
One-pot synthesis of super-bright fluorescent nanogel contrast agents containing a dithiomaleimide fluorophore†
Mathew P.
Robin
a,
Jeffery E.
Raymond
*b and
Rachel K.
O'Reilly
*a
aUniversity of Warwick, Department of Chemistry, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: r.k.o-reilly@warwick.ac.uk; Fax: +44 0 247 652 4112; Tel: +44 0 247 652 3236
bDepartment of Chemistry and Laboratory for Synthetic-Biologic Interactions, Texas A&M University, College Station, Texas 77842-3012, USA. E-mail: jeffery.raymond@mail.chem.tamu.edu
First published on 27th October 2014
Abstract
Fluorescent nanogels with radii ranging from 12–17 nm, high quantum yield, concentration-independent emission, and fluorescent lifetimes of ca. 25 ns have been synthesised in a one-pot process. Solutions demonstrate a concentration independent emission between 0.1–10−3 wt%, allowing for true quantitative imaging where dye emission is a measure of local nanoparticle concentration.
Conceptual insightsFluorescent nanoparticles show a range of benefits over small molecule organic dyes for use in bioimaging and optical sensing; they display superior optical properties including high photostability and brightness, and can also possess greater biocompatibility than conventional organic dyes. Current limitations to the design and fabrication of nanoparticle contrast agents include synthetic complexity, and fluorophore self-quenching within the particle. Non-covalent dye encapsulation within a preformed nanoparticle provides a simple approach to fluorescent labelling, however, dye aggregation within the particles and leaching to the external environment leads to diminished optical properties. Attempts to improve performance by covalently attaching dyes is often synthetically complex, with prevention of intraparticle quenching still more demanding. In the present study we use a fluorophore that is easily incorporated into a methacrylate monomer to demonstrate a simple one-pot route to highly emissive fluorescent nanoparticles. Fluorophore self-quenching is suppressed within these nanogel particles, leading them to outperform commonly used contrast agents. |
Introduction
Fluorescence spectroscopy and microscopy are widely used techniques for the detection and visualisation of compounds containing fluorophores. Fluorescently-labelled nanoparticles possess several features that make them more valuable for this purpose than classical fluorescent organic dyes.1 Organic dyes may have low absorption coefficients, which reduces detection sensitivity, and are susceptible to photobleaching, thus limiting their use to short-term measurements. Furthermore, organic fluorophores often have short emission lifetimes, which can prohibit use in time-resolved measurements, while the toxicity of many organic fluorophores impedes their application to in vitro and in vivo analysis.2 On the contrary, polymer nanoparticles with fluorescent functionality often contain multiple fluorophores leading to brighter emission, while the encapsulation of fluorophores in a macromolecular structure improves dye stability, and can impart biocompatibility to the fluorescently-labelled agent. Dyes can be incorporated either by non-covalent encapsulation, or by covalent attachment, with the latter strategy preferable as it is more efficient and stoichiometrically precise than physical absorption of the fluorophore, and also reduces leaching of the dye from the nanoparticle.1 Covalent fluorophore labelling of polymer nanoparticles can be achieved in a variety of ways. Amphiphilic block copolymers can be labelled with fluorophores (using functional initiators and monomers, or by post-polymerisation functionalisation), with subsequent self-assembly providing fluorescently labelled polymer micelles.3–6 Post-assembly labelling can also be performed, using particles and dyes that are functionalised with complementary reactive groups.7–9 Nanogel particles synthesised by emulsion or mini-emulsion polymerisation,10 can be fluorescently-labelled by several different routes. In contrast to the encapsulation of dye molecules during11 or after12,13 the (mini)emulsion polymerisation the use of functional monomers allows for the covalent attachment of fluorophores,14,15 while post synthesis functionalisation has also been demonstrated.16,17 Fluorescent polymer nanoparticles can also be formed from fluorescent π-conjugated polymers. This can be achieved by performing the step-growth polymerisation in dispersion,18 or by a post-polymerisation route.19,20 Current common strategies for the generation of polymeric fluorescence contrast agents for bioapplications suffer from a variety of complications during characterisation before transition to in vitro and in vivo studies.21 Further difficulties in the generation of polymeric nanotheranostics are the ambiguity of labelling location, particle-to-particle variations in labelling density and the poorly understood effects of particle–dye, dye–dye and dye–solvent interactions on emission response in biologically relevant environments.21,22We have recently reported that the dithiomaleimide (DTM) group is a highly versatile fluorophore which can be simply incorporated into polymers and polymer nanoparticles using functional monomers and initiators for both controlled free radical and ring-opening polymerisation.23–26 We demonstrated that nanoparticles containing the DTM fluorophore could be prepared by amphiphilic block copolymer synthesis using a DTM functional initiator, followed by block copolymer self-assembly in water to produce spherical micelles.25 While the small molecule DTM exhibited all of the typical effects expected of a small dye molecule (quenching, short emission lifetime), when incorporated into the micellar state the DTM does not self-quench. The result was that the DTM-labelled particles showed a concentration independent molar emission and anisotropy, and a long fluorescence lifetime (20 ns). In addition, by using time-domain fluorescence-lifetime imaging (FLIM) in vitro it was possible to resolve assembled and dis-assembled micelles (i.e. unimers) due to a significantly different fluorescent lifetime for the two states.25 In this work we instead chose a simpler synthetic route to DTM functional fluorescent nanoparticles, allowing synthesis in a one-step process. We utilised a recently developed DTM-functional methacrylate monomer,26 in the fabrication of surfactant stabilised nanogel particles by radical cross-linking polymerisation in emulsion.
