
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural aspects of phenylglycines, their biosynthesis and occurrence in peptide natural products
Rashed S.
Al Toma
a,
Clara
Brieke
b,
Max J.
Cryle
*b and
Roderich D.
Süssmuth
*a
aInstitut für Chemie, Technische Universität Berlin, 10623 Berlin, Germany. E-mail: suessmuth@chem.tu-berlin.de
bMax Planck Institute for Medical Research, Department of Biomolecular Mechanisms, 69120 Heidelberg, Germany. E-mail: Max.Cryle@mpimf-heidelberg.mpg.de
First published on 5th May 2015
Abstract
Covering up to February 2015
Phenylglycine-type amino acids occur in a wide variety of peptide natural products, including glycopeptide antibiotics and biologically active linear and cyclic peptides. Sequencing of biosynthesis gene clusters of chloroeremomycin, balhimycin and pristinamycin paved the way for intensive investigations on the biosynthesis of 4-hydroxyphenylglycine (Hpg), 3,5-dihydroxyphenylglycine (Dpg) and phenylglycine (Phg) in recent years. The significance and importance of this type of unusual non-proteinogenic aromatic amino acids also for medicinal chemistry has drawn the attention of many research groups and pharmaceutical companies. Herein structures and properties of phenylglycine containing natural products as well as the biosynthetic origin and incorporation of phenylglycines are discussed.
 Rashed S. Al Toma | Rashed Al Toma, an agricultural engineer from Damascus University in horticulture and plant tissue culture. Between 2006 and 2010 he was a supervising engineer of plant biotechnology laboratory at the national commission for biotechnology in Syria. In 2009 he obtained an MSc in biotechnology with Khalil Al Maarri (Damascus), Wim Vanden Berghe and Guy Haegeman (Gent), where he screened medicinal plants against cancer cells and bacteria. In 2010 he started his PhD with Roderich Süssmuth at TU-Berlin as a BIG-NSE fellow with an Erasmus Mundus fellowship. He studies in vivo incorporation of noncanonical amino acids into lantibiotics and lasso peptides. |
 Clara Brieke | Clara Brieke studied biomedical chemistry at the universities of Mainz and London. For her PhD, she moved to Prof. Heckel's group at the University of Frankfurt focusing on photo-regulation methods and nucleic acid chemistry. In 2012 she joined the group of Dr Cryle at the Max Planck Institute for Medical Research in Heidelberg, where she entered the field of non-ribosomally derived natural products. Her research activities mainly focus on the synthesis of biosynthetic intermediates of glycopeptide antibiotics and the exploration of enzymatic oxidative cross coupling reactions in their biosynthesis. |
 Max J. Cryle | Max Cryle obtained his PhD in organic chemistry from the University of Queensland in Brisbane, Australia in 2006. Following the completion of his PhD he moved to the Max Planck Institute for Medical Research in Heidelberg, Germany as a Cross Disciplinary Fellow of the Human Frontiers Science Program to study Carrier Protein/Cytochrome P450 complexes. In 2011 he was funded through the Emmy Noether program to lead an independent group with a focus on the biosynthesis of glycopeptide antibiotics. His group currently applies a combination of approaches including organic synthesis, biochemistry and structural biology to investigate antibiotic biosynthesis. |
 Roderich D. Süssmuth | Roderich Süssmuth is the Rudolf Wiechert Professor of Biological Chemistry at the Technische Universität Berlin. He obtained his PhD degree in chemistry in 1999 from the Eberhard Karls Universität Tübingen with Günther Jung. In 2000, he was a Feodor-Lynen-Fellow of the Alexander von Humboldt-Foundation with Carlos Barbas III and Richard Lerner. Subsequently he became an Assistant Professor in Tübingen with an Emmy-Noether fellowship granted by the DFG, and then moved in 2004 to the TU Berlin where he is full professor. His research is focused on antibiotics, biosynthetic assembly lines, host–pathogen interactions, medicinal chemistry of peptides, enzymes and enzyme inhibitors. |
1 Introduction
A great many fundamental processes in the cell are based on the interaction of peptides and proteins, which are composed of the repertoire of 20 so-called canonical amino acids (cAA). Generally, the biosynthesis of these peptides and proteins is based on mechanisms of ribosomal synthesis.1 The alphabet of the cAAs has recently been extended by two additional amino acids, selenocysteine (abbreviated as Sec or U, in older publications as Se-Cys) as the 21st proteinogenic amino acid, which is used as a building block for selenoproteins in all kingdoms of life2 and pyrrolysine (Pyl or O) as the 22nd proteinogenic amino acid, which in 2002 was discovered to be used by Methanosarcina barkeri in the active site of a methyl-transferase enzyme. Pyl is encoded by the amber stop codon UAG3–5 and also used by some methanogenic archaea as well as the Gram-positive bacterium Desulfitobacterium hafniense as a part of a methane-producing enzyme complex.6 The structural complexity in the bacterial and fungal world is further increased by posttranslational modifications of the canonical amino acids1 to a multitude of additional amino acid modifications.An alternative biosynthesis route for assembly of peptides is non-ribosomal peptide biosynthesis. In a similar manner to ribosomally synthesized and post-translationally modified peptides (RiPPs) biosynthesis, genes are assembled in gene clusters which harbor a variety of genes coding for non-ribosomal peptide synthetases (NRPSs), amino acid biosynthesis genes, tailoring enzymes, as well as genes for export and resistance. The NRPSs are multimodular giant enzymes, in which one module is typically dedicated to the activation and coupling of one amino acid. Of particular importance are the adenylation (A) domains as essential constituents of these modules. The A-domains perform the recognition and activation of a specific amino acid. The amino acids to be activated can be proteinogenic or non-proteinogenic. In the latter case, the gene cluster or the genome of the producing organism must contain the biosynthesis genes for the corresponding non-proteinogenic amino acid. Structural diversity during non-ribosomal biosynthesis is further attained either by in cis acting domains (methylation, epimerization, cyclization, etc.) or by tailoring enzymes, performing methylations, β-hydroxylations, oxidation reactions and halogenations acting in trans. Methylations have been observed at the β-carbon atom of some amino acids like Asp in friulimicin,7 Glu in daptomycin8 and Phe in hormaomycin.9 Such β-hydroxylations are catalyzed either by nonheme iron-containing α-ketoglutarate-dependent dioxygenases10,11 or cytochrome P450 monooxygenases12,13 and a prominent example is β-hydroxytyrosine in various glycopeptide antibiotics, such as vancomycin.14 Likewise halogenations of proteinogenic amino acids have been described for glycopeptide antibiotics.15
A remarkable family of non-proteinogenic amino acids which are recruited by NRPSs to build up peptide structures are the phenylglycines (Scheme 1), consisting of phenylglycine (Phg), 4-hydroxyphenylglycine (Hpg) and 3,5-dihydroxyphenylglycine (Dpg) as the most important representatives. Phenylglycine-type amino acids occur in various natural products, e.g. in pristinamycins (Phg),16,17 or in almost all glycopeptide antibiotics (Hpg and Dpg). They can formally be regarded as truncated versions of Phe or Tyr which lack the methylene group in contrast to the methylene-extended versions, i.e. the homoamino acids.18 Respective examples are homophenylalanine (homoPhe), in pahayokolides A and B,19 homotyrosine (homoTyr) in cyanopeptolins as well as anabaenopeptins,20 or 3,4-dihydroxyhomotyrosine in echinocandin B.21 In this review we highlight some structural aspects of Phg, Hpg and Dpg, discuss their occurrence in various natural products as well as their biosynthesis, modification and incorporation into peptides.
 | ||
| Scheme 1 Shared biosynthesis scheme for phenylglycines and the aromatic amino acids Phe, Tyr and Trp. While Phg and Hpg originate from chorismate branching off into the biosyntheses of Phe and Tyr, the amino acid Dpg is directly assembled from acetyl-CoA. Detailed insights into the biosynthesis of Phg, Hpg and Dpg are discussed in Section 4. | ||
2 Structure and properties of Phg, Hpg and Dpg
Steric and electronic aspects play an important role for conformation and reactivity of phenylglycine-containing peptides and thus it is pertinent to discuss these influences.Remarkably, in phenylglycines the bulky aromatic sidechain is directly attached to the α-carbon. This is in stark contrast to the proteinogenic aromatic amino acids, which all bear a β-methylene spacer between the α-carbon and the aromatic group. Accordingly, phenylglycines have, assuming that the conformation of the peptide backbone is fixed, strongly restricted degrees of freedom for the aromatic side chain. Furthermore, electronic effects of substituents at the aromatic rings influence reactivity and physical properties. In general, phenylglycines appear more prone to racemization than other α-amino acids.22,23
In the literature, two basic mechanisms of racemization for amino acids are discussed. According to the first mechanism, it is believed that deprotonation occurs at the α-carbon due to acidic nature of the α-proton stabilizing an enolate-like structure, which ultimately leads to racemization upon reprotonation. Alternatively, deprotonation may occur at the amide proton forming an enolate upon rearrangement, leading to racemization.22 Interestingly, Phg, Hpg and Dpg undergo base-catalyzed racemization at the α-position at much higher rate compared to Phe/Tyr and also compared to other proteinogenic amino acids. Experimentally, it was found that Phg has a nine-fold higher rate of racemization compared to alanine.22,23 A 4-hydroxy group substitution on phenylglycine (Hpg) should decelerate the racemization rate, which can be attributed to the mesomeric effect (+M) (Scheme 2) decreasing the acidity of the α-carbon. Electron withdrawing substituents in this position, e.g. a nitro group, should dramatically increase racemization. Due to the m-position, the phenol substituents of Dpg have a significantly reduced M-effect compared to Hpg. Rather −I-effects could induce faster racemization. In addition, one has to consider that the electronic effects of the aromatic sidechain are conformation-depend, i.e. that rotation of the aromatic ring (IV) out of the plane (V) leads to lowered orbital overlap and thus less influence on racemization rate of the α-carbon.
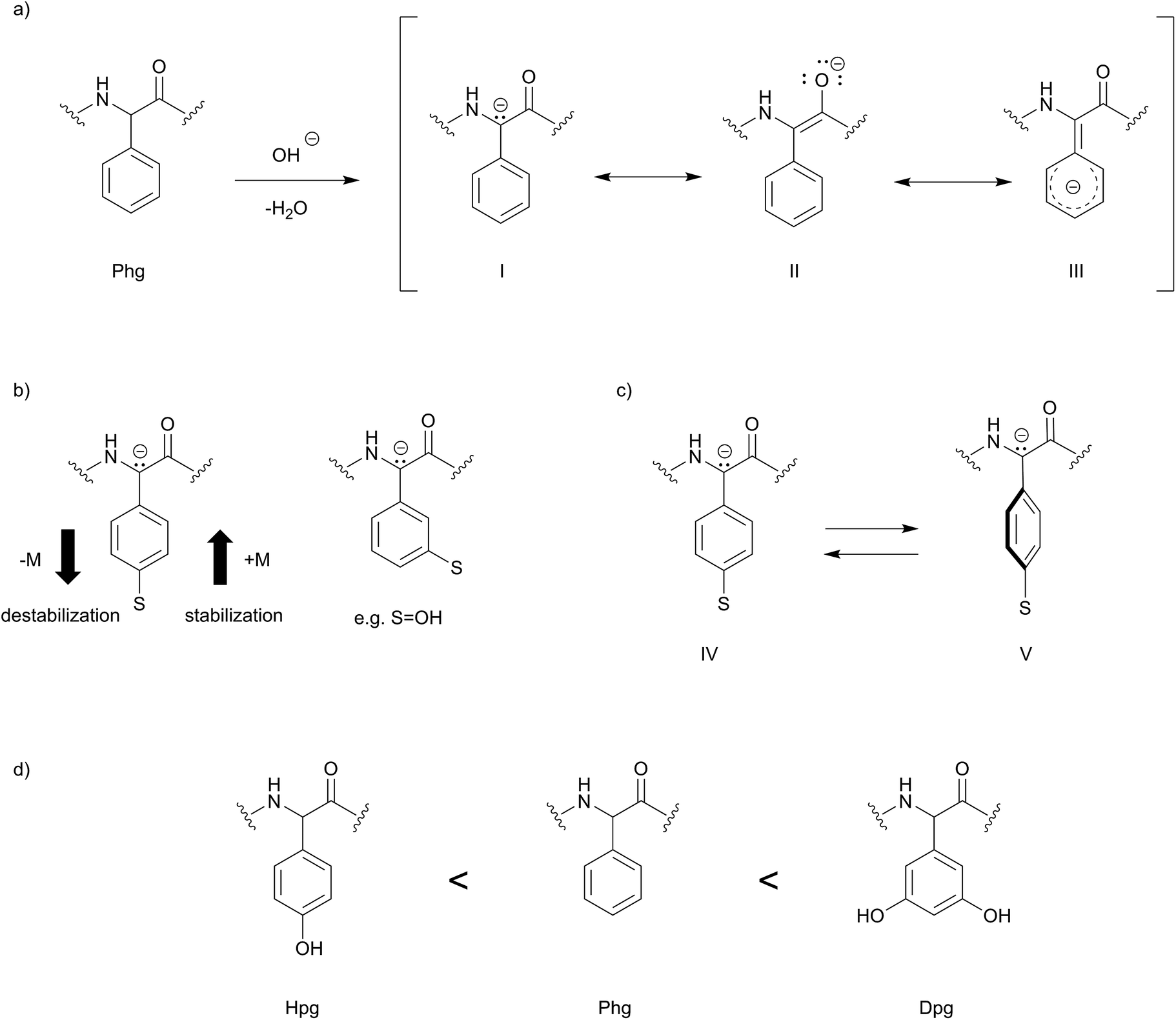 | ||
| Scheme 2 Proposed racemization mechanism of phenylglycines due to stereochemical and conformational influences. (a) The carbanion structure (I) and the enolate structure (II) postulated also for proteinogenic amino acids is further stabilised by delocalisation of the negative charge in the aromatic ring (III); (b) electron donating and electron withdrawing substituents (S) stabilize or destabilize the carbanion structure thus influencing racemisation rates. Substituents in p-position are expected to exert a stronger effect compared to m-substituents; (c) rotation of the aromatic ring (IV) out of plane (V) interrupts communication between π-orbitals of the aromatic ring and is expected to strengthen resistance against racemisation; (d) the rate of racemization increases in the order Hpg < Phg < Dpg. | ||
The racemization-prone nature of phenylglycines is a well-known phenomenon and has been described for synthesis applications during deprotection24 and macrocyclization reactions of amino acids and peptides, which greatly limits the selection of protecting groups, and further requires the careful selection of reaction conditions. This indicates the importance of structural features and of electronic effects on the stability of phenylglycines as well as of peptides derived from them. In addition, electron donating effects from hydroxyl and other substituents on the aromatic ring may influence π–π stacking, as well as capabilities for formation of H-bonds.
