
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biocompatible mannosylated endosomal-escape nanoparticles enhance selective delivery of short nucleotide sequences to tumor associated macrophages
Ryan A.
Ortega
abc,
Whitney J.
Barham
c,
Bharat
Kumar
a,
Oleg
Tikhomirov
c,
Ian D.
McFadden
abc,
Fiona E.
Yull
*c and
Todd D.
Giorgio
*abc
aDepartment of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA. E-mail: todd.d.giorgio@vanderbilt.edu; fiona.yull@vanderbilt.edu
bVanderbilt Institute for Nanoscale Science and Engineering, Vanderbilt University, Nashville, TN 37235, USA
cDepartment of Cancer Biology, Vanderbilt-Ingram Cancer Center, Nashville, Tennessee 37232, USA
First published on 30th October 2014
Abstract
Tumor associated macrophages (TAMs) can modify the tumor microenvironment to create a pro-tumor niche. Manipulation of the TAM phenotype is a novel, potential therapeutic approach to engage anti-cancer immunity. siRNA is a molecular tool for knockdown of specific mRNAs that is tunable in both strength and duration. The use of siRNA to reprogram TAMs to adopt an immunogenic, anti-tumor phenotype is an attractive alternative to ablation of this cell population. One current difficulty with this approach is that TAMs are difficult to specifically target and transfect. We report here successful utilization of novel mannosylated polymer nanoparticles (MnNP) that are capable of escaping the endosomal compartment to deliver siRNA to TAMs in vitro and in vivo. Transfection with MnNP-siRNA complexes did not significantly decrease TAM cell membrane integrity in culture, nor did it create adverse kidney or liver function in mice, even at repeated doses of 5 mg kg−1. Furthermore, MnNP effectively delivers labeled nucleotides to TAMs in mice with primary mammary tumors. We also confirmed TAM targeting in the solid tumors disseminated throughout the peritoneum of ovarian tumor bearing mice following injection of fluorescently labeled MnNP-nucleotide complexes into the peritoneum. Finally, we show enhanced uptake of MnNP in lung metastasis associated macrophages compared to untargeted particles when using an intubation delivery method. In summary, we have shown that MnNP specifically and effectively deliver siRNA to TAMs in vivo.
Introduction
The tumor supportive stroma has been identified as an attractive target for therapeutic intervention in solid tumors. While most tumors exhibit a large degree of cellular heterogeneity, the tumor stroma is potentially more homogenous with respect to local stromal cell phenotype.1 Macrophages, for example, play an important trophic role in tissue development and a growing body of evidence suggests that these trophic roles are recapitulated in the tumor microenvironment.2 Tumor associated macrophages (TAMs) are directly involved in establishing a pro-tumorigenic local microenvironment in many tumor types. TAMs stimulate angiogenesis, promote tumor growth and metastasis, and suppress the normal immune response.3,4 Concordantly, an increase in TAMs at the site of tumor progression is predictive of poor prognosis and survival in mouse models of human breast cancer and in multiple human cancers.5,6TAMs exhibit characteristics that are a blend of the two defined categories of macrophage phenotype. Like the classically immunogenic (M(LPS) or M(LPS + IFNγ)) macrophage, TAMs produce low levels of inflammatory cytokines which results in pro-tumorigenic, smoldering inflammation.7–9 Like the typical tissue remodeling, or alternatively activated macrophage, TAMs break down the surrounding extracellular matrix, secrete growth factors, and inhibit the adaptive immune response.10 Data show that TAMs are a viable therapeutic target in cancer treatment and that ablating these cells can have a powerful anti-tumor effect.3 Rather than ablate these important modulators of immunity, another proposed solution is to target pro-tumor macrophages with a therapeutic agent that can alter their behavior to induce an acute, but strong immunogenic phenotype capable of stimulating anti-tumor immunity.11,12
Recent decades have bought great advances in gene therapy technologies, specifically the emergence of siRNA. siRNAs can be used to inhibit the translation of specific mRNAs without significant off-target side effects by RNA interference (RNAi).13 RNAi results in loss of target protein expression by siRNA binding to complementary mRNA strands, leading to mRNA degradation.14 The clinical translation potential of these nucleotides has been demonstrated by the targeted delivery of therapeutic siRNA in humans.15,16 Another advantage of siRNA is that the degree of knockdown can be tuned to varying degrees of specificity, potency, duration by taking advantage of the transient nature of siRNA inhibition.17 One of the current obstacles therapeutic siRNA faces is the delivery of active siRNA to specific cells types. Free siRNA is rapidly degraded in vivo by circulating RNases In addition, the strong polyanionic charge and significant molecular mass limits cellular entry of unformulated nucleotides. An siRNA delivery vehicle that provides preferential localization to particular tissues and/or target cell types as well as superior nucleotide protection and cellular entry is required for optimal and spatially specific protein knockdown.
Targeted nanoparticles have been used in clinical trials of siRNA therapeutics as a delivery device to reach specific cell populations.18 The generation of a charge-neutral or near-neutral surface of a nanoparticle carrier of siRNA improves biocompatibility, as well as allowing for the potential attachment of a targeting ligand to the surface of the particle, improving cellular specificity. To address this, we have developed and characterized a tri-block polymer nanoparticle capable of targeting TAMs for nucleotide delivery. The core of the particle is comprised of a hydrophobic, pH responsive block that triggers endosomal escape and cytoplasmic delivery of the siRNA.18 One advantage of this core block design is its self-assembly into particles in an aqueous solution due to its tunable hydrophobicity.19 The second block is a poly(DMAEMA) polymer with a polycationic charge that condenses polyanionic oligonucleotides within the particle and serves to carry and protect siRNA for delivery to a target cell. A distal, azide-presenting block serves as a modular platform for further functionalization with targeting ligands or other biomolecules of interest and represents novelty in the nanoparticle synthesis scheme relative to our previous work.
Using ‘click’ chemistry, we previously functionalized the surface of these nanoparticles with a mannose ligand (MnNP) to specifically target TAMs via the mannose receptor, CD206.20 The mannose receptor is highly specific to mature macrophages and has been shown previously to be upregulated on the surface of TAMs.21–23 MnNPs are systematically designed to condense and shield siRNA in the interior of the particle for optimal systemic transport, enter the tumor vasculature via the enhanced permeability and retention (EPR) effect, specifically target TAMs in the tumor microenvironment, and escape the low pH late endosome to deliver functional siRNA into the cytoplasm. In this study, we build upon previously published work to demonstrate that these MnNP are biocompatible in vitro and in vivo at physiologically relevant doses, provide evidence for the efficacy of the CD206-targeting mannose ligand on the surface of the particles, and demonstrate the effective delivery of protected nucleotides to TAMs.
Materials and methods
Materials
For experiments using MnNP to deliver siRNA or short, fluorescently labeled DNA strands, MnNP polymer was reconstituted in sterile PBS at a concentration of 4 mg ml−1 and sonicated for 10 minutes. The MnNP in aqueous solution were used immediately or stored in aliquots at −20 °C. For complexation with the MnNP, all nucleotides were diluted to 50 μM in sterile, nuclease-free water. In order to form MnNP-nucleotide complexes with the optimal N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P ratio as described in our previous work, the 4 mg ml−1 MnNP solution was combined with 50 μM nucleotide solution in a 2:1, vol:vol ratio (160 ng of MnNP polymer per pmol of siRNA). The nucleotides were allowed to complex with the MnNP for 1 hour at room temperature, then used in in vivo and in vitro experiments. Nucleotide loaded, hydroxyl-capped nanoparticles (OHNP) were also formulated using this protocol.
