvent. If necessary, additional purification was carried out by preparative RP-HPLC using a VWR LaPrep system and a Waters Symmetry Prep C18 column (7 μm, 19 × 150 mm). All peptides were lyophilized. The presence of the different elements of the oligomer sequences was validated by 1H-NMR. The purity of the oligomers was investigated by RP-HPLC. Analytical data can be found in the ESI.†
:1 (10 mL g−1 resin) and 1 h incubation time. Fmoc deprotection was accomplished by 4 × 10 min incubation with 20% piperidine in DMF (10 mL g−1 resin). After each coupling and deprotection step a washing procedure comprising 3 × 1 min DMF, 3 × 1 min DCM incubation (10 mL g−1 resin) and a Kaiser59 test were performed. In case of a positive result of the Kaiser test after coupling, the last coupling step was repeated. In case of a negative result after deprotection, the last deprotection step was redone. Symmetrical branching points were introduced using Fmoc-Lys(Fmoc)-OH. Finally, all peptides were cleaved off the resin by incubation with TFA–TIS–H2O 95:2.5:2.5 (10 mL g−1 resin) for 90 min. The cleavage solution was concentrated under reduced pressure and peptides were precipitated in 50 mL pre-cooled MTBE–n-hexane 1:1. All oligomers were purified by size exclusion chromatography using an Äkta purifier system (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) based on a P-900 solvent pump module, a UV-900 spectrophotometrical detector, a pH/C-900 conductivity module, a Frac-950 automated fractionator, a Sephadex G-10 column and 10 mM hydrochloric acid solution–acetonitrile 7:3 as solvent. If necessary, additional purification was carried out by preparative RP-HPLC using a VWR LaPrep system and a Waters Symmetry Prep C18 column (7 μm, 19 × 150 mm). All peptides were lyophilized. The presence of the different elements of the oligomer sequences was validated by 1H-NMR. The purity of the oligomers was investigated by RP-HPLC. Analytical data can be found in the ESI.†
Polyplex formation
pCMVLuc and oligomer at indicated nitrogen/phosphate (N/P) ratios were diluted in separate tubes of equal volumes of 20 mM HEPES buffered 5% glucose pH 7.4 (HBG) each. Only protonatable nitrogens were considered in the N/P calculations. The polycation solution was added to the nucleic acid, mixed vigorously up to 10-times and incubated for 30 min at room temperature.Cell culture
Hepatocellular carcinoma cells (Huh7) were grown in a 50:50 mixture of Dulbecco's modified Eagle's medium (DMEM) and Ham's F12 medium and human prostate cancer cells (DU145) were cultured in RPMI-1640 medium. Both media were supplemented with 10% fetal calf serum (FCS), 4 mM stable glutamine, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin.
Western blots
Huh7 cells (200000 per well) were seeded in 4 mL medium using six-well plates. After 24 h, medium was replaced with 2 mL fresh medium. The transfections were performed with polyplexes containing 5 μg pDNA in a total volume of 500 μL. After 45 min of incubation, the cells were lysed and total protein concentration was determined using a BCA assay. Equal amounts of protein (30 μg) in loading buffer were applied per lane and were separated by SDS-PAGE under reducing conditions, blotted on nitrocellulose membrane and blocked with NET gelatine for 1 h at room temperature. Immunostaining was performed using Met (Santa Cruz Biotechnology, USA), phospho-Met (Cell Signalling, USA), Akt and phospho-Akt antibodies (Cell Signaling, Germany) overnight at 4 °C. After the incubation with the applicable primary antibodies, membranes were washed three times for 15 min with NET gelatine before incubating with the adequate secondary peroxidase antibody for 1 h. When necessary the membranes were stripped in 2% SDS (w/v) with 0.8% (v/v) β-mercaptoethanol in 0.07 M Tris/HCl (pH 6.8) solution for 1 h at 50 °C. After another three washing cycles, the membranes were cut accordingly and the proteins were then visualized using Lumi-Light Western blotting substrate (Roche, Germany).
Cellular internalization
Huh7 cells were seeded into 24-well plates coated with collagen at a density of 50000 cells per well. After 24 h, culture medium was replaced with 400 μL fresh growth medium. pDNA polyplexes (N/P 12) in 100 μL HBG, containing 1 μg pDNA (20% of the nucleic acid was Cy5-labeled) were added to each well and incubated at 37 °C for 45 min. All experiments were performed in triplicates. Subsequently, cells were washed with 500 μL PBS containing 1000 I.U. heparin for 15 min on ice to remove any polyplexes sticking to the cell surface. After additional washing step with PBS only, cells were detached with trypsin/EDTA and taken up in PBS with 10% FCS. Cellular uptake was assayed by excitation of Cy5 at 635 nm and detection of emission at 665 nm. Cells were appropriately gated by forward/sideward scatter and pulse width for exclusion of doublets. DAPI (4′,6-diamidino-2-phenylindole) was used to discriminate between viable and dead cells. Data were recorded by Cyan™ ADP flow cytometer (Dako, Hamburg, Germany) using Summit™ acquisition software (Summit, Jamesville, NY, USA) and analyzed by FlowJo® 7.6.5 flow cytometric analysis software.
In vitro gene transfer
Huh7 cells (8000 per well) were seeded 24 h prior to pDNA delivery in 96-well plates. Transfection efficiency of oligomers was evaluated using 200 ng pCMVLuc per well. All experiments were performed in quintuplicate. Before transfection, medium was replaced with 80 μL fresh medium containing 10% FCS. Polyplexes formed in 20 μL HBG in sterile Eppendorf caps at 25 °C were added to each well and incubated on cells for 45 min at 37 °C, followed by incubation with fresh medium containing endosomolytic agent chloroquine at concentration of 100 μM (for control experiments without chloroquine only fresh medium was added). After 4 h medium was again replaced by fresh medium and cells were further incubated for 20 h. LinPEI at nontoxic optimum N/P 6 with 4 h longer polyplex incubation on cells was used as positive control, HBG buffer was used as negative control. For all experiments 24 h after transfection, cells were treated with 100 μL cell lysis buffer (25 mM Tris, pH 7.8, 2 mM EDTA, 2 mM DTT, 10% glycerol, 1% Triton X-100). Luciferase activity in the cell lysate was measured using a luciferase assay kit (100 μL Luciferase Assay buffer, Promega, Germany) and a Centro LB 960 plate reader luminometer (Berthold Technologies, Germany).Metabolic activity of transfected cells (MTT assay)
The cells were transfected in 96-well plates as described above. At 24 h post transfection, 10 μL of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) were added to each well reaching a final concentration of 0.5 mg mL−1. After an incubation time of 2 h, unreacted dye and medium were removed and the 96-well plates were stored at −80 °C for at least one hour. The purple formazan product was then dissolved in 100 μL DMSO (dimethyl sulfoxide) per well and quantified measuring absorbance using microplate reader (TecanSpectrafluor Plus, Tecan, Switzerland) at 530 nm with background correction at 630 nm. All studies were performed in quintuplicate. The relative cell viability (%) related to control wells treated only with 20 μL HBG was calculated as ([A] test/[A] control) × 100%.Calcein assay
10000 DU145 cells per well were seeded in 8 well chambered μ-Slides (ibiTreat, Ibidi GmbH) 48 h prior to polyplex addition. 20 μl of polyplex solution containing 400 ng pDNA and an oligomer at N/P 12 were added to 200 μl fresh medium. 40 minutes after particle addition, the cell medium was replaced by 300 μl fresh RPMI medium containing 0.5 mg ml−1 calcein. After 20 h incubation time, cells were washed twice with PBS and transferred to a CO2-independent medium containing 10% FCS. The cells were imaged with 488 nm laser excitation by spinning disk confocal microscopy (Nikon TE2000E microscope with Yokogawa CSU10 spinning disk unit, EM-CCD camera (iXon DV884, Andor), Nikon 1.49 NA 100× Plan Apo oil immersion objective) and z-projections of single cells were built. Calcein fluorescence in the cytosol was quantified by digital image analysis in ImageJ. Two threshold values of fluorescence intensity were set to exclude extracellular regions (background) and endosomal compartments (endosome fluorescence) from the quantification. For pixels between the two threshold values, total integrated intensity was calculated (integrated intensity = number of selected pixels × mean grey value of selected pixels).