While the electronic nature and influence of phenylglycines within a peptide context is rather poorly understood, it is apparent that these amino acids play a particular role for both structure and bioactivity. One example for the importance of the Hpg residue was performed at [L-Dap2]ramoplanin A2 aglycon by an alanine scan,25 a commonly used method to systematically replace and hence assess every amino acid position of a peptide by Ala. By means of this method the minimal inhibitory concentration (MIC) decreased 13 to 74 times fold depending on the position of Hpg which had been exchanged with Ala.25 A similar experiment was carried out for the peptide antibiotic feglymycin, where an assessment of contributions of Hpg and Dpg to the antimicrobial activity was performed.26
3 Phenylglycines in peptide natural products
Phenylglycines occur in various forms as constituents of peptide natural products and thus contribute to novel – and even medically exploited – bioactivities. With regard to structural aspects, the number of phenylglycine building blocks varies, from a single occurrence to multiple times in different peptides. Among phenylglycine-containing peptides linear structures, head-to-tail cyclizations or side chain-bridges directly involving phenylglycines have been described. In addition, both enantiomeric forms of phenylglycines, i.e.L or D configuration, are represented in various peptides. In the following sections we will suggest a classification of peptides according to characteristic structural features.3.1 Phg-containing peptides
The amino acid phenylglycine has been reported in the early 1960–70s as a constituent of the peptide antibiotics virginiamycin S27–29 produced by Streptomyces virginiae, streptogramin B (also known as pristinamycin IA and mikamycin IA)30–34 produced by various species of the Streptomyces genus (e.g. Streptomyces diastaticus, and Streptomyces graminofaciens),35 and pristinamycin I28,36–39 produced from Streptomyces pristinaespiralis (Scheme 3). The structure elucidation of these compounds has been performed by identification of hydrolysis products,27,28,40–42 while the configuration of these compounds has been determined by mass spectrometry and X-ray crystallography.28,43 Pristinamycin I, virginiamycin S and streptogramin B are heptameric cyclic peptide antibiotics synthesized by NRPSs28,37 which share the same core structure. Pristinamycin I for example consists of seven amino acids, five of them are noncanonical residues including L-hydroxypicolinic acid (L-Hpa), L-aminobutyric acid (L-Apa), 4-N-N-dimethylamino-L-phenylalanine (L-DAMPA), 4-oxo-L-pipecolic acid (L-Pip) and L-phenylglycine (L-Phg).17,28 Virginiamycin S has exactly the same structure as pristinamycin I with the exception that it contains L-Phe instead of L-DAMPA (Scheme 3). | ||
| Scheme 3 Structures of the Phg-containing peptides: pristinamycin IA/streptogramin B, virginiamycin S1, the bicyclic peptides dityromycin, MBJ-0086 and MBJ-0087. | ||
Virginiamycin S has antimicrobial activities against a broad spectrum of Gram-positive bacteria, and was used as an antibiotic for poultry feeds in order to stimulate growth.44,45 The use of virginiamycin increased from 1995 to 1997 as growth promoter for pigs, which in turn caused an increase in virginiamycin-resistant Enterococcus faecium isolates from 27.3% in 1995 to 66.2% in 1997 in Denmark. In the beginning of 1998, the Danish government banned its use, and the occurrence of virginiamycin-resistance decreased to 33.9% in 2000.46 Similar results have been reported from Norway and Finland.47 Streptogramins (including virginiamycin S and pristinamycin I) inhibit bacterial protein synthesis in Gram-positive bacteria targeting the 23S ribosomal RNA by binding to the P-binding site of the 50S ribosomal subunit. This prevents the elongation of the protein chains by the ribosome, ultimately leading to the premature release of peptides.35
Pristinamycin I exhibits antimicrobial activities against erythromycin-resistant staphylococci and streptococci, as well as against methicillin-resistant Staphylococcus aureus (MRSA). This peptide, which has moderate bacteriostatic effects,17 is synthesized together with the polyunsaturated cyclopeptide/macrolactone pristinamycin II, a streptogramin group A member. The producing strain Streptomyces pristinaespiralis synthesizes both compounds in an approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio.28 Both compounds, pristinamycin I and II, act in a synergistic manner rendering bactericidal affects. In spite of its poor solubility (10−4 mg ml−1 water),28 which limits its usage in intravenous formulations, it is sometimes used as a therapeutic drug in human medicine against multi-resistant Gram-positive bacteria, such as the semisynthetic streptogramin Synercid (Pfizer) which is a mixture of quinupristin (a pristinamycin I derivative) and dalfopristin (a pristinamycin II derivative).29,48 Dalfopristin binds to the 23S ribosomal RNA of the 50S ribosomal subunit and changes its conformation, which enhances the binding of quinopristin 100 fold.17 Pristinamycin I is used as an alternative to rifampicin, fusidic acid or linezolid in the treatment of methicillin-resistant Staphylococcus aureus MRSA, drug-resistant Streptococcus pneumoniae and vancomycin-resistant Enterococcus faecium as well as some Gram-negative bacteria, such as Haemophilus spp.17,49–52
:3 ratio.28 Both compounds, pristinamycin I and II, act in a synergistic manner rendering bactericidal affects. In spite of its poor solubility (10−4 mg ml−1 water),28 which limits its usage in intravenous formulations, it is sometimes used as a therapeutic drug in human medicine against multi-resistant Gram-positive bacteria, such as the semisynthetic streptogramin Synercid (Pfizer) which is a mixture of quinupristin (a pristinamycin I derivative) and dalfopristin (a pristinamycin II derivative).29,48 Dalfopristin binds to the 23S ribosomal RNA of the 50S ribosomal subunit and changes its conformation, which enhances the binding of quinopristin 100 fold.17 Pristinamycin I is used as an alternative to rifampicin, fusidic acid or linezolid in the treatment of methicillin-resistant Staphylococcus aureus MRSA, drug-resistant Streptococcus pneumoniae and vancomycin-resistant Enterococcus faecium as well as some Gram-negative bacteria, such as Haemophilus spp.17,49–52
In addition to the monocyclic peptides mentioned above, a bicyclic peptide named dityromycin is also a Phg-containing peptide (Scheme 3).53,54 Dityromycin is produced by Streptomyces sp. strain AM-2504, and shows antimicrobial activities against Gram-positive bacteria including Bacillus, Clostridium, and Corynebacterium.55 Its structure was elucidated by NMR spectroscopy and FAB-MS, and finally confirmed by fragment synthesis.53 Nevertheless, its stereochemistry has not yet been clarified. The structure of GE82832, synthesized by Streptosporangium cinnabarinum (strain GE82832) is highly related to that of dityromycin and has a mass difference of 2 Da, which might result from an additional yet undetermined position of a double bond.54 Furthermore, GE82832 consists of two bioactive isomeric components (A and B) with the same molecular weight (1286 Da) which may be due to the occurrence of diastereomers.56 Dityromycin and GE82832 inhibit bacterial protein synthesis in vitro and in vivo by targeting the small ribosomal subunit and inhibiting the EF-G-catalyzed translocation of the elongation step.54,57 Recently, two new bicyclic depsipeptides, named MBJ-0086 and MBJ-0087, have been isolated from the culture broth of Sphaerisporangium sp. 3226.58 They have very similar structure to dityromycin and GE82832 peptides (Scheme 3). Their structures were elucidated by NMR spectroscopy and high-resolution-ESI-LC-MS. MBJ-0086 shows a high antimicrobial activity against Bacillus subtilis with an MIC value of 1.1 μM, in comparison to 24 μM for MBJ-0087. Both compounds have no cytotoxicity against human ovarian adenocarcinoma SKOV-3 cells (IC50 > 50 μM) nor antimicrobial activity against Micrococcus luteus (IC50 > 50 μM).58 As the above-mentioned Phg-containing natural products are the only examples of Phg found in nature, this amino acid appears to be the least represented among all phenylglycines.
3.2 Hpg and Dpg-containing peptides
Non-substituted phenylglycines are comparatively rare in peptide natural products, and they do not occur accompanied by other phenylglycines. However, it is quite remarkable that there have been several peptides described with the concomitant presence of Hpg and Dpg. Furthermore, the structural diversity of Hpg- and Dpg-containing peptides is much more pronounced, possibly also due to the H-bonding properties of the phenolic groups, which expands the options for molecular interactions. However, the amino acid 3-hydroxyphenylglycine rarely occurs in nature, and the only example of a known natural product is forphenicine (4-formyl-3-hydroxyphenylglycine), an inhibitor of alkaline phosphatase produced by Actinomyces fulvoviridis var. acarbodicus.59,60 Another rare natural derivative of phenylglycine is m-carboxyphenylglycine, which was isolated from bulbs of Iris tingitana var. Wedgewood.61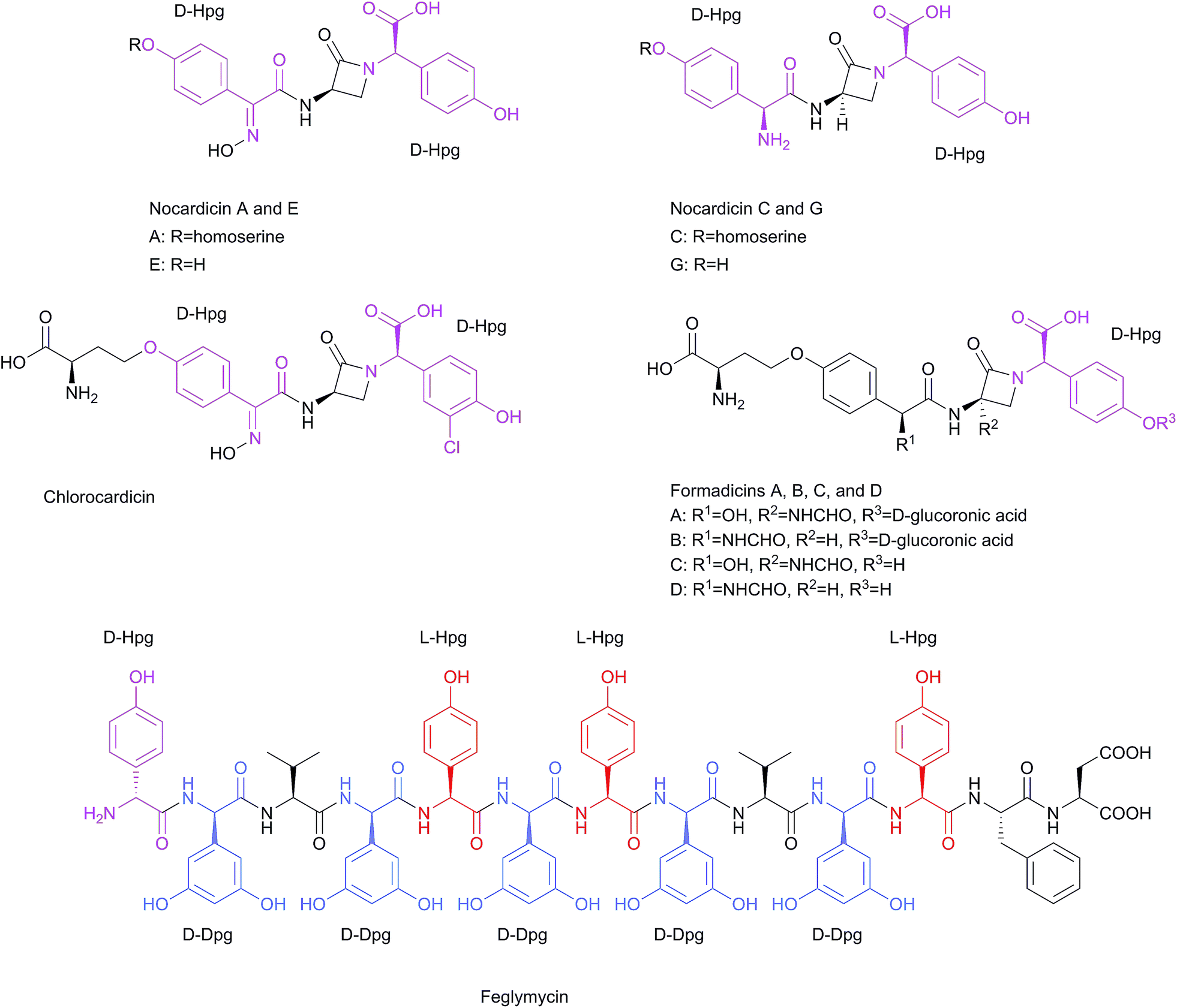 | ||
| Scheme 4 Structures of Hpg- and Dpg-containing linear peptides: nocardicin A, C, E, and G, chlorocardicin, formadicin A–D, and feglymycin. | ||
Some other β-lactam linear peptides, which are structurally-related to nocardicin A are chlorocardicin and the formadicins. Chlorocardicin, was isolated from Streptomyces sp. (SK&F-AAH-873) from a soil sample collected from the root zone of a cactus in Pima County, Arizona.68,69 The structure was elucidated by high resolution FAB-MS and 1H and 13C NMR analysis.69 The only difference to the nocardicin A structure is an additional chlorine atom, resulting in 3-chloro-4-hydroxy phenylglycine instead of the C-terminal Hpg moiety (Scheme 4). The antibacterial activities against Gram-negative bacteria, e.g. Klebsiella pneumoniae were similar to nocardicin A in M9 medium, and eightfold higher in complex media (Müller-Hinton broth MHB; MIC = 25 μg ml−1).68 Chlorocardicin exhibits low activity against Staphylococcus aureus, whereas nocardicin A is not active.68 The formadicins were isolated from Flexibacter alginoliquefaciens sp. nov. YK-498 (ref. 70) in four variants: formadicin A–D with a homologous structure to norcardicin A. Additional structural modifications are a formylamino group at the 3- or 12-position (R2 and R1 respectively in the structure of formadicins in Scheme 4), as well as β-D-glucoronidation at the phenolic group in formadicins A and B.71
Formadicins A and C contain two residues of α,4-dihydroxybenzeneacetic acid instead of the N-terminal D-Hpg residue. Formadicins have antibacterial activities similar to that of nocardicin A, and they exhibit high activity against some Pseudomonas and Proteus species. Generally, the formadicins C and D display higher antimicrobial activities than formacidins A and B.70 A linear peptide which contains both amino acids, Hpg and Dpg, is feglymycin (Scheme 4). The peptide was isolated from Streptomyces DSM 11171 (ref. 72 and 73) and its structure was solved by NMR spectroscopy72 and later by X-ray crystallography.74 It is a 13mer peptide consisting of four Hpg (one D-Hpg and three L-Hpg) and five D-Dpg residues. With exception of the N-terminal D-Hpg the molecule displays a long stretch of alternating D- and L-configured amino acid residues. In the crystal the peptides form a π-helical homodimeric cylinder where the aromatic side chains of Hpg and Dpg point to the outside. The lids of the cylinder are formed by the side chains of Phe.74 While feglymycin shows antibacterial activity only against Gram-positive bacteria it inhibits peptidoglycan biosynthesis enzymes MurA and MurC (E. coli and Staphylococcus aureus) of Gram positive and Gram negative bacteria (Scheme 4).26 Furthermore, feglymycin has been reported to inhibit HIV replication in the low μM range (EC50 = 0.8–3.2 μM) in addition to the inhibition of the formation of syncitia, which results from cell-to-cell transfer between HIV-infected T cells and healthy CD4+ T cells as well as the DC-SIGN-mediated viral transfer to CD4+ T cells. Feglymycin acts as gp120/CD4 binder by interacting with the heavily glycosylated viral envelope protein gp120, as determined by surface plasmon resonance (SPR) spectroscopy, thus inhibiting HIV entry to CD4+ T cells via CCR5 and/or CXCR4 chemokine receptors.75 Finally, the total synthesis of the 13 amino acid antiviral peptide antibiotic feglymycin and its enantiomer was performed by fragment condensation. This study was motivated by the difficulty in assembling a large number of phenylglycines while suppressing racemization and epimerization, respectively. Structure–activity-relationship (SAR) studies with truncated synthetic peptides shed light on structural features relevant to the molecular mode of action of feglymycin.73 A further Ala-scan of feglymycin revealed contributions of amino acid side chains to the activity in antibacterial and antiviral assays.26
The Ala-scan showed a significant decrease in MICs, 4–16 times lower when D-Dpg was exchanged with D-Ala. Interestingly, D-Hpg1 and L-Hpg5 were even more critical for the antimicrobial activities.26
The lipoglycodepsipeptide antibiotic ramoplanin (Scheme 5), formerly called A-16686,76,77 was isolated from the liquid culture of Actinoplanes strain ATCC33076.76,78
 | ||
| Scheme 5 Structures of Hpg-containing cyclic peptide antibiotics, enduracidins A and B, ramoplanin (A1, A2, A3), ramoplanose, and ramoplanin aglycon. | ||
Ramoplanin, produced as a complex of three compounds A1–A3,77,79,80 has excellent antibacterial activities against Gram-positive bacteria (2–10 times more active than vancomycin against 500 strains), particularly against Staphylococci (including methicillin-resistant isolates), vancomycin-resistant Enterococci VRE (MIC = 0.5 μg ml−1), Streptococci, Actinomyces and Gram-positive anaerobes.25,81 It shows excellent therapeutic activities against experimental septicemias in the mouse and exhibits a lack in the cross-resistance with other clinically used antibiotics.76,78 The main mode of ramoplanin action is changing the bacterial peptidoglycan architecture, thus inhibiting bacterial cell wall integrity by interrupting the late stage membrane-associated glycosyltransferase reactions catalyzed by transglycosylases and MurG.81–83 Because of its highly potent antimicrobial activity, ramoplanin reached phase I clinical trials in the early 90ies and since 2001 is in phase III clinical trials for the temporary suppression of VRE gastrointestinal carriage and for the treatment of Clostridium difficile-associated diarrhea. In 2001 the FDA put ramoplanin A2 on a fast-truck status for the prevention of VRE and treatment of C. difficile colitis.80,81,83–85 Ramoplanin A1–A3 structures were elucidated in 1989 by means of chemical degradation, mass spectrometry as well as 1H and 13C NMR studies.77,79 They are macrocyclic peptides consisting of 17 amino acids, 12 of them are non-proteinogenic amino acids, and seven display D-configuration. All ramoplanins contain five 4-Hpg residues, two of them with D-configuration. In addition they contain an L-3-chloro-4-Hpg (L-Chp17) residue (Scheme 5). Ramoplanins A1–A3 only differ in the lipid side chains attached to the N-terminus of Asn1.25,77,79–81
The ramoplanose structure (ramoplanin aglycon) was elucidated in 1991 by means of FAB-MS, amino acid analysis, chiral GC, and NMR spectroscopy86 and subsequently corrected in 1996 by 2D NMR spectroscopy; from these data a 3D structure model has also been derived.81,87 This 3D structure is characterized by two antiparallel β-strands (L-3-OH-Asn2 to D-Hpg7, and D-Orn10 to Gly14) stabilized by six transannular hydrogen bonds and a cluster of hydrophobic aromatic side chains (D-Hpg3, L-Phe9 and L-Chp17). This provides a U-shape topology to the β-sheet with a reverse β-turn formed by L-Thr8 and L-Phe9 at one end and a more flexible connecting loop (L-Leu15 to L-Chp17) at the other end.25,81,87,88 The total synthesis of ramoplanin A2 and ramoplanose was achieved in 2002 by assembly and cyclization of three peptide subunits in a solution phase approach.88 Structure–activity relationship (SAR) studies by Boger and coworkers were performed by an Ala-scan on the derivative [L-Dap2] ramoplanin A2 aglycon.25 This study showed that for Hpg substitutions the MIC decreased between 13 to 74 times upon substitution with Ala, depending on the positions of Hpg within the molecule. Nevertheless, the most critical residue for its activity was D-Orn10 where the MIC decreased three order of magnitude when substituted with Ala.25
The peptide antibiotics enduracidin A and B82,89,90 (Scheme 5) and janiemycin have similar amino acid compositions,91,92 and show similar core structures compared to ramoplanin. Enduracidin A and B were isolated from mycelium of Streptomyces fungidicus B-5477.90 The structure of enduracidin was determined by Hori and coworkers in the early 70s by NMR spectroscopic studies,93–95 while the 3D solution conformation was determined in 2005 by NMR spectroscopy and molecular dynamics.89 Enduracidins are cyclodepsipeptides composed of 17 amino acids, of which 16 form a macrolactam. The N-terminus is acylated with cis,trans-fatty acids (Scheme 5), which is the discriminating feature of enduracidin A and B.89 Apart from significant similarities between enduracidin and ramoplanin the former contains the characteristic amino acid enduracididine (End) at position 10 and 15 and citrulline (Cit9) replacing Phe9, Orn10 and Leu15 in ramoplanin. The structures further differ in D-mannosylation of ramoplanin at Hpg11 as well as in the length of the acyl chain.89 Structural similarities between enduracidin and ramoplanin point to a similar mechanism of action, i.e. binding to the peptidoglycan lipid intermediate and a common bioactive antibiotic pharmacophore.81,82 The total synthesis of enduracidin has not yet been achieved; enduracididin (End) has been synthesized previously by Tsuji and coworkers in 1975.96 Enduracidin shows in vitro and in vivo antibacterial activities against a wide spectrum of Gram-positive bacteria including MRSA causing human urinary tract and skin infections,97 as well as Neisseria gonorhoeae, with a minimal inhibitory concentration (MIC) < 0.05 μg ml−1.98–103 Although enduracidins are known since the 1960s, they have more recently been revisited for their potential as pharmaceutically used antibacterial compounds. In addition, enduracidin hydrochloride has been reported to be used for animal production in order to increase animal weight in pigs and chicken, and to improve feed conversion ratio in these animals.104
Janiemycin is a cell wall biosynthesis inhibitor peptide isolated from Streptomyces macrosporeus. It has a structure related to the peptide antibiotic ramoplanin A2, and shares a similar amino acid composition with enduracidin.92,105,106 Until now a detailed structure analysis of janiemycin has not been performed. Application of janiemycin on Gram-positive and Gram-negative bacteria results in accumulation of lipid intermediates.92,105 Based on amino acid similarities of janiemycin to ramoplanin A2 and to enduracidin, it is proposed that janiemycin might also inhibit transglycosylases and/or MurG by binding to lipid I and/or lipid II.81
The calcium dependent peptide antibiotic CDA (Scheme 6) has first been described by B. Rudd in 1978 as produced by Streptomyces coelicolor A3(2).107 This peptide was called CDA because it kills bacteria only in the presence of calcium ions, where it produces transmembrane channels to conduct monovalent cations.108,109 The CDA structure was elucidated by Jung and co-workers110 by means of amino acid analysis, Edman degradation and 2D NMR spectroscopy. CDA occurs also in various natural subtypes (CDA1b, CDA2a, CDA2b, CDA3a, CDA3b, CDA4a and CDA4b) and their production depends on the culture medium used.111,112 With regard to the structure, CDA is an 11mer cyclic lipopeptide with the C-terminus attached to the side chain of Thr2. An epoxyhexanoyl residue is attached to the N-terminus of Ser1. Further structural characteristics of this molecule are D-4-Hpg at position 6, D-3-hydroxyasparagine at position 9, and four acidic amino acids (L-Asp at positions 4, 5 and 7, and one L-3-methyl-Glu at position 10).110 CDA displays antimicrobial activities against a variety of Gram-positive bacteria, and multicellular bacteria from the Streptomyces genus, except for five strains closely related to the wild type producing organism Streptomyces coelicolor A3(2).108 There are obvious structural similarities of CDA to related lipocyclopeptides such as the marketed antibacterial daptomycin (Cubicin™, injectable form) and to friulimycin.113,114 Main structural similarities are the 10mer peptide macrocycle as a central element. However, neither daptomycin nor friulimycin contain the D-4-Hpg in their structures. Both CDA and friulimycin possess three residues of Asp at position 4, 5 and 7, where Asp4 in friulimycin is β-methylated.114 Daptomycin contains also Asp at position 7, and two other Asp residues but in different positions compared to CDA. Nevertheless, both peptide antibiotics CDA and daptomycin contain L-3-methyl-Glu at the preliminary position in their macrocyclic rings.110,113
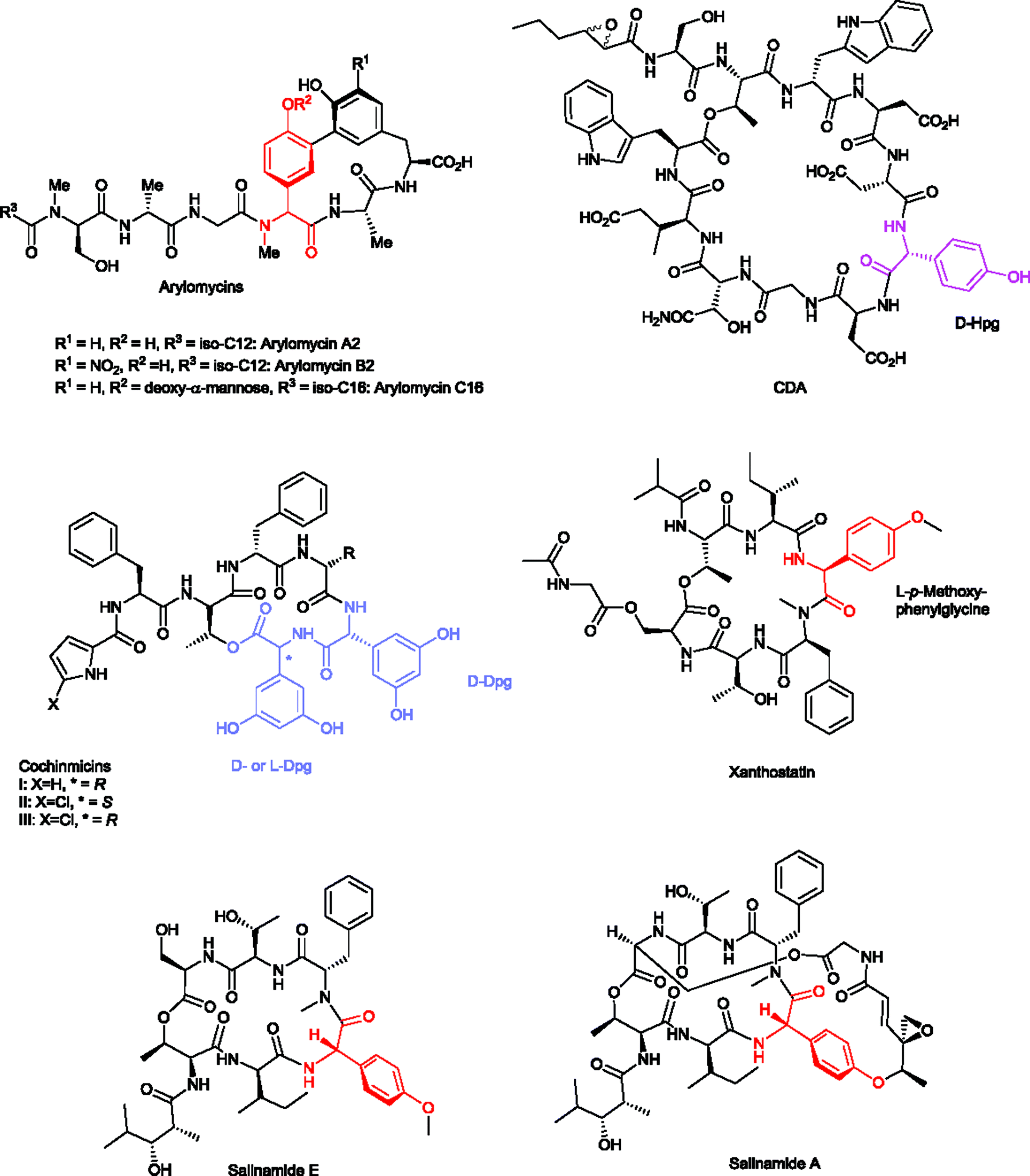 | ||
| Scheme 6 Structures of Hpg- and/or Dpg-containing cyclic peptide antibiotics CDA, cochinmicins I, II, and III, xanthostatin, and salinamides A and E, arylomycins A2, B2, and C16. | ||
The peptide antibiotics arylomycin A and B, isolated and characterized from Streptomyces sp. Tü 6075 by the groups of Fiedler & Jung in 2002,115,116 were shown to bind and inhibit bacterial type I signal peptidase (SPase) in vitro.116–118 The total synthesis of arylomycin has been achieved by Romesberg and coworkers in 2007,119 followed by the synthesis of various derivatives including arylomycin A–C16.119–124 Recently, a new arylomycin derivative (arylomycin A6) was isolated from Streptomyces parvus HCCB10043 and its structure was elucidated by HR-ESI-MS, fatty acid analysis and 1D- and 2D-NMR spectroscopy.125 Together with their synthetic derivatives (e.g. arylomycin C16) (Scheme 6), they contain N-methylated-Hpg in their ring structures and show a remarkably wide spectrum of activities against both Gram-positive and Gram-negative bacteria.120,122,123 The antimicrobial activities (MIC) of natural arylomycin A2 and of synthetic arylomycin C16 against Staphylococcus epidermidis are ∼1.0–0.25 μg ml−1, respectively.119,126
The cyclodepsipeptides cochinmicin I, II and III (Scheme 6), isolated from Microbispora sp. ATCC 55140,127 have been reported as endothelin antagonists. The structures contain two D-Dpg residues (cochinmicin I and III), and L-Dpg and D-Dpg residues (cochinmicin II), respectively.128 The structures were elucidated by mass spectrometry, 1D and 2D NMR spectroscopy in the early 1990s.128
The antagonism of these compounds to the endothelin (ET) receptors could potentially be used for treatment of various diseases caused by elevated levels of endothelin, e.g. cyclosporine-induced nephrotoxicity, myocardial infarction, uremia, diabetes, systemic hypertension, endotoxic shock, cardiac ischemia, post-ischemic renal failure and compromised renal flow.127
The natural product xanthostatin (Scheme 6) is a cycloheptadepsipeptide which was isolated from Streptomyces spiroverticillatus.129 Characteristic features of the structure are a p-methoxyphenylglycine and a macrolactone formed by the sidechain of Thr1 with a modified Ser7 at the C-terminus.130
Remarkably, xanthostatin has a highly specific antimicrobial activity against Xanthomonads and has been described as bacteriostatic against Xanthomonas oryzae (MIC = 2 μg ml−1) and Xanthomonas campestris pv. citri (MIC = 4 μg ml−1). The latter phytopathogenic strain affects citrus plants (‘citrus canker’) causing necrosis on citrus fruits, leafs, and stems, and is considered a plant disease that is difficult to control.131
Salinamides A–F (Scheme 6) are antibacterial and anti-inflammatory depsipeptides isolated from Streptomyces sp. CNB-091. The Streptomyces strain has been isolated from the surface of the jellyfish Cassiopeia xamachana.132–134 Salinamides are macrolactone heptapeptides with the ring formed between the side chain of Thr1 and the C-terminus of Ser7 (salinamide E). The cyclic structure may be extended by another cycle (salinamide A) consisting of an unusual ester in the sidechain of Ser7 and an ether with the OH-group of Hpg3. The total synthesis of salinamide A has been reported by Tan and Ma in 2008.135 Salinamides A and B show moderate antibiotic activities against Gram-positive bacteria and anti-inflammatory activities in a phorbol ester-induced mouse ear edema assay.132 Salinamide A and B exhibit strong transcription inhibition activity by targeting bacterial RNA polymerases of E. coli (Gram-negative) and Staphylococcus aureus (Gram-positive) while they do not inhibit human RNA polymerase I, II and III.136,137
The largest group of antibacterial peptides containing Hpg and Dpg are the glycopeptide antibiotics (GPA). In these peptides the amino acids 4-hydroxyphenylglycine (Hpg) and 3,5-dihydroxyphenylglycine (Dpg) are essential elements of all type-I to type IV glycopeptide antibiotics, since they are involved in side-chain cross-links and thus are crucial for the overall conformation of the molecules.138Vancomycin,139 balhimycin,140 chloroeremomycin141 (all type I), actinoidin (type II),142,143 ristocetin (type III),144,145 and teicoplanin (type IV)146 are the most prominent members of this compound class. Glycopeptide antibiotics have been classified into five types,23 four of them (type I–IV) have antimicrobial activities, while the last one (type V) with complestatin,147 and the anti-HIV antibiotic chloropeptin148 exhibit antiviral activities inhibiting the binding of the human immunodeficiency viral glycoprotein (HIV-1/gp120) to the T-cell CD4 receptor.148,149 Type V GPA kistamicin150 exhibits a moderate antibacterial activity against Gram-positive bacteria but no activity against Gram-negative bacteria.150 Furthermore, kistamicin shows stronger anti-influenza virus activity than ribavirin.151
The antimicrobial activities of type I–IV compounds arise from their highly complex and rigid structures, which perform non-covalent binding of the glycopeptide backbone via five hydrogen bonds to the amide bonds and C-terminal end of the Lys-D-Ala-D-Ala peptide of the peptidoglycan.23,152,153 As the main mode of action, the binding of the substrate prevents subsequent crosslinking of peptidoglycan by transpeptidases. The common structure of type I–IV GPAs is a heptapeptide backbone containing three to five phenylglycine-derivatives (type I or type IV, respectively). The backbone is modified by one biaryl- (A–B ring) and two diarylether side-chain cyclizations (C-O-D and D-O-E rings) of the aromatic amino acids (Scheme 7), forming a cup-shaped cavity to accommodate the Lys-D-Ala-D-Ala peptide.
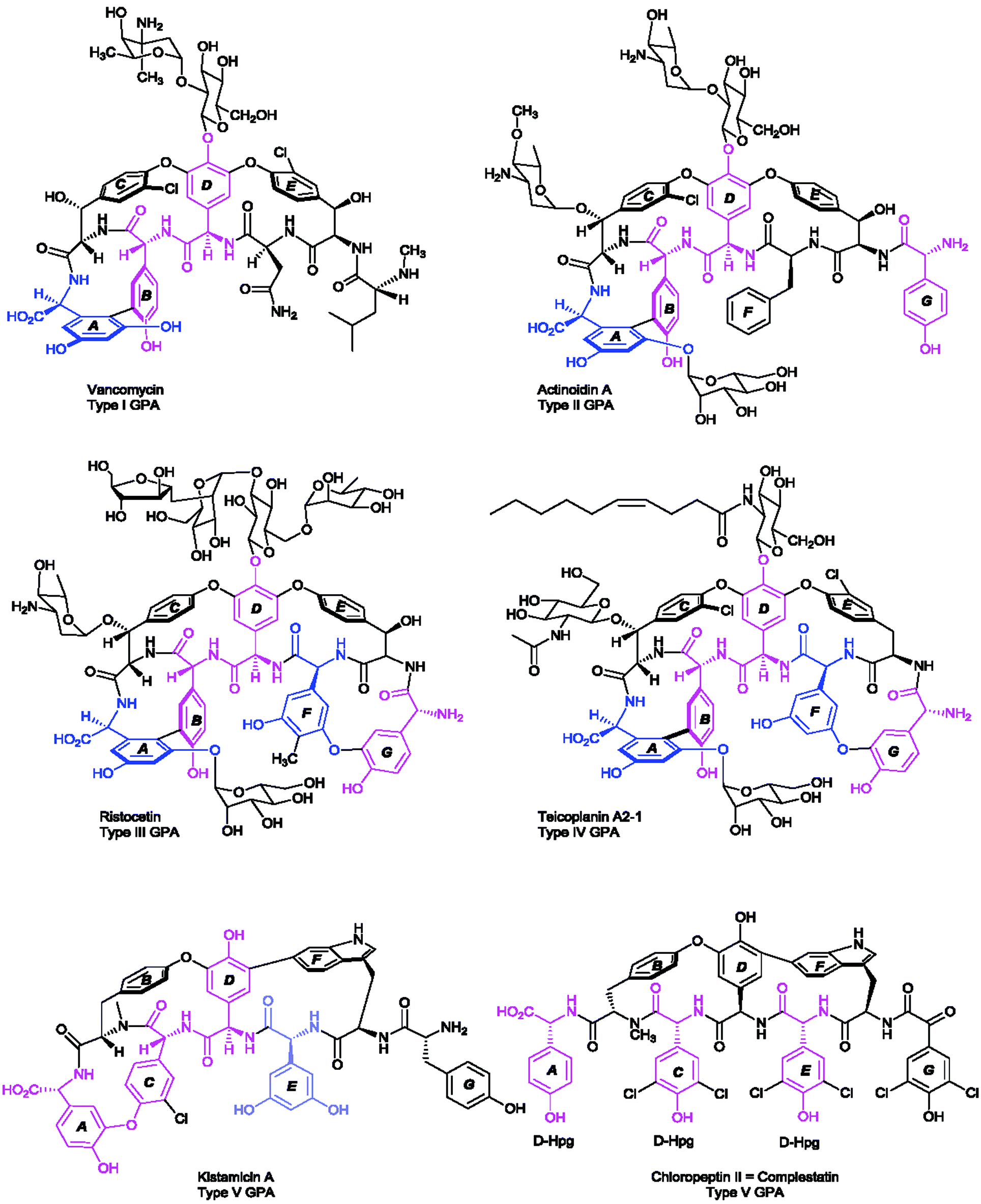 | ||
| Scheme 7 Positions of D-4-hydroxyphenylglycine (Hpg) (coloured pink) and 3,5-dihydroxyphenylglycine (Dpg) (coloured blue) according to the different types of glycopeptide antibiotics (GPA). Due to convention the N-terminus is placed to the right. The rings are named as ring A–G. | ||
These cyclizations are formed by cross-linking of the aromatic amino acids β-hydroxytyrosine (β-Hty), 3-chloro-β-hydroxytyrosine (Cht), tyrosine (Tyr), D-4-hydroxyphenylglycine (Hpg), and L-3,5-dihydroxyphenylglycine (Dpg) (see Section 5.2.1). When Hpg4 forms a DE-biaryl ring with Trp2, type V glycopeptides are obtained that display antiviral activities. Dpg is present in all GPA members of type I–IV and some GPAs of type V, such as kistamicin A & kistamicin B, while Hpg is a constituent of all of these GPAs (Scheme 7). A comprehensive understanding of the mechanistic assembly of glycopeptide antibiotics and their non-proteinogenic building blocks, particularly of Hpg and Dpg as essential components of vancomycin and teicoplanin, might aid in the development of new biologically active drugs. Hence, Hpg and Dpg biosynthesis pathways have been a matter of interest for many research groups over the past 15 years.
4 Biosynthesis of phenylglycines
The biosyntheses of Hpg and Dpg have been intensively investigated over the past years. The sequencing of the chloroeremomycin biosynthesis gene cluster in 1998 by van Wageningen and coworkers was an important step,141 since it allowed first insights into putative genes functions involved into the assembly of these amino acids.Further complete (or partial) sequencing for some other glycopeptides biosynthesis gene clusters such as balhimycin,154 teicoplanin,155 complestatin,156 A47934 (ref. 157) and A40926 (ref. 158) contributed in shedding light on different stages of Hpg and Dpg biosynthesis pathways. Scheme 8 shows the distribution of Phg, Hpg, and/or Dpg biosynthesis genes within some of the most relevant gene clusters of natural products.
 | ||
| Scheme 8 Biosynthesis gene clusters of the important peptide antibiotics illustrating the distribution of biosynthesis genes of Phg (pglA, pglB, pglC, pglD, and pglE; coloured green), Hpg (pdh, hmaS, hmO, and hpgT; coloured red), and Dpg (dpgA, DpgB, dpgC, and dpgD; coloured blue). | ||
4.1 Phg biosynthesis
Although phenylglycine is structurally the simplest member among the phenylglycines, its biosynthesis genes were not identified until 2011.159 The characterization of the pristinamycin biosynthesis gene cluster of Streptomyces pristinaespiralis led to the identification of an operon-like structure of five genes pglA–E.17 These genes exhibited similarities to some of the enzymes involved in Hpg- and Dpg- biosynthesis,17 with overlapping start and stop codons, suggesting that these are translationally coupled.159 Phg is suggested to be synthesised from phenylpyruvate, which is converted into phenylacetyl-CoA by PglB and PglC. These two enzymes are assumed to work together as a pyruvate dehydrogenase-like complex.159This assumption is based on high similarities of the pglB and pglC gene products (57% and 70% respectively) to the α- and β-subunits of pyruvate dehydrogenase from Mycobacterium avium.159,160 The gene expression of pglA gene results in the hydroxylacyl-dehydrogenase PglA which converts phenylacetyl-CoA into benzoylformyl-CoA.
Based on sequence comparisons using Blast data161 PglA was found being related to DpgC of A. balhimycina (47% identity, 62% similarity), a key enzyme in the biosynthesis of Dpg, which converts 3,5-dihydroxyphenylacetyl-CoA into 3,5-dihydroxyphenylglyoxylate (see below Section 4.3).159,162
Due to this similarity, PglA was proposed to have a comparable function to DpgC by catalyzing a similar oxygenation reaction during Phg biosynthesis.159 The product of pglD gene was found to have moderate similarity (53%) to a type II thioesterase of Amycolatopsis mediterranei, suggesting that PglD may have a function as the hydrolytic thioesterase releasing phenylglyoxylate and CoA from benzoylformyl-CoA.159
A basic local alignment of pglE gene product showed 67% similarity to the phenylglycine aminotransferase (Pgat) of Amycolatopsis balhimycina, suggesting that PglE159 may convert benzoylformate into phenylglycine (Scheme 9).159 All biosynthetic assumptions were confirmed by gene insertion mutagenesis studies and L-Phg feeding experiments in S. pristinaespiralis pgl mutants.159 Nevertheless no in vitro protein investigations have been performed to date.
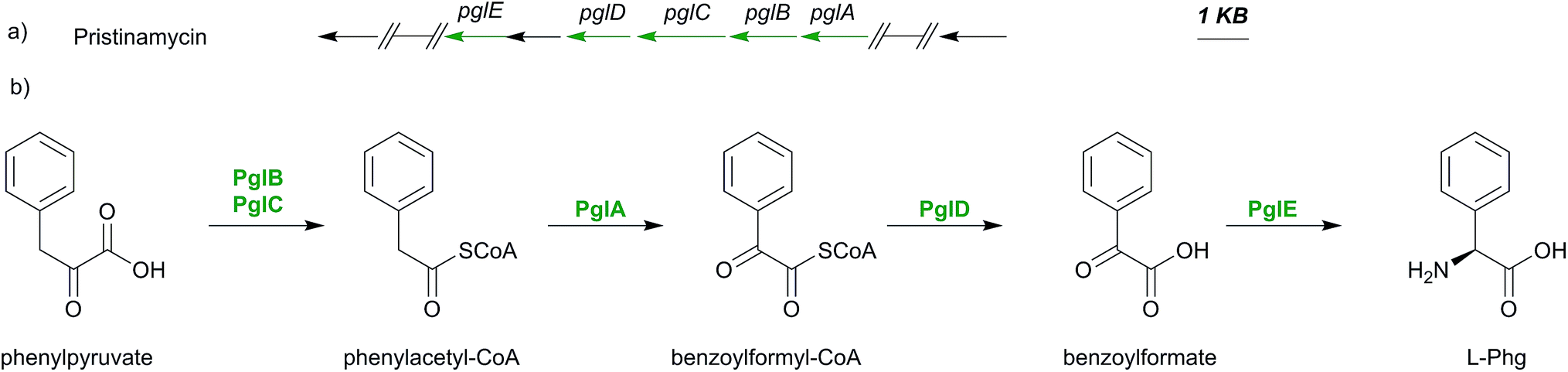 | ||
| Scheme 9 Biosynthesis pathway of L-phenylglycine (Phg): a) portion of the pristinamycin biosynthesis gene cluster (pglA, pglB, pglC, pglD and pglE) from Streptomyces pristinaespiralis representing genes for b) the assembly of phenylglycine starting from phenylpyruvate. | ||
4.2 Hpg biosynthesis
The genes of the L-4-hydroxyphenylglycine (Hpg) biosynthesis pathway were initially assigned based on the data from the chloroeremomycin biosynthesis gene cluster of Amycolatopsis orientalis.163–165 Studies of different glycopeptide antibiotic biosynthesis gene clusters, such as balhimycin, complestatin and calcium-dependent antibiotic (CDA), as well as other compounds like ramoplanin, and nocardicin confirmed essentially identical pathways for Hpg biosynthesis.112,138,154,156,166,167 In early work on the origin of Hpg, [13C]-tyrosine or [2H, 13C]-tyrosine were fed to growing cultures in radioactive labeling studies,168–171 which determined Tyr as a precursor for L-4-hydroxyphenylglycine. Biochemically, prephenate is converted into L-4-hydroxyphenylpyruvate by the action of prephenate dehydrogenase Pdh (Scheme 10).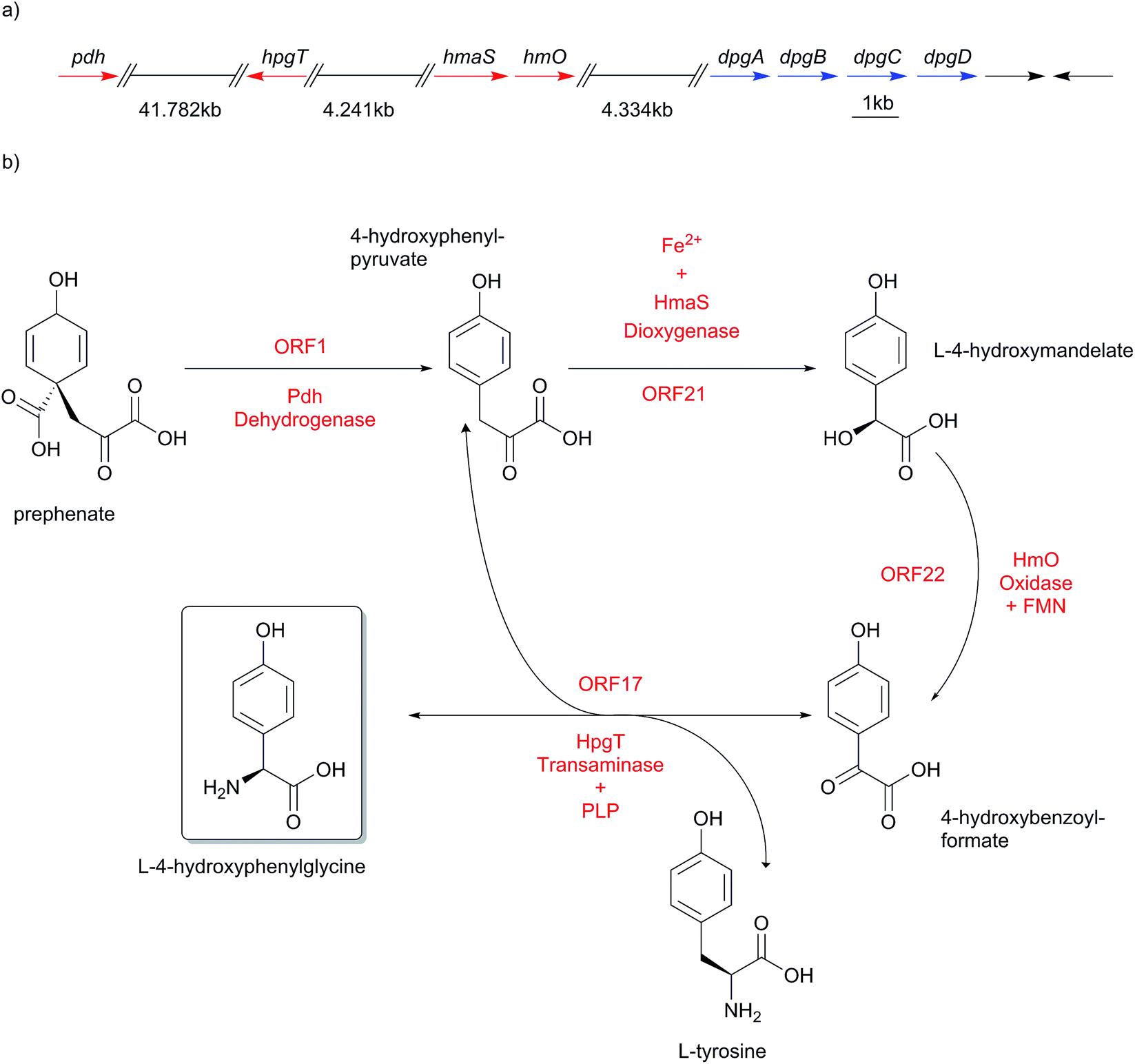 | ||
| Scheme 10 Biosynthesis pathway of L-4-hydroxyphenylglycine (Hpg). (a) Chloroeremomycin biosynthesis gene cluster from Amycolatopsis orientalis with genes for L-4-hydroxyphenylglycine (Hpg) biosynthesis (pdh, hpgT, hmaS and hmO). Numbers indicate ORFs starting from the first gene in the gene cluster. (b) L-4-hydroxyphenylglycine (Hpg) biosynthesis catalytic cycle. Pdh is prephenate dehydrogenase, HmaS is L-4-hydroxymandelate synthase, HmO is L-4-hydroxymandelate oxidase, HpgT is 4-hydroxybenzoylformate/L-4-tyrosine transaminase, PLP is pyridoxal phosphate. | ||
The subsequent step is performed by L-4-hydroxymandelate synthase HmaS, an enzyme closely related to 4-hydroxyphenylpyruvate dioxygenase HppD172 – both members of the α-keto acid-dependent non-heme iron-dependent dioxygenases.173–175 This iron-dependent 4-hydroxyphenylpyruvate decarboxylating hydroxylase catalyzes the conversion of 4-hydroxyphenylpyruvate into L-4-hydroxymandelate by hydroxylating the benzylic position of 4-hydroxyphenylpyruvate (Scheme 11).138,163,164,176,177 The dioxygenation activity of HmaS was confirmed by performing a reaction under [18O2] atmosphere in vitro, whereupon one oxygen atom was incorporated into the carboxyl group and the other oxygen atom into the benzylic hydroxyl group.163 Subsequently L-4-hydroxymandelate oxidase HmO, an FMN dependent enzyme, catalyzes the oxidation of L-4-hydroxymandelate to 4-hydroxybenzoylformate. Interestingly, this enzyme is unable to oxidize the D-isomer of L-4-hydroxymandelate, illustrating its stereospecificity. HmO has amino acid sequence homology with glycolate oxidase (GO, 50% homology, present in the cytosol of the cell) and with mandelate dehydrogenase (MDH, 52% homology, a membrane bound enzyme), which both use FMN as a cofactor.164
 | ||
| Scheme 11 Comparison of the products of the iron-dependent dioxygenases HmaS and HppD starting from the same substrate 4-hydroxyphenylpyruvate. | ||
The reaction sequence is finalized by a transamination reaction catalyzed by L-4-hydroxyphenylglycine transaminase HpgT, which requires pyridoxal phosphate (PLP) as a coenzyme to exert its catalytic activity. HpgT has amino acid sequence homology to various amino acid transaminases. In addition HpgT showed substrate promiscuity by its ability to accepting a variety of amino donors,164 as well as its ability of utilizing both L-4-hydroxyphenylglycine and L-3,5-dichloro-4-hydroxyphenylglycine in the reverse direction in complestatin biosynthesis.138,156 The catalytic activity of HpgT played a critical role in establishing an economical three-enzyme cycle for L-4-hydroxyphenylglycine biosynthesis (Scheme 10), since the reductive amination reaction converts L-4-hydroxybenzoylformate into L-4-hydroxyphenylglycine, and the amino donor co-substrate converts L-Tyr into one equivalent of 4-hydroxyphenylpyruvate, which is returned into the enzymatic cycle to serve as a substrate for HmaS catalyzing a further round of L-4-hydroxyphenylglycine biosynthesis. This also explains why labelling studies suggested L-Tyr as a precursor for L-4-hydroxyphenylglycine biosynthesis in glycopeptide antibiotics.164,168–171
4.3 Dpg biosynthesis
The gene functions of 3,5-dihydroxyphenylglycine (Dpg) biosynthesis pathway (Scheme 12) were assigned in the course of investigating the balhimycin biosynthesis gene cluster from Amycolatopsis mediterranei DSM5908.154,175,178–181 Homologous biosynthesis pathways are found in all other glycopeptide biosynthesis gene clusters, e.g. chloroeremomycin (Scheme 10-a).163–165 BLAST searches for genes with similarities to dpgA from the balhimycin biosynthesis gene cluster revealed a gene encoding for gerberin chalcone synthase 2 (GCHS2), a plant polyketide synthase (PKS) of the chalcone synthase-type (CHS) from Gerbera hybrid (Asteraceae). The enzyme uses acetyl-CoA and malonyl-CoA to synthesize the carbon-skeleton of gerberin, a 2-hydroxy-pyrone plant secondary metabolite.175,182 Significant sequence similarities were particularly found for key amino acids in the active site shown to be required for activity of GCHS2.183 Further similarities of DpgA to other proteins (45–49%) were all chalcone synthase-related PKSs184 such as Streptomyces coelicolor.185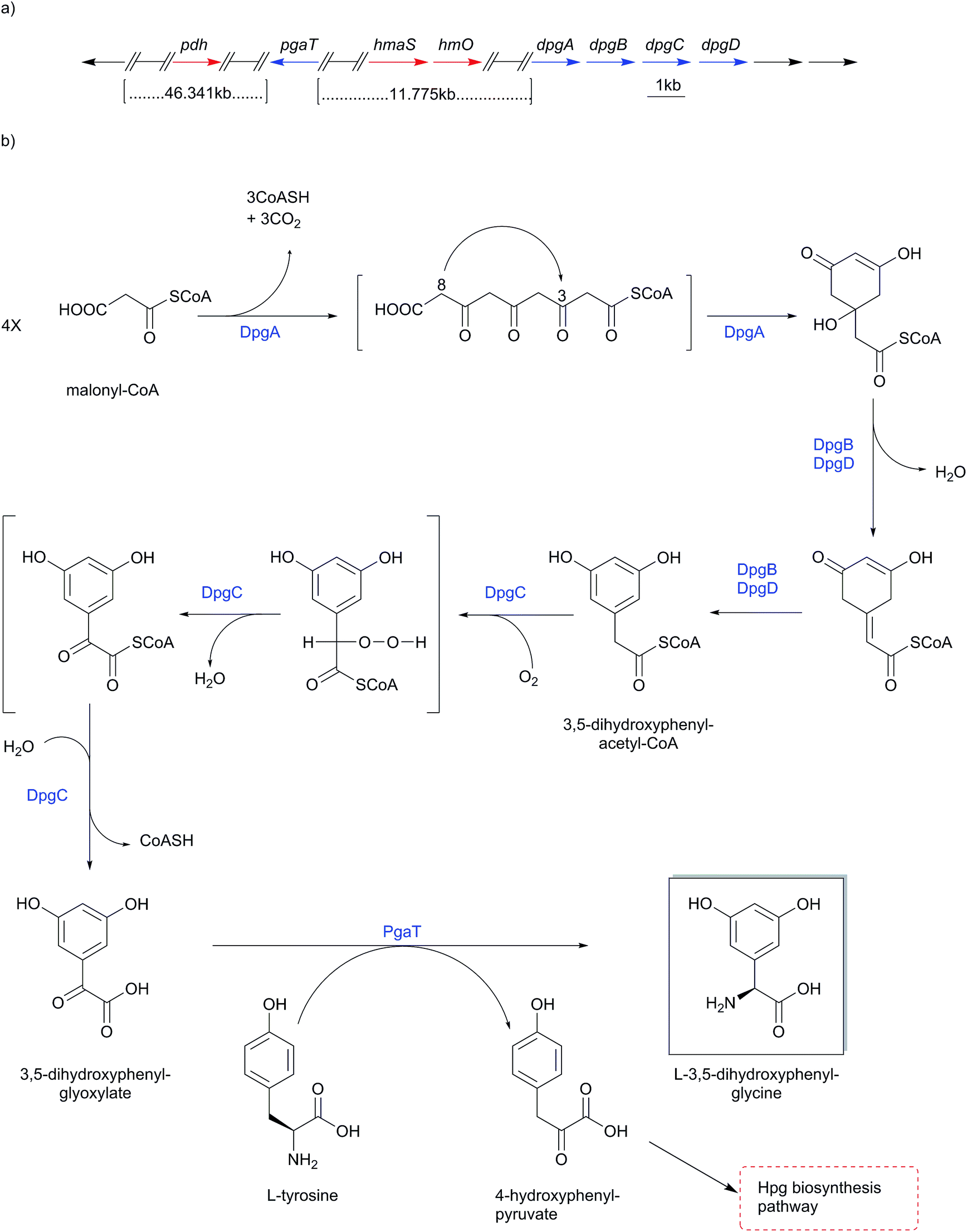 | ||
| Scheme 12 Biosynthesis pathway of 3,5-dihydroxyphenylglycine (Dpg). (a) Balhimycin biosynthesis gene cluster from Amycolatopsis mediterranei DSM5908 indicating genes responsible for 3,5-dihydroxyphenylglycine (Dpg) assembly (dpgA, dpgB, dpgC, dpgD and pgaT). Numbers indicate ORFs starting from the first gene in the gene cluster; (b) catalytic cycle of 3,5-dihydroxyphenylglycine (Dpg) biosynthesis. | ||
Heterologous expression in Streptomyces lividans or in Escherichia coli combined with in vitro studies of DpgA using radioactively labeled acetyl-CoA and malonyl-CoA esters as substrates showed that DpgA is a type III PKS. Accordingly, four molecules of malonyl-CoA are assembled in a decarboxylating Claisen condensation to yield (1,3-dihydroxy-5-oxo-cyclohex-3-enyl)acetyl-CoA, a precursor of 3,5-dihydroxyphenylacetic acid (Scheme 12).175,186,187
In in vitro experiments, DpgA showed the conversion of the above-mentioned precursor into 3,5-dihydroxyphenylacetyl-CoA (DPA-CoA), but in an extremely slow manner that was difficult to detect. However, formation of DPA-CoA was accelerated 17-fold by addition of DpgB. This conversion rate was again doubled by the addition of DpgD to DpgA/DpgB, although the addition of DpgD alone to DpgA did not increase DpgA activity. In total, the formation of DPA-CoA was accelerated 35-fold by the addition of DpgB/DpgD to DpgA.186,187 Hence, DpgB and DpgD likely work as dehydratases on (1,3-dihydroxy-5-oxo-cyclohex-3-enyl)acetyl-CoA to produce DPA-CoA, probably while DpgA is still bound to the CoA-substrate.186,187
In vitro studies on DpgC confirmed its role as a metal- and redox cofactor-independent oxygenase in converting 3,5-dihydroxyphenylacetyl-CoA (DPA-CoA) into 3,5-dihydroxyphenylglyoxylate. This was proven by addition of DpgC to the above mentioned DpgA/DpgB incubation mixture. Likewise 3,5-dihydroxyphenylglyoxylate was also obtained after incubation of synthetic DPA-CoA with DpgC alone. Addition of DpgB and/or DpgD to incubations of DpgC with DPA-CoA did not cause any effect on the CoASH formation rate. Furthermore, DpgC alone was able to yield phenylglyoxylate after incubation with the non-substituted phenylacetyl-CoA (PA-CoA).186,187 Anaerobic incubation of DpgC with DPA-CoA revealed no product formation or detectable loss of substrate. This behavior changed upon admission of air to the reaction vials, revealing molecular oxygen as a likely source for the keto oxygen introduced into 3,5-dihydroxyphenylglyoxylate. Further investigations with mandelyl-CoA as a substrate showed that water can act as a source for the keto oxygen introduced into phenylglyoxylate by DpgC, since the anaerobic incubations did not indicate an absolute requirement for molecular oxygen.186,187 In-frame deletion mutation confirmed phenylglycine aminotransferase (PgaT) to perform transamination of 3,5-dihydroxyphenylglyoxylate to yield 3,5-dihydroxyphenylglycine.175 4-Hydroxyphenylglycine transaminase HpgT was found to be corresponding with PgaT, and have the ability of catalyzing the transamination of L-Hpg and L-Phg from 4-hydroxyphenylglyoxylic acid and phenylglyoxylic acid respectively.164 This substrate promiscuity led to the assumption that both HpgT and PgaT have dual functional activities catalyzing the transamination reaction of L-Hpg and 3,5-dihydroxyphenylglycine (Dpg).175 PgaT uses L-tyrosine as an amino donor for the transamination conversion of 3,5-dihydroxyphenylglyoxylate into 3,5-dihydroxyphenylglycine, yielding 4-hydroxyphenylpyruvate (Scheme 12), which can undergo another cycle of Hpg biosynthesis (Scheme 10).
5 Biosynthesis of phenylglycine-containing peptides
The common element in the biosynthesis of all phenylglycine containing peptides is their origins in non-ribosomal peptide synthesis. This makes the presence of such residues in peptides a characteristic sign of non-ribosomal biosynthetic origins.5.1 Phenylglycine incorporation into non-ribosomal peptides
Based upon the reported non-ribosomal protein sequences available for the natural products discussed above,17,112,120,141,154,156–158,190–195 the A-domain selection pockets for phenylglycines L-Phg, L-Hpg and L-Dpg have been analysed (Scheme 13, prepared using NRPSpredictor-2).196,197 Hpg-activating domains fall into two broad classes (A-domain codes beginning with DIF (Scheme 13, upper left panel) and DAV/L (Scheme 13, upper right panel) respectively) that are typically conserved within a single NRPS assembly line. The exception is GPA biosynthesis, where both types are represented in the NRPS machinery in the Hpg-incorporating modules 4 and 1/5 (Scheme 14).141,154,157,158,190,191 Similar trends are seen for complestatin, although here the first class is found in modules 1, 3, 4 and 5 with the second class only found in module 7.156 In both cases of these Hpg-activating A-domains the fourth residue present is a His, which is presumably involved in binding to the 4-hydroxyl group of Hpg.
 | ||
| Scheme 13 Consensus adenylation selection pockets for phenylglycine residues (excluding ristocetin/ristomycin). Colour code: acidic residues (red), basic residues (blue), amide/alcohol containing residues (orange), cysteine residues (magenta). | ||
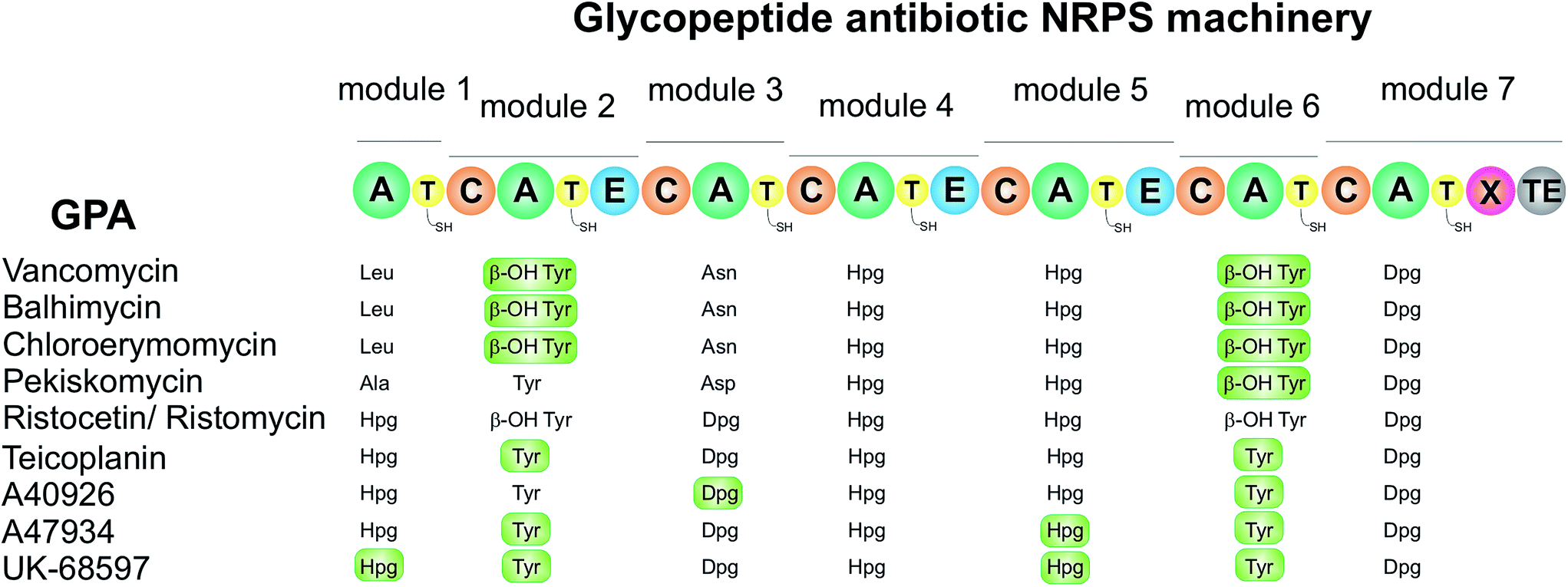 | ||
| Scheme 14 Schematic representation of the NRPS machinery from glycopeptide antibiotic biosynthesis (type I–IV) displaying the amino acids activated by each A-domain of the seven individual modules for the compounds discussed within this review. Halogenation of the residues is indicated by a green box around the relevant residue. Abbreviations: A – adenylation domain, C – condensation domain, T – thiolation (or peptidyl carrier protein (PCP) domain, E − epimerization domain, X – oxygenase recruitment domain, TE – thioesterase domain. | ||
The few examples of Phg activating A-domains characterised show a conserved A-domain code that is similar to that displayed by the Hpg-activating module 4 of GPA NRPS machineries (starting with DIF (Scheme 13, lower left panel)), albeit with the replacement of the His residue with a Leu residue and the subsequent Leu residue with a larger Trp residue.17 Dpg activating A-domains show a selection pocket that maintains the His residue found in Hpg pockets, whilst reducing the general bulk of residues.141,154,157,158,190,191 These pockets also introduce a Thr residue capable of hydrogen bonding and replace the common Phe residue with a Tyr residue; presumably at least one of the His/Tyr/Thr residues are involved in coordinating to the phenol moieties of the Dpg residue (Scheme 13, lower right panel). The similarity of the A-domain codes in these systems argues against the direct activation of modified (e.g. chlorinated) Phg/Hpg/Dpg residues but rather implies their modification following activation by the NRPS machinery and transfer to a carrier protein domain. The same can also be argued for the incorporation of the initial p-hydroxybenzoylformate residue in complestatin biosynthesis, although this needs to be clarified experimentally.156
In the vast majority of cases where the phenylglycine residue incorporated in the peptide is found to be the D-isomer, a corresponding epimerization (E)-domain can be found within the NRPS-module (see Scheme 14).188,198 These domains are structurally related to condensation domains that catalyse peptide bond formation and have been shown to epimerize the residue once it is contained within the peptide, with the exception of the first amino acid of the peptide. Epimerization after peptide bond formation is due to the selectivity of C-domains themselves, which are selective for L-configured amino acids in their donor sites.188 Two notable exceptions where there is a lack of an E-domain in spite of the final incorporation of a D-amino acid can be found for phenylglycine residues: one is found in the first module of GPA-biosynthesis (that incorporates Hpg in type III and IV GPAs) where the final peptide includes a D-amino acid at position 1 although there is no epimerization domain present within the NRPS.199 The second example is found in norcardicin A biosynthesis where epimerization activity of the terminal thioesterase (TE) domain is able to replace an E-domain (see below).67
 | ||
| Scheme 15 Mutasynthesis potential of module 7 (Dpg incorporation) in balhimycin biosynthesis and module 6 (Hpg incorporation) in CDA biosynthesis through gene disruption and amino acid supplementation showing the incorporation potential of these residues by the respective A-domains. | ||
The acceptance of alternative substrates to Hpg has been assessed for calcium dependant antibiotic (CDA) biosynthesis via a similar gene knockout and amino acid supplementation strategy (Scheme 15, right panel).112 In this case, modified CDA compounds were detected upon supplementation with either Phg or 4-fluorophenylglycine whilst 4-chlorophenylglycine and 4-methoxyphenylglycine were not able to be incorporated by the respective A-domain. This suggests tighter steric control during the incorporation of Hpg than Dpg in NRPS biosynthesis, although further examples of Dpg and Hpg activating A-domains would need to be examined to ascertain whether such trends are generally applicable. Finally it is worth mentioning that for all glycopeptides generated by mutasynthesis it was not possible to perform antibacterial profiling due to low production amounts. One possible reason for such reduced production may be that the modulated bioactivity causes problems for the organism by interfering with self-resistance. However, the basic export machinery, e.g. ABC transporters should be available. Production yield is a general problem inherent to many mutasynthesis applications, which needs to be addressed by future biotechnological and biosynthetic engineering efforts.
5.2 Phenylglycine modifying enzymes
Within the product peptides containing phenylglycine residues, many are subject to further modification during peptide assembly or following peptide synthesis. This includes numerous transformations such as alkylation, halogenation, oxidative coupling or glycosylation.In the case of GPA biosynthesis, evidence is clear that these P450-catalysed reactions occur whilst the substrate peptides remain bound to the NRPS-machinery. Extensive in vivo experiments were not only able to imply the NRPS-bound nature of the peptide but could also elucidate both the identity of the enzymes responsible for the individual cross coupling steps (OxyA: D-O-E ring, OxyB: C-O-D ring, OxyC: AB ring, OxyE; F-O-G ring) as well as the order of oxidation for vancomycin-type (OxyB, OxyA, OxyC)205 and teicoplanin-type systems (OxyB, OxyE, OxyA, OxyC; there appears to be more flexibility in this case than in the vancomycin-type system).206In vitro experiments have shown the acceptance of a range of peptide substrates on isolated carrier protein (PCP)-domains for OxyB from the vancomycin system,204,207–211 with the selectivity of OxyB from the teicoplanin system for PCP-bound peptides proving significantly higher and with a reduced turnover yield.212 Other examples of a type-I (chloroerymomycin) and type-IV (A-47934) systems showed virtually no turnover of PCP-bound peptides, raising questions over the true nature of the P450 substrate.213 Recent in vitro data has now shown that the X-domain, a condensation-like domain of previously unknown function present in the final NRPS-module of all glycopeptide antibiotics (see Scheme 14), is able to recruit OxyA–C and thus present the adjacent PCP-bound peptide substrate to the P450 enzymes for oxidation.213 The activity of OxyB with peptide substrates bound to PCP-X constructs is significantly higher in all cases than the PCP alone; an additional point to support the importance of the X-domain was the first demonstration of in vitro activity of a subsequent Oxy enzyme in the cascade (OxyA).213
In arylomycin, the Hpg4 residue of the linear precursor peptide is involved in the formation of an aryl bond with Tyr6 and the enzyme believed responsible for installation of the crosslink is AryC, a member of the Cytochrome P450 superfamily with a high degree of similarity to OxyC from the vancomycin gene cluster (49%).214 An arylomycin producer strain with AryC knocked out was shown to no longer produce crosslinked peptide products, supporting the assignment of the crosslinking function to this enzyme.215
The exact nature of the halogenase substrate (i.e. peptide or amino acid) remains to be clarified, with in vitro and in vivo results from vancomycin221 and balhimycin biosynthesis178,180,201 implicating different substrates. Through the removal of the halogenase from producer strains dechloro forms of the natural products can be isolated: this has been shown for both balhimycin15 and enduracidin.103 A strain producing the GPA A40926 has also been evolved that demonstrates a reduced capacity to produce chlorinated compounds.222 The complementation of a halogenase deletion mutant of the enduracidin producing strain with the halogenase from the ramoplanin gene cluster allowed the isolation of novel halogenated enduracidins, bearing either a single chlorine substituent on Hpg13 or a triple chlorinated compound, with two chlorine atoms on Hpg13 and an additional chlorine substituent on Hpg11.103 This cross complementation does suggest that the target of such halogenase enzymes is the NRPS-bound amino acids: targeting such substrates has precedence in both the hydroxylation of Tyr residues in teicoplanin-type GPA biosynthesis by non-heme iron oxygenases223 as well as the hydroxylation of several hydrophobic residues by one cytochrome P450 in skyllamycin biosynthesis.224–226
In the case of salinamide A, the phenol moiety of the Hpg residue of desmethylsalinamide C is involved in oxidation/cyclisation with the seven-carbon moiety derived from (2E,4E)-4-methylhexa-2,4-dienoyl coenzyme A to afford salinamide A.133,230 The exact mechanistic details have yet to be determined, however it has been postulated that the cyclisation/oxidation proceeds either via initial attack of an epoxide by the Hpg phenol moiety that is followed by dehydration and epoxidation, or via an oxygenase-mediated [2 + 2] cycloaddition/dehydration cascade.230 Formation of salinamides C and E, bearing a methylated Hpg phenol have been described as dead-end products in salinamide biosynthesis, where methylation via AdoMet prevents their final conversion into salinamide A.133
The glycosylation of glycopeptide antibiotics has been extensively characterised as most GPAs bear glycosylations; one important exception is the sulfated compound A47934.157 Glycosylation have been identified to occur on phenylglycine residues Hpg4 and Dpg7 as well as the β-hydroxytyrosine residue at position 6. Whilst the glycosylation at position 6 contains a range of sugar moieties, initial modification of Hpg4 is restricted to glucose or glucosamine and Dpg7 displays a mannose group (teicoplanin, ristocetin, A40926).158,227,228,231,232 The glycosyl transferases responsible for the addition of glycosyl groups have been identified and investigated for chloroerymomycin (GftA, GftB, GftC) and vancomycin (GftD, GftE).234–237 GftB and GftE have been shown to be responsible for the glycosylation of Hpg4 using UDP-glucose. GftE in particular has shown potential for the acceptance of alternate aglycon substrates,234 alternative sugar units235,238 and has been shown to be able to glycosylate A47934 at Hpg4in vivo.235 GftC and GftD were shown to be responsible for further glycosylation of the mono-glycosylated aglycones by 4-epi-vancosamine; GftD was thus able to use an unnatural glycosyl donor and could also catalyse the modification of a teicoplanin mono-glycosylated aglycon.234 Further modification of unnatural mono-glycosylated aglycones produced by GftE was enabled by GftD.238 GftA has been shown to be responsible for the glycosylation of β-hydroxytyrosine residue at position 6, with a substrate preference for the singly glycosylated products of GftB/GftE.236 GftA, GftB and GftD have also been structurally characterised.239–241
The teicoplanin glycosyltransferases tGftA and tGftB can both utilise UDP-N-acetylglucosamine as a donor, with tGftB also able to utilise UDP-glucose and UDP-glucosamine.231,232 The deletion of ORF 9 (with similarity to GftB) from the A40926 producer strain has allowed the isolation of an aglycon with only the mannosyl group present on Dpg7;242 the mannosyl transferase is implicated as ORF 20 (tcp15 in teicoplanin biosynthesis).158,231 Further modifications of the initial glucose243 and second glycosyl moiety attached to Hpg4 have been demonstrated in GPA biosyntheses,244 as well as being exploited to generate novel compounds.245
Glycosyltransferases have been employed in the synthesis of GPAs in vitro: completion of the synthesis of vancomycin and modified forms of vancomycin from synthetic aglycones have exploited the glycosyltransferases GftE and GftD to add the sugar moieties, with GftE first adding a glucose unit to Hpg4.246 The tolerance of these enzymes for aglycon modifications was assessed, revealing that modification of the carboxylic acid moiety was poorly tolerated by GftE, with N-terminal modifications causing a moderate reduction in activity; GftD was less sensitive for these modifications.246 The alteration of the vancomycin backbone amide group of residue Hpg4 to thioamide moieties was more efficiently tolerated by these enzymes.247 The introduction of alkoxyamine-bearing sugars by GftE has also been demonstrated in vitro, allowing neoglycosylation to then produce novel disaccharide substituted aglycones,248 whilst azide-containing sugars introduced using GftE have been used together with a library of alkynes to create a range of modified GPA aglycones using chemoselective Huisgen 1,3-dipolar cycloaddition reactions.249
6 Significance, importance and occurrence in medicinal chemistry
The amino acid 4-hydroxyphenylglycine (Hpg) is a valuable pharmaceutical building block used for production of semisynthetic β-lactam antibiotics, such as the penicillins, cephalosporins, i.e. amoxicillin and cefadroxil.257–259Scheme 16 shows the use of penicillin acylase as a catalyst for the synthesis of β-lactam antibiotics ampicillin, amoxicillin, and cephalexin starting from D-hydroxyphenylglycine amide (D-Hpga) and D-phenylglycine amide (D-Phga) together with 6-aminopenicillanic (6-APA) and 7-aminodesacetoxycephalosporanic (7-ADCA) acid as substrates.260 In addition, D-4-hydroxyphenylglycine (Hpg) has also efficiently been used for the synthesis of polyxamic acid, which is an essential moiety of the antifungal antibiotics polyoxins.261 | ||
| Scheme 16 Synthesis of β-lactam antibiotics ampicillin, amoxicillin and cephalexin starting from D-hydroxyphenylglycine amide (D-Hpga) and D-phenylglycine amide (D-Phga) together with 6-aminopenicillanic (6-APA) and 7-aminodesacetoxycephalosporanic (7-ADCA) acids. This reaction is performed by the mediation of penicillin G acylase as a catalyst. | ||
L-3,5-Dpg was found to be a specific agonist of group I metabotropic glutamate receptors mGluRs262 with significantly low EC50 values.263,264 It has the potency of phosphoinositide hydrolysis stimulation in neonatal (EC50 ∼ 7 μM) and adult (EC50 ∼ 28 μM) rat hippocampus and cerebellum in a dose-dependent manner.264L-3,5-Dpg application causes an agonist response elicitation in non-neuronal cells and Xenopus oocytes expressing recombinant mGlu1a (EC50 ∼ 6–10 μM) or mGlu5 (EC50 ∼ 2 μM) receptor subtypes.262,265–267 The stimulated cAMP levels in the adult can be inhibited by L-3,5-Dpg, while the application of such an amino acid can cause enhancement in the basal cAMP levels in the neonatal brain tissue.264
Interestingly, rac-3,5-Dpg has the ability to stimulate phospholipase C (PLC) but not phospholipase D (PLD), so it works as an antagonist of mGluRs linked to PLD in adult hippocampus but as an agonist in the neonatal brain or astrocyte cultures.268,269 The usage of L-3,5-Dpg as a metabotropic glutamate receptors-group I-agonist induces an elevation in the intracellular calcium concentration [Ca2+]i which plays an important role in controlling neuronal development and necrosis.270 mGluRs activated by L-3,5-Dpg were found to inhibit voltage dependent Ca2+ channels, and to regulate multiple subtypes of the Ca2+ channels including L-, N- and P subtypes, but not the resistant R-subtype.271
The affinity of the high affinity state of D2 receptors for dopamine in striatum was significantly decreased upon the application of L-3,5-Dpg,272 and this application causes an inhibition in the basal and stimulated dopamine release.273 Furthermore, the inhibition of [3H]-acetylcholine ([3H] Ach) release by L-AP4 (an agonist from group III mGluRs) was reversed upon treatment with L-3,5-Dpg in retinal amacrine cells, even the later did not show a significant effect on the ([3H] Ach) release.274
L-3,5-Dpg was reported to enhance or decrease excitatory postsynaptic potentials (EPSP's) in a dose dependent manner, and to have the ability of induction long-term depression (LTD) and long-term potentiation (LTP), which are thought to underpin learning and memory formation.275–279 Some other studies suggested L-3,5-Dpg as a therapeutic agent for epileptogenesis and neuronal injury,280 hypoxia,281 regulation of intestinal motility and secretion,282 antidepression283,284 and in cardiovascular system.285,286
7 Conclusions
Phenylglycines comprise an important and structurally unique class of non-proteinogenic amino acids that occur in natural products but also find broad application in synthetic compounds. The main representatives are the amino acids phenylglycine (Phg), 4-hydroxyphenylglycine (Hpg), and 3,5-dihydroxyphenylglycine (Dpg), which are building blocks for non-ribosomal peptide biosynthesis assembly lines of highly important and clinically used natural products. This includes linear and cyclic peptides, e.g. feglymycin, pristinamycin, arylomycin, CDA, enduracidin, ramoplanin as well as the complete group of glycopeptide antibiotics, like vancomycin and teicoplanin. The unique combination of properties in phenylglycines – restricted side chain flexibility in comparison to the proteinogenic amino acids Tyr or Phe combined with the ability for hydrogen bonding as well as hydrophobic interactions such as π-stacking – makes these amino acids crucial structural and functional elements in these groups of natural products. Interestingly, nature provides two independent solutions for the assembly of phenylglycine-type amino acids: the chorismate pathway (Phg, Hpg) and acetyl-CoA mediated by a chalcone synthase (Dpg). Peptides containing phenylglycine residues assembled on non-ribosomal peptide synthetases undergo a variety of modifications by tailoring enzymes, including those that perform alkylation, oxidation, sulfation, halogenation and glycosylation. Thus, phenylglycines constitute central building blocks of NRPS biosynthesis machineries and futures discoveries of novel peptide natural products containing these amino acids are expected.8 Acknowledgements
This work was supported by the Cluster of Excellence “Unifying Concepts in Catalysis UniCat” funded by the DFG and coordinated by the TU Berlin, and by the StrepSynth project (FP7-KBBE-2013-7-613887) within the 7th Framework program of the European Union. M.J.C. is grateful to the Deutsche Forschungsgemeinschaft (Emmy-Noether Program, CR 392/1-1) for financial support. R. Al Toma gratefully acknowledges support from the EMECW (Erasmus Mundus External Cooperation Window) for a fellowship.9 Notes and references
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC
.
- L. Johansson, G. Gafvelin and E. S. Arner, Biochim. Biophys. Acta, 2005, 30, 1–13 CrossRef PubMed
- G. Srinivasan, C. M. James and J. A. Krzycki, Science, 2002, 296, 1459–1462 CrossRef CAS PubMed
- B. Hao, W. Gong, T. K. Ferguson, C. M. James, J. A. Krzycki and M. K. Chan, Science, 2002, 296, 1462–1466 CrossRef CAS PubMed
- R. S. Al Toma, A. Kuthning, M. P. Exner, A. Denisiuk, J. Ziegler, N. Budisa and R. D. Süssmuth, ChemBioChem, 2014, 11, 201402558 Search PubMed
- Y. Zhang, P. V. Baranov, J. F. Atkins and V. N. Gladyshev, J. Biol. Chem., 2005, 280, 20740–20751 CrossRef CAS PubMed
- F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. E. O'Connor and C. T. Walsh, Nature, 2005, 436, 1191–1194 CrossRef CAS PubMed
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS PubMed
- J. He, N. Magarvey, M. Piraee and L. C. Vining, Microbiology, 2001, 147, 2817–2829 CAS
- J. M. Neary, A. Powell, L. Gordon, C. Milne, F. Flett, B. Wilkinson, C. P. Smith and J. Micklefield, Microbiology, 2007, 153, 768–776 CrossRef CAS PubMed
- M. Strieker, E. M. Nolan, C. T. Walsh and M. A. Marahiel, J. Am. Chem. Soc., 2009, 131, 13523–13530 CrossRef CAS PubMed
-
M. J. Cryle, C. Brieke and K. Haslinger, in Amino Acids, Peptides and Proteins, The Royal Society of Chemistry, 2014, vol. 38, pp. 1–36 Search PubMed
- H. Chen and C. T. Walsh, Chem. Biol., 2001, 8, 301–312 CrossRef CAS
- M. J. Cryle, A. Meinhart and I. Schlichting, J. Biol.
Chem., 2010, 285, 24562–24574 CrossRef CAS PubMed
- O. Puk, P. Huber, D. Bischoff, J. Recktenwald, G. Jung, R. D. Süssmuth, K. H. van Pee, W. Wohlleben and S. Pelzer, Chem. Biol., 2002, 9, 225–235 CrossRef CAS
- V. Blanc, P. Gil, N. Bamas-Jacques, S. Lorenzon, M. Zagorec, J. Schleuniger, D. Bisch, F. Blanche, L. Debussche, J. Crouzet and D. Thibaut, Mol. Microbiol., 1997, 23, 191–202 CAS
- Y. Mast, T. Weber, M. Golz, R. Ort-Winklbauer, A. Gondran, W. Wohlleben and E. Schinko, Microb. Biotechnol., 2011, 4, 192–206 CrossRef CAS PubMed
- D. Seebach, A. K. Beck, S. Capone, G. Deniau, U. Groselj and E. Zass, Synthesis, 2009, 1–32 CrossRef CAS PubMed
- L. Liu, D. W. Bearden and K. S. Rein, J. Nat. Prod., 2011, 74, 1535–1538 CrossRef CAS PubMed
- H. S. Okumura, B. Philmus, C. Portmann and T. K. Hemscheidt, J. Nat. Prod., 2009, 72, 172–176 CrossRef CAS PubMed
- C. Keller-Juslén, M. Kuhn, H. R. Loosli, T. J. Petcher, H. P. Weber and A. von Wartburg, Tetrahedron Lett., 1976, 17, 4147–4150 CrossRef
- M. Sato, T. Tatsuno and H. Matsuo, Yakugaku Zasshi, 1970, 90, 1160–1163 CAS
- K. C. Nicolaou, C. N. Boddy, S. Brase and N. Winssinger, Angew. Chem., Int. Ed. Engl., 1999, 38, 2096–2152 CrossRef
- M. A. Elsawy, C. Hewage and B. Walker, J. Pept. Sci., 2012, 18, 302–311 CrossRef CAS PubMed
- J. Nam, D. Shin, Y. Rew and D. L. Boger, J. Am. Chem. Soc., 2007, 129, 8747–8755 CrossRef CAS PubMed
- A. Hänchen, S. Rausch, B. Landmann, L. Toti, A. Nusser and R. D. Süssmuth, ChemBioChem, 2013, 14, 625–632 CrossRef PubMed
- H. Vanderhaeghe, G. Janssen and F. Compernolle, Tetrahedron Lett., 1971, 12, 2687–2688 CrossRef
- C. Cocito, Microbiol. Rev., 1979, 43, 145–192 CAS
- J. C. Barriere, D. H. Bouanchaud, J. F. Desnottes and J. M. Paris, Expert Opin. Invest. Drugs, 1994, 3, 115–131 CrossRef CAS
- P. Crooy and R. De Neys, J. Antibiot., 1972, 25, 371–372 CrossRef CAS
- C. Cocito, Antibiotics, 1983, 6, 296–332 CAS
- C. Cocito, M. Di Giambattista, E. Nyssen and P. Vannuffel, J. Antimicrob. Chemother., 1997, 39, 7–13 CrossRef CAS PubMed
- D. Vazquez, J. Gen. Microbiol., 1966, 42, 93–106 CrossRef CAS PubMed
- Y. Mast and W. Wohlleben, Int. J. Med. Microbiol., 2014, 304, 44–50 CrossRef CAS PubMed
- T. A. Mukhtar and G. D. Wright, Chem. Rev., 2005, 105, 529–542 CrossRef CAS PubMed
- Y. A. Chabbert and J. F. Acar, Ann. Inst. Pasteur, 1964, 107, 777–790 CAS
-
D. Vazquez, in Mechanism of Action of Antimicrobial and Antitumor Agents, ed. J. Corcoran, F. Hahn, J. F. Snell and K. L. Arora, Springer Berlin Heidelberg, 1975, vol. 3, pp. 521–534 Search PubMed
- J. Preudhomme, A. Belloc, Y. Charpentie and P. Tarridec, C. R. Hebd. Seances Acad. Sci., 1965, 260, 1309–1312 CAS
- J. Preud'homme, P. Tarridec and A. Belloc, Bull. Soc. Chim. Fr., 1968, 2, 585–591 Search PubMed
- G. P. G. Jolies, J. Robert, B. Terlain and J. P. Thomas, Bull. Soc. Chim. Fr., 1965, 8, 2252–2259 Search PubMed
- A. A. Kiryushkin, V. M. Burikov and B. V. Rozynov, Tetrahedron Lett., 1967, 2675–2678 CrossRef CAS
- B. V. Rozynov, V. M. Burikov, I. A. Bogdanova and A. A. Kiryushkin, Zh. Obshch. Khim., 1969, 39, 891–905 CAS
- J. P. Declercq, G. Germain, M. Van Meerssche, S. E. Hull and M. J. Irwin, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3644–3648 CrossRef
- J. D. Yates and P. J. Schaible, Nature, 1962, 194, 183–184 CrossRef CAS
- H. Eyssen and P. de Somer, J. Exp. Med., 1963, 117, 127–138 CrossRef CAS
- F. M. Aarestrup, A. M. Seyfarth, H.-D. Emborg, K. Pedersen, R. S. Hendriksen and F. Bager, Antimicrob. Agents Chemother., 2001, 45, 2054–2059 CrossRef CAS PubMed
- F. M. Aarestrup, H. Kruse, E. Tast, A. M. Hammerum and L. B. Jensen, Microb. Drug Resist., 2000, 6, 63–70 CrossRef CAS
- G. M. Elipoulos, Clin. Infect. Dis., 2003, 36, 473–481 CrossRef CAS PubMed
- P. Weber, Pathol. Biol., 2001, 49, 840–845 CrossRef CAS
- R. Leclercq, C. J. Soussy, P. Weber, N. Moniot-Ville and C. Dib, Pathol. Biol., 2003, 51, 400–404 CrossRef CAS
-
Martindale: The Complete Drug Reference, ed. S. C. Sweetman, Pharmaceutical Press, 2005 Search PubMed
- D. Nathwani, P. G. Davey and C. A. Marwick, Clin. Evid., 2010, 0922 Search PubMed
- T. Teshima, M. Nishikawa, I. Kubota, T. Shiba, Y. Iwai and S. Ōmura, Tetrahedron Lett., 1988, 29, 1963–1966 CrossRef CAS
- L. Brandi, S. Maffioli, S. Donadio, F. Quaglia, M. Sette, P. Milón, C. O. Gualerzi and A. Fabbretti, FEBS Lett., 2012, 586, 3373–3378 CrossRef CAS PubMed
- S. Omura, Y. Iwai, A. Hirano, J. Awaya, Y. Suzuki and K. Matsumoto, Agric. Biol. Chem., 1977, 41, 1827–1828 CrossRef CAS
- L. Brandi, A. Fabbretti, M. Di Stefano, A. Lazzarini, M. Abbondi and C. O. Gualerzi, RNA, 2006, 12, 1262–1270 CrossRef CAS PubMed
- D. Bulkley, L. Brandi, Y. S. Polikanov, A. Fabbretti, M. O'Connor, C. O. Gualerzi and T. A. Steitz, Cell Rep., 2014, 6, 357–365 CrossRef CAS PubMed
- T. Kawahara, M. Itoh, M. Izumikawa, J. Hashimoto, N. Sakata, T. Tsuchida and K. Shin-ya, J. Antibiot., 2015, 68, 67–70 CrossRef CAS PubMed
- T. Yamamoto, K. Kojiri, H. Morishima, H. Naganawa, T. Aoyagi and H. Umezawa, J. Antibiot., 1978, 31, 483–484 CrossRef CAS
- T. Aoyagi, T. Yamamoto, K. Kojiri, F. Kojima, M. Hamada, T. Takeuchi and H. Umezawa, J. Antibiot., 1978, 31, 244–246 CrossRef CAS
- P. Friis and A. Kjaer, Acta Chem. Scand., 1963, 17, 2391–2396 CrossRef CAS PubMed
- M. Hashimoto, T. Komori and T. Kamiya, J. Antibiot., 1976, 29, 890–901 CrossRef CAS
- H. Aoki, H. Sakai, M. Kohsaka, T. Konomi and J. Hosoda, J. Antibiot., 1976, 29, 492–500 CrossRef CAS
- J. Hosoda, T. Konomi, N. Tani, H. Aoki and H. Imanaka, Agric. Biol. Chem., 1977, 41, 2013–2020 CrossRef CAS
- G. M. Salituro and C. A. Townsend, J. Am. Chem. Soc., 1990, 112, 760–770 CrossRef CAS
- C. A. Townsend and B. A. Wilson, J. Am. Chem. Soc., 1988, 110, 3320–3321 CrossRef CAS
- N. M. Gaudelli and C. A. Townsend, Nat. Chem. Biol., 2014, 10, 251–258 CrossRef CAS PubMed
- L. J. Nisbet, R. J. Mehta, Y. Oh, C. H. Pan, C. G. Phelen, M. J. Polansky, M. C. Shearer, A. J. Giovenella and S. F. Grappel, J. Antibiot., 1985, 38, 133–138 CrossRef CAS
- J. A. Chan, E. A. Shultis, J. J. Dingerdissen, C. W. DeBrosse, G. D. Roberts and K. M. Snader, J. Antibiot., 1985, 38, 139–144 CrossRef CAS
- N. Katayama, Y. Nozaki, K. Okonogi, H. Ono, S. Harada and H. Okazaki, J. Antibiot., 1985, 38, 1117–1127 CrossRef CAS
- T. Hida, S. Tsubotani, N. Katayama, H. Okazaki and S. Harada, J. Antibiot., 1985, 38, 1128–1140 CrossRef CAS
- L. Vertesy, W. Aretz, M. Knauf, A. Markus, M. Vogel and J. Wink, J. Antibiot., 1999, 52, 374–382 CrossRef CAS
- F. Dettner, A. Hänchen, D. Schols, L. Toti, A. Nusser and R. D. Süssmuth, Angew. Chem., Int. Ed. Engl., 2009, 48, 1856–1861 CrossRef CAS PubMed
- G. Bunkoczi, L. Vertesy and G. M. Sheldrick, Angew. Chem., Int. Ed. Engl., 2005, 44, 1340–1342 CrossRef CAS PubMed
- G. Férir, A. Hänchen, K. O. François, B. Hoorelbeke, D. Huskens, F. Dettner, R. D. Süssmuth and D. Schols, Virology, 2012, 433, 308–319 CrossRef PubMed
- B. Cavalleri, H. Pagani, G. Volpe, E. Selva and F. Parenti, J. Antibiot., 1984, 37, 309–317 CrossRef CAS
- R. Ciabatti, J. K. Kettenring, G. Winters, G. Tuan, L. Zerilli and B. Cavalleri, J. Antibiot., 1989, 42, 254–267 CrossRef CAS
- R. Pallanza, M. Berti, R. Scotti, E. Randisi and V. Arioli, J. Antibiot., 1984, 37, 318–324 CrossRef CAS
- J. K. Kettenring, R. Ciabatti, G. Winters, G. Tamborini and B. Cavalleri, J. Antibiot., 1989, 42, 268–275 CrossRef CAS
- S. Walker, L. Chen, Y. Hu, Y. Rew, D. Shin and D. L. Boger, Chem. Rev., 2005, 105, 449–476 CrossRef CAS PubMed
- D. G. McCafferty, P. Cudic, B. A. Frankel, S. Barkallah, R. G. Kruger and W. Li, Biopolymers, 2002, 66, 261–284 CrossRef CAS PubMed
- P. Cudic, J. K. Kranz, D. C. Behenna, R. G. Kruger, H. Tadesse, A. J. Wand, Y. I. Veklich, J. W. Weisel and D. G. McCafferty, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7384–7389 CrossRef CAS PubMed
- N. Bionda, J. P. Pitteloud and P. Cudic, Future Med. Chem., 2013, 5, 1311–1330 CrossRef CAS PubMed
- P. Fulco and R. P. Wenzel, Expert Rev. Anti-Infect. Ther., 2006, 4, 939–945 CrossRef CAS PubMed
- D. K. Farver, D. D. Hedge and S. C. Lee, Ann. Pharmacother., 2005, 39, 863–868 CrossRef CAS PubMed
- N. J. Skelton, M. M. Harding, R. J. Mortishire-Smith, S. K. Rahman, D. H. Williams, M. J. Rance and J. C. Ruddock, J. Am. Chem. Soc., 1991, 113, 7522–7530 CrossRef CAS
- M. Kurz and W. Guba, Biochemistry, 1996, 35, 12570–12575 CrossRef CAS PubMed
- W. Jiang, J. Wanner, R. J. Lee, P. Y. Bounaud and D. L. Boger, J. Am. Chem. Soc., 2002, 124, 5288–5290 CrossRef CAS PubMed
- F. Castiglione, A. Marazzi, M. Meli and G. Colombo, Magn. Reson. Chem., 2005, 43, 603–610 CrossRef CAS PubMed
- E. Higashide, K. Hatano, M. Shibata and K. Nakazawa, J. Antibiot., 1968, 21, 126–137 CrossRef CAS
- E. Meyers, F. L. Weisenborn, F. E. Pansy, D. S. Slusarchyk, M. H. Von Saltza, M. L. Rathnum and W. L. Parker, J. Antibiot., 1970, 23, 502–507 CrossRef CAS
- P. E. Linnett and J. L. Strominger, Antimicrob. Agents Chemother., 1973, 4, 231–236 CrossRef CAS
- M. Hori, N. Sugita and M. Miyazaki, Chem. Pharm. Bull., 1973, 21, 1171–1174 CrossRef CAS
- M. Hori, H. Iwasaki, S. Horii, I. Yoshida and T. Hongo, Chem. Pharm. Bull., 1973, 21, 1175–1183 CrossRef CAS
- H. Iwasaki, S. Horii, M. Asai, K. Mizuno, J. Ueyanagi and A. Miyake, Chem. Pharm. Bull., 1973, 21, 1184–1191 CrossRef CAS
- S. Tsuji, S. Kusumoto and T. Shiba, Chem. Lett., 1975, 1281–1284 CrossRef CAS
- M. Peromet, E. Schoutens and E. Yourassowsky, Chemotherapy, 1973, 19, 53–61 CrossRef CAS PubMed
- S. Goto, S. Kuwahara, N. Okubo and H. Zenyoji, J. Antibiot., 1968, 21, 119–125 CrossRef CAS
- K. Tsuchiya, M. Kondo, T. Oishi and T. Yamazaki, J. Antibiot., 1968, 21, 147–153 CrossRef CAS
- M. Kawakami, Y. Nagai, T. Fuji and S. Mitsuhashi, J. Antibiot., 1971, 24, 583–586 CrossRef CAS
- E. Yourassowsky and R. Monsieur, Chemotherapy, 1972, 17, 182–187 CrossRef CAS PubMed
- H. Komatsuzawa, J. Suzuki, M. Sugai, Y. Miyake and H. Suginaka, J. Antimicrob. Chemother., 1994, 33, 1155–1163 CrossRef CAS PubMed
- X. Yin, Y. Chen, L. Zhang, Y. Wang and T. M. Zabriskie, J. Nat. Prod., 2010, 73, 583–589 CrossRef CAS PubMed
- J.-M. Du, L. Li and J.-M. Du, Siliao Yanjiu, 2013, 18–20 CAS
- W. E. Brown, V. Seinerova, W. M. Chan, A. I. Laskin, P. Linnett and J. L. Strominger, Ann. N. Y. Acad. Sci., 1974, 235, 399–405 CrossRef CAS PubMed
- E. Meyers, F. L. Weisenborn, F. E. Pansy, D. S. Slusarchyk, M. H. Von Saltza, M. L. Rathnum and W. L. Parker, J. Antibiot., 1970, 23, 502–507 CrossRef CAS
-
B. A. M. Rudd, Ph.D. thesis, University of East Anglia, Norwich, U.K., 1978
- J. H. Lakey, E. J. Lea, B. A. Rudd, H. M. Wright and D. A. Hopwood, J. Gen. Microbiol., 1983, 129, 3565–3573 CAS
- D. A. Hopwood and H. M. Wright, J. Gen. Microbiol., 1983, 129, 3575–3579 CAS
- C. Kempter, D. Kaiser, S. Haag, G. Nicholson, V. Gnau, T. Walk, K. H. Gierling, H. Decker, H. Zähner, G. Jung and J. W. Metzger, Angew. Chem., Int. Ed. Engl., 1997, 36, 498–501 CrossRef CAS PubMed
- M. Strieker, F. Kopp, C. Mahlert, L.-O. Essen and M. A. Marahiel, ACS Chem. Biol., 2007, 2, 187–196 CrossRef CAS PubMed
- Z. Hojati, C. Milne, B. Harvey, L. Gordon, M. Borg, F. Flett, B. Wilkinson, P. J. Sidebottom, B. A. Rudd, M. A. Hayes, C. P. Smith and J. Micklefield, Chem. Biol., 2002, 9, 1175–1187 CrossRef CAS
- D. Jung, A. Rozek, M. Okon and R. E. W. Hancock, Chem. Biol., 2004, 11, 949–957 CrossRef CAS PubMed
- L. Vertesy, E. Ehlers, H. Kogler, M. Kurz, J. Meiwes, G. Seibert, M. Vogel and P. Hammann, J. Antibiot., 2000, 53, 816–827 CrossRef CAS
- A. Holtzel, D. G. Schmid, G. J. Nicholson, S. Stevanovic, J. Schimana, K. Gebhardt, H.-P. Fiedler and G. Jung, J. Antibiot., 2002, 55, 571–577 CrossRef CAS
- J. Schimana, K. Gebhardt, A. Holtzel, D. G. Schmid, R. Süssmuth, J. Muller, R. Pukall and H.-P. Fiedler, J. Antibiot., 2002, 55, 565–570 CrossRef CAS
- P. Kulanthaivel, A. J. Kreuzman, M. A. Strege, M. D. Belvo, T. A. Smitka, M. Clemens, J. R. Swartling, K. L. Minton, F. Zheng, E. L. Angleton, D. Mullen, L. N. Jungheim, V. J. Klimkowski, T. I. Nicas, R. C. Thompson and S. B. Peng, J. Biol. Chem., 2004, 279, 36250–36258 CrossRef CAS PubMed
- M. Paetzel, J. J. Goodall, M. Kania, R. E. Dalbey and M. G. P. Page, J. Biol. Chem., 2004, 279, 30781–30790 CrossRef CAS PubMed
- T. C. Roberts, P. A. Smith, R. T. Cirz and F. E. Romesberg, J. Am. Chem. Soc., 2007, 129, 15830–15838 CrossRef CAS PubMed
- J. Liu, C. Luo, P. A. Smith, J. K. Chin, M. G. P. Page, M. Paetzel and F. E. Romesberg, J. Am. Chem. Soc., 2011, 133, 17869–17877 CrossRef CAS PubMed
- T. C. Roberts, P. A. Smith and F. E. Romesberg, J. Nat. Prod., 2011, 74, 956–961 CrossRef CAS PubMed
- P. A. Smith, T. C. Roberts and F. E. Romesberg, Chem. Biol., 2010, 17, 1223–1231 CrossRef CAS PubMed
- P. A. Smith, M. E. Powers, T. C. Roberts and F. E. Romesberg, Antimicrob. Agents Chemother., 2011, 55, 1130–1134 CrossRef CAS PubMed
- P. A. Smith and F. E. Romesberg, Antimicrob. Agents Chemother., 2012, 56, 5054–5060 CrossRef CAS PubMed
- M. Rao, W. Wei, M. Ge, D. Chen and X. Sheng, Nat. Prod. Res., 2013, 27, 2190–2195 CrossRef CAS PubMed
- B. Hellmark, M. Unemo, A. Nilsdotter-Augustinsson and B. Soderquist, Clin. Microbiol. Infect., 2009, 15, 238–244 CrossRef CAS PubMed
- Y. K. Lam, D. L. Williams Jr, J. M. Sigmund, M. Sanchez, O. Genilloud, Y. L. Kong, S. Stevens-Miles, L. Huang and G. M. Garrity, J. Antibiot., 1992, 45, 1709–1716 CrossRef CAS
- D. Zink, O. D. Hensens, Y. K. Lam, R. Reamer and J. M. Liesch, J. Antibiot., 1992, 45, 1717–1722 CrossRef CAS
- X. C. Cheng, T. Kihara, H. Kusakabe, R. Fang, Z. Ni, Y. Shen, K. Ko, I. Yamaguchi and K. Isono, Agric. Biol. Chem., 1987, 51, 279–281 CrossRef CAS
- S. Kim, M. Ubukata, K. Kobayashi and K. Isono, Tetrahedron Lett., 1992, 33, 2561–2564 CrossRef CAS
- S.-K. Kim, M. Ubukata and K. Isono, J. Microbiol. Biotechnol., 2003, 13, 998–1000 CAS
- J. A. Trischman, D. M. Tapiolas, P. R. Jensen, R. Dwight, W. Fenical, T. C. McKee, C. M. Ireland, T. J. Stout and J. Clardy, J. Am. Chem. Soc., 1994, 116, 757–758 CrossRef CAS
- B. S. Moore, J. A. Trischman, D. Seng, D. Kho, P. R. Jensen and W. Fenical, J. Org. Chem., 1999, 64, 1145–1150 CrossRef CAS
- H. M. Hassan, D. Degen, K. H. Jang, R. H. Ebright and W. Fenical, J. Antibiot., 2015, 68, 206–209 CrossRef CAS PubMed
- L. Tan and D. Ma, Angew. Chem., Int. Ed. Engl., 2008, 47, 3614–3617 CrossRef CAS PubMed
- S. Miao, M. R. Anstee, K. LaMarco, J. Matthew, L. H. T. Huang and M. M. Brasseur, J. Nat. Prod., 1997, 60, 858–861 CrossRef CAS
- D. Degen, Y. Feng, Y. Zhang, K. Y. Ebright, Y. W. Ebright, M. Gigliotti, H. Vahedian-Movahed, S. Mandal, M. Talaue, N. Connell, E. Arnold, W. Fenical and R. H. Ebright, eLife, 2014, 3 DOI:10.7554/eLife.02451
- B. K. Hubbard and C. T. Walsh, Angew. Chem., Int. Ed. Engl., 2003, 42, 730–765 CrossRef CAS PubMed
- M. H. McCormick, J. M. McGuire, G. E. Pittenger, R. C. Pittenger and W. M. Stark, Antibiot. Annu., 1955, 3, 606–611 Search PubMed
- G. M. Sheldrick, E. Paulus, L. Vertesy and F. Hahn, Acta Crystallogr., Sect. B: Struct. Sci., 1995, 51, 89–98 CrossRef
- A. M. van Wageningen, P. N. Kirkpatrick, D. H. Williams, B. R. Harris, J. K. Kershaw, N. J. Lennard, M. Jones, S. J. Jones and P. J. Solenberg, Chem. Biol., 1998, 5, 155–162 CrossRef CAS
- N. N. Lomakina, M. S. Iurina, M. F. Lavrova and M. G. Brazhnikova, Antibiotiki, 1961, 6, 609–618 CAS
- S. L. Heald, L. Mueller and P. W. Jeffs, J. Antibiot., 1987, 40, 630–645 CrossRef CAS
- W. E. Grundy, A. C. Sinclair, R. J. Theriault, A. W. Goldstein, C. J. Rickher, H. B. Warren Jr, T. J. Oliver and J. C. Sylvester, Antibiot. Annu., 1956, 687–692 Search PubMed
- J. E. Philip, J. R. Schenck and M. P. Hargie, Antibiot. Annu., 1956, 699–705 Search PubMed
- M. R. Bardone, M. Paternoster and C. Coronelli, J. Antibiot., 1978, 31, 170–177 CrossRef CAS
- H. Seto, T. Fujioka, K. Furihata, I. Kaneko and S. Takahashi, Tetrahedron Lett., 1989, 30, 4987–4990 CrossRef CAS
- K. Matsuzaki, H. Ikeda, T. Ogino, A. Matsumoto, H. B. Woodruff, H. Tanaka and S. Omura, Chloropeptins I and II, novel inhibitors against gp120-CD4 binding from Streptomyces sp, J. Antibiot., 1994, 47(10), 1173–1174 CrossRef CAS
- S. B. Singh, H. Jayasuriya, G. M. Salituro, D. L. Zink, A. Shafiee, B. Heimbuch, K. C. Silverman, R. B. Lingham, O. Genilloud, A. Teran, D. Vilella, P. Felock and D. Hazuda, J. Nat. Prod., 2001, 64, 874–882 CrossRef CAS PubMed
- M. N. Preobrazhenskaya and E. N. Olsufyeva, Antiviral Res., 2006, 71, 227–236 CrossRef CAS PubMed
- N. Naruse, O. Tenmyo, S. Kobaru, M. Hatori, K. Tomita, Y. Hamagishi and T. Oki, J. Antibiot., 1993, 46, 1804–1811 CrossRef CAS
- D. H. Williams and B. Bardsley, Angew. Chem., Int. Ed. Engl., 1999, 38, 1172–1193 CrossRef
- R. D. Süssmuth and W. Wohlleben, Appl. Microbiol. Biotechnol., 2004, 63, 344–350 CrossRef PubMed
- S. Pelzer, R. Süssmuth, D. Heckmann, J. Recktenwald, P. Huber, G. Jung and W. Wohlleben, Antimicrob. Agents Chemother., 1999, 43, 1565–1573 CAS
- M. Sosio, A. Bianchi, E. Bossi and S. Donadio, Antonie Van Leeuwenhoek, 2000, 78, 379–384 CrossRef CAS
- H. T. Chiu, B. K. Hubbard, A. N. Shah, J. Eide, R. A. Fredenburg, C. T. Walsh and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8548–8553 CrossRef CAS PubMed
- J. Pootoolal, M. G. Thomas, C. G. Marshall, J. M. Neu, B. K. Hubbard, C. T. Walsh and G. D. Wright, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8962–8967 CrossRef CAS PubMed
- M. Sosio, S. Stinchi, F. Beltrametti, A. Lazzarini and S. Donadio, Chem. Biol., 2003, 10, 541–549 CrossRef CAS
- Y. J. Mast, W. Wohlleben and E. Schinko, J. Biotechnol., 2011, 155, 63–67 CrossRef CAS PubMed
- L. Li, J. P. Bannantine, Q. Zhang, A. Amonsin, B. J. May, D. Alt, N. Banerji, S. Kanjilal and V. Kapur, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12344–12349 CrossRef CAS PubMed
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403–410 CrossRef CAS
- P. F. Widboom, E. N. Fielding, Y. Liu and S. D. Bruner, Nature, 2007, 447, 342–345 CrossRef CAS PubMed
- O. W. Choroba, D. H. Williams and J. B. Spencer, J. Am. Chem. Soc., 2000, 122, 5389–5390 CrossRef CAS
- B. K. Hubbard, M. G. Thomas and C. T. Walsh, Chem. Biol., 2000, 7, 931–942 CrossRef CAS
- T. L. Li, O. W. Choroba, H. Hong, D. H. Williams and J. B. Spencer, Chem. Commun., 2001, 2156–2157 RSC
- J. Recktenwald, R. Shawky, O. Puk, F. Pfennig, U. Keller, W. Wohlleben and S. Pelzer, Microbiology, 2002, 148, 1105–1118 CAS
- M. Gunsior, S. D. Breazeale, A. J. Lind, J. Ravel, J. W. Janc and C. A. Townsend, Chem. Biol., 2004, 11, 927–938 CrossRef CAS PubMed
- S. J. Hammond, M. P. Williamson, D. H. Williams, L. D. Boeck and G. G. Marconi, J. Chem. Soc., Chem. Commun., 1982, 344–346 RSC
- S. J. Hammond, D. H. Williams and R. V. Nielsen, J. Chem. Soc., Chem. Commun., 1983, 116–117 RSC
- S. K. Chung, P. Taylor, Y. K. Oh, C. DeBrosse and P. W. Jeffs, J. Antibiot., 1986, 39, 642–651 CrossRef CAS
- T. I. Nicas and R. D. G. Cooper, Drugs Pharm. Sci., 1997, 82, 363–392 CAS
- J. A. Conrad and G. R. Moran, Inorg. Chim. Acta, 2008, 361, 1197–1201 CrossRef CAS PubMed
- L. Que Jr and R. Y. Ho, Chem. Rev., 1996, 96, 2607–2624 CrossRef PubMed
- L. Serre, A. Sailland, D. Sy, P. Boudec, A. Rolland, E. Pebay-Peyroula and C. Cohen-Addad, Structure, 1999, 7, 977–988 CrossRef CAS
- V. Pfeifer, G. J. Nicholson, J. Ries, J. Recktenwald, A. B. Schefer, R. M. Shawky, J. Schroder, W. Wohlleben and S. Pelzer, J. Biol. Chem., 2001, 276, 38370–38377 CrossRef CAS PubMed
- J. Brownlee, P. He, G. R. Moran and D. H. Harrison, Biochemistry, 2008, 47, 2002–2013 CrossRef CAS PubMed
- P. He and G. R. Moran, J. Inorg. Biochem., 2011, 105, 1259–1272 CrossRef CAS PubMed
- D. Bischoff, S. Pelzer, B. Bister, G. J. Nicholson, S. Stockert, M. Schirle, W. Wohlleben, G. Jung and R. D. Süssmuth, Angew. Chem., Int. Ed. Engl., 2001, 40, 4688–4691 CrossRef CAS
- D. Bischoff, S. Pelzer, A. Holtzel, G. J. Nicholson, S. Stockert, W. Wohlleben, G. Jung and R. D. Süssmuth, Angew. Chem., Int. Ed. Engl., 2001, 40, 1693–1696 CrossRef CAS
- R. D. Süssmuth, S. Pelzer, G. Nicholson, T. Walk, W. Wohlleben and G. Jung, Angew. Chem., Int. Ed. Engl., 1999, 38, 1976–1979 CrossRef
- A. M. Sandercock, E. H. Charles, W. Scaife, P. N. Kirkpatrick, S. W. O'Brien, E. A. Papageorgiou, J. B. Spencer and D. H. Williams, Chem. Commun., 2001, 1252–1253 RSC
- S. Eckermann, G. Schroder, J. Schmidt, D. Strack, R. A. Edrada, Y. Helariutta, P. Elomaa, M. Kotilainen, I. Kilpelainen, P. Proksch, T. H. Teeri and J. Schroder, Nature, 1998, 396, 387–390 CrossRef CAS PubMed
-
S. Pelzer, P. Huber, R. D. Süssmuth, J. Recktenwald, D. Heckmann and W. Wohlleben, International Patent, WO0077182A1, 2000
- B. S. Moore and J. N. Hopke, ChemBioChem, 2001, 2, 35–38 CrossRef CAS
- M. Redenbach, H. M. Kieser, D. Denapaite, A. Eichner, J. Cullum, H. Kinashi and D. A. Hopwood, Mol. Microbiol., 1996, 21, 77–96 CAS
- H. Chen, C. C. Tseng, B. K. Hubbard and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14901–14906 CrossRef CAS PubMed
- C. C. Tseng, S. M. McLoughlin, N. L. Kelleher and C. T. Walsh, Biochemistry, 2004, 43, 970–980 CrossRef CAS PubMed
- G. H. Hur, C. R. Vickery and M. D. Burkart, Nat. Prod. Rep., 2012, 29, 1074–1098 RSC
- G. L. Challis, J. Ravel and C. A. Townsend, Chem. Biol., 2000, 7, 211–224 CrossRef CAS
- M. Sosio, H. Kloosterman, A. Bianchi, P. de Vreugd, L. Dijkhuizen and S. Donadio, Microbiology, 2004, 150, 95–102 CrossRef CAS
- T.-L. Li, F. Huang, S. F. Haydock, T. Mironenko, P. F. Leadlay and J. B. Spencer, Chem. Biol., 2004, 11, 107–119 CAS
- X. Yin and T. M. Zabriskie, Microbiology, 2006, 152, 2969–2983 CrossRef PubMed
- M. Gunsior, S. D. Breazeale, A. J. Lind, J. Ravel, J. W. Janc and C. A. Townsend, Chem. Biol., 2004, 11, 927–938 CrossRef CAS PubMed
- J. J. Banik and S. F. Brady, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17273–17277 CrossRef CAS PubMed
- G. Yim, L. Kalan, K. Koteva, M. N. Thaker, N. Waglechner, I. Tang and G. D. Wright, ChemBioChem, 2014, 15, 2613–2623 CrossRef CAS PubMed