:P ratio as described in our previous work, the 4 mg ml−1 MnNP solution was combined with 50 μM nucleotide solution in a 2:1, vol:vol ratio (160 ng of MnNP polymer per pmol of siRNA). The nucleotides were allowed to complex with the MnNP for 1 hour at room temperature, then used in in vivo and in vitro experiments. Nucleotide loaded, hydroxyl-capped nanoparticles (OHNP) were also formulated using this protocol.
Methods
:vol) FBS and 1% Pen Strep (Gibco) at 37 °C in a 5% CO2 humidified atmosphere.
000 cells per well. TAMs were transfected for 24 hours at 37 °C with MnNP-siRNA complexes with a scrambled siRNA sequence at 10 nM and 50 nM concentrations of siRNA with accompanying MnNP concentrations as described above, with and without 6 hours of TNF-α stimulation (10 ng ml−1) following transfection. A second set TAMs were transfected with the same siRNA using Lipofectamine 2000 (10:1, vol:vol, Lipofectamine:siRNA) (Ambion) for 24 hours with and without TNF-α stimulation. 2 minutes of incubation with Triton x-100 was used as a negative control for cell membrane viability. To stain cell membranes for an exclusion viability assay, samples were incubated with Trypan Blue for 5 minutes, and the number of viable and non-viable cells were counted (over 1000 cells per sample, N = 3 for each condition).
Results and discussion
Particle-siRNA complexation and biocompatibility
MnNPs were synthesized as previously reported.20 The chemical structure of the particles is shown in Fig. 1A. Commercial transfection agents, such as Lipofectamine, possess strong, positive surface charges which facilitate their ability to deliver siRNA and other nucleotides into cells. This same surface charge renders commercial transfection agents unacceptable for in vivo use; they exhibit low biocompatibility due to aggregate formation and cytotoxicity. Strongly cationic structures have the potential to adsorb anionic serum proteins and lose function through aggregation or become opsonized and rapidly cleared from the blood compartment. Therefore, net surface charge of the formulated delivery system is known to be an important determinate of in vivo function. The cationic charges in MnNPs are designed to be largely shielded from surface presentation to optimize their potential for in vivo use. The success of this shielding strategy was confirmed by the moderately positive zeta potential of approximately +20 mV as measured by dynamic light scattering (Fig. 1B) in comparison with Lipofectamine 2000, a commonly used commercial transfection agent with a zeta potential of more than +50 mV. Charge-based considerations are of paramount importance in the context of intravascular administration. Similar considerations are operative for the intraperitoneal and tracheal delivery used in these studies. Thus, the low cationicity MnNPs may optimize successful siRNA delivery to TAMs by the routes of administration considered here.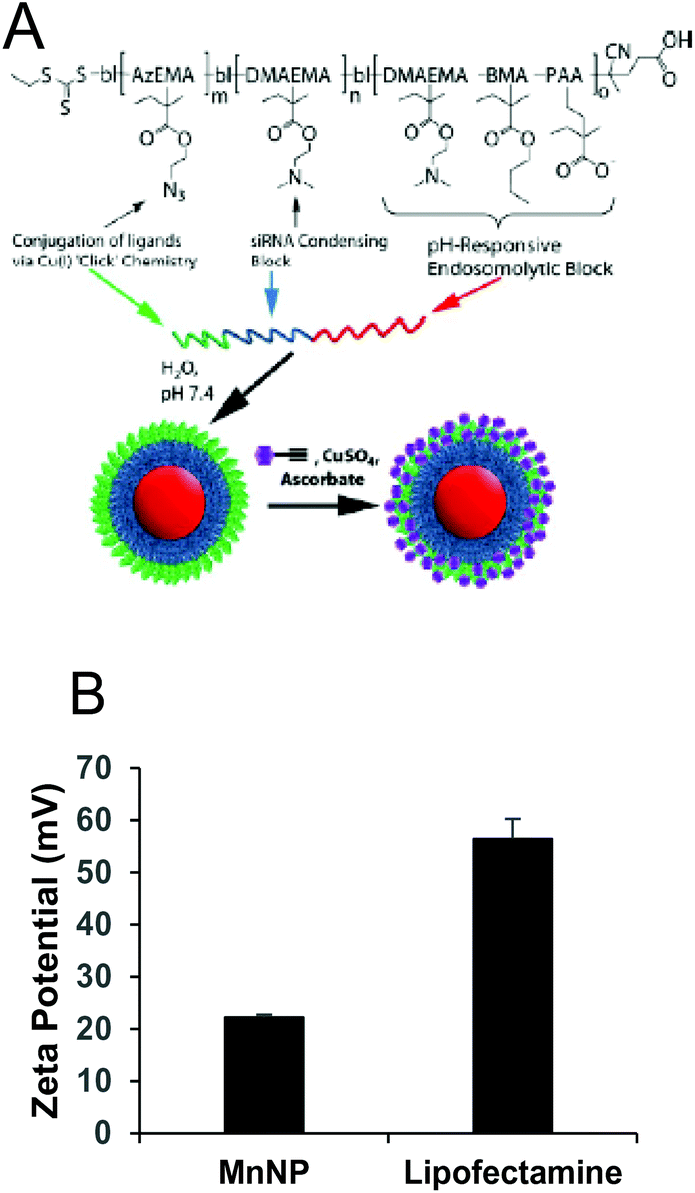 | ||
| Fig. 1 (A) Schematic representation of the mannosylated endosomal escape nanoparticle illustrating the pH-responsive core (red), the siRNA condensing block (blue), the modular ‘clickable’ surface (green), and the mannose surface functionalization (purple). (B) Zeta potential measurements of MnNP show a significantly milder surface charge as compared to commercial transfection agent, Lipofectamine 2000. (P = 0.003, N = 3). | ||
Cationic charge mediates strong interactions with anionic cell membranes to facilitate delivery of cargo in vitro, but also may destabilize cells and be responsible for dose-limiting toxicity. We determined the effect of MnNP on membrane viability in an enriched TAM population as an in vitro measurement of the biocompatibility of the MnNP-siRNA complexes (Fig. 2). siRNA was delivered at low (10 nM) and high (50 nM) concentrations in the presence or absence of a second immunostimulatory cytokine, TNF-α (10 ng ml−1), a common tumor promoter in the tumor microenvironment.32,33 After 24 hours of incubation with MnNP-siRNA there was no significant decrease in TAM membrane viability in samples transfected with MnNP as measured by Trypan Blue staining.34 The high siRNA concentration had a slight effect on TAM membrane viability in the context of the addition of TNF-α, but this effect was not significant. In contrast, in the presence of TNF-α, transfection with the commercial agent, Lipofectamine 2000, resulted in a significant decrease in viability (10–18%). Lipofectamine is a mixture of positively charged lipids; the strongly positive surface charge on the lipid transfection complex results in an efficacious in vitro transfection agent, but significantly decreased biocompatibility in vivo due to strong interactions with the plasma membrane of non-targeted cells.35 MnNP-siRNA complexes possess a mildly positive surface charge, resulting in increased biocompatibility and suitability for in vitro and in vivo transfection. Our studies indicate that uptake of empty MnNPs does not significantly enhance or inhibit the immune response in macrophages and particle samples test below the limit of detection in an LAL endotoxin assay, indicating an endotoxin concentration less than 0.1 EU ml−1.