Results and discussion
Nanoparticle synthesis
The fluorescently labelled DTM-functional particles were formed by radical cross-linking emulsion polymerisation. The nanoparticle scaffold comprised of poly(methyl methacrylate) (PMMA), with crosslinking density (CLD) controlled by copolymerisation of ethyleneglycol dimethacrylate (EGDMA) as detailed in the ESI.† Covalent DTM dye attachment was achieved by copolymerisation of the recently reported monomer dithiomaleimide methacrylate (DTMMA),26 with a variable degree of functionalisation (DoF). Potassium persulfate was used as the radical initiator, and the reaction was performed at 70 °C for 14 h. The reaction solvent was nanopure water (18.2 MΩ cm) with an MMA concentration of 10 g L−1. A high concentration (2 g L−1) of sodium dodecyl sulfate (SDS) was used as the surfactant to target nanogels with radii in the 10–25 nm size range, in accordance with previous work in our group.27,28 (Scheme 1). | ||
| Scheme 1 Synthesis of DTM-labelled nanogels by radical cross-linking emulsion polymerisation. | ||
DoF was varied over three orders of magnitude (for a constant CLD of 0.5 wt%) from 0.3 mol% to 0.0003 mol%, while CLD was also varied (for a constant DoF of 0.03 mol%) as 0.5 wt%, 3 wt% and 10 wt% so that the effect of both dye concentration and mobility in the nanogel core on fluorescence emission could be investigated (Table 1). After the removal of excess surfactant by exhaustive dialysis against water (18.2 MΩ cm), the resultant nanogel particle solutions were characterised by dynamic and static light scattering (DLS and SLS),29 allowing determination of hydrodynamic radius (Rh) and molecular weight (Mw). For each sample, measurements were made with c = 5, 4, 3, 2 & 1 g L−1, at θ = 40°–150° (10° step), and the dn/dc was determined using a differential refractometer. In DLS measurements the autocorrelation functions showed a single mode of decay indicating the presence of a single well dispersed species in solution. From the resultant diffusion coefficient (D), particle Rh was calculated (see ESI, Fig. S2†). Values of Rh were very similar for all samples, ranging from 12.0 nm to 16.5 nm (Table 1 and Fig. 1a). From these measurements, nanogel volume (VNG) could also be calculated, as the volume of a sphere with radius Rh. Particle Mw was measured by SLS, which allowed the average number of DTM units per nanogel (DoFNG) to be calculated from the molar degree of functionalisation (DoF) and Mw (see ESI, Fig. S3†). Knowledge of VNG and DoFNG therefore allowed the local DTM concentration within the nanogels ([DTMNG]) to be derived (see Table 1 for values and ESI for further details†).
| NG1 | NG2 | NG3 | NG4 | NG5 | NG6 | |
|---|---|---|---|---|---|---|
| DoF (mol%) | 0.3 | 0.03 | 0.03 | 0.03 | 0.003 | 0.0003 |
| CLD (wt%) | 0.5 | 0.5 | 3 | 10 | 0.5 | 0.5 |
| R h (nm) | 12.0 | 16.5 | 14.0 | 12.5 | 15.6 | 15.3 |
| M w (Mg mol−1) | 2.4 | 11.6 | 4.8 | 4.0 | 9.7 | 7.8 |
| DoFNG | 71 | 34 | 14 | 12 | 2.8 | 0.23 |
| [DTMNG] (mM) | 16 | 3.0 | 2.0 | 2.3 | 0.30 | 0.025 |
| τ Av,I (ns) | 26.0 | 24.4 | 25.1 | 25.8 | 26.1 | 25.5 |
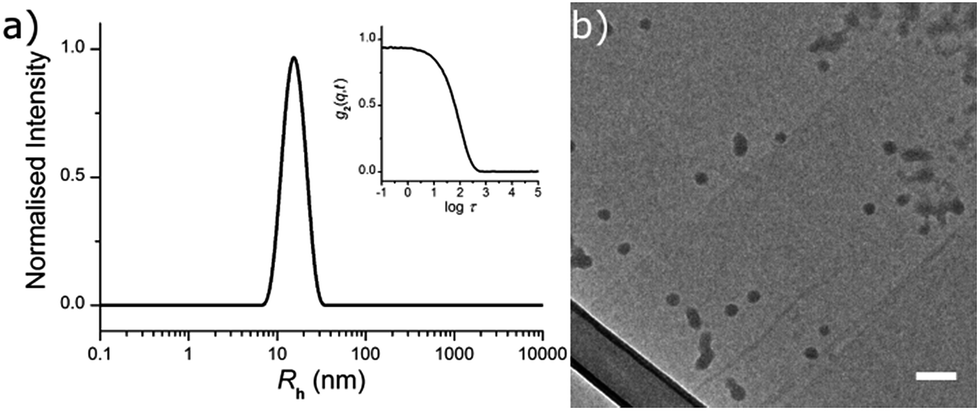 | ||
| Fig. 1 (a) Size distribution obtained by DLS (detection angle of 90°) for a solution of NG2 at 1 g L−1, and the corresponding autocorrelation function (inset); (b) NG2 imaged by TEM on a GO support. The particles showed a tendency to aggregate upon drying (top right corner). Scale bar 100 nm. | ||
Nanogels were also imaged by dry state transmission electron microscopy (TEM), using a graphene oxide (GO) substrate.30 The particles showed a tendency to aggregate when dried to the GO surface (as opposed to their behaviour in solution), however, individual particles were observed to possess a spherical morphology (Fig. 1b).
Steady-state fluorescence
The excitation and emission of the nanogel solutions were characterised by recording a 2D fluorescence spectrum (for example NG1 in Fig. 2a). Excitation maxima are observed at both ca. 275 nm and ca. 415 nm, which have the same corresponding emission maximum at ca. 510 nm. This is in accordance with the fluorescence profile of the small molecule DTM dyes and the DTM labelled polymers and micelles previously reported,24–26 indicating that covalent attachment of the DTM unit into a nanogel particle has not significantly affected the wavelengths of excitation and emission. | ||
| Fig. 2 (a) 2D excitation–emission spectra with a 5 nm step for an aqueous solution of NG1. (b) Molar emission and anisotropy for nanogels with CLD = 0.5 wt% with variable DoF (NG1,2,5,6), and (c) DoF = 0.03 mol% with variable CLD (NG2–4). (d) Molar emissivity vs. concentration for aqueous solutions of NG1 and phloxine B. | ||
The quantum yield (Q) for the aqueous solution of NG1 (which has the highest DTM DoF) was calculated relative to the reference 5-(6)-carboxyfluorescein (5(6)-FAM) at 445 nm and found to be 54% (see ESI for details†). This value is 50-fold higher than the analogous small molecule dithiobutanemaleimide which has Q = 1.1% in methanol.25 The significant difference in Q is attributed to the protection of the DTM fluorophore from solvent/collisional quenching afforded by the nanogel. The molar extinction coefficient (ε) for the aqueous solution of NG1 was measured at the absorption maxima (λabs,max) 405 nm. Using the particle molecular weight calculated by SLS, a value of ε = 1.37 × 105 M−1 cm−1 was obtained with respect to nanogel concentration. Emission brightness can also be calculated as the product of quantum yield and extinction coefficient (Q × ε), which is equal to 4.51 × 104 M−1 cm−1 at the emission maxima (λem,max) 420 nm. Values of ε and brightness can also be calculated based on DTM dye concentration, using the known molarity of DTM-methacrylate used in the nanogel synthesis. In this case ε = 1.89 × 103 M−1 cm−1 per dye unit at λem,max = 405 nm, with the corresponding brightness of 6.24 × 102 M−1 cm−1 per dye unit at 420 nm. While the other nanogel samples (NG2–NG6) displayed bright emission (vide infra), it was not possible to calculate quantum yield and brightness because absorption was too low at 445 nm to obtain accurate measurements as a result of the nanogels' lower DoF.