pDNA polyplex stability in 90% serum via gel shift assay
pDNA (1 μg) and oligomer at N/P 12 were mixed in a total volume of 12.5 μL. Polyplexes were formed by rapid mixing and incubated for 30 min at room temperature. A volume of 112.5 μL of fetal calf serum (FCS) was added to the samples to reach a final concentration of 90% FCS. The samples were then incubated with FCS for 1, 10, 30 or 90 min at 37 °C. Where indicated, 100 IU heparin was added to the polyplexes incubated in serum. Agarose gel (1%) was prepared by dissolving agarose in TBE buffer. After addition of GelRed, the agarose solution was casted into an electrophoresis unit to form a gel. 4 μL loading buffer were added to the samples before they were placed into the sample pockets. Electrophoresis was performed at 120 V for 80 min.Gene transfer in vivo
Animal experiments were carried out using female Rj:NMRI-nu (nu/nu) (Janvier, Le Genest-St-Isle, France). 5 × 106 Huh7 cells were inoculated subcutaneously into the left flank and experiments started approximately 12 days after tumor cell injection when the tumors reached the adequate size (about 500–700 mm3). Tumor size was monitored with a caliper and determined by formula a × b2/2 (a = longest side of the tumor; b = widest side vertical to a). For intratumoral administration, polyplexes containing 50 μg pCMVLuc (approximately 2.5 μg g−1 body weight) at N/P 12 in HBG in total volume of 60 μL were applied and mice were sacrificed after 24 h. Systemic gene transfer in tumor bearing mice was conducted using polyplexes containing 80 μg pCMVLuc (approximately 4 μg g−1 body weight) at N/P 12 in HBG in total volume of 200 μL. Polyplexes were injected into the tail vein and animals were sacrificed 48 h after application. Tumors and/or organs were dissected and homogenized in cell culture lysis reagent using a tissue and cell homogenizer (FastPrep®-24, MP Biomedicals, USA). The samples were then centrifuged at 3000g at 4 °C for 10 min to separate insoluble cell components. Luciferase activity was determined in the supernatant using a Centro LB 960 luminometer (Berthold, Germany). All animal procedures were approved and controlled by animal experiments ethical committee of Regierung von Oberbayern, District Government of Upper Bavaria, Germany, and carried out according to the guidelines of the German law of protection of animal life.Quantitative RT-PCR for determination of intratumoral pDNA
For pDNA quantification by real-time PCR (RT-PCR) in tumors, polyplexes were administered as described above. Total DNA was isolated according to manufacturer's instructions using peqGOLD guanidinisothiocyanate/phenol method (Peqlab, Germany). Quantitative RT-PCR was then performed on a LightCycler 480 system (Roche) using UPL Probe #84 (Roche) and Probes Master (Roche). The following primer sequences were used: reverse primer 5′-CCC CGT AGA AAA GAT CAA AGG-3′ and forward primer 5′-GCT GGT AGC GGT GGT TTT T-3′. The pDNA dilution series were run in parallel to allow the absolute quantification.Ethidium bromide (EtBr) exclusion assay
Oligomer solution was added at increasing N/P ratios to 10 μg pDNA in 1 mL HBG containing 0.4 μg EtBr. After each addition the EtBr fluorescence was measured at the excitation wavelength λex = 510 nm and emission wavelength λem = 590 nm using a Cary Eclipse spectrophotometer (Varian, Germany). A solution of 0.4 μg EtBr in 1 mL HBG presented the blank value. Maximal fluorescence intensity was set 100% for the EtBr solution containing free nucleic acid (10 μg) and decrease in fluorescence was measured after stepwise addition of oligomer solution.Transmission electron microscopy
A carbon coated 200 mesh copper grid (Plano GmbH, Wetzlar, Germany) was activated by mild plasma cleaning. Afterwards, one drop (10 μL) of the polyplex solution at N/P 12 prepared as described above was placed on the grid. Excess liquid was blotted off using filter paper until the grid was almost dry. Subsequently, the copper grid was incubated with 10 μL of a 1% phosphotungstic acid solution (PTA) (Science Services, Germany), air-dried as before and analyzed immediately using a FEI Titan 80–300 operated at 80 kV.Statistical analysis
Statistical significance was analyzed by Student's one-tailed t-test. Significance levels are indicated with star symbols: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.Results and discussion
c-Met targeted gene transfer with sequence-defined oligomers
As c-Met presents an encouraging target receptor in cancer therapy, we applied two phage display library derived c-Met binding peptides (cMBP1 and 2) and evaluated their suitability for gene delivery. The first peptide herein designated as cMBP1 (YLFSVHWPPLKA) was previously demonstrated to specifically bind to c-Met, thereby competing with its natural ligand HGF and inhibiting tumor cell proliferation in vitro. Its potential as a diagnostic agent for tumor imaging was verified in experiments with radioiodinated cMBP1.60 Kim et al. utilized another peptidic c-Met binding ligand, here called cMBP2 (KSLSRHDHIHHH). They reported the use of the peptide and accordant conjugates as molecular probes for radio- and near-infrared fluorescence imaging of tumors by targeting the c-Met receptor.53,54 For the first time, we evaluated the two c-Met binding ligands cMBP1 and cMBP2 in terms of nucleic acid delivery. Solid-phase supported synthesis was utilized for the assembly of targeted and shielded oligomers (Fig. 1). By this method, the targeting ligand (cMBP1 or cMBP2) can be directly attached to the multifunctional oligomers within a single solid-phase synthesis, providing high-precision conjugates. RP-HPLC analysis confirmed a high grade of the compounds. The identity of the sole peptidic ligands has been verified by mass spectrometry and the presence of the individual elements within the oligomer sequences has been validated by 1H-NMR. The first-generation targeted oligomers (Fig. 1-I) being the most suitable for ligand evaluation consist of a monodisperse polyethylene glycol (PEG) moiety with 24 oxyethylene units for the reduction of unspecific interactions with blood components, lysine as a branching point, polycationic core comprising four repeating units of the novel artificial amino acids succinoyl-tetraethylene pentamine (Stp) for nucleic acid packaging, endosomal buffering and endosomal escape,21,22 as well as an N-terminal cysteine residue at the end of each polycationic arm for redox-sensitive polyplex stabilization.12,26,61,62 The resulting cMBP1-targeted conjugate was denoted as oligomer #1 and the cMBP2-containing conjugate as oligomer #2. A non-targeted alanine oligomer (#3) was constructed as a control. Structures and sequences are displayed in Fig. 1. | ||
| Fig. 1 Structures and topologies, sequences, internal library identification numbers (compound ID) of the synthesized oligomers and their assigned numbers. A, K, H and C represent the α-amino acids in a one-letter-code. α- and ε-amines of branching lysines are indicated. L stands for the targeting ligand or the corresponding control. | ||