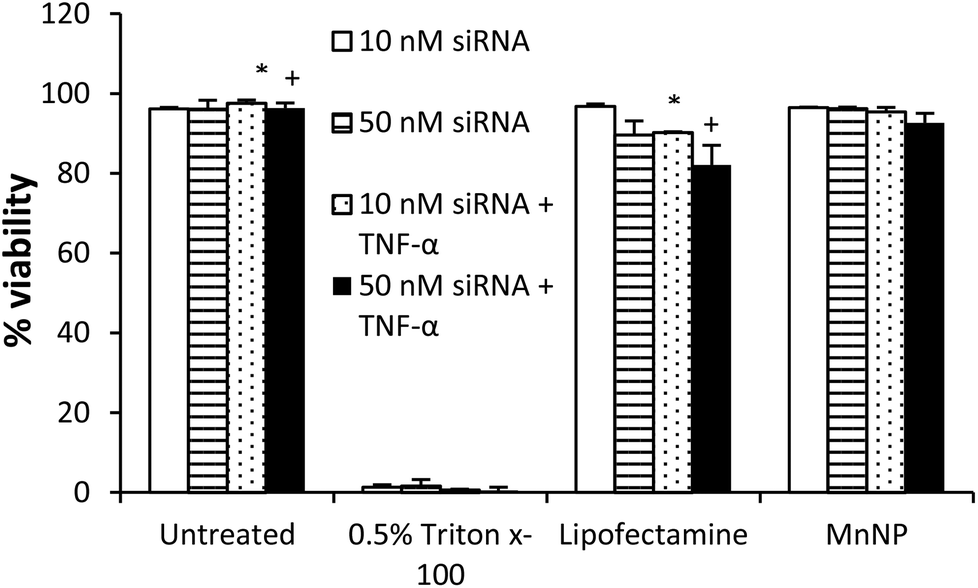 | ||
| Fig. 2 Transfection of TAMs in vitro with MnNP-siRNA complexes results in no significant loss in membrane viability, even with secondary stimulation from 10 ng ml−1 TNF-α stimulation for 6 hours. There is significant loss in viability associated with the use of Lipofectamine as a transfection agent under these conditions. (*, +: P < 0.05 by T-test, N = 3 for each condition). | ||
To test in vivo biocompatibility, wild type FVB mice were intravenously (i.v.) injected with MnNP-scrambled siRNA complexes at 5 mg kg−1 once daily for 3 consecutive days. This route of administration was chosen for the biocompatibility study because systemic administration is expected to potentially induce any off target, negative effects more strongly than other, more spatially contained administration routes such as intraperitoneal injection or intratracheal delivery. 24 hours after the final injection, blood serum was analyzed for alanine transaminase (ALT) and aspartate transaminase (AST) levels as an indicator of hepatic function and blood urea nitrogen (BUN) and serum creatinine (CREAT) as an indicator of renal function. No significant change in liver or kidney function was observed following 3 sequential doses every 24 hours with MnNP, consistent with a lack of acute, organ level toxicity (Table 1). Average serum creatinine was slightly lower than the normal range, but the difference is not large enough to indicate renal damage.36 Since we anticipate that MnNPs will clear the vasculature primarily through liver/spleen/reticuloendothelial mechanisms and that molecular components from disassembled micelles (if any) will be sufficiently small for renal excretion, the lack of significant dysregulation in liver and kidney characteristics suggests both nontoxicity and serum stability. Also by utilizing spatially confined routes of administration many of the complications potentially associated with systemic delivery of nanoparticles can be mitigated.
| Mouse number | |||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | Average | σ | Normal | |
| ALT (U L−1) | 37 | 49 | 73 | 105 | 66 | 30 | 26–120 |
| AST (U L−1) | 96 | 86 | 213 | 140 | 133.8 | 57.8 | 69–191 |
| BUN (mg dL−1) | 21 | 23 | 27 | 23 | 23.5 | 2.51 | 19–34 |
| CREAT (mg dL−1) | 0.4 | 0.3 | 0.3 | 0.4 | 0.35 | 0.05 | 0.4–0.6 |
MnNPs enhance delivery of fluorescently labeled siRNA to TAMs ex vivo
Spontaneously arising murine mammary tumors were harvested from 12 week old mice containing a mammary epithelium targeted polyoma middle T oncogene (PyMT).37Ex vivo TAMs were transfected with FAM-labeled siRNA for 2 or 6 hours with either MnNP, hydroxyl-capped (non-targeted) endosomal escape nanoparticles (OHNP), or Lipofectamine (Fig. 3) and intracellular FAM fluorescence was used as an indicator of nanoparticle uptake in the transfected TAMs.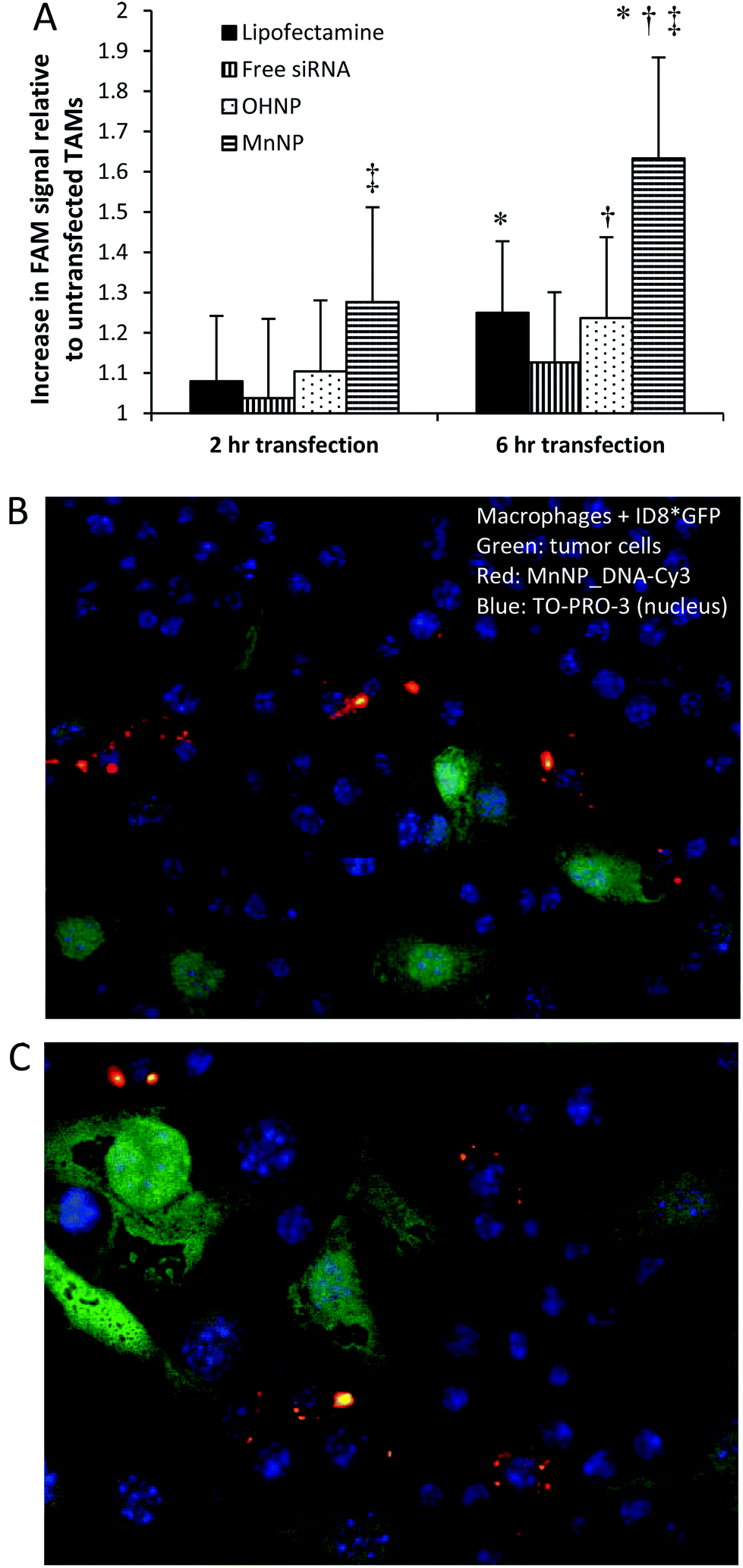 | ||
| Fig. 3 (A) In vitro transfection of TAMs isolated from murine mammary PyMT tumors with MnNP results in greater uptake of FAM labeled siRNA as compared to other transfection agents. MnNP creates a significantly greater increase in FAM fluorescence in the TAMs from the 2 h to the 6 h time point as compared to Lipofectamine or the non-targeted OHNP (ANOVA, P < 0.05) (*,†, ‡: P < 0.05, by T-test) and the high rate of change of FAM fluorescence in these cells indicates a rapid burst effect in delivery using MnNP. The two non-targeted agents have similar delivery efficacies. (B & C) Wild type macrophages and ID8 ovarian tumor cells (green) in co-culture with Cy3 labeled MnNP (red). Particles cane be seen in macrophages, but not in the tumor cells. | ||
After 6 hours of transfection, TAMs exposed to MnNP-siRNA_FAM had significantly higher FAM fluorescence than TAMs transfected with Lipofectamine (1.6-fold increase in FAM fluorescence vs. 1.25-fold increase). Furthermore, fluorescence measured in TAMs transfected with the non-targeted OHNP was not significantly different than the fluorescence measured in TAMs transfected with the highly cationic Lipofectamine (also possessing no specificity for TAMs). For all samples, siRNA delivery increased from 2 to 6 hours, with the greatest increase occurring in the MnNP transfected samples. The rate of MnNP uptake by the TAMs is also increased at the 2 hour time point compared to the untargeted transfection agents. We observed a burst effect in delivery during the first few hours in the MnNP transfected samples indicating rapid, mannose-mediated uptake. This burst effect was not observed with the other transfection agents, which had more steady rates of delivery following the first hours of transfection.