Steady-state anisotropy (r) was measured for the solutions of nanogels (note that it was not possible to measure r for NG6 (lowest DoF) due to overlap of the Raman scattering peak of water with the DTM emission at this low emission intensity). In all cases anisotropy was high (r > 0.2) indicating that the DTM fluorophore had been successfully incorporated into a macromolecular structure (Fig. 2b and c). In comparison, we have previously shown that analogous small molecule DTM dyes have r ca. 0 in solution.25
The steady-state emission spectra (λex = 420 nm, λem = 510 nm) for nanogel samples with constant CLD = 0.5 wt% (NG1,2,5,6) showed that molar emission increased slightly by a factor of 4, as DoF (and also [DTMNG]) decreased by a factor of 1000 (Fig. 2b). The similarity in molar emission between NG1 (DoF = 0.3 mol%) and NG6 (DoF = 0.0003 mol%) shows that even at the highest DoF there is little intraparticle dye self-quenching, as light scattering measurements showed that NG6 contained on average less than one fluorophore per particle (Table 1). Varying the CLD from 10 wt% to 0.5 wt% for nanogels with DoF = 0.03 mol% (NG2–4) was found to have almost no effect on molar emission (Fig. 2b), with values of 2.37 × 108 ± 0.16 × 108, suggesting that reducing the core mobility (by increasing the CLD) is not required to prevent dye self-quenching (Fig. 2c).
For the nanoparticles with the highest dye loading (NG1, DoF = 0.3 wt%) a study of molar emission as a function of concentration was performed. Between 0.1–10−3 wt% molar emission remained constant, with only a slight decrease (ca. 10%) between 1–0.1 wt%. This feature would allow the DTM functional nanoparticles to be used as a quantitative imaging agent where dye emission is a true measure of local nanogel concentration. In contrast, the commonly used commercially available water soluble dye phloxine B shows a ×105-fold decrease in molar emission between 1–0.01 wt%, due to fluorophore aggregation and self-quenching (Fig. 2d). While polymeric nanoparticles which emit in the near infrared region show many benefits (reduced photon scattering and background cellular autofluorescence) over visible region emitters for in vivo imaging,31,32 the similarity of the DTM emission to FITC33 (λexca. 520 nm) and the high local dye concentrations and quantum yield achievable with these DTM nanogels would not preclude their potential application in vivo.
Time-resolved fluorescence spectroscopy
Solution state fluorescence lifetime was measured for the nanogel samples, using time-correlated single photon counting (TCSPC). All samples showed very similar emission decay (Fig. 3a), with an intensity averaged fluorescence lifetime (τAv,I) of ca. 25 ns obtained by fitting to a multi-exponential decay (details in ESI†). For comparison, we have previously reported that the solution state average fluorescence lifetime of small molecule DTM dyes is ca. 1–2 ns, whereas diblock copolymer micelles containing the DTM fluorophore at the core–corona interface had τAv,I = 20 ns when assembled into spherical micelles.25 These values of τAv,I for the DTM functional nanogels are significantly above that of cellular autofluorescence which is caused by endogenous fluorescent biomolecules such as keratin, collagen, flavins, metal-free porphyrins and NAD(P)H coenzymes, and is typically on the order of 2–3 ns.34 This means that the DTM containing nanogels synthesised here would be ideal as in vitro fluorescence-lifetime imaging microscopy (FLIM) contrast agents, as we have previously demonstrated for DTM functional polymer micelles.25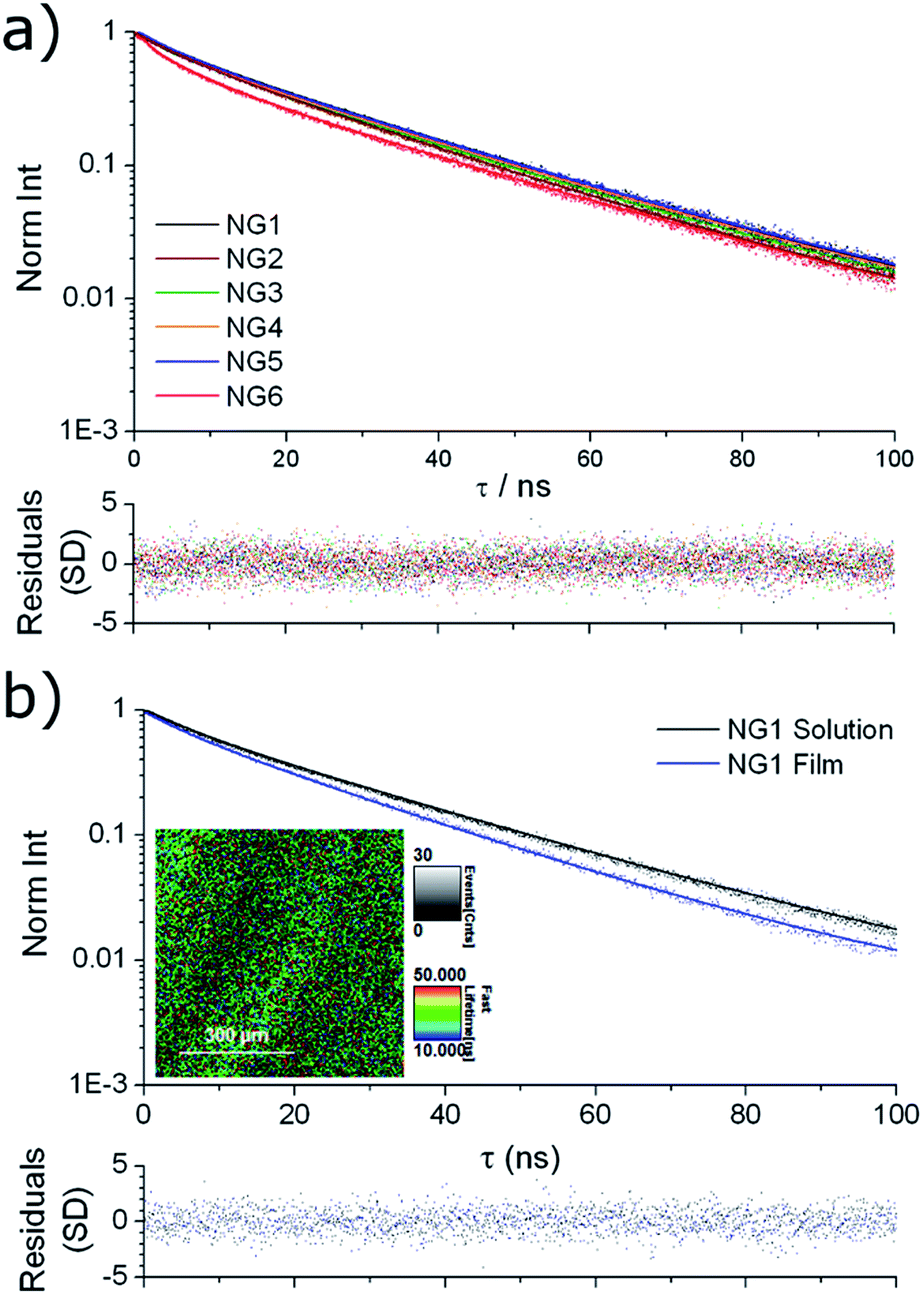 | ||
| Fig. 3 Fluorescence lifetime decay spectra (points) with fitting (line) and residuals (bottom) for (a) NG1–6 in solution; (b) NG1 in solution and dried to a film, with FLIM image of the film (inset). | ||
The fluorescence lifetime of thin films prepared by drying the nanogel solutions to a glass slide (investigated using FLIM) provided further information about the nature of the dye incorporation into the nanogel particles. In all cases near identical fluorescence decay was observed when comparing nanogel solutions and films. For example, the comparison for NG1 is shown in Fig. 3b, with fitting of the decays indicating τAv,I = 23.4 ns for the nanogel films, and τAv,I = 26.0 ns for the nanogel solutions. The similarity in the fluorescence intensity decay between nanogel particles either in solution or dehydrated, indicates that encapsulation within a nanogel provides excellent protection to the DTM fluorophore's excited state from collisional quenching with the surrounding solvent. The result is a drastically increased fluorescence lifetime when compared to solutions of the small molecule DTM dyes.
The data presented here demonstrates that covalent attachment of the DTM fluorophore inside a PMMA nanogel virtually eliminates both self-quenching caused by dye aggregation, and collisional quenching from the surrounding solvent. Given the quasi-encapsulated nature of the nanogels and the macromolecular architecture, it is reasonable to conclude that the resistance to quenching in these systems is related to both long range superstructural isolation and shorter range volume exclusion due to polymeric bonding. When considering the high concentrations of fluorophore in the particle cores (up to 16 mM) the lack of self-quenching is quite remarkable, as most dyes or dye/polymer blends would be ‘off-switched’ in the solid state at these concentrations. This highlights the advantage of a covalent dye attachment, as opposed to non-covalent encapsulation, which is facilitated by the ease with which DTM-functional monomers can be synthesised and polymerised.
Conclusions
A simple one-pot procedure has been used to synthesise aqueous solutions of nanogel particles labelled with a DTM fluorophore. By simply varying the ratio of reagents, the loading of DTM as well as the cross-linking density can be easily tailored, with particle radii in the range 12–17 nm. Covalent attachment of the DTM unit to the nanogel particles provides protection to the fluorophore from solvent-quenching and self-quenching, leading to very bright emission and significant fluorescence lifetimes. Furthermore, the fluorescent nanogels display a concentration independent molar emission profile up to very high concentration (0.1 wt%). When used as contrast agents these fluorescent nanogels could therefore be applied for true quantitative imaging, and for fluorescence-lifetime imaging microscopy. We are excited by the potential to expand this simple synthetic approach to prepare fluorescent particles with further functionality, for example response to an external stimulus (e.g. temperature, pH), using the vast library of functional and responsive monomers that have been previously utilised in emulsion polymerisation.Acknowledgements
We thank the EPSRC for funding, and the IAS at the University of Warwick for an Early Career Fellowship (MPR). Equipment used in this research was funded in part through Advantage West Midlands (AWM) Science City Initiative and in part by the ERDF. We thank Dr Anaïs Pitto-Barry for TEM analysis.Notes and references
- V. Sokolova and M. Epple, Nanoscale, 2011, 3, 1957–1962 RSC.
- M. J. Ruedas-Rama, J. D. Walters, A. Orte and E. A. H. Hall, Anal. Chim. Acta, 2012, 751, 1–23 CrossRef CAS PubMed.
- J. Hu, L. Dai and S. Liu, Macromolecules, 2011, 44, 4699–4710 CrossRef CAS.
- Z. M. Hudson, D. J. Lunn, M. A. Winnik and I. Manners, Nat. Commun., 2014, 5, 3372 Search PubMed.
- G. Marcelo, T. J. V. Prazeres, M.-T. Charreyre, J. M. G. Martinho and J. P. S. Farinha, Macromolecules, 2009, 43, 501–510 CrossRef.
- Y. Yamamoto, K. Yasugi, A. Harada, Y. Nagasaki and K. Kataoka, J. Controlled Release, 2002, 82, 359–371 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, K. L. Wooley and C. J. Hawker, Chem. Mater., 2005, 17, 5976–5988 CrossRef.
- G. Sun, M. Y. Berezin, J. Fan, H. Lee, J. Ma, K. Zhang, K. L. Wooley and S. Achilefu, Nanoscale, 2010, 2, 548–558 RSC.
- J. Liu, H. Liu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 899–912 CrossRef CAS.
- N. Sanson and J. Rieger, Polym. Chem., 2010, 1, 965–977 RSC.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS PubMed.
- C. Yuan, K. Raghupathi, B. C. Popere, J. Ventura, L. Dai and S. Thayumanavan, Chem. Sci., 2014, 5, 229–234 RSC.
- M. Li, Z. Tang, H. Sun, J. Ding, W. Song and X. Chen, Polym. Chem., 2013, 4, 1199–1207 RSC.
- J. Chen, P. Zhang, G. Fang, P. Yi, X. Yu, X. Li, F. Zeng and S. Wu, J. Phys. Chem. B, 2011, 115, 3354–3362 CrossRef CAS PubMed.
- S. Zhou, X. Min, H. Dou, K. Sun, C.-Y. Chen, C.-T. Chen, Z. Zhang, Y. Jin and Z. Shen, Chem. Commun., 2013, 49, 9473–9475 RSC.