All three oligomers decorated with cMBP1, cMBP2 or alanine control were able to fully complex pDNA already at the low N/P (protonatable nitrogens of oligomer/phosphate in the nucleic acid backbone) ratios (Fig. S1†). Initially cell association studies were performed on hepatocellular carcinoma (Huh7) and prostate cancer (DU145) cell lines both displaying high receptor expression (Fig. S2†). In both cell lines cMBP2-containing polyplexes (#2) displayed very efficient cell binding (>90% of cells), being superior to cMBP1-targeted polyplexes (#1) and far higher than for the alanine control oligomer (#3) (Fig. S3A, B†). The internalized cMBP1- (#1), cMBP2- (#2) and non-targeted (#3) polyplexes were further imaged by fluorescence microscopy. The greatest intracellular uptake of labeled pDNA was again observed in the case of the cMBP2-containing polyplexes (Fig. S3C†). The c-Met-targeting effect was then finally confirmed by pDNA transfections on both cell lines (Fig. S3D, E†). However, as previously demonstrated by Martin et al.,39 for analogous PEGylated carriers endosomal escape represents one of the greatest intracellular hurdles in the delivery, and their transfection efficacy is dependent on the addition of chloroquine.63,64 This endosomotropic agent accumulates in endosomes by protonation, triggers osmotic swelling and thereby promotes release of entrapped polyplexes. Alongside, cytotoxicity studies were performed showing no negative effects of applied polyplexes on the cell viability, apart from a minor cytotoxicity caused by the chloroquine incubation (Fig. S3F, G†). As cMBP2 ligand showed a better delivery effect than cMBP1 on both tested cell lines, this more potent ligand was applied in the further studies and its specificity was analyzed in more detail. For this reason, four scrambled sequences chosen by random computer-supported permutation (cMBP2sc1 (#4): LHHHDRKSSIHH, cMBP2sc2 (#5): KSHHRDHIHLHS, cMBP2sc3 (#6): HHSIHRLHHKSD and cMBP2sc4 (#7): RKIHHHLHSHSD) were synthesized and conjugated to the same initial oligomer structure (Fig. 1-I). In cell association studies the four scrambled sequence-decorated polyplex types hardly bound to the cell surface of the Huh7 (Fig. S4B†) and DU145 (Fig. S4C†) cells. Together with the low association of the alanine control polyplexes, this confirms the ligand dependent cellular interaction and the sequence specificity of the cMBP2 ligand.
Moreover, c-Met activation causes an increased cell proliferation and invasion65 and could therewith lead to unwanted side effects of cMBP-mediated gene delivery. Therefore, in the next step possible HGFR/c-Met receptor activation after polyplex administration was investigated. As the binding of its natural ligand hepatocyte growth factor (HGF) causes the c-Met phosphorylation and downstream signaling, this was investigated by Western blot. Receptor activation could only be observed with the HGF positive control. None of the polyplexes, regardless of the ligand, mediated any receptor phosphorylation or activation of the c-Met downstream protein Akt (Fig. 2). Similar was observed by Mickler et al.43 for a short peptide ligand GE11 targeting the EGFR. GE11 polyplexes were demonstrated to bind the EGFR but not to induce the receptor signaling which resulted in a slower uptake as compared to EGF. This non-mitogenic alternative actin-dependent pathway was discussed as crucial for future cancer therapies not stimulating the cancer cell proliferation. Accordingly, no induction of cell proliferation was observed upon transfection with cMBP1 or cMBP2-targeted polyplexes (Fig. S5†).
 | ||
| Fig. 2 Lack of c-Met receptor activation. Huh7 cells were treated either with cMBP1-targeted (lane 1), cMBP2-containing (lane 2) or alanine control (lane 3) polyplexes at N/P 12. HBG buffer (lane 4) was used as a negative control and natural ligand hepatocyte growth factor (HGF) (lane 5) as a positive control. After 45 min incubation, total cell lysates were subjected to Western blot analysis and incubated with A) p-Met and Met and B) P-Akt and Akt antibodies. | ||
Carrier optimization: incorporation of histidines, extension of PEG chain or implementation of additional polycationic arms
Due to the chloroquine dependence of the initial oligomers (Fig. 1-I), histidines were implemented into the oligomer backbone before each (oligoethanamino) amide building block of the polycationic arms as well as prior to the lysine branching point, bringing forth the cMBP2-containing oligomer #8 and its non-targeted alanine counterpart #9 (Fig. 1-II). The fine-tuning of the buffering capacity has previously been demonstrated to be crucial for the gene transfer potency of related oligoamino nucleic acid carriers.19 Histidines, based on their imidazole ring with the pKa ∼ 6 can be protonated in acidifying endosomes and can thus greatly ameliorate the endosomal escape of the polyplexes.19,66,67Apart from modifications of the cationic core, the PEG chain offers another option to alter the properties of the targeted polyplexes. Hydrophilic polymers such as PEG have been verified as indispensable for nanoparticle surface shielding against unintended interactions with biological surfaces, inhibition of activation of the complement system and prolongation of blood circulation. The length of the PEG spacer not only influences the accessibility of the targeting ligand to the tumor tissue, but can also alter the polyplex biodistribution. Several studies have reported favorable effects with an increasing PEG length, whereas other studies have favored shorter PEG chains which emphasizes that the shielding needs to be adjusted individually to the carrier and peptidic ligand.68–72 For this purpose, cMBP2-containing (#10) and alanine control (#11) oligomers having an additional second PEG moiety of 24 oxyethylene units as well as histidines in the cationic backbone were synthesized (Fig. 1-III). Another control oligomer contains alanines replacing the backbone histidines (#12, Fig. 1-IV).
In cellular internalization experiments the histidine-enriched polyplexes (#8, #9; Fig. 1-II) were first compared to the polyplexes formed with the initial oligomers without histidines (#2, #3; Fig. 1-I). A pronounced targeting effect was observed for both cMBP2-containing polyplexes #8 and #2 (Fig. 3A). The histidine implementation, as expected, caused no significant change in the cellular uptake. Both types of alanine control polyplexes (#3 and #9) displayed practically no cellular uptake. Furthermore, the cellular internalization of polyplexes formed with the oligomers containing two consecutive PEG24 chains was analyzed (Fig. 3B) to investigate the effect of an extended PEG chain. Both cMBP2-containing PEG48 oligomers (#10 and #11) showed high cellular internalization in vitro, comparable to the targeted PEG24 polyplexes before in Fig. 3A. The non-targeted oligomer (#12) once more displayed very low cellular internalization.