- M. Röttig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W362–W367 CrossRef PubMed
- C. Rausch, T. Weber, O. Kohlbacher, W. Wohlleben and D. H. Huson, Nucleic Acids Res., 2005, 33, 5799–5808 CrossRef CAS PubMed
- S. A. Samel, P. Czodrowski and L.-O. Essen, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1442–1452 CAS
- J. W. Trauger and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3112–3117 CrossRef CAS
- S. Weist, C. Kittel, D. Bischoff, B. Bister, V. Pfeifer, G. J. Nicholson, W. Wohlleben and R. D. Süssmuth, J. Am. Chem. Soc., 2004, 126, 5942–5943 CrossRef CAS PubMed
- D. Bischoff, B. Bister, M. Bertazzo, V. Pfeifer, E. Stegmann, G. J. Nicholson, S. Keller, S. Pelzer, W. Wohlleben and R. D. Süssmuth, ChemBioChem, 2005, 6, 267–272 CrossRef CAS PubMed
- M. J. Cryle, J. E. Stok and J. J. De Voss, Aust. J. Chem., 2003, 56, 749–762 CrossRef CAS
- A. N. Holding and J. B. Spencer, ChemBioChem, 2008, 9, 2209–2214 CrossRef CAS PubMed
- N. Geib, K. Woithe, K. Zerbe, D. B. Li and J. A. Robinson, Bioorg. Med. Chem. Lett., 2008, 18, 3081–3084 CrossRef CAS PubMed
- E. Stegmann, S. Pelzer, D. Bischoff, O. Puk, S. Stockert, D. Butz, K. Zerbe, J. Robinson, R. D. Süssmuth and W. Wohlleben, J. Biotechnol., 2006, 124, 640–653 CrossRef CAS PubMed
- B. Hadatsch, D. Butz, T. Schmiederer, J. Steudle, W. Wohlleben, R. Süssmuth and E. Stegmann, Chem. Biol., 2007, 14, 1078–1089 CrossRef CAS PubMed
- P. C. Schmartz, K. Wölfel, K. Zerbe, E. Gad, E. S. El Tamany, H. K. Ibrahim, K. Abou-Hadeed and J. A. Robinson, Angew. Chem., Int. Ed. Engl., 2012, 51, 11468–11472 CrossRef CAS PubMed
- K. Woithe, N. Geib, O. Meyer, T. Wortz, K. Zerbe and J. A. Robinson, Org. Biomol. Chem., 2008, 6, 2861–2867 CAS
- K. Woithe, N. Geib, K. Zerbe, D. B. Li, M. Heck, S. Fournier-Rousset, O. Meyer, F. Vitali, N. Matoba, K. Abou-Hadeed and J. A. Robinson, J. Am. Chem. Soc., 2007, 129, 6887–6895 CrossRef CAS PubMed
- K. Zerbe, K. Woithe, D. B. Li, F. Vitali, L. Bigler and J. A. Robinson, Angew. Chem., Int. Ed. Engl., 2004, 43, 6709–6713 CrossRef CAS PubMed
- C. Brieke, V. Kratzig, K. Haslinger, A. Winkler and M. J. Cryle, Org. Biomol. Chem., 2015, 13, 2012–2021 CAS
- K. Haslinger, E. Maximowitsch, C. Brieke, A. Koch and M. J. Cryle, ChemBioChem, 2014, 15, 2719–2728 CrossRef CAS PubMed
- K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015 DOI:10.1038/nature14141
- W.-T. Liu, R. D. Kersten, Y.-L. Yang, B. S. Moore and P. C. Dorrestein, J. Am. Chem. Soc., 2011, 133, 18010–18013 CrossRef CAS PubMed
- X. Jin, M. Rao, W. Wei, M. Ge, J. Liu, D. Chen and Y. Liang, Biotechnol. Lett., 2012, 34, 2283–2289 CrossRef CAS PubMed
- G. Yim, M. N. Thaker, K. Koteva and G. Wright, J. Antibiot., 2014, 67, 31–41 CrossRef CAS PubMed
- A. Hornung, M. Bertazzo, A. Dziarnowski, K. Schneider, K. Welzel, S.-E. Wohlert, M. Holzenkämpfer, G. J. Nicholson, A. Bechthold, R. D. Süssmuth, A. Vente and S. Pelzer, ChemBioChem, 2007, 8, 757–766 CrossRef CAS PubMed
- B. Bister, D. Bischoff, G. J. Nicholson, S. Stockert, J. Wink, C. Brunati, S. Donadio, S. Pelzer, W. Wohlleben and R. D. Süssmuth, ChemBioChem, 2003, 4, 658–662 CrossRef CAS PubMed
- O. Puk, D. Bischoff, C. Kittel, S. Pelzer, S. Weist, E. Stegmann, R. D. Süssmuth and W. Wohlleben, J. Bacteriol., 2004, 186, 6093–6100 CrossRef CAS PubMed
- S. Weist, B. Bister, O. Puk, D. Bischoff, S. Pelzer, G. J. Nicholson, W. Wohlleben, G. Jung and R. D. Süssmuth, Angew. Chem., Int. Ed. Engl., 2002, 41, 3383–3385 CrossRef CAS
- P. C. Schmartz, K. Zerbe, K. Abou-Hadeed and J. A. Robinson, Org. Biomol. Chem., 2014, 12, 5574–5577 CAS
- F. Beltrametti, A. Lazzarini, C. Brunati, A. Marazzi, S. Jovetic, E. Selva and F. Marinelli, J. Antibiot., 2003, 56, 773–782 CrossRef CAS
- S. Stinchi, L. Carrano, A. Lazzarini, M. Feroggio, A. Grigoletto, M. Sosio and S. Donadio, FEMS Microbiol. Lett., 2006, 256, 229–235 CrossRef CAS PubMed
- K. Haslinger, C. Brieke, S. Uhlmann, L. Sieverling, R. D. Süssmuth and M. J. Cryle, Angew. Chem., Int. Ed. Engl., 2014, 53, 1–6 CrossRef PubMed
- S. Uhlmann, R. D. Süssmuth and M. J. Cryle, ACS Chem. Biol., 2013, 8, 2586–2596 CrossRef CAS PubMed
- S. Pohle, C. Appelt, M. Roux, H.-P. Fiedler and R. D. Süssmuth, J. Am. Chem. Soc., 2011, 133, 6194–6205 CrossRef CAS PubMed
- A. W. Truman, M. J. Kwun, J. Cheng, S. H. Yang, J.-W. Suh and H.-J. Hong, Antimicrob. Agents Chemother., 2014, 58, 5687–5695 CrossRef CAS PubMed
- M. Spohn, N. Kirchner, A. Kulik, A. Jochim, F. Wolf, P. Muenzer, O. Borst, H. Gross, W. Wohlleben and E. Stegmann, Antimicrob. Agents Chemother., 2014, 58, 6185–6196 CrossRef PubMed
- J. J. Banik, J. W. Craig, P. Y. Calle and S. F. Brady, J. Am. Chem. Soc., 2010, 132, 15661–15670 CrossRef CAS PubMed
- B. S. Moore and D. Seng, Tetrahedron Lett., 1998, 39, 3915–3918 CrossRef CAS
- M. Sosio, H. Kloosterman, A. Bianchi, V. P. de, L. Dijkhuizen and S. Donadio, Microbiology, 2004, 150, 95–102 CrossRef CAS
- T.-L. Li, F. Huang, S. F. Haydock, T. Mironenko, P. F. Leadlay and J. B. Spencer, Chem. Biol., 2004, 11, 107–119 CAS
- T. C. Roberts, M. A. Schallenberger, J. Liu, P. A. Smith and F. E. Romesberg, J. Med. Chem., 2011, 54, 4954–4963 CrossRef CAS PubMed
- H. C. Losey, M. W. Peczuh, Z. Chen, U. S. Eggert, S. D. Dong, I. Pelczer, D. Kahne and C. T. Walsh, Biochemistry, 2001, 40, 4745–4755 CrossRef CAS PubMed
- P. J. Solenberg, P. Matsushima, D. R. Stack, S. C. Wilkie, R. C. Thompson and R. H. Baltz, Chem. Biol., 1997, 4, 195–202 CrossRef CAS
- W. Lu, M. Oberthür, C. Leimkuhler, J. Tao, D. Kahne and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4390–4395 CrossRef CAS PubMed
- M. Oberthür, C. Leimkuhler, R. G. Kruger, W. Lu, C. T. Walsh and D. Kahne, J. Am. Chem. Soc., 2005, 127, 10747–10752 CrossRef PubMed
- H. C. Losey, J. Jiang, J. B. Biggins, M. Oberthür, X.-Y. Ye, S. D. Dong, D. Kahne, J. S. Thorson and C. T. Walsh, Chem. Biol., 2002, 9, 1305–1314 CrossRef CAS
- A. M. Mulichak, H. C. Losey, C. T. Walsh and R. M. Garavito, Structure, 2001, 9, 547–557 CrossRef CAS
- A. M. Mulichak, W. Lu, H. C. Losey, C. T. Walsh and R. M. Garavito, Biochemistry, 2004, 43, 5170–5180 CrossRef CAS PubMed
- A. M. Mulichak, H. C. Losey, W. Lu, Z. Wawrzak, C. T. Walsh and R. M. Garavito, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9238–9243 CrossRef CAS PubMed
- M. Sosio, S. Stinchi, F. Beltrametti, A. Lazzarini and S. Donadio, Chem. Biol., 2003, 10, 541–549 CrossRef CAS
- M. N. Thaker, W. Wang, P. Spanogiannopoulos, N. Waglechner, A. M. King, R. Medina and G. D. Wright, Nat. Biotechnol., 2013, 31, 922–927 CrossRef CAS PubMed
- J.-Y. Ho, Y.-T. Huang, C.-J. Wu, Y.-S. Li, M.-D. Tsai and T.-L. Li, J. Am. Chem. Soc., 2006, 128, 13694–13695 CrossRef CAS PubMed
- Y.-C. Liu, Y.-S. Li, S.-Y. Lyu, L.-J. Hsu, Y.-H. Chen, Y.-T. Huang, H.-C. Chan, C.-J. Huang, G.-H. Chen, C.-C. Chou, M.-D. Tsai and T.-L. Li, Nat. Chem. Biol., 2011, 7, 304–309 CrossRef CAS PubMed
- A. Nakayama, A. Okano, Y. Feng, J. C. Collins, K. C. Collins, C. T. Walsh and D. L. Boger, Org. Lett., 2014, 16, 3572–3575 CrossRef CAS PubMed
- A. Okano, A. Nakayama, A. W. Schammel and D. L. Boger, J. Am. Chem. Soc., 2014, 136, 13522–13525 CrossRef CAS PubMed
- P. Peltier-Pain, K. Marchillo, M. Zhou, D. R. Andes and J. S. Thorson, Org. Lett., 2012, 14, 5086–5089 CrossRef CAS PubMed
- X. Fu, C. Albermann, J. Jiang, J. Liao, C. Zhang and J. S. Thorson, Nat. Biotechnol., 2003, 21, 1467–1469 CrossRef CAS PubMed
- R. Shi, C. Munger, L. Kalan, T. Sulea, G. D. Wright and M. Cygler, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11824–11829 CrossRef CAS PubMed
- S. S. Lamb, T. Patel, K. P. Koteva and G. D. Wright, Chem. Biol., 2006, 13, 171–181 CrossRef CAS PubMed
- R. Shi, S. S. Lamb, S. Bhat, T. Sulea, G. D. Wright, A. Matte and M. Cygler, J. Biol. Chem., 2007, 282, 13073–13086 CrossRef CAS PubMed
- N. M. Gaudelli, D. H. Long and C. A. Townsend, Nature, 2015, 520, 383–387 CrossRef CAS PubMed
- A. M. Reeve, S. D. Breazeale and C. A. Townsend, J. Biol. Chem., 1998, 273, 30695–30703 CrossRef CAS PubMed
- B. A. Wilson, S. Bantia, G. M. Salituro, A. M. Reeve and C. A. Townsend, J. Am. Chem. Soc., 1988, 110, 8238–8239 CrossRef CAS
- W. L. Kelly and C. A. Townsend, J. Am. Chem. Soc., 2002, 124, 8186–8187 CrossRef CAS PubMed
- A. Bruggink, E. C. Roos and E. de Vroom, Org. Process Res. Dev., 1998, 2, 128–133 CrossRef CAS
- R. V. Ulijn, L. De Martin, P. J. Halling, B. D. Moore and A. E. Janssen, J. Biotechnol., 2002, 99, 215–222 CrossRef CAS
- I. Aranaz, N. Acosta and A. Heras, Enzyme Microb. Technol., 2006, 39, 215–221 CrossRef CAS PubMed
- M. I. Youshko, H. M. Moody, A. L. Bukhanov, W. H. Boosten and V. K. Svedas, Biotechnol. Bioeng., 2004, 85, 323–329 CrossRef CAS PubMed
- F. Matsuura, Y. Hamada and T. Shiori, Tetrahedron Lett., 1994, 35, 733–736 CrossRef CAS
- K. Wisniewski and H. Car, CNS Drug Rev., 2002, 8, 101–116 CrossRef CAS PubMed
- P. J. Conn and J. P. Pin, Annu. Rev. Pharmacol. Toxicol., 1997, 37, 205–237 CrossRef CAS PubMed
- D. D. Schoepp, J. Goldsworthy, B. G. Johnson, C. R. Salhoff and S. R. Baker, J. Neurochem., 1994, 63, 769–772 CrossRef CAS
- I. Brabet, S. Mary, J. Bockaert and J. P. Pin, Neuropharmacology, 1995, 34, 895–903 CrossRef CAS
- M. A. Desai, J. P. Burnett, N. G. Mayne and D. D. Schoepp, Mol. Pharmacol., 1995, 48, 648–657 CAS
- I. Ito, A. Kohda, S. Tanabe, E. Hirose, M. Hayashi, S. Mitsunaga and H. Sugiyama, NeuroReport, 1992, 3, 1013–1016 CrossRef CAS PubMed
- J.-M. Servitja, R. Masgrau, E. Sarri and F. Picatoste, J. Neurochem., 1999, 72, 1441–1447 CrossRef CAS
- D. E. Pellegrini-Giampietro, S. A. Torregrossa and F. Moroni, Br. J. Pharmacol., 1996, 118, 1035–1043 CrossRef CAS PubMed
- K. Maiese, I. Ahmad, M. Tenbroeke and J. Gallant, J. Neurosci. Res., 1999, 55, 472–485 CrossRef CAS
- R. J. Sayer, J. Neurophysiol., 1998, 80, 1981–1988 CAS
- S. Ferre, P. Popoli, R. Rimondini, R. Reggio, J. Kehr and K. Fuxe, Neuropharmacology, 1999, 38, 129–140 CrossRef CAS
- M. T. Taber and H. C. Fibiger, J. Neurosci., 1995, 15, 3896–3904 CAS
- O. L. Caramelo, P. F. Santos, A. P. Carvalho and C. B. Duarte, J. Neurosci. Res., 1999, 58, 505–514 CrossRef CAS
- S. M. Fitzjohn, Z. A. Bortolotto, M. J. Palmer, A. J. Doherty, P. L. Ornstein, D. D. Schoepp, A. E. Kingston, D. Lodge and G. L. Collingridge, Neuropharmacology, 1998, 37, 1445–1458 CrossRef CAS
- S. M. Fitzjohn, A. J. Irving, M. J. Palmer, J. Harvey, D. Lodge and G. L. Collingridge, Neurosci. Lett., 1996, 203, 211–213 CrossRef CAS
- K. M. Huber, M. S. Kayser and M. F. Bear, Science, 2000, 288, 1254–1257 CrossRef CAS
- M. J. Palmer, A. J. Irving, G. R. Seabrook, D. E. Jane and G. L. Collingridge, Neuropharmacology, 1997, 36, 1517–1532 CrossRef CAS
- R. Schnabel, I. C. Kilpatrick and G. L. Collingridge, Br. J. Pharmacol., 2001, 132, 1095–1101 CrossRef CAS PubMed
- A. I. Sacaan and D. D. Schoepp, Neurosci. Lett., 1992, 139, 77–82 CrossRef CAS
- U. H. Schroder, T. Opitz, T. Jager, C. F. Sabelhaus, J. Breder and K. G. Reymann, Neuropharmacology, 1999, 38, 209–216 CrossRef CAS
- H.-Z. Hu, J. Ren, S. Liu, C. Gao, Y. Xia and J. D. Wood, Br. J. Pharmacol., 1999, 128, 1631–1635 CrossRef CAS PubMed
- K. P. Lesch, C. S. Aulakh, B. L. Wolozin and D. L. Murphy, Pharmacol. Toxicol., 1992, 71, 49–60 CAS
- A. Pilc, P. Branski, A. Palucha, K. Tokarski and M. Bijak, Eur. J. Pharmacol., 1998, 349, 83–87 CrossRef CAS
- X. C. Li, P. M. Beart, J. A. Monn, N. M. Jones and R. E. Widdop, Br. J. Pharmacol., 1999, 128, 823–829 CrossRef CAS PubMed
- T. Tsuchihashi, Y. Liu, S. Kagiyama, K. Matsumura, I. Abe and M. Fujishima, Brain Res. Bull., 2000, 52, 279–283 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2015 |