Without the aid of transfection complexes, a small amount of free siRNA is taken up by the TAMs. This is likely due to phagocytosis of the siRNA molecules by the macrophages following binding of serum proteins to the siRNA. It is important to note that while unmodified siRNA may be taken up by cells in a non-specific manner both in vitro and in vivo, siRNA delivered in this fashion exhibits little to no activity. The transfection efficacy of the untargeted nucleotide, OHNP and Lipofectamine, are similar. Previously, we have demonstrated enhanced siRNA delivery to murine bone marrow derived macrophages (BMDMs) ex vivo by MnNPs to be mannose dependent. Results of this study, with TAMs, are consistent with earlier findings confirming MnNP recognition by CD206 in BMDMs.20
To confirm that the MnNPs are taken up by macrophages rather than tumor cells in the tumor environment, we co-cultured wild type macrophages with and GFP expressing, ID8 ovarian tumor cell line. MnNP were complexed with a Cy3 labeled DNA sequence with a base pair arrangement matching the scrambled siRNA sequence used previously. After overnight incubation with the MnNP_DNA-Cy3 complexes, confocal imaging of the co-cultured cells (Fig. 3b & c) shows that there is no uptake of the MnNP by the ID8 tumor cells (green) and the Cy3 labeled MnNP (red) are visible in the macrophages. The images show high intensity Cy3 signal in large volumes of the macrophages’ cytoplasm, likely indicative of MnNP clusters or aggregates inside the cells following release from the endosomes. The images also show more diffuse, punctate Cy3 signal, possibly showing the early stages of particle uptake in intact endosomes.
MnNPs deliver fluorescently labeled nucleotides to murine TAMs in vivo
In order to test the in vivo efficacy of the TAM-targeted MnNP, we delivered MnNP carrying fluorescently-tagged, scrambled DNA strands to murine tumors in several tumor models. PyMT mice have spontaneously developed palpable mammary tumors with dimensions ≥1 cm at 12 weeks and the tumors have a necrotic core with significant immune cell infiltration and TAM population.37,38 Due to the poorly vascularized nature of this tissue, cells and fluid entering the tumor are retained in the local microenvironment longer than they would be retained in normal tissue.39,40 We directly injected MnNP-DNA conjugates into the center of the tumor for a well contained depot of particles that would co-localize with TAMs (Fig. 4A). We observed no adverse effects of multiple, direct MnNP injections.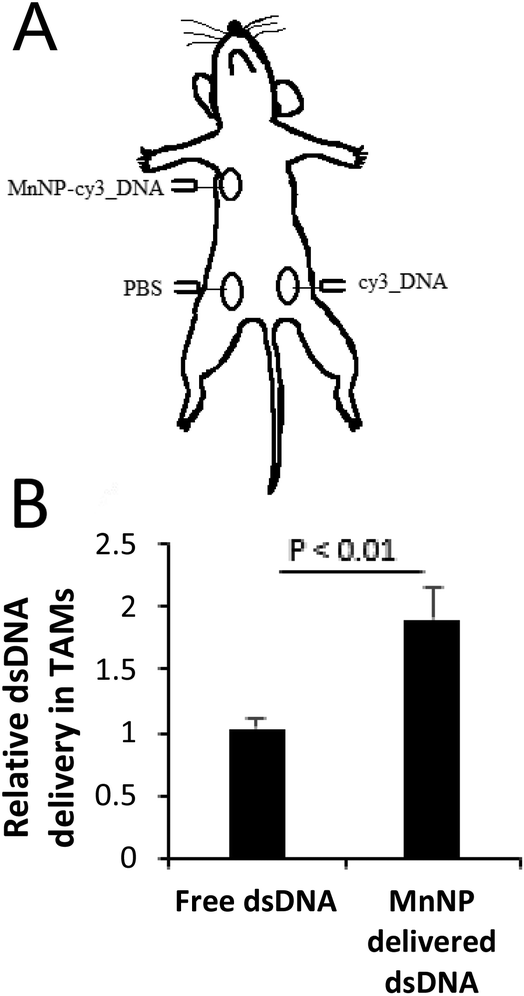 | ||
| Fig. 4 (A) Diagram of mammary tumor injection scheme for PyMT mice. Three isolated tumors from each mouse (N = 3) were selected based on uniformity of size and shape. The tumors were injected 3 times, 24 hours apart with MnNP-DNA_Cy3 complexes, free DNA_Cy3, or PBS into the center of each selected tumor. The TAMs were isolated from each tumor and analyzed for cy3 fluorescence. (B) There was a statistically significant increase (T-test for significance) in Cy3 fluorescence in TAMs from tumors that received MnNP-DNA_Cy3 as compared to non-specific DNA_Cy3 phagocytosis. | ||
Formulation with MnNPs significantly increased DNA_Cy3 delivery to TAMs in murine breast tumors (Fig. 4B). Intratumoral injection of a fluorescently labeled DNA mimic of siRNA resulted in nearly 2-fold greater TAM uptake compared to unformulated control DNA_Cy3 administered to another mammary tumor in the same mouse. These data suggest that the MnNP formulation is capable of interacting with TAMs in a persistent way, noting that this study was carried out as three injections, each separated by 24 hours. This demonstrates that injecting MnNPs directly into a primary tumor can successfully deliver nucleic acid material to TAMs. The direct injection approach avoids the potential issues associated with intravascular administration of MnNPs.