 | ||
| Fig. 3 Influence of cMBP2 ligand, histidine addition and enhanced PEG chain length. (A) Cellular internalization of the Cy5-pDNA polyplexes (N/P 12) of the oligomers with one PEG24 chain analyzed by flow cytometry after 45 min incubation at 37 °C followed by removal of extracellularly bound polyplexes; cMBP2-containing polyplexes are presented in red (#2 and #8), alanine controls in black (#3 and #9). Initial oligomer structures are displayed with a solid line, histidine modified oligomers with a dotted line. HBG treated cells are presented in grey. Logarithmic X-scale represents Cy5 fluorescence of polyplexes internalized into Huh7 cells. (B) Cellular internalization of the polyplexes (N/P 12) formed with oligomers comprising two consecutive PEG24 chains; cMBP2-containing polyplexes are presented in red (dotted line for the oligomer with the histidine (#10) spacers and solid line for the oligomer with the alanine (#11) spacers), alanine control (#12) polyplexes in black. (C) Luciferase reporter gene expression in Huh7 cells with (pattern) or without (no pattern) chloroquine and (D) cell viability assay performed in parallel. LinPEI was used as positive control, HBG treated cells as background. Cell viability was calculated as percentage to cells treated with HBG. Data are presented as mean value (±SD) out of quintuplicate. | ||
In contrast to cell uptake, the luciferase gene transfer studies revealed an immense influence of histidines on pDNA transfection efficiency. In the absence of the endosomolytic chloroquine (no pattern), the histidine-modified cMBP2-containing oligomer #8 displayed a greatly enhanced gene transfer (100-fold) as compared to the original structure without the histidines (#2). In the presence of chloroquine (patterned bar) the transfection efficiency remained unchanged. These data confirmed a markedly improved endosomal buffering and endosomal escape of the histidine-equipped targeted polyplexes. The cMBP2- and histidine-containing oligomer with a second PEG24 chain #10 led to reduced transfection efficiency as compared to the less shielded #8 polyplexes (Fig. 3C). Replacing the histidines in the backbone with alanine spacers (#11) led to comparably efficient gene transfer upon chloroquine incubation, which vastly diminished in the absence of chloroquine. The alanine control with two PEG24 units (#12) mediated only a minor luciferase expression. The MTT assay (Fig. 3D) showed no reduction in cell viability for any of the polyplexes. A minor cytotoxicity was again observed only due to the presence of chloroquine. An improved endosomal escape based on implementation of histidines was further confirmed in a calcein assay. Spinning disk confocal microscopy revealed a greatly enhanced calcein release for histidine-enriched (#10) polyplexes as compared to their control (#11) polyplexes (Fig. 4).
 | ||
| Fig. 4 Calcein intracellular release evaluated by spinning disk confocal microscopy. Representative images of cells transfected with (A) histidines-modified (#10) and (B) alanine control (#11) cMBP2 polyplexes in medium containing 0.5 mg ml−1 calcein. (C) Quantification of cytosolic calcein release by digital image processing (n = 22 for #10 and n = 21 for #11). | ||
Apart from implementation of the functional moieties within each polycationic arm, addition of extra polycationic arms and design of more highly branched oligomers has previously been indicated as favorable for gene delivery by non-targeted conjugates.21,22 Therefore, as the next step in optimization, further modifications of the cationic part of the carrier were made. A cMBP2- and histidine-containing oligomer was synthesized having four instead of two polycationic arms in the backbone, and designated as oligomer #13 and its alanine counterpart as #14 (Fig. 1-V). Moreover, in these two highly branched targeted oligomers, the Stp building block was replaced by the novel Sph building block19,21 having one additional diaminoethane repeat per unit (Fig. 1) and thus a longer continuous ethanamino motif, as this might lead to more favorable protonation profiles of the carrier within the endolysosomal pH range.19 However, an increment in the number of polycationic arms did not provide a benefit in the case of cMBP2-containing polyplexes in vitro (Fig. S6†).
Confirmation of cMBP2-mediated targeting in vivo
The histidine-enriched 2-arm oligomer #8 with one PEG24 unit yielded the most promising cellular uptake and gene transfer in vitro and it was thus selected for the first in vivo studies. An additional PEG24 chain did not show favorable effects in vitro (Fig. 3), yet it might be beneficial in the in vivo situation. Therefore, also the histidine-enriched oligomer with two PEG24 units #10 was chosen for further experiments and compared to its analog #8. Prior to in vivo experiments, the polyplex stability in serum was evaluated. All polyplexes, regardless of the ligand or extent of shielding, seem to remain stable in 90% FCS during the investigated time frame and remained stable also after addition of highly negatively charged heparin (Fig. 5A).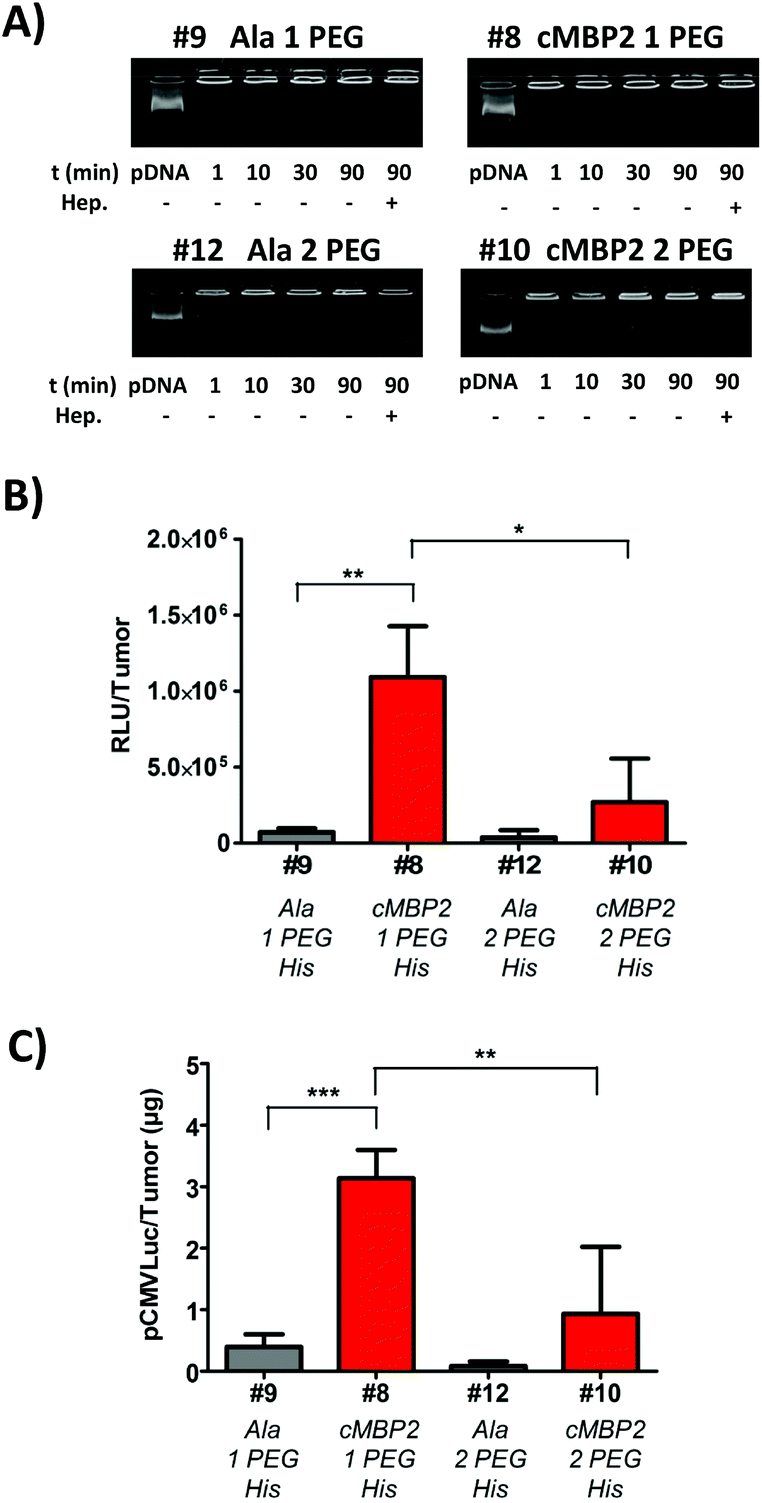 | ||
| Fig. 5 In vivo confirmation of cMBP2 targeting. (A) Stability of polyplexes in serum. pDNA binding of oligomers #9, #8, #12 and #10 in the presence of 90% fetal bovine serum (FCS) analyzed by means of an agarose gel shift assay. Polyplexes at N/P 12 were incubated at room temperature for 30 min in order to allow polyplex formation. Next, FCS was added and polyplexes were further incubated for 90, 30, 10 or 1 min. As indicated (last lane), heparin was added to the polyplexes after incubation in serum for 90 min. (B) Luciferase gene expression at 24 h after intratumoral administration of pCMVLuc polyplexes at N/P 12 into Huh7 tumor-bearing mice. Luciferase gene expression is presented as relative light units per tumor (RLU/tumor; N = 5, mean ± SEM)). Lysis buffer RLU values were subtracted. The Huh7 tumor weights were 387 ± 146 mg. (C) Quantification of pCMVLuc detected in tumors at 24 h after intratumoral injection of cMBP2-targeted and alanine control polyplexes (N/P 12) with either one or two PEG24 chains as determined with qPCR (N = 4, mean ± SEM). | ||