- D. A. Heller, Y. Levi, J. M. Pelet, J. C. Doloff, J. Wallas, G. W. Pratt, S. Jiang, G. Sahay, A. Schroeder, J. E. Schroeder, Y. Chyan, C. Zurenko, W. Querbes, M. Manzano, D. S. Kohane, R. Langer and D. G. Anderson, Adv. Mater., 2013, 25, 1449–1454 CrossRef CAS PubMed.
- S. Zhou, H. Dou, Z. Zhang, K. Sun, Y. Jin, T. Dai, G. Zhou and Z. Shen, Polym. Chem., 2013, 4, 4103–4112 RSC.
- M. C. Baier, J. Huber and S. Mecking, J. Am. Chem. Soc., 2009, 131, 14267–14273 CrossRef CAS PubMed.
- L. Feng, C. Zhu, H. Yuan, L. Liu, F. Lv and S. Wang, Chem. Soc. Rev., 2013, 42, 6620–6633 RSC.
- K. Landfester, R. Montenegro, U. Scherf, R. Güntner, U. Asawapirom, S. Patil, D. Neher and T. Kietzke, Adv. Mater., 2002, 14, 651–655 CrossRef CAS.
- T. P. Gustafson, Y. H. Lim, J. A. Flores, G. S. Heo, F. Zhang, S. Zhang, S. Samarajeewa, J. E. Raymond and K. L. Wooley, Langmuir, 2014, 30, 631–641 CrossRef CAS PubMed.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- M. P. Robin, M. W. Jones, D. M. Haddleton and R. K. O'Reilly, ACS Macro Lett., 2012, 1, 222–226 CrossRef CAS.
- M. P. Robin, P. Wilson, A. B. Mabire, J. K. Kiviaho, J. E. Raymond, D. M. Haddleton and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 2875–2878 CrossRef CAS PubMed.
- M. P. Robin, A. B. Mabire, J. C. Damborsky, E. S. Thom, U. H. Winzer-Serhan, J. E. Raymond and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 9518–9524 CrossRef CAS PubMed.
- M. P. Robin and R. K. O'Reilly, Chem. Sci., 2014, 5, 2717–2723 RSC.
- A. Lu, D. Moatsou, D. A. Longbottom and R. K. O'Reilly, Chem. Sci., 2013, 4, 965–969 RSC.
- B. L. Moore, D. Moatsou, A. Lu and R. K. O'Reilly, Polym. Chem., 2014, 5, 3487–3494 RSC.
- J. P. Patterson, M. P. Robin, C. Chassenieux, O. Colombani and R. K. O'Reilly, Chem. Soc. Rev., 2014, 43, 2412–2425 RSC.
- J. P. Patterson, A. M. Sanchez, N. Petzetakis, T. P. Smart, T. H. Epps III, I. Portman, N. R. Wilson and R. K. O'Reilly, Soft Matter, 2012, 8, 3322–3328 RSC.
- D. Ding, J. Liu, G. Feng, K. Li, Y. Hu and B. Liu, Small, 2013, 9, 3093–3102 CrossRef CAS PubMed.
- G. Hong, Y. Zou, A. L. Antaris, S. Diao, D. Wu, K. Cheng, X. Zhang, C. Chen, B. Liu, Y. He, J. Z. Wu, J. Yuan, B. Zhang, Z. Tao, C. Fukunaga and H. Dai, Nat. Commun., 2014, 5, 4206 Search PubMed.
- K. Wang, S. Purushotham, J.-Y. Lee, M.-H. Na, H. Park, S.-J. Oh, R.-W. Park, J. Y. Park, E. Lee, B. C. Cho, M.-N. Song, M.-C. Baek, W. Kwak, J. Yoo, A. S. Hoffman, Y.-K. Oh, I.-S. Kim and B.-H. Lee, J. Controlled Release, 2010, 148, 283–291 CrossRef CAS PubMed.
- S. Seidenari, F. Arginelli, C. Dunsby, P. M. W. French, K. König, C. Magnoni, C. Talbot and G. Ponti, PLoS One, 2013, 8, e70682 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of experimental protocol, light scattering analysis, quantum yield and brightness calculations, and TCSPC analysis. See DOI: 10.1039/c4mh00167b |
| This journal is © The Royal Society of Chemistry 2015 |