MnNPs facilitated a similar increase in in vivo delivery of DNA_Cy5 to TAMs in a murine ovarian tumor model (Fig. 5A).41 In this case, the targeting ligand on the particles was tested by comparing delivery facilitated by the mannosylated particles to delivery facilitated by untargeted, hydroxyl-capped particles. The solid tumors in this implanted ovarian tumor model contain a significant TAM population and the ascites fluid generated contains a large amount of blood and immune cell infiltrate composed largely of TAMs.28,42–45 Evidence specifically supports mannose interaction of the MnNPs with TAMs in peritoneal tumors as DNA_Cy5 fluorescence is significantly elevated following MnNP delivery in comparison with identical, but non-mannosylated, hydroxyl-capped NPs. This result confirms the persistence of DNA_Cy5 delivery over two intraperitoneal injections spaced 24 hours apart. The environment of the ovarian tumors is somewhat unique in that prior to extremely late stages of progression, tumors are confined within the peritoneal compartment. This data provides evidence that an approach in which MnNPs are injected directly into the peritoneal cavity can successfully deliver nucleic acid material to TAMs without the potential complications associated with intravascular delivery.
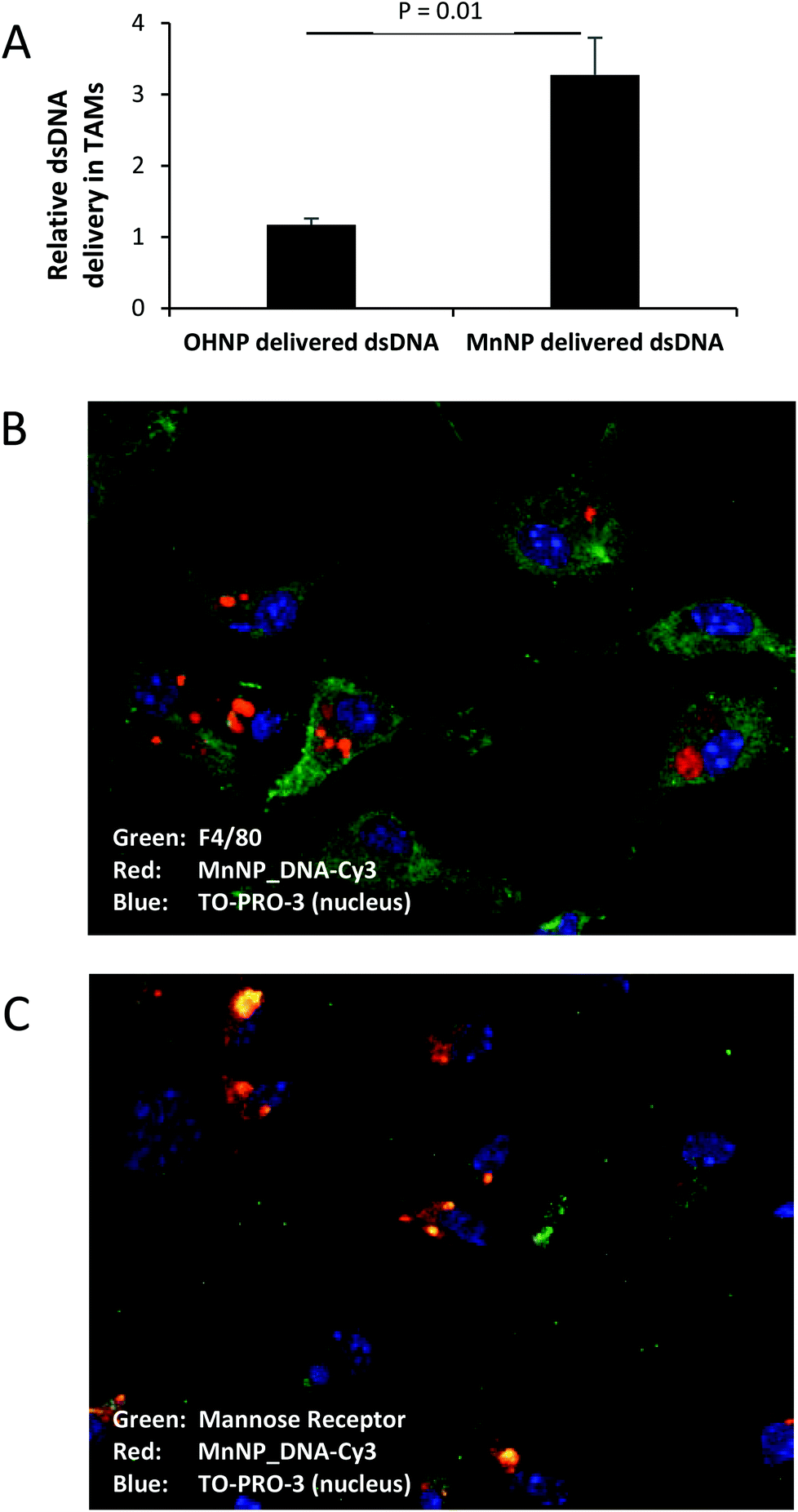 | ||
| Fig. 5 (A) Cy5 fluorescence is significantly increased in murine ovarian tumor TAMs when Cy5-labeled DNA is delivered with mannosylated nanoparticles in vivo as compared to labeled DNA delivered with non-targeted nanoparticles (T-test for significance, n = 3 mice). (B & C) Ex vivo ascites TAMs incubated with MnNP-DNA_Cy3 for 24 hours show uptake and retention of fluorescently labeled nucleotides. (Blue TO-PRO-3 nuclear stain) (B) Green, F4/80 labeling on the cell membrane confirms that the adhered cells are macrophages. Red, Cy3 labeling can be seen retained in the cytoplasm of the macrophages. (C) The mannose receptor (green) can be seen on the cell surface and co-localized with Cy3 fluorescence (red: DNA-Cy3, yellow: co-localization). | ||
Fig. 5B and C show confocal images of ex vivo ovarian ascites TAMs labeled for macrophage marker F4/80 (Fig. 5B, green) or alternatively activated macrophage marker and MnNP target, mannose receptor (Fig. 5C, green).23,30 Nanoparticle fluorescence patterns show punctate staining indicative of endosomes containing small numbers of MnNP as well as larger aggregate staining consistent with cytoplasmic release of the MnNP contents. These images confirm that the MnNP are taken up and retained by TAMs. Furthermore, MnNP are internally co-localized with the mannose receptor, as seen by the green-red overlap (yellow) in Fig. 5C. Unbound mannose receptor can be seen at the cell surface. The mannose receptor facilitates endosomal uptake of bound ligands (in this case the MnNP) and the bond ligand–receptor complex is transported into the cytoplasm inside the resultant endosome.
The third in vivo model used to demonstrate MnNP targeting of TAMs was a model in which PyMT mammary tumor cells are injected via the tail vein to establish tumors in the lungs (Fig. 6A). Lung metastases are one of the most common, potentially deadly sites for breast cancer metastases.46 The murine tail vein metastasis model simulates primary mammary tumor cells that have seeded into the lungs and established metastatic growth. DNA_Cy5 carrying nanoparticles were delivered directly into the metastasis bearing lungs via intubation. This delivery method was chosen for multiple reasons: not only are the nanoparticles spatially contained within the lungs, but intubation is a clinically relevant delivery method that results in longer lung retention of the delivered therapeutic compared to systemic delivery.47 Though we've chosen to model delivery to breast cancer lung metastases in our studies, intubation would be applicable for MnNP delivery to primary lung cancer as well.
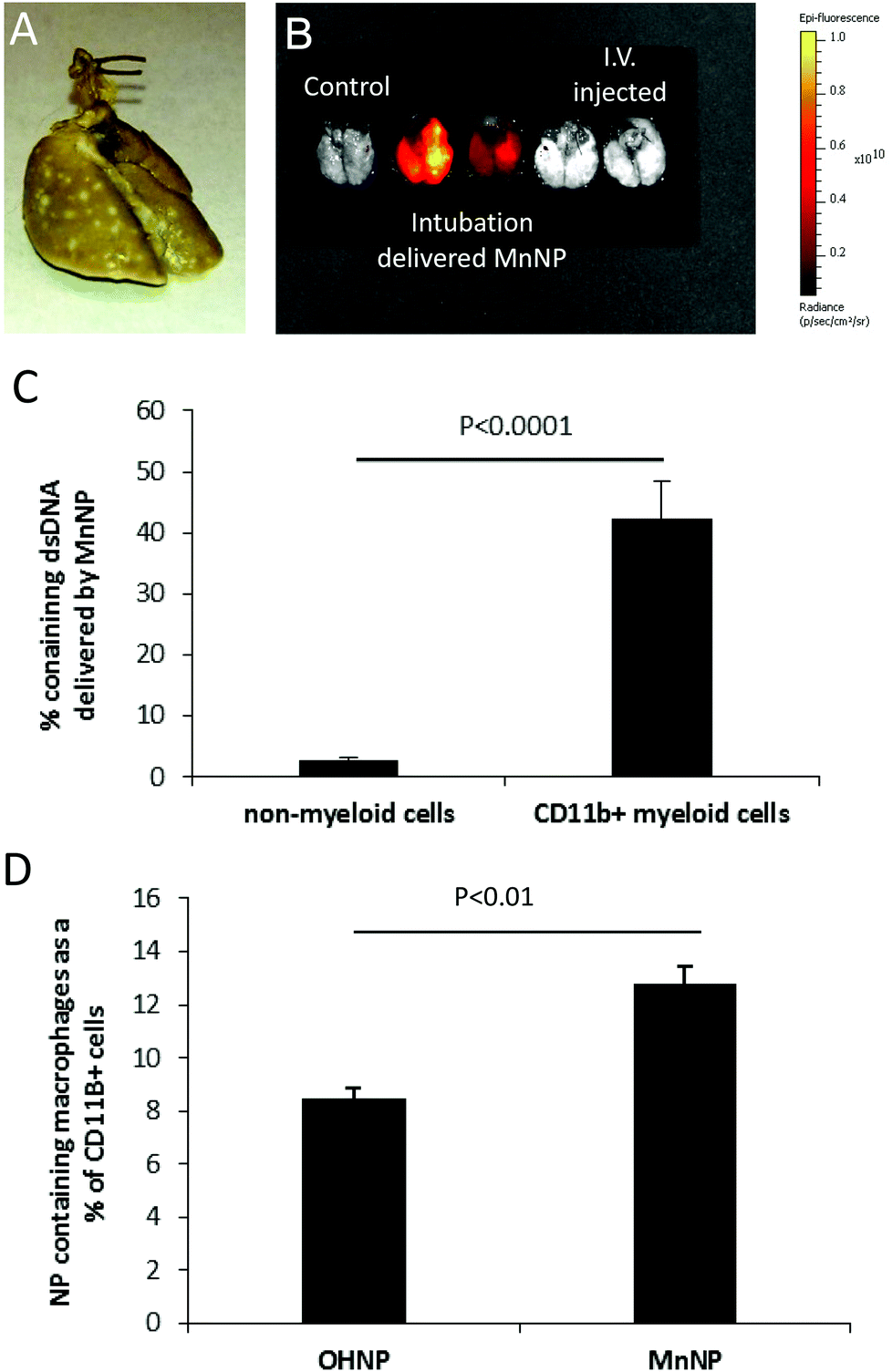 | ||
| Fig. 6 MnNP have enhanced uptake in TAMs associated with mammary lung metastases using an intubation delivery model. (A) Metastases are visualized in the lungs ex vivo following inflation with Bouin's fixative. Tumors appear white against the normal lung, which stains yellow. (B) IVIS imaging of lungs ex vivo shows intubation administration of MnNP results in more than 10 times the Cy5 fluorescence vs. intravenous administration of MnNP as visualized 6 hours after delivery. (C) Flow cytometry reveals minimal uptake of MnNP (approximately 2%) in non-myeloid cells. There is significantly more uptake of MnNP in CD11b+ myeloid cells. (D) MnNP show significantly more uptake in macrophages than OHNP. | ||
Fig. 6B shows that 6 hours after administration, MnNP delivered via intubation are detected in the lungs in greater amounts than MnNP delivered i.v. This image also demonstrates that delivery via intubation is capable of perfusing the MnNP solution throughout the entire lung. Flow cytometry analysis performed 24 hours after particles were delivered via intubation shows that approximately 40% of CD11b+ myeloid cells contain DNA_Cy5 formulated with MnNPs (Fig. 6C). This population is primarily composed of macrophages and granulocytes with some dendritic cells (DCs) and natural killer (NK) cells. DNA_Cy5 was detected in a relatively small number of non-myeloid cells, on the order of 2% in the same samples of lung homogenate. Furthermore, DNA_Cy5 uptake by CD45+/CDllb+/Gr1− macrophages is significantly increased in this model through formulation with MnNP as compared with DNA_Cy5 formulated with untargeted OHNP (Fig. 6D). DNA_Cy5 uptake is not enhanced by mannose presentation on the NP corona in PMN cells and monocytes (CD45+/CDllb+/Gr1+); there is no significant difference in DNA_Cy5 delivery between MnNP and OHNP formulations with these cell types. These results are consistent with strong, mannose-dependent delivery to mannose receptor expressing macrophages and weak, non-specific delivery (presumably by phagocytosis) to PMN cells and monocytes. Intubation enables direct access to lung tumor TAMs for NPs, avoiding NP dilution and other challenges resulting from intravascular administration. These data demonstrate that mannose decoration of the endosomal escape nanoparticle mediates preferential delivery of NPs to TAMs but not non-myeloid cell types which are co-localized with the TAMs in this model system.
The design of siRNA delivery vehicles intended for intravascular administration leading to tissue-specific accumulation is an enormous, multidimensional challenge. Such a construct must sequester and protect siRNA, avoid adverse effects resulting from interactions with serum proteins and the formed elements of the blood, possess sufficient blood compartment half-life for robust extravasation and delivery in the target tissues and offer a mechanism for preferential retention. Nonspecific toxicity must also be low. In addition, once localized, the carrier must facilitate cellular entry and endosomal escape to provide optimal conditions for effective intracellular RNAi. Despite the significant and broad potential clinical applications of RNAi, these barriers limit practical use of siRNA in humans.
Many potential clinical uses of siRNA may be approachable from other, non-intravascular, routes of administration. In this study, we assessed the capacity of an advanced nanoscale delivery system, designed for siRNA protection, preferential interaction with cells displaying CD206 and endosomal escape to facilitate efficient RNAi in models with clinical relevance using methods that avoid intravascular administration.