Subsequent in vivo experiments were conducted in subcutaneous Huh7 tumor-bearing mice and polyplexes were injected intratumorally. The results of luciferase gene transfer (Fig. 5B) were in accordance with in vitro studies. A significant cMBP2 targeting effect was successfully demonstrated in the case of #8 polyplexes, with a 15-fold higher gene expression than with alanine #9 control polyplexes. The oligomer #10 with higher PEG content displayed a lower gene expression but still with a targeting ligand effect compared with its alanine control #12 (7-fold lower expression). Alongside, as a further confirmation of cMBP2 targeting in vivo, quantitative polymerase chain reaction was performed enabling the quantification of the plasmid amount retained in tumors after local polyplexes application. In line with in vivo luciferase gene transfer studies the highest amount of plasmid was retained in tumors with the cMBP2-containing PEG24 oligomer (#8), being significantly (almost 10-fold) higher as compared to its non-targeted analog #9 and (>3-fold) to the cMBP2-PEG48 containing polyplexes #10 (Fig. 5C).
Intravenous application of c-Met-directed polyplexes
As the intratumoral studies using cMBP2- and histidine-containing oligomer #8 displayed promising results, this oligomer was consequently chosen for further in vivo studies. This time the polyplexes were injected intravenously. Based on enhanced permeability and retention (EPR) effect, the polyplexes accumulate in tumors at much higher concentrations than in normal tissues. This can be attributed to rapid angiogenesis and poor vasculature of tumors.73 Still, incorporation of targeting ligands is necessary for active targeting to tumor tissues. Mice were sacrificed two days after #8 and control #9 polyplex administrations. The subsequent analysis of luciferase expression in tumors of the mice treated with the cMBP2-decorated polyplexes (#8) showed only moderate expression levels in various organs and did not reveal any significant targeting effect over the non-targeted polyplexes (Fig. 6A). These surprising results pointed out to additional requirements of systemic delivery in comparison to cell culture or regional intratumoral delivery. The evaluated oligomers all contain a high content of PEG (same number of ethylene glycol units as protonatable Stp nitrogens already in the case of PEG24 oligomers) which had revealed imperfect pDNA compaction in a previous in vitro study.39 As shown above in vitro and upon intratumoral administration, the increase to an even 2-fold higher PEG content was found as unfavorable. | ||
| Fig. 6 Improving gene transfer after intravenous administration by combination of 2-arm PEGylated cMBP2-containing targeting oligomer #8 (or alanine control oligomer #9) with non-PEGylated 3-arm compacting oligomer #15. (A) In vivo gene expression at 48 h after i.v. administration of #8 and #9 polyplexes at N/P 12 into Huh7 tumor bearing mice (N = 5, mean ± SEM) in tumor, lung and liver. (B) EtBr exclusion assay comparing single oligomer polyplexes of #8 and combination polyplexes of #8 plus #15 at the adequate ratio (oligomer #8 at 70% and oligomer #15 at 30% of the total N/P). (C) Transmission electron microscopy images of polyplexes (N/P 12) formed with single oligomer #8 (above) and with the combination of oligomers #8 and #15 (below). (D) Cellular internalization comparing the cellular uptake of the Cy5-labeled pDNA polyplexes formed with the cMBP2-containing (#8 + #15) and alanine control (#9 + #15) polyplexes. (E) Serum stability of combination polyplexes (total N/P 12) formed with cMBP2 or alanine control oligomers analyzed at different serum incubation times by agarose gel shift assay. Where indicated, heparin was added to polyplexes after incubation in serum for 90 min. (F) In vivo gene expression in tumor, lung and liver at 48 h after i.v. administration of the combination polyplexes at N/P 12 into Huh7 tumor bearing mice (N = 5, mean ± SEM). Luciferase gene expression is presented as relative light units per organ or tumor (RLU/organ). Lysis buffer RLU values were subtracted. Liver weight was around 1.6 g, lung weight around 230 mg and Huh7 tumor weight 452 ± 189 mg. | ||
Therefore, for further optimization in vivo, the opposite direction was taken towards enhanced dimension of the polycationic oligomer core. Thus, the polyplexes formed with the PEGylated 4-arm oligomers #13 and #14 (Fig. 1-V), although they showed no improvement in vitro (Fig. S6†), were subjected to the intravenous administration studies. Encouragingly, these more polycationic oligomers indeed triggered a significant cMBP2 targeting-dependent gene expression in the distant tumor (Fig. S7†), though expression levels were moderate and similar as in other organs (liver and lung). In order to further improve cMBP2 targeting also upon intravenous administration, an alternative approach was considered for optimizing the nanoparticles. Instead of directly tuning the chemical oligomer structure, the desired two nanoparticle functions, pDNA compaction on the one hand, targeting and surface shielding on the other hand, were distributed between two different sequence-defined oligomers with or without PEG-content. As shown before, the PEG chains presumably not only shield the surface of the nanoparticles, but also interfere with the condensation process between the pDNA double strands. PEG-free analogs however were shown to mediate effective compaction of pDNA into rod- or toriod-like structures.39 For this purpose, a novel compacting 3-arm oligomer #15 (Fig. 1-VI) was synthesized being devoid PEG but having three oligocationic arms of the repeating Stp units with alternating histidines and terminal cysteines that are supposed to disulfide-crosslink with the terminal cysteines of the targeted PEGylated oligomer. This new apparently compacting oligomer #15 was mixed with the PEGylated cMBP2-containing 2-arm oligomer #8 at an optimized 30:70 cationic ratio to reach the total N/P ratio of 12 (oligomer #15 at N/P 3.6 and oligomer #8 at N/P 8.4) prior to polyplex formation with the pDNA. The resulting combination polyplexes were first evaluated for their pDNA compaction ability and compared to polyplexes formed with only oligomer #8. The decreased fluorescence in the EtBr exclusion assay confirmed an increased pDNA compaction (Fig. 6B). The gel-shift assays at lower N/P ratios as well indicated an improved pDNA complexation for combination polyplexes (Fig. S8A, B†). Moreover, their maintained redox disassembly based on incorporated cysteines was demonstrated with an addition of a reducing agent TCEP (Fig. S8C†). Transmission electron microscopy images revealed profound change in nanoparticles shape when the oligomer #8 was mixed with the non-PEGylated oligomer #15. The single oligomer #8 polyplexes (Fig. 6C, above) formed rather longer worm-like structures of several hundred nanometers, whereas the novel combination polyplexes (Fig. 6C, below) led to preferred formation of either approx. 50 nm round-shaped toroidal nanoparticles or 100–150 nm short nanorods. Next, the preservation of the cMBP2 target-specificity for such bi-oligomeric polyplexes needed to be analyzed, as the co-addition of 30% positively charged non-shielded oligomer might reduce the effects of surface shielding and targeting. The cellular internalization studies using Cy5-labeled pDNA revealed a far higher cellular uptake of the cMBP2-containing combination polyplexes (#8 + #15) as compared to their alanine control analogs (#9 + #15) (Fig. 6D), although the uptake was somewhat lower as compared to the cellular internalization of the single oligomer #8 polyplexes (Fig. 3A). Nevertheless, the receptor-specificity ligand dependence was well maintained; since the alanine control combination polyplexes exhibited practically no cellular uptake (Fig. 6D). The enhanced green fluorescent protein (EGFP) expression studies in vitro using flow cytometry showed superior protein expression for targeted combination polyplexes over single-oligomeric polyplexes (Fig. S9†). Prior to in vivo experiments, the stability of combination polyplexes in serum was evaluated. The gel shift cargo compaction studies in 90% FCS confirmed their integrity in serum for a time period of at least 90 min (Fig. 6E). Combination polyplexes displayed prolonged stability in serum versus single-oligomeric polyplexes (Fig. S10†). Size measurements in serum also pointed out to improved stability for bi-oligomeric particles (Fig. S11†). Compacted polyplexes were then injected intravenously in the subcutaneous Huh7 tumor-bearing mice. Remarkably and in sharp contrast to #8 polyplexes (Fig. 6A), with the cMBP2-containing (#8 + #15) polyplexes a greatly increased luciferase expression was achieved in the tumor (Fig. 6F) exceeding the signal of the non-targeted compacted control (#9 + #15) polyplexes by 22-fold and, excitingly, of the cMBP2-equipped oligomer (#8) polyplexes by 35-fold (Fig. 6A). Notably, the luciferase expression in the tumor was up to 50-fold higher than in the lung or liver (Fig. 6F). Gene transfer in other organs such as spleen or kidney was negligible (data not shown). In addition, qPCR disclosed the highest amount of residual pDNA in tumor for the combination (#8 + #15) polyplexes (Fig. S12†).
To investigate whether this co-addition effect is specific for the added compacting oligomer #15 or it can be achieved also by the addition of another non-shielded oligomer; a different oligomer combination was evaluated in polyplex formation. Instead of 3-arm oligomer #15, a 4-arm polycationic oligomer comprising Sph building blocks (Fig. 1-VII) was combined with the PEGylated targeting oligomer #8 to the total N/P ratio of 12 as above. This combination resulted in only minor additional decrease in EtBr fluorescence as compared to the single oligomer #8 (Fig. S13A†) and slightly lower cellular uptake of cMBP2-containing polyplexes (Fig. S13B†) as compared to the 3-arm oligomer combination (Fig. 6D). The stability in serum was again confirmed (Fig. S13C†). The intravenous application of these combination polyplexes again led to a significant cMBP2 targeting effect (Fig. S13D†), though with a lower luciferase expression in tumor and, interestingly, increased gene transfer in the lung. Apparently, the co-formulation of non-shielded compacting oligomers can significantly alter the nanoparticle compaction and shape. Interestingly, only intravenous in vivo studies and not in vitro transfections or local injections were able to disclose the advantage of polyplex compaction; this appears as important requirement to improve targeted gene transfer in vivo.
Conclusions
For the first time the proto oncogene c-Met/hepatocyte growth factor receptor which is over-expressed in many solid tumors was applied for tumor-targeted non-viral gene delivery. A selected c-Met-binding peptide called cMBP2 was confirmed as a potent and very promising targeting ligand. Incorporating this peptide, novel c-Met directed nanocarriers were developed for efficient gene delivery in vitro and, notably, successful c-Met targeted systemic gene transfer in vivo. The study was based on a precise way of assembling sequence-defined (oligoethanamino) amides. A recent combination of solid-phase supported peptide and polymer synthesis13,18,20,22 enabled easy modifications and implementation of various peptidic and artificial functional groups. This provides an optimal tool for determination of structure–activity relationships and optimization of gene carriers. The carriers were thus readily functionalized with c-Met targeting peptides, PEG for shielding against unintended interactions with biological surfaces, and cysteines for additional polyplex stabilization via bioreversible disulfide bond formation. Further optimization with endosomal escape promoting histidines yielded a targeting oligomer which displayed high luciferase expression in tumor upon locoregional administration. For intravenous administration, a new form of pDNA polyplexes, containing both a pDNA compacting oligomer and the surface-shielding c-Met targeting oligomer, was formulated for successful systemic and receptor-mediated gene transfer into distant tumors. The designed c-Met directed polyplexes emphasize the importance of each functional moiety and the proper relation of the polycationic part in relation to the shielding part within the nanoparticle. Moreover, the clear-cut functional findings with precise oligomers present a very useful springboard for further chemical evolution of biocompatible receptor-targeted nucleic acid carriers.Acknowledgements
This work was partly supported by the DFG Cluster “Nanosystems Initiative Munich” (to CB and EW), SFB 824 (to SM and EW) and the China Scholarship Council (CSC to DH). We thank Wolfgang Rödl for technical support, Michael Günther for scientific discussions, Olga Brück for skillful assistance and Markus Kovac for keeping the animals.Notes and references
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- P. Erbacher, T. Bettinger, E. Brion, J. L. Coll, C. Plank, J. P. Behr and J. S. Remy, J. Drug Targeting, 2004, 12, 223–236 CrossRef CAS PubMed.
- P. Kos, C. Scholz, E. E. Salcher, A. Herrmann and E. Wagner, Pharm. Nanotechnol., 2013, 1, 269–281 CrossRef CAS.
- C. Dufès, I. F. Uchegbu and A. G. Schätzlein, Adv. Drug Delivery Rev., 2005, 57, 2177–2202 CrossRef PubMed.
- A. Kwok, G. A. Eggimann, J. L. Reymond, T. Darbre and F. Hollfelder, ACS Nano, 2013, 7, 4668–4682 CrossRef CAS PubMed.
- D. A. Modi, S. Sunoqrot, J. Bugno, D. D. Lantvit, S. Hong and J. E. Burdette, Nanoscale, 2014, 6, 2812–2820 RSC.
- K. A. Howard, U. L. Rahbek, X. Liu, C. K. Damgaard, S. Z. Glud, M. O. Andersen, M. B. Hovgaard, A. Schmitz, J. R. Nyengaard, F. Besenbacher and J. Kjems, Mol. Ther., 2006, 14, 476–484 CrossRef CAS PubMed.
- M. Lavertu, S. Methot, N. Tran-Khanh and M. D. Buschmann, Biomaterials, 2006, 27, 4815–4824 CrossRef CAS PubMed.
- T. Luhmann, M. Rimann, A. G. Bittermann and H. Hall, Bioconjugate Chem., 2008, 19, 1907–1916 CrossRef CAS PubMed.
- M. Meyer, C. Dohmen, A. Philipp, D. Kiener, G. Maiwald, C. Scheu, M. Ogris and E. Wagner, Mol. Pharm., 2009, 6, 752–762 CrossRef CAS PubMed.
- Y. Liu, L. Feng, T. Liu, L. Zhang, Y. Yao, D. Yu, L. Wang and N. Zhang, Nanoscale, 2014, 6, 3231–3242 RSC.
- Q. R. Chen, L. Zhang, S. A. Stass and A. J. Mixson, Nucleic Acids Res., 2001, 29, 1334–1340 CrossRef CAS PubMed.
- L. Hartmann and H. G. Börner, Adv. Mater., 2009, 21, 3425–3431 CrossRef CAS PubMed.
- P. Kos and E. Wagner, Chim. Oggi – Chem. Today, 2013, 31, 6–10 CAS.
- H. Uchida, K. Miyata, M. Oba, T. Ishii, T. Suma, K. Itaka, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2011, 133, 15524–15532 CrossRef CAS PubMed.
- V. Russ, T. Frohlich, Y. Li, A. Halama, M. Ogris and E. Wagner, J. Gene Med., 2010, 12, 180–193 CAS.
- C. C. Lee, Y. Liu and T. M. Reineke, Bioconjugate Chem., 2008, 19, 428–440 CrossRef CAS PubMed.
- L. Hartmann, S. Hafele, R. Peschka-Suss, M. Antonietti and H. G. Borner, Chemistry, 2008, 14, 2025–2033 CrossRef CAS PubMed.
- U. Lachelt, P. Kos, F. M. Mickler, A. Herrmann, E. E. Salcher, W. Rodl, N. Badgujar, C. Brauchle and E. Wagner, Nanomedicine, 2014, 10, 35–44 CrossRef PubMed.
- D. Schaffert, N. Badgujar and E. Wagner, Org. Lett., 2011, 13, 1586–1589 CrossRef CAS PubMed.
- E. E. Salcher, P. Kos, T. Frohlich, N. Badgujar, M. Scheible and E. Wagner, J. Controlled Release, 2012, 164, 380–386 CrossRef CAS PubMed.
- D. Schaffert, C. Troiber, E. E. Salcher, T. Frohlich, I. Martin, N. Badgujar, C. Dohmen, D. Edinger, R. Klager, G. Maiwald, K. Farkasova, S. Seeber, K. Jahn-Hofmann, P. Hadwiger and E. Wagner, Angew. Chem., Int. Ed., 2011, 50, 8986–8989 CrossRef CAS PubMed.
- P. M. Carlson, J. G. Schellinger, J. A. Pahang, R. N. Johnson and S. H. Pun, Biomater. Sci., 2013, 1, 736–744 RSC.
- A. del Pozo-Rodriguez, S. Pujals, D. Delgado, M. A. Solinis, A. R. Gascon, E. Giralt and J. L. Pedraz, J. Controlled Release, 2009, 133, 52–59 CrossRef CAS PubMed.
- Y. W. Won, H. A. Kim, M. Lee and Y. H. Kim, Mol. Ther., 2010, 18, 734–742 CrossRef CAS PubMed.
- C. Scholz, P. Kos, L. Leclercq, X. Jin, H. Cottet and E. Wagner, ChemMedChem, 2014, 9, 2104–2110 CrossRef CAS PubMed.
- L. Wang, W. Su, Z. Liu, M. Zhou, S. Chen, Y. Chen, D. Lu, Y. Liu, Y. Fan, Y. Zheng, Z. Han, D. Kong, J. C. Wu, R. Xiang and Z. Li, Biomaterials, 2012, 33, 5107–5114 CrossRef CAS PubMed.
- T. Merdan, J. Callahan, H. Petersen, K. Kunath, U. Bakowsky, P. Kopeckova, T. Kissel and J. Kopecek, Bioconjugate Chem., 2003, 14, 989–996 CrossRef CAS PubMed.
- J. Li, D. Cheng, T. Yin, W. Chen, Y. Lin, J. Chen, R. Li and X. Shuai, Nanoscale, 2014, 6, 1732–1740 RSC.
- J. Cheng, B. A. Teply, I. Sherifi, J. Sung, G. Luther, F. X. Gu, E. Levy-Nissenbaum, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Biomaterials, 2007, 28, 869–876 CrossRef CAS PubMed.
- E. M. Kim, H. J. Jeong, I. K. Park, C. S. Cho, H. B. Moon, D. Y. Yu, H. S. Bom, M. H. Sohn and I. J. Oh, J. Controlled Release, 2005, 108, 557–567 CrossRef CAS PubMed.
- K. S. Kim, Y. Lei, D. B. Stolz and D. Liu, Gene Ther., 2007, 14, 704–708 CrossRef CAS PubMed.
- C. Dohmen, D. Edinger, T. Frohlich, L. Schreiner, U. Lachelt, C. Troiber, J. Radler, P. Hadwiger, H. P. Vornlocher and E. Wagner, ACS Nano, 2012, 6, 5198–5208 CrossRef CAS PubMed.
- R. Weissleder, K. Kelly, E. Y. Sun, T. Shtatland and L. Josephson, Nat. Biotechnol., 2005, 23, 1418–1423 CrossRef CAS PubMed.
- C. Y. Zhang, P. Kos, K. Muller, W. Schrimpf, C. Troiber, U. Lachelt, C. Scholz, D. C. Lamb and E. Wagner, J. Controlled Release, 2014, 180, 42–50 CrossRef CAS PubMed.
- Y. Ping, Q. Hu, G. Tang and J. Li, Biomaterials, 2013, 34, 6482–6494 CrossRef CAS PubMed.
- R. P. Harbottle, R. G. Cooper, S. L. Hart, A. Ladhoff, T. McKay, A. M. Knight, E. Wagner, A. D. Miller and C. Coutelle, Hum. Gene Ther., 1998, 9, 1037–1047 CrossRef CAS PubMed.
- T. Y. Lee, C. T. Lin, S. Y. Kuo, D. K. Chang and H. C. Wu, Cancer Res., 2007, 67, 10958–10965 CrossRef CAS PubMed.
- I. Martin, C. Dohmen, C. Mas-Moruno, C. Troiber, P. Kos, D. Schaffert, U. Lachelt, M. Teixido, M. Gunther, H. Kessler, E. Giralt and E. Wagner, Org. Biomol. Chem., 2012, 10, 3258–3268 CAS.
- M. E. Hernandez, J. D. Rembao, D. Hernandez-Baltazar, R. A. Castillo-Rodriguez, V. M. Tellez-Lopez, Y. M. Flores-Martinez, C. E. Orozco-Barrios, H. A. Rubio, A. Sanchez-Garcia, J. Ayala-Davila, M. L. Arango-Rodriguez, L. Pavon, T. Mejia-Castillo, P. Forgez and D. Martinez-Fong, Nanomedicine, 2014, 10, 745–754 CrossRef CAS PubMed.
- P. Kos, U. Lachelt, D. He, Y. Nie, Z. Gu and E. Wagner, J. Pharm. Sci., 2015, 104, 464–475 CrossRef CAS PubMed.
- M. F. Wolschek, C. Thallinger, M. Kursa, V. Rossler, M. Allen, C. Lichtenberger, R. Kircheis, T. Lucas, M. Willheim, W. Reinisch, A. Gangl, E. Wagner and B. Jansen, Hepatology, 2002, 36, 1106–1114 CrossRef CAS PubMed.
- F. M. Mickler, L. Mockl, N. Ruthardt, M. Ogris, E. Wagner and C. Brauchle, Nano Lett., 2012, 12, 3417–3423 CrossRef CAS PubMed.
- J. T. Koepsel, E. H. Nguyen and W. L. Murphy, Integr. Biol., 2012, 4, 914–924 RSC.
- T. A. Martin and W. G. Jiang, Anticancer Agents Med. Chem., 2010, 10, 2–6 CrossRef CAS.
- B. Peruzzi and D. P. Bottaro, Clin. Cancer Res., 2006, 12, 3657–3660 CrossRef CAS PubMed.
- R. M. Lu, Y. L. Chang, M. S. Chen and H. C. Wu, Biomaterials, 2011, 32, 3265–3274 CrossRef CAS PubMed.
- M. J. Vosjan, J. Vercammen, J. A. Kolkman, M. Stigter-van Walsum, H. Revets and G. A. van Dongen, Mol. Cancer Ther., 2012, 11, 1017–1025 CrossRef CAS PubMed.
- E. S. Mittra, H. Fan-Minogue, F. I. Lin, J. Karamchandani, V. Sriram, M. Han and S. S. Gambhir, Clin. Cancer Res., 2013, 19, 5711–5721 CrossRef CAS PubMed.
- X. Chen, G. Ding, Q. Gao, J. Sun, Q. Zhang, L. Du, Z. Qiu, C. Wang, F. Zheng, B. Sun, J. Ni, Z. Feng and J. Zhu, PLoS One, 2013, 8, e63093 CAS.
- R. Heukers, I. Altintas, S. Raghoenath, E. De Zan, R. Pepermans, R. C. Roovers, R. Haselberg, W. E. Hennink, R. M. Schiffelers, R. J. Kok and P. M. van Bergen en Henegouwen, Biomaterials, 2014, 35, 601–610 CrossRef CAS PubMed.
- T. H. Nguyen, N. Loux, I. Dagher, C. Vons, K. Carey, P. Briand, M. Hadchouel, D. Franco, J. Jouanneau, R. Schwall and A. Weber, Cancer Gene Ther., 2003, 10, 840–849 CrossRef CAS PubMed.
- E. M. Kim, E. H. Park, S. J. Cheong, C. M. Lee, D. W. Kim, H. J. Jeong, S. T. Lim, M. H. Sohn, K. Kim and J. Chung, Nucl. Med. Biol., 2009, 36, 371–378 CrossRef CAS PubMed.
- E. M. Kim, E. H. Park, S. J. Cheong, C. M. Lee, H. J. Jeong, D. W. Kim, S. T. Lim and M. H. Sohn, Bioconjugate Chem., 2009, 20, 1299–1306 CrossRef CAS PubMed.
- C. Lin, C.-J. Blaauboer, M. M. Timoneda, M. C. Lok, M. van Steenbergen, W. E. Hennink, Z. Zhong, J. Feijen and J. F. J. Engbersen, J. Controlled Release, 2008, 126, 166–174 CrossRef CAS PubMed.
- P. M. Klein and E. Wagner, Antioxid. Redox Signaling, 2014, 21, 804–817 CrossRef CAS PubMed.
- P. M. Klein, K. Muller, C. Gutmann, P. Kos, A. Krhac Levacic, D. Edinger, M. Hohn, J. C. Leroux, M. A. Gauthier and E. Wagner, J. Controlled Release, 2014 DOI:10.1016/j.jconrel.2014.12.035.
- C. Plank, K. Zatloukal, M. Cotten, K. Mechtler and E. Wagner, Bioconjugate Chem., 1992, 3, 533–539 CrossRef CAS.
- E. Kaiser, R. L. Colescott, C. D. Bossinger and P. I. Cook, Anal. Biochem., 1970, 34, 595–598 CrossRef CAS.
- P. Zhao, T. Grabinski, C. Gao, R. S. Skinner, T. Giambernardi, Y. Su, E. Hudson, J. Resau, M. Gross, G. F. Vande Woude, R. Hay and B. Cao, Clin. Cancer Res., 2007, 13, 6049–6055 CrossRef CAS PubMed.
- C. Troiber, D. Edinger, P. Kos, L. Schreiner, R. Klager, A. Herrmann and E. Wagner, Biomaterials, 2013, 34, 1624–1633 CrossRef CAS PubMed.
- B. Newland, A. Aied, A. V. Pinoncely, Y. Zheng, T. Zhao, H. Zhang, R. Niemeier, E. Dowd, A. Pandit and W. Wang, Nanoscale, 2014, 6, 7526–7533 RSC.
- W. Zauner, A. Kichler, W. Schmidt, K. Mechtler and E. Wagner, Exp. Cell Res., 1997, 232, 137–145 CrossRef CAS.
- J. Cheng, R. Zeidan, S. Mishra, A. Liu, S. H. Pun, R. P. Kulkarni, G. S. Jensen, N. C. Bellocq and M. E. Davis, J. Med. Chem., 2006, 49, 6522–6531 CrossRef CAS PubMed.
- L. J. Appleman, J. Clin. Oncol., 2011, 29, 4837–4838 CrossRef CAS PubMed.
- P. Midoux, C. Pichon, J. J. Yaouanc and P. A. Jaffres, Br. J. Pharmacol., 2009, 157, 166–178 CrossRef CAS PubMed.
- S. T. Chou, Q. Leng, P. Scaria, J. D. Kahn, L. J. Tricoli, M. Woodle and A. J. Mixson, Biomacromolecules, 2013, 14, 752–760 CrossRef CAS PubMed.
- J. F. Stefanick, J. D. Ashley, T. Kiziltepe and B. Bilgicer, ACS Nano, 2013, 7, 2935–2947 CrossRef CAS PubMed.
- S. Khargharia, K. Kizzire, M. D. Ericson, N. J. Baumhover and K. G. Rice, J. Controlled Release, 2013, 170, 325–333 CrossRef CAS PubMed.
- S. Mishra, P. Webster and M. E. Davis, Eur. J. Cell Biol., 2004, 83, 97–111 CrossRef CAS PubMed.
- M. Hamidi, A. Azadi and P. Rafiei, Drug Delivery, 2006, 13, 399–409 CrossRef CAS PubMed.
- C. E. Nelson, J. R. Kintzing, A. Hanna, J. M. Shannon, M. K. Gupta and C. L. Duvall, ACS Nano, 2013, 7, 8870–8880 CrossRef CAS PubMed.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189–207 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nr06556e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |