
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selenium, selenoproteins and neurodegenerative diseases
Bárbara Rita
Cardoso
*ab,
Blaine R.
Roberts
a,
Ashley I.
Bush†
 a and
Dominic J.
Hare†
*acd
a and
Dominic J.
Hare†
*acd
aThe Florey Institute of Neuroscience and Mental Health, The University of Melbourne, 30 Royal Parade, Parkville, Victoria 3052, Australia. E-mail: barbara.rita@florey.edu.au; Tel: +61 450 633 537
bFaculty of Pharmaceutical Sciences, Department of Food and Experimental Nutrition, University of São Paulo, São Paulo, Brazil
cElemental Bio-imaging Facility, University of Technology Sydney, Thomas Street, Broadway, New South Wales 2007, Australia. E-mail: dominic.hare@uts.edu.au; Tel: +61 3 9035 9549
dExposure Biology, Frank R. Lautenberg Environmental Health Sciences Laboratory, Department of Preventive Medicine, Icahn School of Medicine at Mount Sinai, New York, USA
First published on 13th May 2015
Abstract
It is unsurprising that our understanding of the role of selenium in neurological function is somewhat immature, considering its relatively recent discovery as an essential element to human health. Selenocysteine, the 21st amino acid, is the defining feature of the 25 selenoprotein-encoding genes so far discovered within the human genome. The low abundance of these proteins in the brain belies the integral role they play in normal neurological function, from well-characterised antioxidant activity in the periphery to poorly understood mechanisms that modulate mitochondrial function and response to brain pathology. Selenium has been identified as playing a role in several neurodegenerative disorders, including Alzheimer's and Parkinson's disease, though its function as a ‘cause or effect’ of disease process remains unclear. This review discusses selenium metabolism in detail, specifically with regard to the role it plays within the central nervous system, and examines the most current literature investigating how selenium may be involved in chronic diseases of the central nervous system.
 Bárbara Rita Cardoso | Bárbara Rita Cardoso is a nutritionist, and holds a Masters degree in Human Applied Nutrition. She obtained her PhD in Food Science from the University of São Paulo in 2014. She joined the Florey Institute of Neuroscience and Mental Health in 2014 to investigate the association between selenium status and cognition as part of The Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL). |
 Blaine R. Roberts | Blaine Roberts is the head of the Neuroproteomics Laboratory at the Florey Institute of Neuroscience and Mental Health. He received his PhD in 2007 from Oregon State University before moving to Australia in 2008 to pursue the evolving field of metalloproteomics and its application to neurodegenerative diseases. |
 Ashley I. Bush | Ashley Bush heads the Oxidation Biology Unit at the Florey Institute of Neuroscience and Mental Health, is Professor of Neuroscience at The University of Melbourne, NHMRC Australia Fellow, co-director of biomarker development for the Australian Imaging Biomarker Lifestyle Study of Ageing (AIBL), Chief Scientific Officer of the Cooperative Research Center for Mental Health, and has staff appointments at the Massachusetts General Hospital. |
 Dominic J. Hare | Dominic Hare is a Chancellors Postdoctoral Fellow at the University of Technology, Sydney, Head of the Analytical Neurochemistry Development Group at the Florey Institute of Neuroscience and Mental Health, and an Adjunct Assistant Professor at the Icahn School of Medicine at Mount Sinai in New York. He received his PhD at UTS in 2009, and works primarily in studying the roles of metals in neurodegeneration. |
Introduction
The observation that selenium (Se) prevented necrotic liver damage in mice by Schwarz and Foltz in 1957 was the first direct experimental evidence that this element was essential to normal health.1 Almost twenty years later, Flohe et al.2 confirmed the importance of Se by identifying it as an essential cofactor of glutathione peroxidase (GPx). Twenty-five mammalian selenoprotein-encoding genes have since been identified,3 all containing selenocysteine (Sec) residues, which are analogous to cysteine (Cys) with a selenol moiety replacing the thiol group.4 From these genes, it is estimated that more than 30 unique selenoproteins are synthesised in mammals,5 and it is likely more will be characterised in the future. Selenoproteins have diverse roles throughout the body, acting as antioxidants, modulators of immune function, detoxification agents for heavy metals and xenobiotics, and participants in thyroid hormone metabolism.6Selenium is essential to human health, and the antioxidant activity (or lack thereof) of specific selenoproteins is of particular interest in diseases of the central nervous system. The brain is the most metabolically active of all organs in the body, and although it represents only 2% of the total mass, cerebral metabolism accounts for about 20% of the oxygen and 25% of the glucose consumed by the human body.7 In concert with this high metabolic activity, the high concentration of polyunsaturated fatty acids in the brain makes it more vulnerable to peroxidation. A selective vulnerability within certain neurons to oxidative stress may arise from deficiencies in antioxidant enzyme activity,8 which is a likely upstream event in the pathogenesis of several neurodegenerative diseases,9 including Alzheimer's disease (AD),10 Parkinson's disease (PD)11 and other neurodegenerative disorders. Selenoproteins, with their inherent antioxidant activity, thus play essential roles as part of the free radical defence system, though the function of these proteins is not necessarily restricted to this singular classification. This review summarises the current data and important concepts concerning selenoproteins on brain function and the association between Se and diseases of the brain.
Selenoprotein synthesis
Dietary Se first must be converted to selenide (Se2−), which serves as the donor for the incorporation of Se into selenoproteins. Inorganic Se forms, such as selenite (SeO32−), are converted to Se2−via the glutathione–glutaredoxin and thioredoxin pathways, while organic forms are metabolised to Se2− by selenocysteine lyase (Scly) or trans-selenation (analogous to trans-sulfuration).12The incorporation of Se into proteins was described in detail by Turanov et al.,13 recently reviewed by Labunskyy et al.,14 and is summarised in Fig. 1. After conversion of dietary Se to Se2−, selenophosphate (SePO33−) is produced in a reaction catalysed by selenophosphate synthase 2.4 Selenocysteine (Sec) is incorporated into proteins via its own unique tRNA, (tRNA[Ser]Sec). tRNA[Ser]Sec decodes the UGA codon as Sec instead of a stop codon, as is typical of other amino acids, and thus prevents premature termination. This is possible due to the presence of a Sec insertion sequence (SECIS) element, a specific cis-acting stem-loop structure, in the 3′-untranslated region (UTR) of selenoprotein mRNA that works as a platform for RNA-binding proteins. SECIS forms complexes with two trans-acting elements: the specific elongation factor (EFsec) and the SECIS binding protein 2 (SBP2).15 EFsec interacts with SBP2 to recruit tRNA[Ser]Sec and mediates insertion of Sec into nascent protein chains in response to the UGA codon.15 Among the three distinct domains of SBP2, the Sec incorporation domain and a COOH-terminal RNA-binding domain are specifically involved in SECIS binding. Additional SECIS binding proteins have been identified, including ribosomal L30, which is part of the basal Sec insertion machinery and has a similar RNA-binding domain to SBP2;16 a eukaryotic initiation factor (eIF4a3) that links Se status and differential selenoprotein expression,17 and nucleolin, which demonstrates a high affinity for SECIS, though its complete function is not currently known.18 Recently, Bröcker et al.19 used Escherichia coli formate dehydrogenase mutants to reassign 64 codons encoding a critical Sec residue, finding that 58 of these were able to site-specifically incorporate Sec. At 15 sense codons selenoprotein synthesis could be promoted compared to the 3 stop codons. This study provides critical new information about how selenoprotein production can be manipulated through genetic recoding, and provides exciting new opportunities for additional studies to further elucidate the mechanisms involved in selenoprotein synthesis, which remains a relatively new frontier in molecular biology.
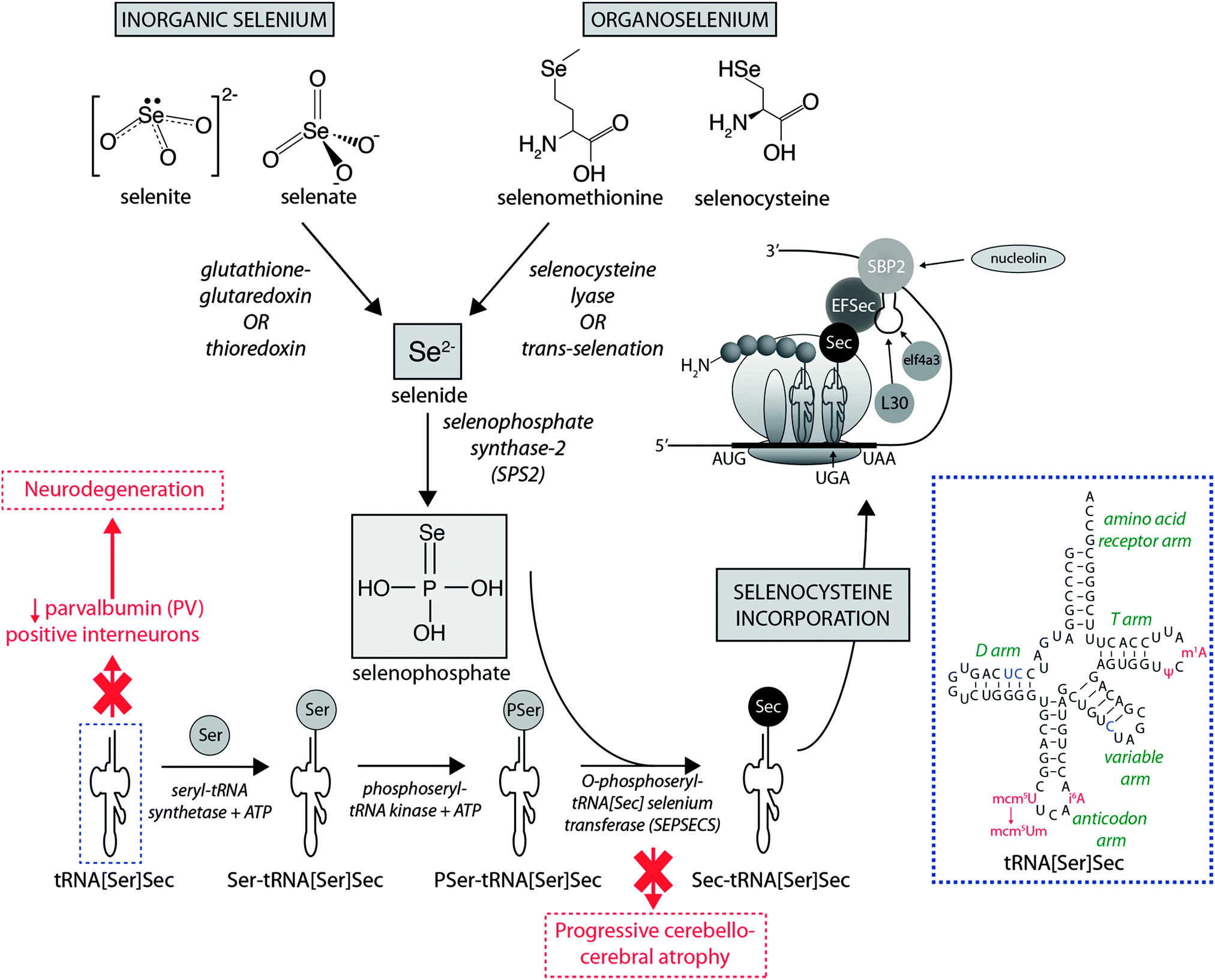 | ||
| Fig. 1 Selenoprotein synthesis pathway. Dietary Se intake, as either inorganic or organic species is metabolised to Se2−, and the SeH3PO3, which is incorporated into proteins via a unique tRNA pathway, where a seryl (Ser) residue is replaced with selenocysteine (Sec) prior to integration into mammalian proteins. Deficiencies in key metabolic factors involved in selenoprotein synthesis have been associated with specific neurological disorders, marked here in red. The inset in the dashed blue box is the simplified cloverleaf structure of mammalian tRNA[Sec] (adapted from Birringer et al.196), showing variable (in blue) and modifiable (in red) bases. Abbreviations: Ψ = pseudouridine; EFSec = Sec elongation factor; eIF4a3 = eukaryotic initiation factor; i6A = N6-isopentenyladenosine; L30 = ribosomal protein L30; mcm5U = 5-methoxycarbonylmethyluridine; m1A = 1-methyladenosine; mcm5Um = 5-methoxycarbonylmethyluridine-2′-O-methylribose; SBP2 = Sec insertion sequence (SCIS) binding protein-2. | ||
It is known that selenoprotein synthesis is critical for viability of life, as shown by tRNA[Ser]Sec (Trsp) knockout mice, which present an embryonically lethal phenotype. However, Trsp null mice allow tissue-specific manipulation of tRNA[Ser]Sec, and this model has been used to investigate the importance of selenoprotein synthesis to brain metabolism. Wirth et al.20 showed that inactivation of selenoprotein synthesis in Trsp null neurons abolished the developmental appearance of parvalbumin-positive (PV) interneuron cells, characterising progressive neurodegeneration. This was later confirmed by additional studies that demonstrated this damage was restricted to specific brain areas, such as the somatosensory cortex and hippocampus, confirming the essential requirement of selenoprotein synthesis for maintenance of CNS metabolism.21,22 In humans, two mutations in the gene encoding O-phosphoseryl-tRNA[Sec]:selenocysteinyl-tRNA[Sec] synthase (SepSecS) were associated with progressive cerebellocerebral atrophy (PCCA), an autosomal-recessive phenotype of nondysmorphic profound mental retardation, severe spasticity and microencephaly.23 These gene alterations abrogate SepSecS activity and thus block the synthesis of tRNA[Ser]Sec, and subsequent Se incorporation into selenoproteins.