This study is the first to demonstrate mannose-mediated preferential siRNA delivery to TAMs in vivo. Ex vivo studies inform three subsequent in vivo treatment approaches, each employing local/regional routes of administration that avoid the significant obstacles and design challenges associated with intravascular administration. Some additional advantages common to NP delivery approaches that are confined to local/regional tissues is avoidance of the liver delivery and rapid urinary clearance that are significant limiters for successful intravascular administration of siRNA delivery vehicles. Importantly, the characterizations obtained in ex vivo experiments with TAMs are predictive of potential in vivo efficacy.
We demonstrated that formulation of an siRNA surrogate into the MnNP construct enables preferential delivery to TAMs following intratumoral injection. Many characteristics likely contribute to this result, including the slightly positive zeta potential of the MnNP relative to free polynucleotide and protection against enzymatic degradation. In vivo stability and biocompatibility of the MnNP is also inferred from this result, consistent with previous studies and confirmed explicitly in this work by a more rigorous, intravascular study. Intratumoral administration has potential as an adjuvant therapy for primary, recurrent breast cancers, but likely is more suitable for other cancers that can be individually identified and present near the skin surface, such as head and neck cancers or melanoma.
MnNPs administered intraperitoneally effectively deliver nucleotide payloads to TAMs in the distributed tumor burden associated with ovarian cancers. This study provides the clearest evidence that mannosylation is responsible for TAM selectivity in vivo, in agreement with previous work and the known surface display of CD206 on macrophages, especially pro-tumor TAMs. Multiple doses of MnNPs were well tolerated. Spatial confinement of the MnNP dose in the peritoneal cavity presumably enhances the opportunity for interaction with ovarian TAMs relative to intravascular dosing through greater proximity and longer persistence. The ability of MnNPs to preferentially localize in lung metastasis TAMs following intubation delivery implies transport of the nanomaterials from the alveolar side of the tissue into the tumors. Mannose-dependent interaction of MnNPs with TAMs is demonstrated from a different perspective in this study relative to the ovarian results in that flow cytometry confirms both strong nucleotide delivery to TAMs and lack of delivery to non-myeloid cells. All animals tolerated the intubation and delivery of MnNPs, despite a brief, acute response to the significant fluid burden administered to the lungs in both experimental and control groups. MnNPs reformulated as a more concentrated suspension will be explored to minimize the acute response through minimization of the liquid volume required, although careful attention must be devoted to the consequent stability of MnNPs in a more concentrated solution.
Conclusions
These studies demonstrate the biocompatibility of MnNP both in vitro and in vivo, and provide evidence for enhanced TAM-targeting generated through the use of mannose as a targeting ligand on the particle surfaces. Here we provide evidence that these MnNP produce no significant toxicity when used in vivo. Incubating TAMs with MnNP did not significantly decrease cell membrane viability and repeated, treatments in adult mice creates no acute kidney or liver damage. Furthermore, we showed that MnNP are effective at delivering fluorescently-labeled nucleotides to TAMs in spontaneously formed, primary mammary tumors. Additionally, mannose-targeting on the surface of the MnNP results in greater delivery of labeled nucleotides to ovarian tumor TAMs compared to non-targeted, hydroxyl-capped nanoparticles with the same core structure.Future studies will aim to further develop MnNP for biological applications and eventual clinical use, specifically utilizing MnNP to deliver active siRNAs to TAMs in vivo to knock down specific target proteins in key transcriptional pathways essential for creating the TAM phenotype. Potential targets include proteins of the NF-κB or JAK-STAT transcriptional pathways, both of which have been implicated in generating the TAM phenotype. By manipulating these pathways with MnNP-delivered siRNA, it may be possible to mitigate pro-tumor contributions from TAMs or to activate an immunogenic, anti-tumor phenotype in this macrophage population. Immune modulation could be confirmed by direct cytotoxicity assay and an analysis of changes in tumor cytokine levels.
Acknowledgements
The work performed in this study was funded in part by a Collaborative Idea Award through the Department of Defense CDMRP Breast Cancer Research Program (W81XWH-11-1-0242, W81XWH-11-1-0344) and by donations from Mr Chris Hill via the Anglo-American Charity. The ovarian tumor experiment was partially funded by a Translational Pilot Award through the Department of Defense CDMRP Ovarian Cancer Research Program (W81XWH-11-1-0509). Nanoparticle characterization was performed at the Vanderbilt Institute of Nanoscale Science and Engineering, using facilities renovated under National Science Foundation Grant ARI-R2 DMR-0963361. Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). Analysis of blood serum enzyme levels was performed at the Vanderbilt Translational Pathology shared resource. The authors would like to thank Andrew Wilson and Dineo Khabele from the Vanderbilt Ingram Cancer Center for instruction in setting up the ovarian tumor model and for providing the ID8 cells for that experiment. We would also like to thank Rinat Zaynagetdinov and Wei Han, Division of Allergy, Pulmonary and Critical Care Medicine, Vanderbilt University Medical Center, for advice and instruction for performing the rapid adhesion isolation of TAMs and for advice and assistance regarding intratracheal delivery of nanoparticles and the following flow cytometry. We would like to acknowledge Hongmei Li and Shann Yu for their work in the design and synthesis of mannosylated endosomal escape polymers. Finally, we would like to thank Kellye Kirkbride, Vanderbilt Department of Biomedical Engineering, for her assistance with manuscript editing.Notes and references
- Y. Mao, E. T. Keller, D. H. Garfield, K. Shen and J. Wang, Cancer Metastasis Rev., 2013, 32, 303–315 CrossRef PubMed.
- J. W. Pollard, Nat. Rev. Immunol., 2009, 9, 259–270 CrossRef CAS PubMed.
- J. Condeelis and J. W. Pollard, Cell, 2006, 124, 263–266 CrossRef CAS PubMed.
- B. Z. Qian and J. W. Pollard, Cell, 2010, 141, 39–51 CrossRef CAS PubMed.
- L. S. Ojalvo, W. King, D. Cox and J. W. Pollard, Am. J. Pathol., 2009, 174, 1048–1064 CrossRef CAS PubMed.
- M. Heusinkveld and S. H. van der Burg, J. Transl. Med., 2011, 9, 216 CrossRef CAS PubMed.
- L. M. Coussens and Z. Werb, Nature, 2002, 420, 860–867 CrossRef CAS PubMed.
- F. R. Greten, L. Eckmann, T. F. Greten, J. M. Park, Z. W. Li, L. J. Egan, M. F. Kagnoff and M. Karin, Cell, 2004, 118, 285–296 CrossRef CAS PubMed.
- P. J. Murray, J. E. Allen, S. K. Biswas, E. A. Fisher, D. W. Gilroy, S. Goerdt, S. Gordon, J. A. Hamilton, L. B. Ivashkiv, T. Lawrence, M. Locati, A. Mantovani, F. O. Martinez, J. L. Mege, D. M. Mosser, G. Natoli, J. P. Saeij, J. L. Schultze, K. A. Shirey, A. Sica, J. Suttles, I. Udalova, J. A. van Ginderachter, S. N. Vogel and T. A. Wynn, Immunity, 2014, 41, 14–20 CrossRef CAS PubMed.
- A. Mantovani, S. Sozzani, M. Locati, P. Allavena and A. Sica, Trends Immunol., 2002, 23, 549–555 CrossRef CAS PubMed.
- W. Han, M. Joo, M. B. Everhart, J. W. Christman, F. E. Yull and T. S. Blackwell, Am. J. Physiol. Lung Cell. Mol. Physiol., 2009, 296, L320–L327 CrossRef CAS PubMed.
- R. Zaynagetdinov, T. P. Sherrill, V. V. Polosukhin, W. Han, J. A. Ausborn, A. G. McLoed, F. B. McMahon, L. A. Gleaves, A. L. Degryse, G. T. Stathopoulos, F. E. Yull and T. S. Blackwell, J. Immunol., 2011, 187, 5703–5711 CrossRef CAS PubMed.
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS PubMed.
- A. Fire, S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS PubMed.
- M. E. Davis, Mol. Pharm., 2009, 6, 659–668 CrossRef CAS PubMed.
- M. E. Davis, J. E. Zuckerman, C. H. Choi, D. Seligson, A. Tolcher, C. A. Alabi, Y. Yen, J. D. Heidel and A. Ribas, Nature, 2010, 464, 1067–1070 CrossRef CAS PubMed.
- A. D. Judge, M. Robbins, I. Tavakoli, J. Levi, L. Hu, A. Fronda, E. Ambegia, K. McClintock and I. MacLachlan, J. Clin. Invest., 2009, 119, 661–673 CAS.
- A. J. Convertine, D. S. Benoit, C. L. Duvall, A. S. Hoffman and P. S. Stayton, J. Controlled Release: Off. J. Controlled Release Soc., 2009, 133, 221–229 CAS.
- H. Li, S. S. Yu, M. Miteva, C. E. Nelson, T. Werfel, T. D. Giorgio and C. L. Duvall, Adv. Funct. Mater., 2013, 23, 3040–3052 CrossRef CAS PubMed.
- S. S. Yu, C. M. Lau, W. J. Barham, H. M. Onishko, C. E. Nelson, H. Li, C. A. Smith, F. E. Yull, C. L. Duvall and T. D. Giorgio, Mol. Pharm., 2013, 10, 975–987 CrossRef CAS PubMed.
- P. Allavena, M. Chieppa, G. Bianchi, G. Solinas, M. Fabbri, G. Laskarin and A. Mantovani, Clin. Dev. Immunol., 2010, 2010, 547179 CAS.
- Y. Luo, H. Zhou, J. Krueger, C. Kaplan, S. H. Lee, C. Dolman, D. Markowitz, W. Wu, C. Liu, R. A. Reisfeld and R. Xiang, J. Clin. Invest., 2006, 116, 2132–2141 CAS.
- P. D. Stahl and R. A. Ezekowitz, Curr. Opin. Immunol., 1998, 10, 50–55 CrossRef CAS PubMed.
- L. A. Dethloff and B. E. Lehnert, J. Leukocyte Biol., 1988, 43, 80–90 CAS.
- M. A. Schonhegrad and P. G. Holt, J. Immunol. Methods, 1981, 43, 169–173 CrossRef CAS.
- J. B. Yee and J. C. Hutson, Biol. Reprod., 1983, 29, 1319–1326 CrossRef CAS PubMed.
- W. Xu, N. Schlagwein, A. Roos, T. K. van den Berg, M. R. Daha and C. van Kooten, Eur. J. Immunol., 2007, 37, 1594–1599 CrossRef CAS PubMed.
- A. J. Wilson, W. Barham, J. Saskowski, O. Tikhomirov, L. Chen, H. J. Lee, F. Yull and D. Khabele, J. ovarian res., 2013, 6, 63 CrossRef PubMed.
- L. Connelly, W. Barham, H. M. Onishko, L. Chen, T. P. Sherrill, T. Zabuawala, M. C. Ostrowski, T. S. Blackwell and F. E. Yull, Breast Cancer Res., 2011, 13, R83 CrossRef CAS PubMed.
- R. Zaynagetdinov, T. P. Sherrill, P. L. Kendall, B. H. Segal, K. P. Weller, R. M. Tighe and T. S. Blackwell, Am. J. Respir. Cell Mol. Biol., 2013, 49, 180–189 CrossRef CAS PubMed.
- D. Pilling, T. Fan, D. Huang, B. Kaul and R. H. Gomer, PLoS One, 2009, 4, e7475 Search PubMed.
- P. Szlosarek, K. A. Charles and F. R. Balkwill, Eur. J. Cancer, 2006, 42, 745–750 CrossRef CAS PubMed.
- P. W. Szlosarek and F. R. Balkwill, Lancet Oncol., 2003, 4, 565–573 CrossRef CAS PubMed.
- W. Strober, Current protocols in immunology, ed. J. E. Coligan, et al., 2001 Search PubMed, Appendix 3, Appendix 3B.
- R. Mauisse, D. D. Semir, H. Emamekhoo, B. Bedayat, A. Abdolmohammadi, H. Parsi and D. C. Gruenert, BMC Biotechnol., 2010, 10 Search PubMed.
- F. C. Brosius, 3rd, C. E. Alpers, E. P. Bottinger, M. D. Breyer, T. M. Coffman, S. B. Gurley, R. C. Harris, M. Kakoki, M. Kretzler, E. H. Leiter, M. Levi, R. A. McIndoe, K. Sharma, O. Smithies, K. Susztak, N. Takahashi, T. Takahashi and C. Animal Models of Diabetic Complications, J. Am. Soc. Nephrol., 2009, 20, 2503–2512 CrossRef PubMed.
- E. Y. Lin, J. G. Jones, P. Li, L. Zhu, K. D. Whitney, W. J. Muller and J. W. Pollard, Am. J. Pathol., 2003, 163, 2113–2126 CrossRef PubMed.
- R. D. Leek, R. J. Landers, A. L. Harris and C. E. Lewis, Br. J. Cancer, 1999, 79, 991–995 CrossRef CAS PubMed.
- K. Greish, J. Drug Targeting, 2007, 15, 457–464 CrossRef CAS PubMed.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189–207 CrossRef CAS PubMed.
- J. Greenaway, R. Moorehead, P. Shaw and J. Petrik, Gynecol. Oncol., 2008, 108, 385–394 CrossRef CAS PubMed.
- T. Hagemann, J. Wilson, F. Burke, H. Kulbe, N. F. Li, A. Pluddemann, K. Charles, S. Gordon and F. R. Balkwill, J. Immunol., 2006, 176, 5023–5032 CrossRef CAS.
- E. Schutyser, S. Struyf, P. Proost, G. Opdenakker, G. Laureys, B. Verhasselt, L. Peperstraete, I. Van de Putte, A. Saccani, P. Allavena, A. Mantovani and J. Van Damme, J. Biol. Chem., 2002, 277, 24584–24593 CrossRef CAS PubMed.
- A. Sica, A. Saccani, B. Bottazzi, S. Bernasconi, P. Allavena, B. Gaetano, F. Fei, G. LaRosa, C. Scotton, F. Balkwill and A. Mantovani, J. Immunol., 2000, 164, 733–738 CrossRef CAS.
- M. J. Turk, D. J. Waters and P. S. Low, Cancer Lett., 2004, 213, 165–172 CrossRef CAS PubMed.
- A. T. Berman, A. D. Thukral, W. T. Hwang, L. J. Solin and N. Vapiwala, Clin. Breast Cancer, 2013, 13, 88–94 CrossRef PubMed.
- O. B. Garbuzenko, M. Saad, S. Betigeri, M. Zhang, A. A. Vetcher, V. A. Soldatenkov, D. C. Reimer, V. P. Pozharov and T. Minko, Pharm. Res., 2009, 26, 382–394 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |