
Tabtoxinine-β-lactam is a “stealth” β-lactam antibiotic that evades β-lactamase-mediated antibiotic resistance†‡§
Kathryn M.
Hart
a,
Margaret
Reck
b,
Gregory R.
Bowman
*a and
Timothy A.
Wencewicz
*b
aDepartment of Biochemistry and Molecular Biophysics, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110, USA. E-mail: bowman@biochem.wustl.edu; Fax: +314 362 7183; Tel: +314 362 7433
bDepartment of Chemistry, Washington University in St. Louis, One Brookings Drive, St. Louis, MO 63130, USA. E-mail: wencewicz@wustl.edu; Fax: +314 935 6530; Tel: +314 935 7247
First published on 7th September 2015
Abstract
Tabtoxinine-β-lactam (TβL) is a phytotoxin produced by plant pathogenic strains of Pseudomonas syringae. Unlike the majority of β-lactam antibiotics, TβL does not inhibit transpeptidase enzymes but instead is a potent, time-dependent inactivator of glutamine synthetase, an attractive and underexploited antibiotic target. TβL is produced by P. syringae in the form of a threonine dipeptide prodrug, tabtoxin (TβL-Thr), which enters plant and bacterial cells through dipeptide permeases. The role of β-lactamases in the self-protection of P. syringae from tabtoxin has been proposed, since this organism produces at least three β-lactamases. However, using in vitro and cellular assays and computational docking we have shown that TβL and TβL-Thr evade the action of all major classes of β-lactamase enzymes, thus overcoming the primary mechanism of resistance observed for traditional β-lactam antibiotics. TβL is a “stealth” β-lactam antibiotic and dipeptide prodrugs such as tabtoxin from P. syringae represent a novel antibiotic therapeutic strategy for treating multi-drug resistant Gram-negative pathogens expressing high levels of β-lactamase enzymes.
Introduction
Antibiotic resistance represents one of the greatest global threats to human health.1 Nearly every major class of approved antibiotics used today has some level of documented resistance, and the rate of observed resistance in clinical settings is on a steady rise.2,3 In light of the abundance of multi-drug resistant pathogens and the slim antibiotic drug pipeline, there is a desperate need for new antibiotic classes based on new chemical structures that act on new biological targets.4Natural products continue to be a productive source of novel antibiotic structures, and many of these antibiotics reported in the literature are underexplored as potential therapeutics. One such molecule is tabtoxinine-β-lactam (TβL; Fig. 1a), which is a monocyclic hydroxy-β-lactam antibiotic produced by plant pathogenic strains of Pseudomonas syringae.5,6 Unlike all other known β-lactam antibiotics, such as penicillin, TβL does not inhibit transpeptidase enzymes but instead inhibits the enzyme glutamine synthetase.7–9 This unusual mechanism of action makes TβL an attractive antibiotic because it can escape certain types of pre-existing resistance such as those derived from transpeptidases that have evolved insensitivity.4,10
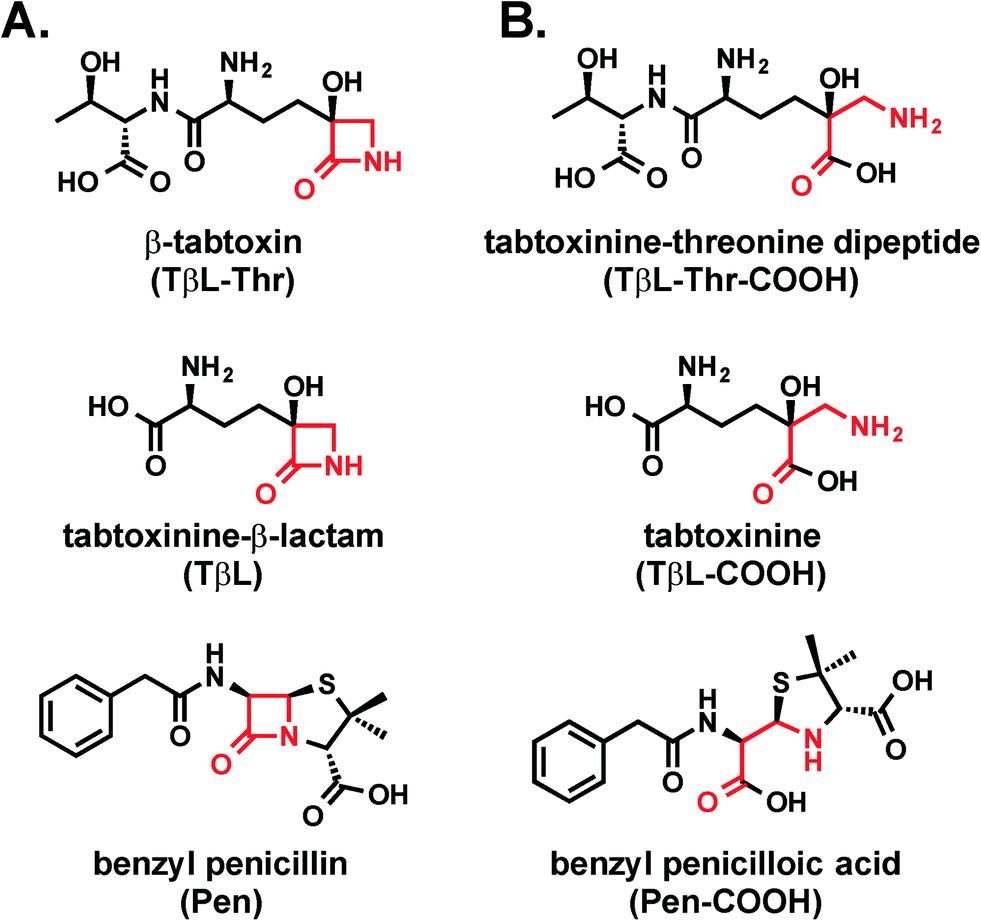 | ||
| Fig. 1 (A) Structures of the β-lactam antibiotics β-tabtoxin (TβL-Thr), tabtoxinine-β-lactam (TβL), and benzyl penicillin (Pen). (B) Structures of β-lactam hydrolysis products tabtoxinine–threonine dipeptide (TβL-Thr-COOH), tabtoxinine (TβL-COOH), and benzyl penicilloic acid (Pen-COOH). | ||
Although TβL acts on a different biological target than common β-lactam antibiotics, it is important to determine if TβL can escape another predominant mechanism of resistance: β-lactamase enzymes. β-Lactamase enzymes are responsible for the vast majority of β-lactam resistance found in Gram-negative bacterial pathogens.11 Since TβL possesses the “enchanted” β-lactam ring found in the penicillins (Fig. 1), it is possible that existing β-lactamase-mediated resistance could render this novel compound ineffective. Here, we use a combination of biochemical and cellular assays to assess whether TβL is a β-lactamase substrate, a critical aspect to its viability as a candidate for antibiotic development.
Results and discussion
TβL-Thr and TβL are not substrates for TEM-1 and CTX-M-9 β-lactamases
Tabtoxinine-β-lactam (TβL) derives from the dipeptide tabtoxin (TβL-Thr; Fig. 1a), which enters plant and bacterial cells via membrane-embedded dipeptide permeases before peptidase cleavage reveals TβL as a cytoplasmic glutamine synthetase inhibitor.12,13 In addition to the prodrug formulation of tabtoxin, producing strains of P. syringae self-protect from the antibiotic through adenylation of glutamine synthetase14 and coexpression of biosynthetic genes (tabABC; tblSCDEF) with a dedicated efflux pump (tblR).15 Another unlinked gene, ttr, has been shown to encode for an acetyl transferase that regioselectively acetylates the α-amino group of TβL to confer resistance to the pathogen.13 The role of β-lactamases in the self-protection of P. syringae from tabtoxin has been proposed since this organism produces at least three β-lactamases.16 The possibility is intriguing given the presence of the β-lactam ring in the TβL structure.Both TβL and TβL-Thr could encounter β-lactamases in the periplasm or cytoplasm of bacterial cells, so we tested both compounds as substrates for TEM-1 and CTX-M-9 class A serine β-lactamases using HPLC and LCMS assays. Both TβL and TβL-Thr lack a chromophore, are highly polar and degrade to the more stable δ-lactam isomers (TδL and TδL-Thr; ESI§ Schemes S1 and S2). Therefore, we used a previously optimized quenching protocol for β-lactamase reactions using fluorenylmethyloxycarbonyl (Fmoc) tagging of all primary amines.13 This enabled good chromatographic separation of Fmoc tagged products (FmocTβL, FmocTβL-Thr, bisFmocTβL-COOH, and bisFmocTβL-Thr-COOH) for HPLC and LCMS analysis. Benzyl penicillin (Pen) was included as the positive control and was rapidly degraded to the corresponding benzyl penicilloic acid (Pen-COOH) by both TEM-1 and CTX-M-9 (Fig. 2a and d and ESI§ Fig. S2).
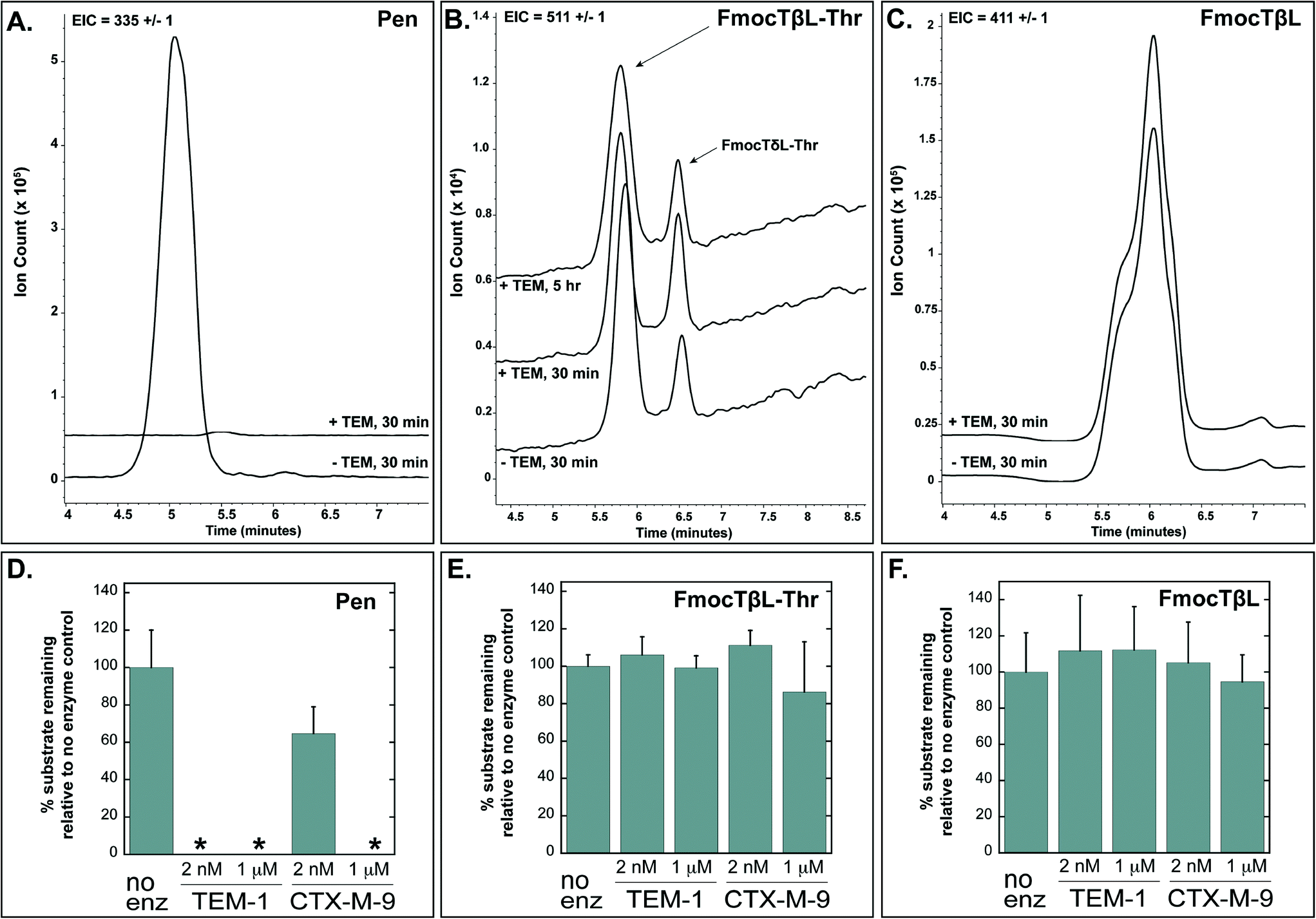 | ||
| Fig. 2 Benzyl penicillin (Pen) is hydrolyzed by TEM-1 and CTX-M-9 β-lactamases while TβL-Thr dipeptide and TβL are unaffected. Panels A, B, and C show LCMS traces with extracted ion counts (EIC) for the substrate [M + H]+ ion. Overlapping traces are aligned on the time axis and offset by 10% on the ion count axis. Dipeptide TβL-Thr and TβL were Fmoc tagged. Panels D, E, and F depict quantified HPLC data in bar graph form, which shows the percent of substrate remaining after treatment with the indicated enzyme relative to a no enzyme negative control. The 2 nM enzyme reactions were quenched after 30 minutes, and the 1 μM enzyme reactions were carried out to 5 hours. The small peak at 6.5 min in panel B represents the δ-lactam isomer of tabtoxin (TδL-Thr) which is known to from TβL-Thr with defined kinetics in aqueous solutions.13 Error bars are standard deviations. *The absence of bars for several penicillin samples in panel D indicates that no substrate was detectable after treatment with enzyme. | ||
Both TβL and TβL-Thr dipeptide proved to be stable towards purified TEM-1 and CTX-M-9 β-lactamases at enzyme concentrations of 2 nM and 1 μM and time points taken out to 5 hours. There was no measurable drop in TβL or TβL-Thr levels in the presence of the β-lactamases as judged by both HPLC (Fig. 2e and f and ESI§ Fig. S3 and S4) and LCMS analysis (Fig. 2b and c). In contrast to the Pen control, which is consumed by 2 nM TEM-1 in <30 min, the β-lactam rings of TβL and TβL-Thr are stable to β-lactamase hydrolysis under these experimental conditions.
TβL-Thr maintains antibiotic activity against E. coli overexpressing β-lactamases
To explore the stability of TβL and TβL-Thr towards TEM-1 and CTX-M-9 β-lactamases in bacterial cells, we determined minimum inhibitory concentrations (MICs) against E. coli expressing these enzymes on inducible plasmids. The dipeptide TβL-Thr was used for this study because TβL alone does not show activity against wild type E. coli strains (MIC values > 128 μM against control strain E. coli ATCC 25922), likely because it cannot permeate the cell membrane. TβL-Thr, on the other hand, enters E. coli cells via dipeptide permeases. While it shows no inhibitory activity towards glutamine synthetase, the dipeptide serves as a prodrug for the active glutamine synthetase inhibitor TβL, which is the ultimate cause of halted bacterial growth.13 All cellular assays were performed in glutamine-free minimal media. Bacteria growth in glutamine-free media requires the enzyme glutamine synthetase to make glutamine from glutamate. Addition of glutamine to minimal media abolishes all growth inhibitory activity of TβL-Thr, consistent with TβL acting as a glutamine antimetabolite.7,17TβL-Thr shows similar efficacy to benzyl penicillin (MIC = 4–8 μM) against the parent E. coli strain lacking β-lactamase, but is not as potent as ampicillin (MIC = 0.5 μM). Strains expressing TEM-1 and CTX-M-9 β-lactamases are also susceptible to TβL-Thr and with similar MIC values (8–16 μM) to the parent strain (Table 1). β-Lactamases are typically exported to the periplasmic space to degrade β-lactams targeting the transpeptidases residing there. Our construct of TEM-1 was so efficiently transported, that a colorimetic activity assay using nitrocefin substrate revealed the vast majority of enzyme was actually in the culture media (Table 2), which is consistent with previous purification methods used for this construct.18 This was not the case for CTX-M-9, which remained in the periplasm, but we wanted to further test the effects of localization. TβL's mechanism of action against glutamine synthetase occurs in the cytosol, so we tested constructs of β-lactamase lacking the necessary export leader sequence confining the enzymes to the cytoplasm to ensure colocalization with the active form of the drug (TβL). TEM-1 and CTX-M-9 showed similar levels of susceptibility (MIC = 4 μM) to TβL-Thr regardless of their cellular location (Table 1). We also tested another extended spectrum TEM variant, TEM-64, because like CTX-M-9 it has extended activity against third-generation cephalosporins,19,20 and contains amino acid substitutions known to contribute to hydrolysis of the monobactam aztreonam, which is more structurally related to the monobactam TβL.21 TEM-64 offered no additional protection (MIC = 16 μM), demonstrating that TβL-Thr escapes both the broad-spectrum β-lactamase TEM-1 and extended-spectrum β-lactamases, TEM-64 and CTX-M-9. Overall, the cellular assay results are consistent with the in vitro assays, which showed the enzymes were unable to hydrolyze TβL-Thr or TβL, and further support the conclusion that TβL-Thr evades at least one mechanism of antibiotic resistance observed in natural and clinical environments.
| Phenotypeb | β-Lactamase classc | MIC (μM, μg mL−1) | ||||
|---|---|---|---|---|---|---|
| Gentamicin | β-lactams | |||||
| Ampicillin | Benzyl penicillin | Tabtoxin | ||||
| a MIC determination was repeated six times. Values are most commonly observed concentration with an error of ± one well. b β-Lactamase phenotypic classification was developed by Bush and Jacoby:11 2b, similar activity against penicillins and early cephalosporins; 2be, activity against extended-spectrum cephalosporins and monobactams; 2df, activity against cloxacillin or oxacillin and carbapenems; 2f, activity against carbapenems, cephamycins, and oxyimino-β-lactams; 3a, broad-spectrum activity against carbapenems but not monobactams. Classifications for these enzymes are taken from the Lahey Clinic database at www.lahey.org. c β-Lactamase structural classification was developed by Jacoby and Munoz-Price.52 d ND: not determined. A complete list of strains, plasmids, inducible genes/markers, and origin/reference is provided in the ESI, Table S1. | ||||||
| E. coli BL21(DE3) | 1.4, 0.8 | 0.5, 0.2 | 4, 1.3 | 8, 2.3 | ||
| +TEM-1 | 2b | A | 1.4, 0.8 | >128, >45 | >128, >43 | 4, 1.2 |
| +TEM-1, no leader sequence | — | A | 1.4, 0.8 | >128, >45 | >128, >43 | 16, 4.6 |
| +TEM-64 | 2be | A | 1.4, 0.8 | >128, >45 | >128, >43 | 16, 4.6 |
| +CTX-M-9 | 2be | A | 1.4, 0.8 | 128, 45 | 128, 43 | 4, 1.2 |
| +CTX-M-9, no leader sequence | — | A | 1.4, 0.8 | 128, 45 | 128, 43 | 8, 2.3 |
| E. coli DH5α | 0.25, 0.1 | 2, 0.7 | NDd | 0.5, 0.14 | ||
| +TEM-1 | 2b | A | 1, 0.6 | >128, >45 | ND | 0.25, 0.07 |
| +TEM-24 | 2be | A | 0.5, 0.3 | >128, >45 | ND | 0.0625, 0.02 |
| +CTX-M-15 | 2be | A | 1, 0.6 | >128, >45 | ND | 0.125, 0.04 |
| +SHV-5 | 2be | A | <0.25, <0.1 | >128, >45 | ND | <0.031, 0.009 |
| +SHV-12 | 2be | A | 0.5, 0.3 | >128, >45 | ND | 0.25, 0.07 |
| +OXA-24/40 | 2df | D | 0.5, 0.3 | >128, >45 | ND | 2, 0.58 |
| +KPC-3 | 2f | A | 0.5, 0.3 | >128, >45 | ND | 0.5, 0.14 |
| +NDM-1 | 3a | B | 1, 0.6 | >128, >45 | ND | 0.5, 0.14 |
| +VIM-2 | 3a | B | 1, 0.6 | >128, >45 | ND | 2, 0.58 |
| Intracellular | Extracellular | |
|---|---|---|
| a Expression was detected using nitrocefin assay of culture media (extracellular) and cell lysate (intracellular). | ||
| E. coli BL21(DE3) | − | − |
| +TEM-1 | − | + |
| +TEM-1, no leader sequence | + | − |
| +TEM-64 | + | − |
| +CTX-M-9 | + | − |
| +CTX-M-9, no leader sequence | + | − |
To explore the effectiveness of TβL-Thr against E. coli expressing other major classes of β-lactamases we turned to an isogenic E. coli library expressing β-lactamases on inducible plasmids.22 The E. coli library contains representative β-lactamase members from major classes of serine (SHV-5, SHV-12, TEM-1, TEM-24, CTX-M-15, OXA-24/40, KPC-3) and metallo β-lactamases (NDM-1 and VIM-2) found in the clinic.11 TβL-Thr was effective at halting the growth of all β-lactamase expressing E. coli in minimal media with MICs ranging from 0.05–8 μM. We judge this MIC range to be significant relative to the dramatic increase in MIC observed for ampicillin (MIC = 0.25 μM against E. coli DH5α; MIC > 128 μM against E. coli DH5α expressing any β-lactamases) when challenged with a β-lactamase (Table 1). We also note that TβL-Thr is not bactericidal to E. coli under these conditions. Normal growth of all E. coli strains treated with TβL-Thr is recovered upon reinoculation of nutrient broth containing glutamine with cells from all wells of MIC plates treated with a range of 0.06 μM up to 128 μM. This result indicates that TβL-Thr is bacteriostatic against E. coli, which could be a general trend for glutamine synthetase inhibition as an antibiotic strategy. Also noteworthy, we observed that TβL-Thr has glutamine-dependent bacteriostatic activity against both Gram-negative bacteria (E. coli) and Gram-positive bacteria (S. aureus) in minimal medias.
Throughout our MIC studies we consistently recorded higher MICs for TβL-Thr against E. coli BL21(DE3), a strain optimized for protein overexpression, compared to other laboratory E. coli strains such as DH5α a control pathogenic strain of E. coli ATCC 25922, a clinical isolate. The underlying reason for this observation is unknown, but it could be a result of E. coli BL21(DE3) being deficient in Lon and OmpT proteases, which might catalyze the intracellular hydrolysis of TβL-Thr dipeptide to the active form TβL.23 This mechanism would suggest that dipeptidases, or lack thereof, might provide a means for TβL resistance.24
TβL-Thr and TβL bind TEM-1 β-lactamase in an unproductive conformation
To gain further insight on why TEM-1 does not recognize TβL-Thr or TβL as substrates, the molecules were docked against TEM-1's active site to generate models of potential binding interactions. The docked structures reveal that TβL-Thr and TβL lack critical interactions in the active site, and overall score poorly relative to the known substrate benzyl penicillin (Fig. 3b and c). Mechanistic studies of TEM-1 hydrolysis of β-lactams highlight the significance of several key interactions between the substrate and active site residues (Fig. 3a). A basic pocket formed by residues R244 and K234 is filled by the substrate's negatively-charged substituent off the β-lactam ring, which positions the substrate in close proximity of two catalytically important serine residues.25,26 S130 is within hydrogen bonding distance of the β-lactam nitrogen and is involved in protonating this nitrogen via a hydrogen-bonded cluster that also includes K234.27,28 The β-lactam ring is broken via nucleophilic attack by the S70 side chain to form an acyl-enzyme intermediate. Unlike known substrates of TEM-1 and transpeptidases, neither TβL-Thr nor TβL contain a carboxylate or other negatively charged functional group extending from the β-lactam ring nitrogen. According to the Tipper–Strominger hypothesis traditional β-lactam antibiotics act as substrate mimics of the C-terminal D-Ala–D-Ala moiety of the bacterial peptidoglycan (PG) peptidyl stem, which contains a carboxylate in an analogous position.29 β-Lactamase enzymes are believed to have evolved from transpeptidase enzymes, which explains the similar substrate specificity of these enzymes.30 Most PBPs and β-lactamases strongly prefer substrates with a C-terminal charged carboxylate.31 The absence of this key interaction may contribute to their inability to act as substrate for β-lactamases or transpeptidases. In fact, the docking studies suggest that the carboxylate of TβL-Thr, which in contrast to known substrates is at the opposite end of the molecule from the β-lactam ring, (Fig. 3b) binds in the basic pocket in lieu of a carboxylate substituent. As a result, the β-lactam ring is situated too far (~8 Å) from the nucleophilic Ser70 to react and form the acyl-enzyme intermediate.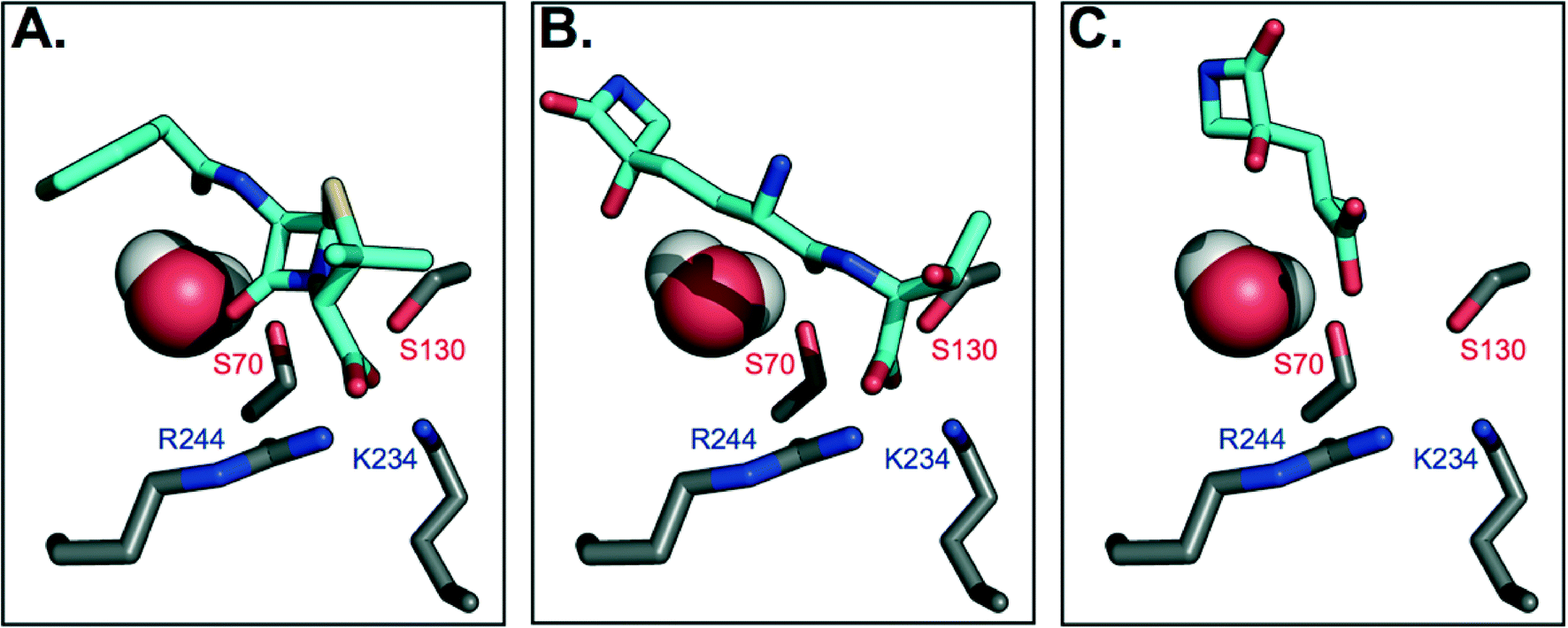 | ||
| Fig. 3 Molecular docking models of TβL and TβL-Thr to TEM-1 active site predict unproductive binding. Carbon atoms are shown in gray (protein) and cyan (antibiotic), and all other atoms are colored according to CPK conventions. The active site water is shown in space-filling mode. (A) Benzyl penicillin binds to the active site via electrostatic interactions between the carboxylate and basic residues R244 and K234, which position the β-lactam ring for nucleophilic attack by S70. (B) TβL-Thr binds with its C-terminus positioned in the basic pocket, which places its β-lactam ring distant from S70. (C) TβL binds even further away from the active site residues. See also ESI§ Fig. S5 for additional poses. | ||
The highest probability pose for TβL (Fig. 3c) also situates the β-lactam ring far from the active site residues. Unlike benzyl penicillin and TβL-Thr, the top five TβL poses looked quite different from one another, possibly due to its small size relative to the size of the active site (ESI§ Fig. S5). For this reason, we considered that each of the top five poses could reflect potential binding modes; however, none of these poses were any more promising in terms of being oriented for catalysis. The second most probable pose (ESI§ Fig. S5c) situates TβL closer to the active site residues, but the orientation of the β-lactam ring is flipped 180° relative to the benzyl penicillin-bound structure, which prevents the necessary hydrogen bonding with S170. Overall, TβL-Thr and TβL score poorly relative to the known substrate benzyl penicillin when docked to TEM-1's active site. Furthermore, the models reveal that even the lowest energy binding poses situate both TβL-Thr and TβL in orientations that are unproductive for catalysis. These studies are consistent with our experimental data showing that TβL-Thr and TβL are simply not recognized as substrates of TEM β-lactamases, allowing them to stealthily evade these clinically important resistance factors.
TβL is a stealth β-lactam antibiotic
TβL is a stealth β-lactam antibiotic at multiple levels. First, it evades inactivation by β-lactamases as shown in this work. Second, it will be unaffected by mutations in transpeptidases or target swapping (as for penicillin-binding protein (PBP) 2a PBP2a β-lactam resistance in MRSA32), since it acts on the enzyme glutamine synthetase. Third, it likely does not activate the pathways that upregulate β-lactamase expression and transpeptidase swapping in Gram-positive or Gram-negative bacteria, respectively: β-lactamase and transpeptidase expression in MRSA is tightly regulated via the bla/mec operons while the amp operon controls β-lactamase expression in E. coli. The bla/mec genes are upregulated by cell surface sensors that are structurally homologous to β-lactamases and that bind and hydrolyze β-lactam antibiotics to initiate the signaling cascade.33 The amp genes are upregulated by hydrolytic PG fragments released via action of lytic transglycosylases after PG is damaged by β-lactam antibiotics.34 Since transcriptional activation relies on structural recognition and turnover of β-lactam antibiotics or metabolites resulting from β-lactam inhibition of transpeptidases, TβL is unlikely to trigger the increased expression of these genes that underlie resistance. This aspect of overcoming resistance is perhaps not important to TβL directly, because it is unaffected by the gene products (i.e. β-lactamases) at high levels, but it is important if combination therapies with β-lactam antibiotics were to be pursued. We cannot directly rule out the possibility of TβL or TβL-Thr inhibiting PBPs. A study by Pratt and coworkers challenged the importance of the C-terminal carboxyl group for inhibition of certain PBPs. Pratt and coworkers found that penems, cephalosporins, and D-Ala–D-Ala mimetics with charged carboxylate or neutral amide C-termini and even descarboxy analogs are all comparably reactive towards class B1 PBPs including B. subtilis PBP3 and S. aureus PBP2a.35 Profiling of PBPs36 and cell morphology analysis37 with TβL and TβL-Thr would provide insight into the possible targeting of PBPs. However, our observations that TβL and TβL-Thr are both unaffected by β-lactamases and inhibit the growth of Gram-negative and Gram-positive in a glutamine-dependent bacteriostatic manner strongly indicate that glutamine synthetase is the primary target. Overall, TβL overcomes the most common resistance mechanisms observed for traditional β-lactam antibiotics.38The transpeptidase targets of β-lactam antibiotics, like penicillin, are located in the periplasm at the site of the PG cell wall. Under β-lactam challenge, resistant bacteria upregulate the cytoplasmic expression of β-lactamases, such as TEM-1, with a leader peptide sequence that signals export and cleavage of the leader peptide by signal peptidases located in the inner lipid membrane. The now periplasmic β-lactamase is in the perfect place to protect transpeptidases from β-lactam antibiotics through rapid, catalytic hydrolysis of the β-lactam ring. β-Lactamase enzymes are often found in the extracellular space where they can inactivate β-lactam antibiotics in the infection microenvironment prior to porin-mediated diffusion across the outer membrane (Fig. 4a). In the case of TβL-Thr (Fig. 4b), the dipeptide diffuses across the E. coli outer lipid membrane through dipeptide permeases. Free TβL can be liberated in the periplasm via peptidase action and either TβL or TβL-Thr dipeptide can be transported into the cytoplasm, perhaps with the assistance of a transporter in the major facilitator superfamily (MFS).39,40 Once in the cytoplasm, TβL is free to inhibit glutamine synthetase. TβL and TβL-Thr dipeptide are unaffected by the presence of β-lactamases in the extracellular space, periplasm, and cytoplasm.
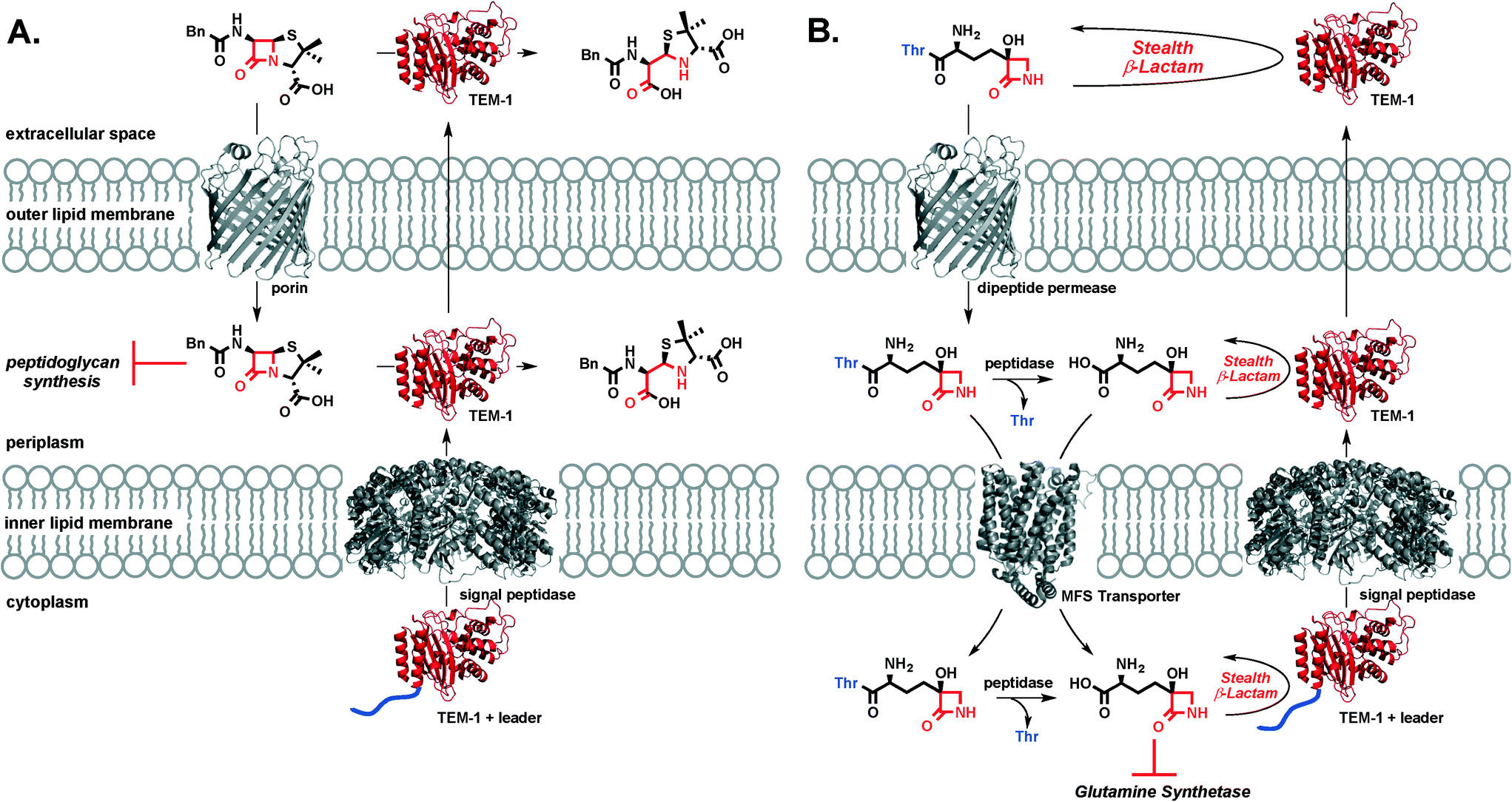 | ||
| Fig. 4 (A) Benzyl penicillin is inactivated by TEM-1 β-lactamase (red) excreted to the periplasm after cleavage of the signal peptide (blue) by a signal peptidase. Benzyl penicillin is also inactivated by TEM-1 in the extracellular space excreted by an unknown mechanism. (B) Tabtoxin (TβL-Thr) enters E. coli cells via dipeptide permeases. Peptidase cleavage in the periplasm can release TβL and either TβL-Thr or TβL can enter the periplasm with the help of a major facilitator superfamily (MFS) transporter. Once in the cytoplasm, TβL is free to inhibit glutamine synthetase. At all stages (extracellular, periplasmic, and cytoplasmic), TβL-Thr and TβL are unaffected by the presence of β-lactamases such as TEM-1. | ||
Overcoming existing antibiotic resistance mechanisms is essential for any new antibiotic in the discovery phase. Our work shows that the most widespread and broad-spectrum β-lactamases are not capable of inactivating TβL or TβL-Thr. These compounds will no doubt encounter other types of resistance mechanisms (glutamine synthetase adenylation, efflux, exclusion, acetylation as mentioned in earlier sections) if pursued as antibiotic therapeutics, but this work mitigates concerns about existing β-lactamases. This work confirms that dipeptide prodrug inhibitors of glutamine synthetase, such as tabtoxin from Pseudomonas syringae, are promising new bacteriostatic agents for halting E. coli growth with excellent potency against strains expressing all major classes of β-lactamases (Table 1). We are currently investigating dipeptide prodrugs of glutamine synthetase inhibitors TβL, glufosinate,43 and methionine sulfoximine43 for the treatment of urinary tract infections (UTIs) caused by uropathogenic E. coli (UPEC), which account for 75% of uncomplicated UTIs and 65% of complicated UTIs in the United States.41 Antibiotic resistance is rampant among UPECs with plasmid-encoded extended spectrum β-lactamases being of particular concern.41 Glutamine synthetase is an attractive target for other types of infectious diseases, including Mycobacterium tuberculosis with many recent efforts focused on developing glutamine synthetase inhibitors as antibacterial agents.42–45 β-Lactamase production in M. tuberculosis is also a rising concern. Tabtoxin represents a new structural class of antibiotics acting on glutamine synthetase that overcomes β-lactamase resistance and presents new opportunities for antibiotic development against multi-drug resistant, β-lactamase expressing bacterial pathogens.