In vivo studies have shown that Cys can replace Sec in the Sec biosynthetic organisation, generating Cys-tRNA[Ser]Sec and the subsequent insertion of Cys in some selenoproteins.24 This is possible due to either substitution of Se2− by sulfide (S2−) in the SPS2 catalysed reaction; or substitution of SePO33− by thiophosphates in the reaction with the O-phosphoseryl-tRNA[Ser]Sec intermediate during Sec-tRNA synthesis. The same study showed that the incorporation of Cys in selenoproteins is dependent of Se availability to the cells. Tobe et al.25 showed that some antibiotics could also interfere with Sec insertion in murine EMT6 cells. Cells treated with doxycycline, chloramphenicol and G418 caused lower Sec insertion in thioredoxin reductase 1 (Trxr1), and GPx1 and 4. However, Sec was also exchanged by arginine and tryptophan in addition to Cys. The specific mechanisms that are associated with mis-insertion of amino acids in place of Sec and how they are associated with changes in selenoproteins activity need to be clarified, but these reports show another way selenoprotein hierarchy is potentially regulated, which may also include as-yet undetermined environmental factors.
Selenoprotein P and brain selenium hierarchy
Selenoprotein expression is modulated by Se availability, and a hierarchy is observed in response to dietary Se intake. In cases of Se deficiency the synthesis of some proteins is maintained at a higher level than that of others, suggesting that insufficient available Se results in competition between transcripts of different selenoproteins.26 Iodothyronine deiodinase 1 is atop this hierarchy and is comparatively the least affected by restricted dietary Se; glutathione peroxidases GPx2 and GPx4 are less vulnerable to Se deficiency than GPx1 and GPx3; and selenoprotein P (SelP) is in an intermediary position on this selenoprotein transcription ladder.27Some mechanisms underlying Se regulation of the selenoprotein transcriptome have been identified. During Se deficiency, non-essential selenoproteins are down regulated via nonsense-mediated mRNA decay, which decodes the UGA-Sec codon as a termination codon causing RNA degradation.28,29 Also, Se levels directly correlate with the degree of Um34 methylation (the single methyl group differentiating the two tRNA[Ser]Sec isoforms30), one step of maturation of tRNA[Ser]Sec which can be altered as a result of some mutations. The Um34-containing isoform of tRNA[Ser]Se; 5-methoxycarbonylmethyl-2′-O-methyluridine (mcm5Um), is recruited for efficient expression of the stress-related selenoproteins; and the non-Um34-containing isoform, 5-methoxycarbonylmethyluridine (mcm5U), is not required specifically. The distinct use of these tRNA[Ser]Sec isoforms shows that this mechanism contributes to Se regulation for the purpose of selenoprotein synthesis.28
The hypothesis that explains the hierarchy between selenoprotein syntheses suggests that they are classified according to their importance to the body. However, it is important to note that Se concentration is highly variable within the periphery (i.e. liver, kidneys, etc.), though in the brain Se levels remain relatively stable by comparison during periods of deficiency.31 Additionally, the ability of organs outside the CNS to retain Se is also variable. The CNS, together with reproductive and endocrine organs, has the highest priority for Se both uptake and retention during Se deficiency, although the Se concentration in the CNS is low in comparison with other organs.32 Though interrelated, supply and retention are not exclusively linked, and it is thus a testament to the importance of Se in brain function that rigorous mechanisms to ensure adequate Se levels are available in times of dietary shortage are in place. The brain's top position on a tissue-specific grading further indicates that Se is essential for its normal metabolism.33 This is primarily due to the high expression of apolipoprotein E receptor-2 (apoER2) at the blood–brain barrier (see below) to facilitate Se uptake and strong SelP-apoER2 interaction within the CNS providing an adequate pool of available Se. Selenium deficient rats presented approximately 29% lower Se levels in the brain compared to animals with a sufficient Se supply, which is not nearly as dramatic as the 99% and 92% decrease in Se concentration in the liver and kidneys of deficient animals.34 Se distribution is heterogeneous among different brain regions, and grey matter appears to have preference for greater uptake. Imaging using a 75Se radiotracer combined with autoradiographic localisation and proteomic analysis to profile Se levels in different brain regions of rats fed a Se-replete diet revealed that Se was enriched in choroid plexus, putamen, parietal inferior lobule and occipital cortex.35 This observation has been reflected in human brains, where the highest Se concentration occurs in grey matter, with the putamen, parietal inferior lobule and occipital cortex in particular, while the lowest levels are found in the cerebellum and medulla.36
The mechanism that maintains Se levels in the brain at the expense of other organs, even during times of deprivation, is mainly orchestrated by SelP, which is recognised as the most important Se supply to different tissue types. The role of SelP to deliver Se to cells is facilitated by the presence of ten SECIS forms: one is situated in the larger N-terminal region, which acts as an enzyme; and nine are located in a Sec-rich C-terminal domain, providing a mechanism for transporting the highly reactive Se atom safely, incorporated into the protein as Sec.37 Deletion of the conserved, non-SECIS containing 3′UTR of SelP does not affect the efficiency of incorporation of multiple Sec residues into SelP in vitro whilst still remaining sensitive to ambient Se levels, indicating that the 3′UTR are not involved in Se incorporation per se.38 The structure of plasma SelP remains unresolved, and there is some contention as to the number of circulating isoforms of the protein. Ballihaut et al.39 recently reported three isoforms in a human plasma standard reference material, two of which incorporated Se, suggesting the third was in fact a truncated variant.
Burk et al.40 recently proposed the mechanism by which Se is delivered to the brain (represented in Fig. 2a). SelP from plasma is endocytosed following docking with apoER2 at the blood–brain barrier (BBB) in brain capillary endothelial cells (BCECs) and choroid plexus epithelial cells, which facilitates Se transport, in an as-yet unknown chemical form, into the brain. Selenium is then delivered to brain cells via SelP synthesised by astrocytes on the abluminal side of BCECs, composing both the main transport mechanism and distinct pool of Se. Direct transport of SelP to neurons from BCECs has not been excluded. Neurons regulate Se levels via apoER2-mediated uptake of SelP,41 which has obvious implications for AD due its role in neurodevelopment and synaptic function. In the periphery, apoER2 is widely expressed in the body and is also involved in Se uptake in myoblasts42 and testis cells.43 In the CNS, apoER2 is also expressed in choroid plexus and on PV interneurons in the hippocampus, inferior colliculus, medial septum, red nucleus, reticular thalamus and cerebellum.40,44 ApoER2 expression is regulated by physiological factors related to developmental stage; fetal brain mRNA ApoER2 are more than 9 times greater than that observed in adult brain, and apparently not influenced by Se status.40 Although deletion of apoER2 gene does not affect the whole-body Se levels, apoER2−/− and SelP−/− mice have lower Se levels in brain,45,46 and this may be associated with uptake of Se during specific developmental windows.40 In this study, fetal apoER2−/− mice brains contain 75% less Se levels compared to age-matched apoER2+/+ mice, decreasing to 39% when reaching adulthood. Moreover, apoER2−/− mice had no SelP in BCECs or choroid plexus cells at the BBB, suggesting a key role of ApoER2 in brain Se uptake, and also the presence of an unknown redundancy mechanism for Se transport. It remains unclear if other members of the lipoprotein receptor family may participate of SelP uptake, since Burk et al.47 observed an alternative mechanism that does not appear to depend directly on selenoproteins participating on the transfer of Se from pregnant dam to the fetus, and it is unclear if this mechanism is conserved in the postnatal period.
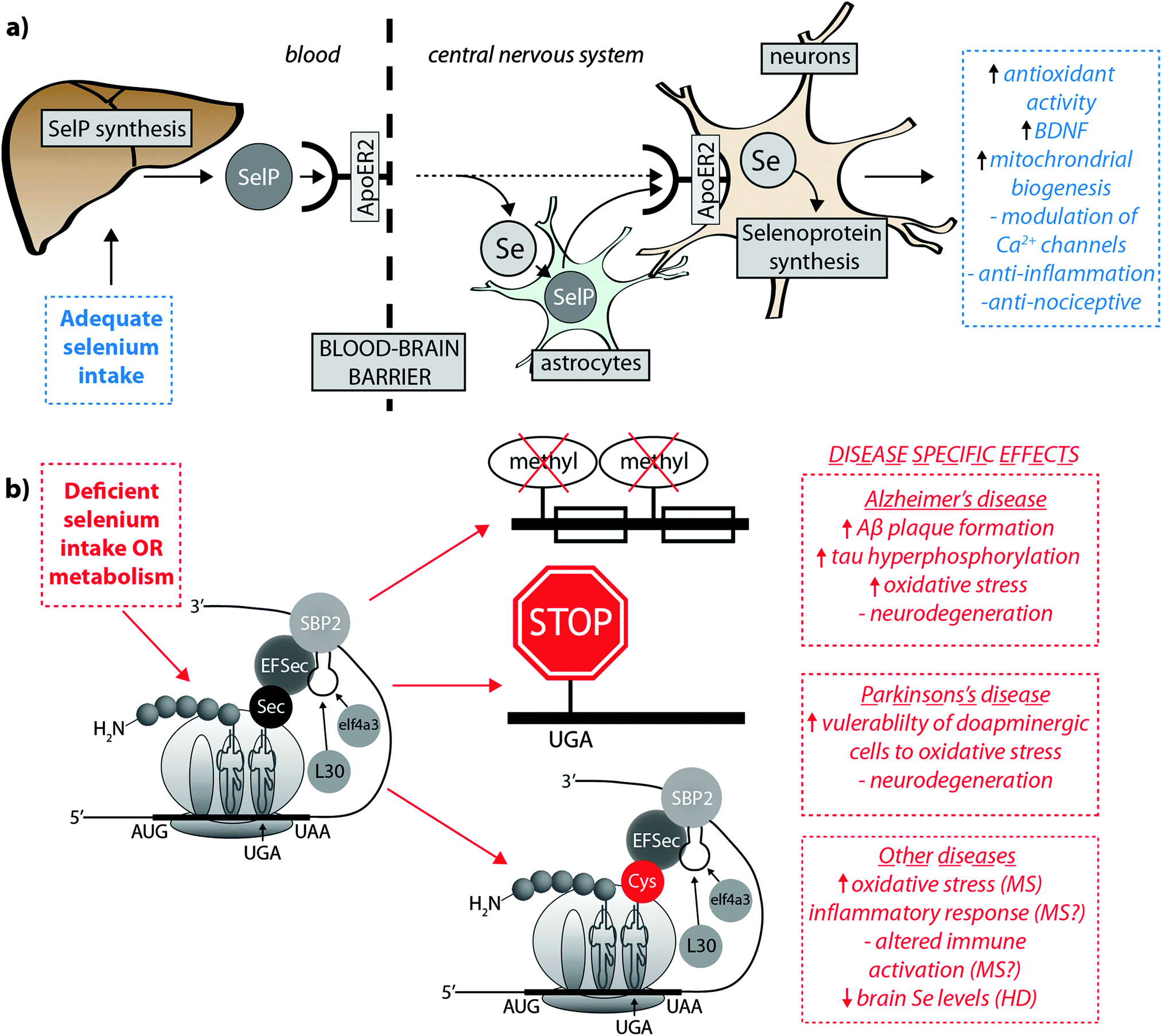 | ||
| Fig. 2 (a) SelP, the master protein driving Se bioavailability is synthesised in the liver from both inorganic and organoselenium compounds accessible through dietary sources. At the blood–brain barrier, SelP releases Se into the CNS via the ApoER2 receptor, which is in turn incorporated into newly – formed SelP in astrocytes (figure adapted from Burk et al.35), or is transported directly to neurons (dashed arrow). SelP is made available to neurons via the same membrane-bound ApoER2, where additional selenoproteins essential to neurological function (blue box) are biosynthesised. (b) In cases of deficient Se intake Um34 methylation is downregulated, causing premature termination of protein synthesis via failure of tRNA[Ser]Sec to decode the UGA codon as Sec instead of a stop codon, and misincorpoartion of selenocysteine (Sec) as cysteine (Cys; in red). These three pathological features may have downstream effects on a range of neurological disorders (red boxes). | ||
Though primarily expressed in astrocytes, neurons have also been identified as a source of endogenous SelP48,49 in all the areas of the brain, though particularly high expression was identified in the putamen and substantia nigra.50 In contrast to apoER2−/− animals, SelP−/− mice present generalised Se deficiency, and in brain this gene deletion causes severe neurological dysfunction.47,51SelP−/− mice showed decreased density of PV interneurons in different brain areas, most marked in the inferior colliculus. This was associated with higher oxidative damage and a behavioural phenotype, characterised by impairments in contextual fear extinction, latent inhibition and sensorimotor gating. These data suggest that besides the effect on PV-expressing neurons causing increased vulnerability to oxidative damage, the behavioural deficits resulting from SelP deletion are likely associated with impaired GABAergic function of the inferior colliculus.44 Both SelP−/− and apoER2−/− mice, when maintained on a Se-deficient diet, are more vulnerable to neuronal damage and eventual neurodegeneration. In this way, Burk et al.40 reported that extremely Se-deficient apoER2−/− and SelP−/− mice displayed neurological dysfunction, while the littermate wild type mice did not show equivalent disruption, despite a decrease in brain Se levels. In another important study,46 progressive neurodegeneration was evaluated in brains of SelP−/− postnatal mice maintained on a Se-deficient diet after weaning. Higher lipid peroxidation and decreased dendrite length and spine density was observed, and the areas more susceptible to neurodegeneration during this post-weaning developmental window were the forebrain bundle, somatosensory cortex and lateral striatum. In the hippocampus decreased dendrite length and spine density was also present, which might be associated with the disrupted hippocampal-dependent learning and alteration to synaptic transmission observed in these mice.
As Scly is essential for breakdown of Sec into L-analine and Se2−, it has been proposed that this enzyme plays important role in Se metabolism in different tissue, and interacts with SelP to maintain adequate selenoprotein function in the brain. Although Scly−/− mice do not demonstrate neurological dysfunction,51 deletion of both Scly and SelP exacerbates the neurological phenotype of SelP−/− mice, which display impaired motor coordination and locomotion compared with SelP null mice alone. Moreover, Scly−/−SelP−/− mice have lower expression of Trxr1, GPx1 and selenoprotein M (SelM) than SelP−/− mice, showing that interplay of Scly and SelP helps maintain selenoprotein levels within the brain.52
Selenium and other selenoproteins: role in brain function
Selenium appears to have a multifaceted role in the nervous system, with a number of studies showing its importance to normal brain physiological function. The antioxidant activity of selenoproteins in the CNS is well established, and lower levels of Se has been associated with brain injury.53 Selenium treatment as organic selenomethionine and inorganic SeO32− resulted in a similar increased activity of antioxidant enzymes, scavenging of reactive oxygen species (ROS) and reduced protein carbonyl content of patulin-induced brain damaged rats,54 and reduced the pro-oxidant effects of glyceryl trinitrate in a rat model of migraine headaches.55 Administration of SeO32− to suckling pups whose dams had methimazole-induced hypothyroidism decreased total oxidant status and reduced degenerative changes in the granular cell layer of the dental gyrus,56 and increased levels of brain-derived neurothophic factor (BDNF) in the hippocampus and cerebellum of both hypothyroid and control groups.57 In a rat model of postnatal protein malnutrition oral Se and zinc (Zn) supplementation increased not only antioxidant enzyme activity, but also corrected the neurobehavioral deficits caused by a protein-deficient diet.58Besides the role of Se as essential component of the antioxidant system in the brain, studies have also shown that Se can act in response to oxidative stress through regulation of Ca2+ channels and mitochondrial biogenesis. Mice dorsal ganglion root cells treated with Se showed reduced Ca2+ release induced by H2O2 when compared with non-treated cells, with corresponding decreases in apoptosis and lipid peroxidation levels, and increased levels of GPx and glutathione (GSH) activity.59 In a traumatic brain injury animal model exhibiting oxidative stress, increased apoptosis and activation of transient receptor potential vanilloid 1 (TRVP1) cation channels, Se treatment modulated Ca2+ entry through TRVP1 in neurons, reduced levels of ROS, prevented apoptosis, and reduced activity of caspases 3 and 9. Since brain injury induces Ca2+ influx and mitochondrial dysfunction in neurons, these data show that Se can increase cell viability not only as an antioxidant, but also by modulating apoptotoic pathways and mitochondrial function.60 Studies with murine hippocampal HT22 cells treated with SeO32− clearly showed improved mitochondrial function, even after glutamate exposure and hypoxia. This was associated with activation of complexes I, II + III and IV, and increased mitochondrial respiratory rate. Selenium administration increased levels of peroxisome proliferator-activated receptor γ-coactivator-1α (PGC-1α) and nuclear respiratory factor 1 (NRF1), two important nuclear transcriptional factors associated with mitochondrial biogenesis. This role of Se appears to be mediated through activation of the cyclic adenosine monophosphate response element-binding protein (CREB) and activated protein kinase (AKT) cycles.61–63
Synthetic organoselenium compounds have been studied as potential antioxidant therapies due their lower toxicity compared to inorganic forms of Se. Diphenyl diselenide [(PhSe)2] mimics GPx activity, producing both beneficial or harmful effects, depending on dose and route of administration.64 Acute treatment of rat hippocampal slices with (PhSe)2 modified various proteins involved in glutamate signaling damage to astrocytes.65 Conversely, (PhSe)2 demonstrated a neuroprotective activity in animal models of autoimmune encephalomyelitis and hypothyroidism due its antioxidant and anti-inflammatory role.66,67 A derivate of (PhSe)2, m-trifluoromethyl (m-CF3-PhSe)2 and the Se-containing salicylic acid derivative 5–5′-diselanediylbis-(2-hydroxybenzoic acid) both exhibit antinociceptive action by interaction with serotoninergic system, as well as an anti-inflammatory role in mice.68,69 Selenium supplementation as a promoter of antioxidant activity does appear to be somewhat dependent on the chemical form; 3-methyl-1-pheny-2-(phenylseleno)oct-2-en-1-one was less effective as an antioxidant compared to SeO42−, though it did reduce adenosine deaminase activity, suggesting a potential immunomodulatory property.54 Musik et al.70 examined two other Se-containing molecules; (4-(o-tolyl-)-selenosemicarbazide of 2-chlorobenzoic acid; 3-(2-chlorobenzoylamino-)-2-(o-tolylimino-)-4-methyl-4-selenazoline) reporting differential impacts on brain antioxidant mechanisms, including GPx and superoxide dismutase-1 (SOD-1). These contradictory results regarding Se supplementation on brain function are likely due to the variety of chemical structures, dose, route of administration and the animal models used. More targeted studies regarding the role and the safety of these compounds are required before they can be considered as potential antioxidant therapeutics in humans.
The apparent neuroprotective role of Se supplementation in the brain is not only directly associated with antioxidant effects of organic and inorganic Se species, but also suggests a role in de novo selenoprotein synthesis, as demonstrated by increased activity of certain selenoproteins following Se treatment (Fig. 2). Methamphetamine treatment decreased levels of GPx1 and GPx4 in SH-SY5Y cells; an effect that was arrested with addition of SeO32− 24 hours after methamphetamine exposure.71 In their patulin-induced brain damaged rats, Song et al.72 showed that, although both SeO32− and selenomethionine were able to offer some degree of increased antioxidant activity, the organic form of Se was more efficient in increasing the expression and activity of TrxR, GPx1 and GPx4.
The GPx family of enzymes are expressed in both neurons and glia,73,74 and are recognised as one of the most important mechanisms protecting against ROS-induced damage.75 Among these isoenzymes, GPx4 protects cells against lipid hydroperoxide damage76 and is the most expressed GPx in brain, found in neurons of cerebellum, hippocampus and hypothalamus.77 Following brain injury, GPx4 is also observed in reactive astrocytes, suggesting that this GPx may have an important role in the protective cellular response to stress-induced oxidative damage and apoptosis.78 Autooxidation of dopamine to the reactive dopamine quinone causes a decrease in GPx4 activity by covalently modifying this mitochondrial enzyme,79 suggesting that antioxidant properties of GPx4 are restricted by dopamine breakdown in neurons. Wirth et al.20 showed that mice with a neuron-specific deletion of GPx4 had mild neurological dysfunction, and that GPx4 deficiency may contribute to dysfunction in other selenoproteins expressed in the brain, including TrxR, SelM and selenoprotein W (SelW).
TrxR is abundant throughout the CNS. In the mouse brain TrxR1 (the cytosolic isoform) is abundantly expressed in glial cells of the cerebellar white matter and TrxR2 (the mitochondrial isoform) is observed mainly in the cell bodies of neurons localised in the Purkinje and molecular cell layers in the cerebellum.80 This selenoenzyme plays an important role in maintaining the redox balance inside cells. It forms, together with thioredoxin (Trx), a disulfide reductase system.81 Mitozo et al.75 showed that TrxR inhibition resulted in increased DNA damage and poor cell viability following a H2O2 challenge in neuronal C6 cells, though it did not impede the ability of N2a cells to dispose of peroxides. However, specific inhibitors used to estimates the relative role of the Trx2, glutathione system and catalase for hydrogen peroxide detoxification in different preparations from rat brain showed that Trx2-system contributes to over 60% of the intra-mitochondrial H2O2 turnover, whereas the glutathione system contributes only about 20% to H2O2 detoxification reactions in the rat hippocampus.82 Recently, Cohen-Kutner et al.,83 using two thioredoxin mimetic peptides (NAc-Cys-Pro Cys-amide; CB3 and Ac-Cys-Gly-Pro-Cys-amide; CB4) as a treatment for oxidative stress in SH-SY5Y human neuroblastoma cells, observed anti-inflammatory and antioxidant effects of both peptides, as well as reduced apoptosis markers. The potentially protective role of TrxR is also important for dopaminergic cells, which exhibit high levels of oxidative stress when undergoing parkinsonian degeneration. In cells exposed to 6-hydroxydopamine (6-OHDA) and paraquat, inhibition of TrxR potentiated mitochondrial dysfunction, decreased maximal and reserve respiratory capacity, and increased H2O2 levels and cell death through oxidative stress. Also, TrxR2 deficient cells showed decreased basal mitochondrial oxygen consumption rates.84 Immunoreactivity of TrxR2 was increased in the spinal neurons and hippocampus of aged dogs, suggesting that this selenoenzyme is important for maintaining neuronal homeostasis following the increase in oxidative stress experienced during normal ageing.85 Human astrocytomas, which are the most common form of gliomas, were found to express TrxR in a manner that correlated with both tumour grading and Ki-67 labelling index (a marker of cell proliferation), with both quantitative reverse transcription polymerase chain reaction and immunostaining confirming higher TrxR levels correspond to tumour severity.86 Though no clear causative link between TrxR activity and astrocytoma proliferation has been identified, this association with cancer progression and TrxR expression is a tantalising research direction considering that, besides its antioxidant role, the TrxR class of enzymes contributes to several physiological and biochemical functions, including cell growth, maintenance and programmed death.
Most selenoproteins have the conserved Cys-X-X-Sec (or CXXU) redox motif, including selenoproteins H, W, T and O. Other selenoproteins include Cys-X-X-Cys (CXXC) in TxrRs, Cys-X-Sec (CXU) in selenoprotein-15,87 Sec-X-X-Cys (UXXC) in SelP,88 as well as Sec-X-X-Ser (UXXS) and Sec-X-X-Thr (UXXT), identified in a range of prokaryotic selenoproteins with mammalian homologs.89 Their role it is not well elucidated, though it is thought they act as thiol–disulfide oxidoreductases that participate in the formation of disulfide bonds.37,90–94 SelM has an endoplasmatic reticulum (ER)/Golgi-directing signaling peptide with a short N-terminal extension, the CXXU motif, and a thioredoxin-like domain in the middle.95 It is characterised by a two-layer α/β/α sandwich with a βαβββα secondary structure pattern, and an ER retention signal tetrapeptide in the C-terminal.96,97 In the mouse brain, SelM expression is highest in the olfactory bulb and cerebellum, with intermediate levels found in the hypothalamus and brain stem and low expression in the hippocampus and cerebral cortex.98 The precise role of SelM is not well understood; some studies using mice overexpressing this protein have shown that it promotes antioxidant activity in brain, in this case increasing the activity of other antioxidant enzymes including SOD-1 and GPx, which could be enhanced with further dietary supplementation of Se.99–102 SelM knockdown primary cortical neurons show disturbed Ca2+ homeostasis compared to controls.99 Pitts et al.98 observed that the deletion of SelM gene in mice did not result in morphological changes to the brain, nor did it result in motor or cognitive deficits and anxiety-like behavior. However, these mice presented an obesity phenotype accompanied by higher leptin levels and elevated hypothalamic leptin resistance when compared with wild type controls. As SelM is expressed in high quantities in the hypothalamus, the authors hypothesised that deletion of this gene adversely affected hypothalamic thyrodoxin balance and cause metabolic dysfunction, indicating that SelM plays an important role in maintaining redox balance in brain areas associated with high metabolic output, though further investigation is obviously required to confirm this supposition.
Yim et al.100 showed that the association between SelM overexpression and Se treatment via intraperitoneal injection contributed to the activation of ERK MAPK signaling pathway, an important mechanism involved in the modulation of protein phosphorylation. Additionally, hippocampal mouse cells and C8-D1A cerebellar cells overexpressing SelM exhibited decreased cytosolic Ca2+ in response to oxidative stress, without affecting basal Ca2+ levels, suggesting that SelM can modulate the apoptotic pathway by mediating Ca2+ release from internal cell stores.99 Another proposed mechanism for the role of SelM in providing neuroprotection was the observation that SelM interacts with galectin-1,103 a β-galactoside-binding lectin associated with modulation of microglia activation in neuroinflammation, neuronal myelination and neuronal stem cell proliferation.104,105 Additionally, it was recently shown that overexpression of SelM could change the expression of other proteins; Kim et al.102 showed that, when compared with wild type controls, mice overexpressing SelM upregulated expression of several different genes, including creatine-kinase B-type and LDH-B, essential for energy homeostasis; the E3 ubiquitin-protein ligase RING1 that participates in cellular defense against environmental insult; eIF-4H, which facilitates the binding of initiator rRNA to ribosomes, regulating protein synthesis; and SytXV, that regulates Ca2+-dependent membrane trafficking, and thus is crucial for maintenance of adequate neurotransmitter release. On the other hand, these mice demonstrated downregulation of CENP-N, which mediates mitotic progression, assembly of kinetochore proteins and chromosome segregation; dihydropyrimidinase-related protein 2, that plays important role in neurite guidance; and proteasome subunit K. This study strongly suggests that the protective role of SelM may in fact be carried out indirectly through the modulation of different genes that regulate cell viability.
SelW is abundant in brain of chickens,106,107 and is highly stable in rat brain even during Se-deficiency. In vivo, where Se supply is entirely dictated by the media in which cells are grown, SelW expression is highly dependent on ambient Se concentrations. Low levels (10−9 mol L−1) of Se yielded the highest levels of SelW mRNA expression, which progressively increased over time, along with better general embryonic chicken neuronal health. High levels of Se (up to 10−5 mol L−1) were obviously neurotoxic, with SelW mRNA expression suppressed.108 This clearly indicates that neurons depend on an external Se pool to which it can draw from, and has a place lower on the Se brain hierarchy. It is more likely that delivery of Se from astrocytes as SelP to endogenous neuronal apoE2 receptors is the more controlled mechanism of neuronal Se uptake, as opposed to direct translocation of SelP immediately following BBB transit. This study, though clearly useful in investigating what, if any, buffering capacity neurons have for excessive Se, does however highlight a very pertinent point when considering in vitro studies of Se (or any other low-level essential element) metabolism: what conclusions can appropriately be drawn when the multicellular interplay that typically regulates Se hierarchy are removed and the system is viewed in isolation? As such, these studies should be also considered somewhat in vacuo; in this case purely as an example of how excessive Se levels impact upon SelW expression and neurotoxicity.
In murine brain tissue, SelW is expressed in axons and dendrites of neurons in several nuclei, mainly the cortex, hippocampus and cerebellum. It is highly abundant in apical dendrites of most pyramidal neurons in the hippocampus, cingulate cortex and piriform cortex, and is also expressed in synapses.51,109,110 However, as observed for other selenoproteins, SelW expression is dependent on the master regulator SelP, as SelP deleted mice present a reduced level of SelW mRNA.110 SelW is present in survival motor neuron protein (SMN) complexes, the major components of the spliceosome (the machinery that carries out pre-mRNA splicing).111 This particularly interesting study showed that SelW is in part dependent of SMN-complexes due to its neuronal transport along neurites, since SelW was decreased by 17% in neurites of SMN-depleted NSC-34 cells. The role of SelW in the brain remains unclear: its redox motif suggests that this selenoprotein acts as antioxidant in different cells, though in muscle cell cultures depletion of SelW was compensated for by alternate antioxidant proteins, suggesting its major function is not antioxidant activity alone.112 Also, because SelW is highly expressed in brain areas associated with synaptic plasticity, Raman et al.110 suggested that it is essential for maintaining redox homeostasis and mediates proper synaptic adaptation and development.
Several selenoproteins are not highly expressed in brain, such as selenoproteins S (SelS) and H (SelH), though recent studies are attempting to determine their role in CNS, suggesting that whilst they exist in low abundance, they may be essential participants in normal brain function. Speciation of selenoproteins is analytically challenging, and in situ hybridisation often lacks the necessary sensitivity to probe selenoprotein RNA where expression is very low. Selenoprotein S appears to have a neuroprotective effect, and is found mainly in neurons, sparingly in astrocytes; and animal studies have reported that it is typically confined to the hippocampus and cortex.113–115 SelS expression is induced in reactive astrocytes as part of an activation program triggered by astrogliosis-inducing stimuli.114 As a result, SelS overexpression decreased ER-stress and IL-6 release in astrocytes. These results were confirmed by Liu et al.,115 who observed increased SelS expression after 3–7 days of ischemic reperfusion, suggesting that SelS modulates astrocyte function by reducing the synthesis and release of inflammatory cytokines and limiting degradation of unfolded proteins. Considering that previous studies have shown that SelS has an important anti-inflammatory role in different tissue types,116,117 other work regarding the role and therapeutic potential of this selenoprotein in brain are encouraged. In the same way, although SelH is only moderately expressed in the brain,118 overexpression in neuronal cells improved mitochondrial biogenesis and functional performance. Overexpression of SelH in HT-22 neurons increased the respiratory function of mitochondria and preserved UVB stress-induced mitochondrial membrane potential depolarisation. The study of neurons either overexpressing SelH or with the gene ablated revealed that this selenoprotein improved mitochondrial biogenesis signaling via PGC-1α, NRF1 and mitochondrial transcription factor A (Tfam), likely mediated by modulation of CREB dependent PGC-1α activation.61,62 The protective role of SelH is also possibly linked to its capacity to maintain genomic stability, protecting the cells against replicative senescence under chronic oxidative stress.119 According to this hypothesis, besides its direct antioxidant role arising from the presence of the CXXU motif, which prevents ribosomal DNA oxidation in the nucleoli, SelH may induce de novo synthesis of GSH and phase II detoxification enzymes. Although this theory was not evaluated in brain cells, it does pose one possibly important role of this selenoprotein in neurodegenerative diseases associated with ageing. The examples described here exemplify the ways by which selenoproteins can exert a neuroprotective role, even though they are expressed at relatively low levels.
Selenium in brain diseases
Alzheimer's disease
Alzheimer's disease (AD) is a neurodegenerative condition characterised by progressive loss of memory and cognition, and compromising of daily activities. Its key pathological features are neuron death, loss of synaptic connections, extracellular deposition of beta-amyloid (Aβ) protein aggregates (plaques), and intracellular precipitation of hyperphosphorylated tau protein, which manifest as neurofibrillary tangles.120–122 Oxidative stress is a central component of AD pathogenesis, with ROS adversely affecting mitochondrial function, synaptic transmission, axonal transport, and stimulating neuroinflammation.123–125Studies in humans have reported negative correlations between cognitive decline and Se levels.126 Selenium status in the elderly with mild cognitive impairment (MCI) and AD compared to healthy cognitively intact controls revealed a decrease in the Se content of erythrocytes that correlated with a decrease in cognitive function. However, plasma Se was not significantly different between healthy and MCI participants.127 As plasma is a marker of recent exposure and erythrocytes tend to report longer-term Se status, these data suggest that chronic deficiency correlates with cognition decline. González-Domínguez et al.128 reported lower Se levels in serum from AD patients in comparison to MCI elderly, although both groups were not considered deficient. Even in “mild” AD patients (defined as a Mini-Mental State Examination [MMSE] score ≥20) Se levels in plasma were reported to be lower compared to healthy age-matched elderly subjects.129 These data are in agreement with previous studies that suggest that a lack of Se might increase the risk of dementia.127,130 Supplementation with compounds containing Se has shown potential for stimulating cognitive improvement, although this effect cannot be directly attributed to Se alone.131,132 Cardoso et al.133 showed that the daily intake of one Brazil nut over 6 months, which provides about 288 μg of Se, was associated with cognitive performance improvement assessed by constructional praxis and animal naming; both subtests of the CERAD battery. These data are promising; though as Brazil nuts contain other antioxidant components, more studies are required to elucidate the precise effects of selenium intake on cognition via this dietary source.
Animal models and in vitro studies have also provided some insight into the role of Se on AD pathogenesis. A Se deficient diet was associated with increased Aβ plaque formation in the brains of Tg2576 transgenic mice.134 Sodium selenate treatment reduced tau phosphorylation in both cell culture and tau transgenic animal models. In cells, the treatment antagonised the protein phosphatase 2A inhibitor OA,135 activated the protein phosphatase 2A and dephosphorylated tau.136 This treatment prevented and reversed memory and motor deficits; neurofibrillary tangles formation and neurodegeneration in transgenic animals with tau dysfunction. Although SeO32− appears to be more toxic to cells, the treatment with this inorganic Se compound to streptozotocin toxin model of mental impairment improved tolerance to oxidative stress and prevented cognitive decline.137 In hippocampal and cortical cells, it was associated with lower activity of α- and β-secretases and corresponding reduced production of Aβ1–40 and Aβ1–42.138 Treatment of triple-transgenic AD mice expressing mutant forms of human APPswe, PS1M146V, and tauP301L with selenomethionine resulted in reduced total and phosphorylated tau, lower inflammatory biomarkers and improvement in cognition.139 Organoselenium compounds also display interesting effects in improving AD pathology in murine models: p,p′-methoxyl-diphenyl diselenide treatment to streptozotocin-exposed mice slowed memory decline, reduced oxidative stress and normalised acetyl-cholinesterase activity.140 Selenium-containing 8-hydroxyquinoline derivates bound to Cu2+, Fe2+ and Zn2+ inhibited aggregation and disassembled Cu2+-induced aggregates of Aβ.141
Some studies have shown an association between certain selenoproteins and pathological features of AD. In the post-mortem brain SelP was found to colocalise with senile plaques and neurofibrillary tangles,142 and was elevated in the choroid plexus and cerebrospinal fluid of AD patients.143 Considering that oxidative stress is a hallmark of AD, these data could suggest that SelP might act by protecting neurons against the hallmark lesions damage of the disease via its prominent antioxidant role, or by transporting Se to the synthesis of other antioxidant selenoproteins. SelP encodes two His-rich regions that act as high-affinity binding sites for transition metals, potentially blocking metal-mediated Aβ1–42 aggregation and subsequent ROS generation.144,145 This protein also appears to modulate tau aggregation, with Du et al.146 showing that the His-rich domain of SelP inhibited aggregation and disassembled formed aggregates of tau induced by the presence of Cu+/Cu2+. These two His-rich regions associate with the acidic tail of α-tubulin via a charge–charge interaction. This suggests that SelP possibly participates in microtubule events, which are associated with maintenance of cell polarity, intracellular transport and cell division and migration, although the exact role of SelP within microtubules has yet to be elucidated.147 Overexpression of SelM, as well as Se treatment contributes to activation of ERK signalling, which induces a decrease in tau phosphorylation, α-secretase and γ-secretase activity and an increase in β-secretase.100 Selenium treatment to SelM-overexpressing mice resulted in significant ROS inhibition, reduced mitochondrial damage, Aβ aggregation;101 and increased γ-secretase activity.102
Parkinson's disease
Parkinson's disease (PD) is a neurodegenerative disorder characterised by loss of pigmented dopaminergic neurons in the substantia nigra (SN) pars compacta together with the presence of intraneuronal proteinacious inclusions of α-synculein termed Lewy bodies. Several mechanisms have been proposed that induce toxicity inside the cytoplasm of neurons and also in the extracellular space, including α-synuclein/neuromelanin pathology,148 ceruloplasmin dysfunction,149 copper and cuproprotein dyshomeostasis11 and an iron/dopamine-mediated oxidative stress cycle.150 Dopaminergic neurons are particularly vulnerable to oxidative damage, potentially due to their propensity to accumulate iron with age,151,152 and thus oxidative stress presents a central role on PD pathogenesis,153–155 and a logical target of selenoprotein antioxidant activity.An association between Se status and PD pathology is not clear. Shahar et al.156 measured plasma levels in 1,012 elderly (≥65 years of age) Italian subjects and were able to associate performance-based assessments of coordination with circulating Se concentrations. Although there was no measurable association between plasma Se and PD, older subjects demonstrated a significant association between circulating Se levels and decreased performance in neurological coordination tests. In other smaller PD cohorts (<100 PD subjects) no difference in plasma Se levels compared to healthy controls have been identified.157,158
As noted by Shahar et al.,156 causal influence between Se and PD pathology is difficult to ascertain, particularly considering motor function is often related to malnutrition, and thereby circulating Se levels. Dietary Se may indeed be such a confounding factor that any possible causal, or even indicative relationship between circulating Se and PD (or any other chronic illness, for that matter) impossible to elucidate. Selenium in primary food sources is heavily dependent on the Se content of soil, which is highly variable around the world; soil Se levels in parts of Scandinavia, New Zealand and a wide belt of land extending from Siberia through north-eastern China are notably low, and are particularly replete in the US Midwest and Amazon regions of the Americas.159 In contrast to Shahar's comments that PD pathology may cause malnutrition and Se deficiency, a large cohort of PD patients and controls from eastern China, (238 PD; 302 controls) reported that plasma Se (and iron) levels were significantly increased in patients with idiopathic PD.160 Clearly, epidemiological studies with a higher statistical power are required to confirm or deny a possible association between circulating Se and PD. Additionally, it is advisable that several different biological matrices (e.g. erythrocytes, hair, nails etc.) should be used to evaluate Se levels. This would provide a more comprehensive overview of Se levels, and could potentially be used to identify times of exposure. Further, speciation of selenoproteins, as opposed to measuring total Se levels provides another avenue to identify specific Se functionality and its possible role in disease process.
Several studied have suggested a Se-deficient diet may contribute to dopaminergic cell vulnerability,161,162 particularly prior to exposure to parkinsonian neurotoxins.163 Conversely, Se pretreatment somewhat mitigated 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced depletion of striatal dopamine in a dose dependent manner, though only a high dose of 3 mg kg−1 of Se (as SeO32−) produced a statistically significant effect.164 In a paraquat animal model of PD when treated with SeO32− at a low dose (in drinking water at 11.8 μg L−1) presented normalisation of motor activity when co-administered with the toxin.165
SelP is localised within cell bodies, axons and presynaptic terminals of SN neurons, and in PD patients this selenoprotein co-localises within Lewy bodies.50 Like SelP, GPx4 is also reduced in the SN of the PD brain, but is increased relative to cell density in surviving neurons.166 Hauser et al.79 showed that GPx4 is covalently modified by dopamine quinone, an oxidised and reactive metabolite of dopamine, and that this deleterious product of neurotransmitter breakdown may result in degradation and reduced activity of this antioxidant selenoprotein. More studies are necessary to elucidate the precise mechanisms by which GPx metabolism is altered during PD progression.
Other neurodegenerative diseases
A number of other progressive neurological disorders have been studied with regard to a possible association with Se metabolism and selenoprotein activity. Multiple sclerosis (MS) is one such chronic, progressive, inflammatory disease affecting the CNS with a possible link to Se. Although the aetiology of MS remains elusive, it is believed that disease onset and development depend on a genetic and environmental interaction. The pathogenesis of this disease is characterised by abnormal immune response against self-antigens, resulting in inflammation and massive neurodegeneration characterised by marked demyelination. Additionally, infiltration of pro-inflammatory and autoreactive leukocytes into the brain initiates a cascade of events including blood–brain-barrier disruption, microglial activation and excitotoxicity; all potentiating oligodendrocyte death and axon damage.167–169 There is growing evidence that the involvement of oxidative stress in MS pathogenesis as a crucial factor in initiating and perpetuating mechanisms involved in neurodegeneration. Glutathione peroxidase activity, as well other antioxidant enzymes, is decreased in MS patients when compared to the control subjects.170,171 In a proteomic study, among a set of 44 proteins that discriminated experimental autoimmune encephalomyelitis (EAE) rats from control groups, GPx3 was increased in EAE animals.172 Selenium levels tend to decrease in MS, as observed in a cohorts of Iranian173 and Polish patients.174 Complementing the suggestive data regarding a role of Se in MS pathogenesis, Chanaday et al.66 observed that treatment with diphenyl diselenide to the EAE animal model reduced signs of the disease phenotype, partly by preventing the reduction in body weight that accompanies the disease progression and decreased the time and the severity of symptoms. In these animals, the treatment also improved immune system function by diminishing the extent of infiltrating macrophages and controlling mononuclear cell proliferation.Amyotrophic lateral sclerosis (ALS) is another degenerative disease, in this case characterised by motor neuron loss in the brain and spinal cord, which leads to muscle atrophy, weakness and fasciculation.175 Although the cause of ALS is unknown, several studies indicate a link between genetic and environmental factors, including excessive exposure to inorganic Se. This association was first suggested in 1977, when researchers observed a higher prevalence of ALS in a known seleniferous area.176 In a northern Italian locale, where some inhabitants consumed drinking water with unusually high inorganic Se content, a higher risk of ALS was observed, with dose–response relation.177,178 ALS patients presented higher SeO32− levels and lower organoselenium compounds, particularly SelP, in CSF compared with healthy control subjects.179 This finding is something of a curiosity, as SeO32− is not typically found at high levels in biological fluids; conversion to non-redox active forms occurs rapidly through a reaction with GSH, as SeO32− is toxic,180 though improved analytical technology has identified the presence of SeO32− at low ng mL−1 concentrations in urine181 and serum.182 It should be noted that the same method used in this study previously failed to identify SeO32− in CSF samples.183 Selenite is, however, toxic, though paradoxically it has been shown to offer some degree of neuroprotection against lead neurotoxicity in Caenorhabditis elegans,184 though this may result from conversion to other selenoproteins rather than activity of the SeO32− itself. Selenium in excess has neurotoxic effects to motor neuronal cells in vitro. Human neuroblastoma SKNBE cells were more vulnerable to Se toxicity when compared with kidney and prostatic cells when treated with organic and inorganic Se, even at a low dose (0.1 μM).185 Selenite induced degeneration of cholinergic and GABAergic neurons and impaired locomotor activity in Caenorhabditis elegans.186–188 The elevated SeO32− in CSF of ALS patients may be due to excessive exposure to this compound from environmental sources, alteration of the body's detoxification capacity or even genetic modifications.
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease caused by increased repetition of CAG trinucleotide in exon 1 of the Huntington gene, resulting in polyQ-expanded sequence in N-terminal region of the Huntington protein. This genetic alteration makes the Huntington protein more prone to aggregation, which compromises proteasomal activity, impairs mitochondrial function, increases oxidative stress and contributes to neurodegeneration.189–191 Clinically, HD is characterised by chorea, psychiatric disturbance and cognitive decline.192,193 Although Se levels in plasma were not different between HD patients and healthy controls, HD patients presented a decrease in regional brain Se content; specifically in the putamen, dorso-lateral prefrontal cortex, primary visual cortex, cingulate gyrus and cerebellum.194 Treatment of N171-82Q HD mice with SeO32− significantly protected against decreased brain weight and reduced Huntington protein aggregation and oxidised glutathione.194 In another HD mice model (3-nitropropionic acid exposure) treatment with ((Z)-2,3-bis(4-chlorophenylselanyl)prop-2-en-1-ol)(bis selenide) improved locomotor activity and motor coordination, restored succinate dehydrogenase enzyme activity and protected against oxidative stress.195 Although there are few studies investigating Se in HD, these findings suggest that Se may be a potential avenue of exploration with regard to prevention and treatment of HD, and more studies are therefore encouraged.
Conclusions
Selenium has an essential role in the brain; not only due to its direct antioxidant role, but also via its participation in maintaining redox balance, mitochondrial dynamics, regulation of Ca2+ channels and modulation of neurogenesis. The relevance of Se in the pathogenesis of different neurological diseases has been shown by several studies in humans, animal models and cell culture that all associate Se deficiency with a disturbance in brain metabolism. It is important to note that the strong hierarchical nature of Se regulation in the brain, and its ability to remain somewhat efficiently isolated from Se deficiency in the periphery, and thereby in vitro studies that control ambient Se levels in a manner not reflective of in vivo Se metabolism are suited only to investing a specific selenoprotein in isolation, as opposed to the interplay between the 30+ known selenoproteins in toto. Future research is required to fully elucidate the role of different selenoproteins in brain cells, and in turn this will provide potential uses of selenoproteins as biomarkers of normal or pathogenic biological processes in CNS. As recognition of the importance Se has in maintaining normal brain function, studies examining the benefits of Se supplementation are emerging, but literature is still unclear regarding the precise effects of different forms and doses of Se. Thus, additional experiments investigating the potential neurotoxicity of Se overload are also required to better assess the safe range of Se exposures.Notes and references
- K. Schwarz and C. M. Foltz, J. Am. Chem. Soc., 1957, 79, 3292 CrossRef CAS.
- L. Flohe, W. Günzler and H. Schock, FEBS Lett., 1973, 32, 132–134 CrossRef CAS.
- G. V. Kryukov, S. Castellano, S. V. Novoselov, A. V. Lobanov, O. Zehtab, R. Guigó and V. N. Gladyshev, Science, 2003, 300, 1439–1443 CrossRef CAS PubMed.
- M. Roman, P. Jitaru and C. Barbante, Metallomics, 2014, 6, 25–54 RSC.
- J. Gromadzińska, E. Reszka, K. Bruzelius, W. Wsowicz and B. Åkesson, Eur. J. Nutr., 2008, 47, 29–50 CrossRef PubMed.
- D. H. Holben and A. M. Smith, J. Am. Diet. Assoc., 1999, 99, 836–843 CrossRef CAS.
- M. Bélanger, I. Allaman and P. J. Magistretti, Cell Metab., 2011, 14, 724–738 CrossRef PubMed.
- X. Wang and E. K. Michaelis, Front. Aging Neurosci., 2010, 2, 12 Search PubMed.
- A. Federico, E. Cardaioli, P. Da Pozzo, P. Formichi, G. N. Gallus and E. Radi, J. Neurol. Sci., 2012, 322, 254–262 CrossRef CAS PubMed.
- W. R. Markesbery, Free Radical Biol. Med., 1997, 23, 134–147 CrossRef CAS.
- K. M. Davies, S. Bohic, A. Carmona, R. Ortega, V. Cottam, D. J. Hare, J. P. M. Finberg, S. Reyes, G. M. Halliday, J. F. B. Mercer and K. L. Double, Neurobiol. Aging, 2014, 35, 858–866 CrossRef CAS PubMed.
- M. Navarro-Alarcon and C. Cabrera-Vique, Sci. Total Environ., 2008, 400, 115–141 CrossRef CAS PubMed.
- A. A. Turanov, A. V. Lobanov, D. L. Hatfield and V. N. Gladyshev, Nucleic Acids Res., 2013, 41, 6952–6959 CrossRef CAS PubMed.
- V. M. Labunskyy, D. L. Hatfield and V. N. Gladyshev, Physiol. Rev., 2014, 94, 739–777 CrossRef CAS PubMed.
- J. N. Gonzalez-Flores, N. Gupta, L. W. DeMong and P. R. Copeland, J. Biol. Chem., 2012, 287, 38936–38945 CrossRef CAS PubMed.
- A. L. Bifano, T. Atassi, T. Ferrara and D. M. Driscoll, BMC Mol. Biol., 2013, 14, 12 CrossRef CAS PubMed.
- M. E. Budiman, J. L. Bubenik, A. C. Miniard, L. M. Middleton, C. A. Gerber, A. Cash and D. M. Driscoll, Mol. Cell, 2009, 35, 479–489 CrossRef CAS PubMed.
- A. C. Miniard, L. M. Middleton, M. E. Budiman, C. A. Gerber and D. M. Driscoll, Nucleic Acids Res., 2010, 38, 4807–4820 CrossRef CAS PubMed.
- M. J. Bröcker, J. M. L. Ho, G. M. Church, D. Söll and P. O'Donoghue, Angew. Chem., Int. Ed., 2014, 53, 319–323 CrossRef PubMed.
- E. K. Wirth, M. Conrad, J. Winterer, C. Wozny, B. A. Carlson, S. Roth, D. Schmitz, G. W. Bornkamm, V. Coppola, L. Tessarollo, L. Schomburg, J. Köhrle, D. L. Hatfield and U. Schweizer, FASEB J., 2010, 24, 844–852 CrossRef CAS PubMed.
- E. K. Wirth, B. S. Bharathi, D. Hatfield, M. Conrad, M. Brielmeier and U. Schweizer, Biol. Trace Elem. Res., 2014, 158, 203–210 CrossRef CAS PubMed.
- S. Seeher, B. A. Carlson, A. C. Miniard, E. K. Wirth, Y. Mahdi, D. L. Hatfield, D. M. Driscoll and U. Schweizer, Biochem. J., 2014, 462, 67–75 CrossRef CAS PubMed.
- O. Agamy, B. Ben Zeev, D. Lev, B. Marcus, D. Fine, D. Su, G. Narkis, R. Ofir, C. Hoffmann, E. Leshinsky-Silver, H. Flusser, S. Sivan, D. Söll, T. Lerman-Sagie and O. S. Birk, Am. J. Hum. Genet., 2010, 87, 538–544 CrossRef CAS PubMed.
- X.-M. Xu, A. A. Turanov, B. A. Carlson, M.-H. Yoo, R. A. Everley, R. Nandakumar, I. Sorokina, S. P. Gygi, V. N. Gladyshev and D. L. Hatfield, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21430–21434 CrossRef CAS PubMed.
- R. Tobe, S. Naranjo-Suarez, R. A. Everley, B. A. Carlson, A. A. Turanov, P. A. Tsuji, M.-H. Yoo, S. P. Gygi, V. N. Gladyshev and D. L. Hatfield, J. Biol. Chem., 2013, 288, 14709–14715 CrossRef CAS PubMed.
- L. Schomburg and U. Schweizer, Biochim. Biophys. Acta, Gen. Subj., 2009, 1790, 1453–1462 CrossRef CAS PubMed.
- R. Sunde, in Selenium – Its molecular biology and role in human health, ed. D. L. Hatfield, M. Berry and V. N. Gladyshev, Springer, 2012, pp. 137–152 Search PubMed.
- M. T. Howard, B. A. Carlson, C. B. Anderson and D. L. Hatfield, J. Biol. Chem., 2013, 288, 19401–19413 CrossRef CAS PubMed.
- A. Seyedali and M. J. Berry, RNA, 2014, 20, 1248–1256 CrossRef CAS PubMed.
- J. Y. Kim, B. A. Carlson, X.-M. Xu, Y. Zeng, S. Chen, V. N. Gladyshev, B. J. Lee and D. L. Hatfield, Biochem. Biophys. Res. Commun., 2011, 409, 814–819 CrossRef CAS PubMed.
- B. A. Zachara, H. Pawluk, E. Bloch-Boguslawska, K. M. Sliwka, J. Korenkiewicz, Z. Skok and K. Ryć, Arch. Environ. Health, 2001, 56, 461–466 CrossRef CAS PubMed.
- D. Behne and W. Wolters, J. Nutr., 1983, 113, 456–461 CAS.
- M. Haratake, K. Koga, M. Inoue, T. Fuchigami and M. Nakayama, Metallomics, 2011, 3, 1019–1026 RSC.
- M. Kühbacher, J. Bartel, B. Hoppe, D. Alber, G. Bukalis, A. U. Bräuer, D. Behne and A. Kyriakopoulos, J. Neurochem., 2009, 110, 133–142 CrossRef PubMed.
- M. Kühbacher, J. Bartel, D. Alber, G. Bukalis and H. Kühn, 2014, http://www.laborjournal.de/editorials/ed424/Ms421_v425_web.pdf, accessed 11 May 2015.
- P. Ramos, A. Santos, N. R. Pinto, R. Mendes, T. Magalhães and A. Almeida, Biol. Trace Elem. Res., 2014, 163, 89–96 CrossRef PubMed.
- L. R. Ferguson, N. Karunasinghe, S. Zhu and A. H. Wang, Mutat. Res., 2012, 733, 100–110 CrossRef CAS PubMed.
- S. P. Shetty, R. Shah and P. R. Copeland, J. Biol. Chem., 2014, 289, 25317–25326 CrossRef CAS PubMed.
- G. Ballihaut, L. E. Kilpatrick, E. L. Kilpatrick and W. C. Davis, Metallomics, 2012, 4, 533–538 RSC.
- R. F. Burk, K. E. Hill, A. K. Motley, V. P. Winfrey, S. Kurokawa, S. L. Mitchell and W. Zhang, FASEB J., 2014, 28, 3579–3588 CrossRef CAS PubMed.
- A. E. Clatworthy, W. Stockinger, R. H. Christie, W. J. Schneider, J. Nimpf, B. T. Hyman and G. W. Rebeck, Neuroscience, 1999, 90, 903–911 CrossRef CAS.
- S. Kurokawa, K. E. Hill, W. H. McDonald and R. F. Burk, J. Biol. Chem., 2012, 287, 28717–28726 CrossRef CAS PubMed.
- G. E. Olson, V. P. Winfrey, S. K. NagDas, K. E. Hill and R. F. Burk, J. Biol. Chem., 2007, 282, 12290–12297 CrossRef CAS PubMed.
- M. W. Pitts, A. V. Raman, A. C. Hashimoto, C. Todorovic, R. A. Nichols and M. J. Berry, Neuroscience, 2012, 208, 58–68 CrossRef CAS PubMed.
- I. Masiulis, T. A. Quill, R. F. Burk and J. Herz, Biol. Chem., 2009, 390, 67–73 CrossRef CAS PubMed.
- S. W. Caito, D. Milatovic, K. E. Hill, M. Aschner, R. F. Burk and W. M. Valentine, Brain Res., 2011, 1398, 1–12 CrossRef CAS PubMed.
- R. F. Burk, G. E. Olson, K. E. Hill, V. P. Winfrey, A. K. Motley and S. Kurokawa, FASEB J., 2013, 27, 3249–3256 CrossRef CAS PubMed.
- H. Steinbrenner, L. Alili, E. Bilgic, H. Sies and P. Brenneisen, Free Radical Biol. Med., 2006, 40, 1513–1523 CrossRef CAS PubMed.
- M. Scharpf, U. Schweizer, T. Arzberger, W. Roggendorf, L. Schomburg and J. Köhrle, J. Neural Transm., 2007, 114, 877–884 CrossRef CAS PubMed.
- F. P. Bellinger, A. V. Raman, R. H. Rueli, M. T. Bellinger, A. S. Dewing, L. A. Seale, M. A. Andres, J. H. Uyehara-Lock, L. R. White, G. W. Ross and M. J. Berry, J. Parkinson's Dis., 2012, 2, 115–126 CAS.
- A. V. Raman, M. W. Pitts, A. Seyedali, A. C. Hashimoto, L. A. Seale, F. P. Bellinger and M. J. Berry, Genes, Brain Behav., 2012, 11, 601–613 CrossRef CAS PubMed.
- C. N. Byrns, M. W. Pitts, C. A. Gilman, A. C. Hashimoto and M. J. Berry, J. Biol. Chem., 2014, 289, 9662–9674 CrossRef CAS PubMed.
- K.-M. Fang, F.-C. Cheng, Y.-L. Huang, S.-Y. Chung, Z.-Y. Jian and M.-C. Lin, Biol. Trace Elem. Res., 2013, 152, 66–74 CrossRef CAS PubMed.
- P. Bitencourt, L. Belle, G. Bonfanti, L. Cargnelutti, K. D. Bona, P. Silva, F. Abdalla, R. Zanette, R. Guerra, C. Funchal and M. Moretto, Hum. Exp. Toxicol., 2013, 32, 942–949 CAS.
- M. Nazıroğlu, Ö. Çelik, A. C. Uğuz and A. Bütün, Biol. Trace Elem. Res., 2015, 164, 72–79 CrossRef PubMed.
- I. Ben Amara, H. Fetoui, F. Guermazi and N. Zeghal, Int. J. Dev. Neurosci., 2009, 27, 719–726 CrossRef CAS PubMed.
- A. S. Abedelhaffez and A. Hassan, Acta Physiol. Hung., 2013, 100, 197–210 CrossRef CAS PubMed.
- O. L. Adebayo, G. A. Adenuga and R. Sandhir, Nutr. Neurosci., 2014, 17, 268–278 CrossRef CAS PubMed.
- A. C. Uğuz and M. Nazıroğlu, Neurochem. Res., 2012, 37, 1631–1638 CrossRef PubMed.
- M. Nazıroğlu, N. Şenol, V. Ghazizadeh and V. Yürüker, Cell. Mol. Neurobiol., 2014, 34, 895–903 CrossRef PubMed.
- N. Mendelev, S. L. Mehta, S. Witherspoon, Q. He, J. Z. Sexton and P. A. Li, Mitochondrion, 2011, 11, 76–82 CrossRef CAS PubMed.
- S. L. Mehta, N. Mendelev, S. Kumari and P. Andy Li, Int. J. Biochem. Cell Biol., 2013, 45, 604–611 CrossRef CAS PubMed.
- S. Kumari, S. L. Mehta and P. A. Li, PLoS One, 2012, 7, e39382 CAS.
- A. S. de Freitas and J. B. T. Rocha, Neurosci. Lett., 2011, 503, 1–5 CrossRef PubMed.
- A. P. Ardais, G. G. Viola, M. S. Costa, F. Nunes, G. A. Behr, F. Klamt, J. C. F. Moreira, D. O. Souza, J. B. T. Rocha and L. O. Porciúncula, Toxicol. Sci., 2010, 113, 434–443 CrossRef CAS PubMed.
- N. L. Chanaday, A. F. de Bem and G. A. Roth, Neurochem. Int., 2011, 59, 1155–1162 CrossRef CAS PubMed.
- G. Roseni Mundstock Dias, R. Medeiros Golombieski, R. de Lima Portella, G. Pires do Amaral, F. Antunes Soares, J. B. Teixeira da Rocha, C. Wayne Nogueira and N. Vargas Barbosa, Neuroendocrinology, 2014, 100, 45–59 CrossRef CAS PubMed.
- C. A. Brüning, B. M. Gai, S. M. Soares, F. Martini and C. W. Nogueira, Pharmacol., Biochem. Behav., 2014, 125, 15–20 CrossRef PubMed.
- P. M. Chagas, S. G. Rosa, M. H. M. Sari, C. E. S. Oliveira, R. F. S. Canto, S. C. A. da Luz, A. L. Braga and C. W. Nogueira, Pharmacol., Biochem. Behav., 2014, 118, 87–95 CrossRef CAS PubMed.
- I. Musik, M. Kiełczykowska and J. Kocot, BioMetals, 2013, 26, 763–771 CrossRef CAS PubMed.
- S. M. Barayuga, X. Pang, M. A. Andres, J. Panee and F. P. Bellinger, Neurotoxicology, 2013, 37, 240–246 CrossRef CAS PubMed.
- E. Song, C. Su, J. Fu, X. Xia, S. Yang, C. Xiao, B. Lu, H. Chen, Z. Sun, S. Wu and Y. Song, Life Sci., 2014, 109, 37–43 CrossRef CAS PubMed.
- T. Garcia, J. L. Esparza, M. R. Nogués, M. Romeu, J. L. Domingo and M. Gómez, Hippocampus, 2010, 20, 218–225 CAS.
- S. Zhang, C. Rocourt and W.-H. Cheng, Mech. Ageing Dev., 2010, 131, 253–260 CrossRef CAS PubMed.
- P. A. Mitozo, L. F. de Souza, G. Loch-Neckel, S. Flesch, A. F. Maris, C. P. Figueiredo, A. R. S. dos Santos, M. Farina and A. L. Dafre, Free Radical Biol. Med., 2011, 51, 69–77 CrossRef CAS PubMed.
- M.-H. Yoo, X. Gu, X.-M. Xu, J. Y. Kim, B. A. Carlson, A. D. Patterson, H. Cai, V. N. Gladyshev and D. L. Hatfield, Antioxid. Redox Signaling, 2010, 12, 819–827 CrossRef CAS PubMed.
- M. W. Pitts, C. N. Byrns, A. N. Ogawa-Wong, P. Kremer and M. J. Berry, Biol. Trace Elem. Res., 2014, 161, 231–245 CrossRef CAS PubMed.
- N. E. Savaskan, A. Borchert, A. U. Bräuer and H. Kuhn, Free Radical Biol. Med., 2007, 43, 191–201 CrossRef CAS PubMed.
- D. N. Hauser, A. A. Dukes, A. D. Mortimer and T. G. Hastings, Free Radical Biol. Med., 2013, 65, 419–427 CrossRef CAS PubMed.
- J. R. Godoy, M. Funke, W. Ackermann, P. Haunhorst, S. Oesteritz, F. Capani, H. P. Elsasser and C. H. Lillig, Biochim. Biophys. Acta, 2011, 1810, 2–92 CrossRef CAS PubMed.
- D. Silva-Adaya, M. E. Gonsebatt and J. Guevara, Oxid. Med. Cell. Longevity, 2014, 2014, 1–13 CrossRef PubMed.
- A. P. Kudin, B. Augustynek, A. K. Lehmann, R. Kovács and W. S. Kunz, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1901–1906 CrossRef CAS PubMed.
- M. Cohen-Kutner, L. Khomsky, M. Trus, H. Ben-Yehuda, J. M. Lenhard, Y. Liang, T. Martin and D. Atlas, Redox Biol., 2014, 2, 447–456 CrossRef CAS PubMed.
- P. Lopert, B. J. Day and M. Patel, PLoS One, 2012, 7, e50683 CAS.
- J. H. Ahn, J. H. Choi, J. M. Song, C. H. Lee, K.-Y. Yoo, I. K. Hwang, J. S. Kim, H.-C. Shin and M. H. Won, Exp. Gerontol., 2011, 46, 946–952 CrossRef CAS PubMed.
- H. Esen, F. Erdi, B. Kaya, B. Feyzioglu, F. Keskin and L. S. Demir, J. Neuro-Oncol., 2014, 121, 451–458 CrossRef PubMed.
- L. V. Papp, J. Lu, A. Holmgren and K. K. Khanna, Antioxid. Redox Signaling, 2007, 9, 775–806 CrossRef CAS PubMed.
- R. F. Burk and K. E. Hill, Annu. Rev. Nutr., 2005, 25, 215–235 CrossRef CAS PubMed.
- Y. Zhang and V. N. Gladyshev, PLoS Genet., 2008, 4, e1000095 Search PubMed.
- A. Dikiy, S. V. Novoselov, D. E. Fomenko, A. Sengupta, B. A. Carlson, R. L. Cerny, K. Ginalski, N. V. Grishin, D. L. Hatfield and V. N. Gladyshev, Biochemistry, 2007, 46, 6871–6882 CrossRef CAS PubMed.
- F. Musiani, S. Ciurli and A. Dikiy, J. Proteome Res., 2011, 10, 968–976 CrossRef CAS PubMed.
- M. E. Moustafa and H. A. Antar, Biochem. Genet., 2012, 50, 736–747 CrossRef CAS PubMed.
- F. Li, P. B. Lutz, Y. Pepelyayeva, E. S. J. Arnér, C. A. Bayse and S. Rozovsky, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6976–6981 CrossRef CAS PubMed.
- S.-J. Han, B. C. Lee, S. H. Yim, V. N. Gladyshev and S.-R. Lee, PLoS One, 2014, 9, e95518 Search PubMed.
- S. Guariniello, G. Colonna, R. Raucci, M. Costantini, G. Di Bernardo, F. Bergantino, G. Castello and S. Costantini, Biochim. Biophys. Acta, Proteins Proteomics, 2014, 1844, 447–456 CrossRef CAS PubMed.
- A. D. Ferguson, V. M. Labunskyy, D. E. Fomenko, D. Araç, Y. Chelliah, C. A. Amezcua, J. Rizo, V. N. Gladyshev and J. Deisenhofer, J. Biol. Chem., 2006, 281, 3536–3543 CrossRef CAS PubMed.
- J. L. Martin, Structure, 1995, 3, 245–250 CrossRef CAS.
- M. W. Pitts, M. A. Reeves, A. C. Hashimoto, A. Ogawa, P. Kremer, L. A. Seale and M. J. Berry, J. Biol. Chem., 2013, 288, 26121–26134 CrossRef CAS PubMed.
- M. A. Reeves, F. P. Bellinger and M. J. Berry, Antioxid. Redox Signaling, 2010, 12, 809–818 CrossRef CAS PubMed.
- S. Y. Yim, K. R. Chae, S. B. Shim, J. T. Hong, J. Y. Park, C. Y. Lee, H. J. Son, Y. Y. Sheen and D. Y. Hwang, Int. J. Mol. Med., 2009, 24, 91–96 CAS.
- P. Chen, R.-R. Wang, X.-J. Ma, Q. Liu and J.-Z. Ni, Int. J. Mol. Sci., 2013, 14, 4385–4399 CrossRef CAS PubMed.
- Y. Kim, J. S. Goo, I. Y. Kim, J. E. Kim, M. H. Kwak, J. Go, S. Shim, J. T. Hong, D. Y. Hwang and J. K. Seong, Int. J. Mol. Med., 2014, 34, 1688–1698 CAS.
- X. Qiao, J. Tian, P. Chen, C. Wang, J. Ni and Q. Liu, Int. J. Mol. Sci., 2013, 14, 22233–22245 CrossRef PubMed.
- S. C. Starossom, I. D. Mascanfroni, J. Imitola, L. Cao, K. Raddassi, S. F. Hernandez, R. Bassil, D. O. Croci, J. P. Cerliani, D. Delacour, Y. Wang, W. Elyaman, S. J. Khoury and G. A. Rabinovich, Immunity, 2012, 37, 249–263 CrossRef CAS PubMed.
- K. Kajitani, Y. Kobayakawa, H. Nomaru, T. Kadoya, H. Horie and Y. Nakabeppu, NeuroReport, 2014, 25, 171–176 CrossRef CAS PubMed.
- J.-L. Li, H.-F. Ruan, H.-X. Li, S. Li, S.-W. Xu and Z.-X. Tang, Mol. Biol. Rep., 2010, 38, 4015–4022 CrossRef PubMed.
- B.-R. Ou, M.-J. Jiang, C.-H. Lin, Y.-C. Liang, K.-J. Lee and J.-Y. Yeh, BioMetals, 2011, 24, 323–333 CrossRef CAS PubMed.
- J.-L. Li, H.-X. Li, S. Li, X.-J. Gao, S.-W. Xu and Z.-X. Tang, BioMetals, 2012, 25, 459–468 CrossRef CAS PubMed.
- I. J. Cajigas, G. Tushev, T. J. Will, S. T. Dieck, N. Fuerst and E. M. Schuman, Neuron, 2012, 74, 453–466 CrossRef CAS PubMed.
- A. V. Raman, M. W. Pitts, A. Seyedali, A. C. Hashimoto, F. P. Bellinger and M. J. Berry, Brain Behav., 2013, 3, 562–574 CrossRef PubMed.
- F. Rage, N. Boulisfane, K. Rihan, H. Neel, T. Gostan, E. Bertrand, R. Bordonne and J. Soret, RNA, 2013, 19, 1755–1766 CrossRef CAS PubMed.
- W. Xiao-Long, Y. Chuan-Ping, X. Kai and Q. Ou-Jv, Biochemistry, 2010, 75, 201–207 Search PubMed.
- Y. Zhang, Y. Zhou, U. Schweizer, N. E. Savaskan, D. Hua, J. Kipnis, D. L. Hatfield and V. N. Gladyshev, J. Biol. Chem., 2008, 283, 2427–2438 CrossRef CAS PubMed.
- N. Fradejas, M. Del Carmen Serrano Pérez, P. Tranque and S. Calvo, Glia, 2011, 59, 959–972 CrossRef PubMed.
- L. X. Liu, X. Y. Zhou, C. S. Li, L. Q. Liu, S. Y. Huang and S. N. Zhou, J. Neurol. Sci., 2013, 34, 1671–1678 CrossRef PubMed.
- A. A. Turanov, V. A. Shchedrina, R. A. Everley, A. V. Lobanov, S. H. Yim, S. M. Marino, S. P. Gygi, D. L. Hatfield and V. N. Gladyshev, Biochem. J., 2014, 462, 555–565 CrossRef CAS PubMed.
- X. A. Du, H. M. Wang, X. X. Dai, Y. Kou, R. P. Wu, Q. Chen, J. L. Cao, X. Y. Mo and Y. M. Xiong, Osteoarthritis and Cartilage, 2015, 23, 210–216 CrossRef CAS PubMed.
- S. V. Novoselov, G. V. Kryukov, X.-M. Xu, B. A. Carlson, D. L. Hatfield and V. N. Gladyshev, J. Biol. Chem., 2007, 282, 11960–11968 CrossRef CAS PubMed.
- R. T. Y. Wu, L. Cao, B. P. C. Chen and W.-H. Cheng, J. Biol. Chem., 2014, 289, 34378–34388 CrossRef CAS PubMed.
- P. H. Reddy, Brain Res., 2011, 1415, 136–148 CrossRef CAS PubMed.
- P. Mao and P. H. Reddy, Biochim. Biophys. Acta, Mol. Basis Dis., 2011, 1812, 1359–1370 CrossRef CAS PubMed.
- Y. Yoshiyama, V. M. Y. Lee and J. Q. Trojanowski, J. Neurol., Neurosurg. Psychiatry, 2013, 84, 784–795 CrossRef PubMed.
- R. H. Swerdlow, J. M. Burns and S. M. Khan, Biochim. Biophys. Acta, Mol. Basis Dis., 2014, 1842, 1219–1231 CrossRef CAS PubMed.
- A. Picco, M. C. Polidori, M. Ferrara, R. Cecchetti, D. Arnaldi, M. Baglioni, S. Morbelli, P. Bastiani, I. Bossert, G. Fiorucci, A. Brugnolo, M. E. Dottorini, F. Nobili and P. Mecocci, Eur. J. Nucl. Med. Mol. Imaging, 2013, 41, 764–775 CrossRef PubMed.
- S. Haider, S. Saleem, T. Perveen, S. Tabassum, Z. Batool, S. Sadir, L. Liaquat and S. Madiha, Age, 2014, 36, 1291–1302 CrossRef CAS PubMed.
- B. R. Cardoso, V. Silva Bandeira, W. Jacob-Filho and S. M. Franciscato Cozzolino, J. Trace Elem. Med. Biol., 2014, 28, 422–426 CAS.
- B. R. Cardoso, T. P. Ong, W. Jacob-Filho, O. Jaluul, M. I. D. A. Á. Freitas and S. M. F. Cozzolino, Br. J. Nutr., 2010, 103, 803–806 CrossRef CAS PubMed.
- R. González-Domínguez, T. García-Barrera and J. L. Gómez-Ariza, BioMetals, 2014, 27, 539–549 CrossRef PubMed.
- M. G. M. Olde Rikkert, F. R. Verhey, J. W. C. Sijben, F. H. Bouwman, P. L. J. Dautzenberg, M. Lansink, W. M. W. Sipers, D. Z. B. van Asselt, A. M. J. van Hees, M. Stevens, B. Vellas and P. Scheltens, J. Alzheimer's Dis., 2014, 41, 261–271 CAS.
- H. Vural, H. Demirin, Y. Kara, I. Eren and N. Delibas, J. Trace Elem. Med. Biol., 2010, 24, 169–173 CAS.
- P. Scheltens, P. J. G. H. Kamphuis, F. R. J. Verhey, M. G. M. Olde Rikkert, R. J. Wurtman, D. Wilkinson, J. W. R. Twisk and A. Kurz, Alzheimer's Dementia, 2010, 6, 1–10.e11 CrossRef CAS PubMed.
- E. Kesse-Guyot, L. Fezeu, C. Jeandel, M. Ferry, V. Andreeva, H. Amieva, S. Hercberg and P. Galan, Am. J. Clin. Nutr., 2011, 94, 892–899 CrossRef CAS PubMed.
- B. R. Cardoso, D. Apolinário, V. da Silva Bandeira, A. L. Busse, R. M. Magaldi, W. Jacob-Filho and S. M. F. Cozzolino, Eur. J. Nutr., 2015, 1–10, DOI:10.1007/s00394-014-0829-2.
- M. Haratake, S. Yoshida, M. Mandai, T. Fuchigami and M. Nakayama, Metallomics, 2013, 5, 479–483 RSC.
- J. van Eersel, Y. D. Ke, X. Liu, F. Delerue, J. J. Kril, J. Götz and L. M. Ittner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13888–13893 CrossRef CAS PubMed.
- N. M. Corcoran, D. Martin, B. Hutter-Paier, M. Windisch, T. Nguyen, L. Nheu, L. E. Sundstrom, A. J. Costello and C. M. Hovens, J. Clin. Neurosci., 2010, 17, 1025–1033 CrossRef CAS PubMed.
- T. Ishrat, K. Parveen, M. M. Khan, G. Khuwaja, M. B. Khan, S. Yousuf, A. Ahmad, P. Shrivastav and F. Islam, Brain Res., 2009, 1281, 117–127 CrossRef CAS PubMed.
- A.-R. Gwon, J.-S. Park, J.-H. Park, S.-H. Baik, H.-Y. Jeong, D.-H. Hyun, K. W. Park and D.-G. Jo, Neurosci. Lett., 2010, 469, 391–395 CrossRef CAS PubMed.
- G. Song, Z. Zhang, L. Wen, C. Chen, Q. Shi, Y. Zhang, J. Ni and Q. Liu, J. Alzheimer's Dis., 2014, 41, 85–99 CAS.
- S. Pinton, C. A. Brüning, C. E. Sartori Oliveira, M. Prigol and C. W. Nogueira, J. Nutr. Biochem., 2013, 24, 311–317 CrossRef CAS PubMed.
- Z. Wang, Y. Wang, W. Li, F. Mao, Y. Sun, L. Huang and X. Li, ACS Chem. Neurosci., 2014, 5, 952–962 CrossRef CAS PubMed.
- F. P. Bellinger, Q. P. He, M. T. Bellinger and Y. Lin, J. Alzheimer's Dis., 2008, 15, 465–472 CAS.
- R. H. L. H. Rueli, A. C. Parubrub, A. S. T. Dewing, A. C. Hashimoto, M. T. Bellinger, E. J. Weeber, J. H. Uyehara-Lock, L. R. White, M. J. Berry and F. P. Bellinger, J. Alzheimer's Dis., 2015, 44, 379–383 CAS.
- X. Du, H. Li, Z. Wang, S. Qiu, Q. Liu and J. Ni, Metallomics, 2013, 5, 861–870 RSC.
- X. Du, Z. Wang, Y. Zheng, H. Li, J. Ni and Q. Liu, Inorg. Chem., 2014, 53, 1672–1678 CrossRef CAS PubMed.
- X. Du, Y. Zheng, Z. Wang, Y. Chen, R. Zhou, G. Song, J. Ni and Q. Liu, Inorg. Chem., 2014, 53, 11221–11230 CrossRef CAS PubMed.
- X. Du, S. Qiu, Z. Wang, R. Wang, C. Wang, J. Tian and Q. Liu, Int. J. Mol. Sci., 2014, 15, 10199–10214 CrossRef PubMed.
- G. M. Halliday, A. Ophof, M. Broe, P. H. Jensen, E. Kettle, H. Fedorow, M. I. Cartwright, F. M. Griffiths, C. E. Shepherd and K. L. Double, Brain, 2005, 128, 2654–2664 CrossRef PubMed.
- S. Ayton, P. Lei, J. A. Duce, B. X. W. Wong, A. Sedjahtera, P. A. Adlard, A. I. Bush and D. I. Finkelstein, Ann. Neurol., 2013, 73, 554–559 CrossRef CAS PubMed.
- D. J. Hare, P. Lei, S. Ayton, B. R. Roberts, R. Grimm, J. L. George, D. P. Bishop, A. D. Beavis, S. J. Donovan, G. McColl, I. Volitakis, C. L. Masters, P. A. Adlard, R. A. Cherny, A. I. Bush, D. I. Finkelstein and P. A. Doble, Chem. Sci., 2014, 5, 2160 RSC.
- L. Zecca, A. Stroppolo, A. Gatti, D. Tampellini, M. Toscani, M. Gallorini, G. Giaveri, P. Arosio, P. Santambrogio and R. G. Fariello, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9843 CrossRef CAS PubMed.
- D. Hare, S. Ayton, A. Bush and P. Lei, Front. Aging Neurosci., 2013, 5, 34 Search PubMed.
- C. Venkateshappa, G. Harish, R. B. Mythri, A. Mahadevan, M. M. S. Bharath and S. K. Shankar, Neurochem. Res., 2011, 37, 358–369 CrossRef PubMed.
- G. S. Gaki and A. G. Papavassiliou, NeuroMol. Med., 2014, 16, 217–230 CrossRef CAS PubMed.
- A. Recasens and B. Dehay, Front. Neuroanat., 2014, 8, a159 Search PubMed.
- A. Shahar, K. V. Patel, R. D. Semba, S. Bandinelli, D. R. Shahar, L. Ferrucci and J. M. Guralnik, Mov. Disord., 2010, 25, 1909–1915 CrossRef PubMed.
- S. Younes-Mhenni, M. Aissi, N. Mokni, A. Boughammoura-Bouatay, S. Chebel, M. Frih-Ayed, A. Kerkeni, M. Bost, G. Chazot, M. T. Sfar and M. H. Sfar, Tunis. Med., 2013, 91, 402–405 CAS.
- L. K. Mischley, J. Allen and R. Bradley, J. Neurol. Sci., 2012, 318, 72–75 CrossRef CAS PubMed.
- G. F. Combs, Br. J. Nutr., 2001, 85, 517–547 CrossRef CAS.
- H.-W. Zhao, J. Lin, X.-B. Wang, X. Cheng, J.-Y. Wang, B.-L. Hu, Y. Zhang, X. Zhang and J.-H. Zhu, PLoS One, 2013, 8, e83060 Search PubMed.
- M. L. Vizuete, V. Steffen, A. Machado and J. Cano, Eur. J. Pharmacol., 1994, 270, 183–187 CAS.
- H.-C. Kim, W.-K. Jhoo, E.-J. Shin and G. Bing, Brain Res., 2000, 862, 247–252 CrossRef CAS.
- D. J. Hare, P. A. Adlard, P. A. Doble and D. I. Finkelstein, Metallomics, 2013, 5, 91–109 RSC.
- H. A. Khan, Neurochem. Int., 2010, 57, 489–491 CrossRef CAS PubMed.
- J. H. Ellwanger, P. Molz, D. R. Dallemole, A. Pereira Dos Santos, T. E. Muller, L. Cappelletti, M. Goncalves da Silva, S. I. Rech Franke, D. Pra and J. A. Pegas Henriques, Nutrition, 2015, 31, 359–365 CrossRef CAS PubMed.
- F. P. Bellinger, M. T. Bellinger, L. A. Seale, A. S. Takemoto, A. V. Raman, T. Miki, A. B. Manning-Boğ, M. J. Berry, L. R. White and G. W. Ross, Mol. Neurodegener., 2011, 6, 8 CrossRef CAS PubMed.
- R. Gandhi, A. Laroni and H. L. Weiner, J. Neuroimmunol., 2010, 221, 7–14 CrossRef CAS PubMed.
- B. Broux, P. Stinissen and N. Hellings, Crit. Rev. Immunol., 2013, 33, 283–306 CrossRef CAS.
- G. G. Ortiz, F. P. Pacheco-Moisés, M. Á. Macías-Islas, L. J. Flores-Alvarado, M. A. Mireles-Ramírez, E. D. González-Renovato, V. E. Hernández-Navarro, A. L. Sánchez-López and M. A. Alatorre-Jiménez, Arch. Med. Res., 2014, 45, 687–697 CrossRef CAS PubMed.
- S. Ljubisavljevic, I. Stojanovic, T. Cvetkovic, S. Vojinovic, D. Stojanov, D. Stojanovic, V. Bojanic, D. Stokanovic and D. Pavlovic, NeuroImmunoModulation, 2014, 21, 13–20 CrossRef CAS PubMed.
- I. Tasset, E. Agüera, F. Sánchez-López, M. Feijóo, A. I. Giraldo, A. H. Cruz, F. Gascón and I. Túnez, Clin. Biochem., 2012, 45, 440–444 CrossRef CAS PubMed.
- T. Rosenling, M. P. Stoop, A. Attali, H. van Aken, E. Suidgeest, C. Christin, C. Stingl, F. Suits, P. Horvatovich, R. Q. Hintzen, T. Tuinstra, R. Bischoff and T. M. Luider, J. Proteome Res., 2012, 11, 2048–2060 CrossRef CAS PubMed.
- M. Mehrpour, A. Kyani, M. Tafazzoli, F. Fathi and M. T. Joghataie, Magn. Reson. Chem., 2013, 51, 102–109 CAS.
- K. Socha, J. Kochanowicz, E. Karpińska, J. Soroczyńska, M. Jakoniuk, Z. Mariak and M. H. Borawska, Nutr. J., 2014, 13, 62 CrossRef PubMed.
- L. P. Rowland and N. A. Shneider, N. Engl. J. Med., 2001, 344, 1688–1700 CrossRef CAS PubMed.
- A. W. Kilness and F. H. Hichberg, JAMA, J. Am. Med. Assoc., 1977, 237, 2843–2844 CrossRef CAS.
- M. Vinceti, F. Bonvicini, K. J. Rothman, L. Vescovi and F. Wang, Environ. Health, 2010, 9, 77 CrossRef CAS PubMed.
- M. Vinceti, D. Guidetti, M. Pinotti, S. Rovesti, M. Merlin, L. Vescovi, M. Bergomi and G. Vivoli, Epidemiology, 1996, 7, 529–532 CrossRef CAS PubMed.
- M. Vinceti, N. Solovyev, J. Mandrioli, C. M. Crespi, F. Bonvicini, E. Arcolin, E. Georgoulopoulou and B. Michalke, Neurotoxicology, 2013, 38, 25–32 CrossRef CAS PubMed.
- C. M. Weekley and H. H. Harris, Chem. Soc. Rev., 2013, 42, 8870–8894 RSC.
- S. Kokarnig, A. Tsirigotaki, T. Wiesenhofer, V. Lackner, K. A. Francesconi, S. A. Pergantis and D. Kuehnelt, J. Trace Elem. Med. Biol., 2015, 29, 83–90 CAS.
- Ò. Palacios, J. R. Encinar, D. Schaumlöffel and R. Lobinski, Anal. Bioanal. Chem., 2006, 384, 1276–1283 CrossRef PubMed.
- B. Michalke and A. Berthele, J. Anal. At. Spectrom., 2011, 26, 165–170 RSC.
- W.-H. Li, Y.-C. Shi, I.-L. Tseng and V. H.-C. Liao, PLoS One, 2013, 8, e62387 CAS.
- T. Maraldi, M. Riccio, L. Zambonin, M. Vinceti, A. De Pol and G. Hakim, Neurotoxicology, 2011, 32, 180–187 CrossRef CAS PubMed.
- K. L. Morgan, A. O. Estevez, C. L. Mueller, B. Cacho-Valadez, A. Miranda-Vizuete, N. J. Szewczyk and M. Estevez, Toxicol. Sci., 2010, 118, 530–543 CrossRef CAS PubMed.
- A. O. Estevez, K. L. Morgan, N. J. Szewczyk, D. Gems and M. Estevez, Neurotoxicology, 2014, 41, 28–43 CrossRef CAS PubMed.
- A. O. Estevez, C. L. Mueller, K. L. Morgan, N. J. Szewczyk, L. Teece, A. Miranda-Vizuete and M. Estevez, Neurotoxicology, 2012, 33, 1021–1032 CrossRef CAS PubMed.
- J. P. Vonsattel, R. H. Myers, T. J. Stevens, R. J. Ferrante, E. D. Bird and E. P. Richardson Jr., J. Neuropathol. Exp. Neurol., 1985, 44, 559–577 CrossRef CAS PubMed.
- A. Bush, W. Pettingell, G. Multhaup, M. D. Paradis, J. Vonsattel, J. Gusella, K. Beyreuther, C. Masters and R. Tanzi, Science, 1994, 265, 1464–1467 CAS.
- T.-C. Ju, H.-M. Chen, Y.-C. Chen, C.-P. Chang, C. Chang and Y. Chern, Biochim. Biophys. Acta, Mol. Basis Dis., 2014, 1842, 1668–1680 CrossRef CAS PubMed.
- A. D. Ha and V. S. C. Fung, Curr. Opin. Neurol., 2012, 25, 491–498 CrossRef PubMed.
- J. Paulsen, R. Ready, J. Hamilton, M. Mega and J. Cummings, J. Neurol., Neurosurg. Psychiatry, 2001, 71, 310–314 CrossRef CAS.
- Z. Lu, E. Marks, J. Chen, J. Moline, L. Barrows, M. Raisbeck, I. Volitakis, R. A. Cherny, V. Chopra, A. I. Bush, S. Hersch and J. H. Fox, Neurobiol. Dis., 2014, 71C, 34–42 CrossRef PubMed.
- C. F. Bortolatto, C. R. Jesse, E. A. Wilhelm, P. M. Chagas and C. W. Nogueira, Neurotoxic. Res., 2012, 23, 214–224 CrossRef PubMed.
- M. Birringer, S. Pilawa and L. Flohé, Nat. Prod. Rep., 2002, 19, 693–718 RSC.
Footnote |
| † Co-senior authors. |
| This journal is © The Royal Society of Chemistry 2015 |