Conclusions
We have demonstrated that TβL and its prodrug, TβL-Thr, evade the action of all major classes of β-lactamase enzymes. Neither drug is degraded by the NDM-1 metallo β-lactamase or TEM/CTX-M serine β-lactamases, either in cellular or cell-free in vitro assays. These compounds appear to maintain their bacteriostatic activity even in the face of this common mode of resistance to β-lactam drugs because they are not recognized as substrates for β-lactamases. This surprising ability to evade β-lactamases may derive, in part, from the missing anionic substituent adjacent to the β-lactam nitrogen found in all β-lactam antibiotics. Therefore, TβL is a “stealth” β-lactam that evades clinically relevant β-lactamases. This work validates that formulations of TβL are potentially novel antibiotic options for killing multi-drug resistant Gram-negative pathogens expressing high levels of β-lactamase enzymes.Experimental
Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g, 4 °C) the pellet was resuspended in an equal volume of ice-cold 5 mM MgSO4 and stirred for 10 minutes at 4 °C. Centrifugation (20 min at 10000 × g, 4 °C) produced a supernatant which contained the periplasmic fraction of the cells. The pH of the periplasmic fraction was adjusted to 20 mM sodium acetate, pH 5.5 using concentrated buffer, and the sample was purified over a UNOsphere Rapid S column (BioRad) and eluted with a 50 CV gradient from 0 to 0.6 M NaCl. Peak fractions were then pooled, concentrated and buffer exchanged into 20 mM Tris, pH 8.0. Each protein's purity and molecular weight were confirmed by SDS-PAGE and electrospray mass spectrometry (ESI§ Fig. S1), for which proteins were diluted 2× in 50% acetonitrile/0.1% formic acid and infused using the nanomate Triversa (Advion) into the LTQ-Orbitrap Velos. The data was acquired using the Orbitrap at 100000 resolution. Protein concentrations were determined in Edelhoch buffer using extinction coefficients calculated based on the number of tryptophan and tyrosine residues.47
000 × g, 4 °C) the pellet was resuspended in an equal volume of ice-cold 5 mM MgSO4 and stirred for 10 minutes at 4 °C. Centrifugation (20 min at 10000 × g, 4 °C) produced a supernatant which contained the periplasmic fraction of the cells. The pH of the periplasmic fraction was adjusted to 20 mM sodium acetate, pH 5.5 using concentrated buffer, and the sample was purified over a UNOsphere Rapid S column (BioRad) and eluted with a 50 CV gradient from 0 to 0.6 M NaCl. Peak fractions were then pooled, concentrated and buffer exchanged into 20 mM Tris, pH 8.0. Each protein's purity and molecular weight were confirmed by SDS-PAGE and electrospray mass spectrometry (ESI§ Fig. S1), for which proteins were diluted 2× in 50% acetonitrile/0.1% formic acid and infused using the nanomate Triversa (Advion) into the LTQ-Orbitrap Velos. The data was acquired using the Orbitrap at 100000 resolution. Protein concentrations were determined in Edelhoch buffer using extinction coefficients calculated based on the number of tryptophan and tyrosine residues.47
E. coli DH5α strains containing plasmids with the various β-lactamase genes were grown overnight at 37 °C in LB broth containing 15 μg mL−1 tetracycline. Fresh LB broth (100 mL) in baffle flasks containing 15 μg mL−1 tetracycline was inoculated with 100 μL of overnight culture. Flasks were incubated with shaking at 37 °C until OD600 reached about 0.4. At this point 50 μL of a 1 M sterile IPTG solution was added to each flask to induce protein expression. Flasks were then incubated with shaking at 20 °C for four hours before being diluted in minimal media for use on 96-well plates. E. coli BL21(DE3) strains containing plasmids with the various β-lactamase genes were grown overnight at 37 °C in LB broth containing 50 μg mL−1 kanamycin and 1 mM IPTG.
Antibacterial activity of the compounds was determined by measuring their minimum inhibitory concentrations (MIC90's) using the broth microdilution method according to the Clinical and Laboratory Standards Institute (CLSI, formerly the NCCLS) guidelines.48 Each well of a 96-well microtiter plate was filled with 50 μL of sterile minimal media broth. Each test compound was dissolved in water making a 20 mM solution, then diluted with sterile minimal media broth to 512 μM. Exactly 50 μL of the compound solution was added to the first well of the microtiter plate and 2-fold serial dilutions were made down each row of the plate. Exactly 50 μL of bacterial inoculum from overnight LB/IPTG cultures in minimal media (5 × 105 CFU mL−1) was then added to each well giving a total volume of 100 μL minimal media per well and a compound concentration gradient of 128–0.0625 μM. The plate was incubated at 37 °C for 24 h and then each well was examined for bacterial growth. The MIC90 was recorded as the lowest compound concentration (μM) required to inhibit 90% of bacterial growth as judged by turbidity of the culture media relative to a row of wells filled with a water standard. Gentamicin was included in a control row at a concentration gradient of 128–0.0625 μM.
000 different configurations, and charges were added with molcharge using the AM1BCC method.50 The receptor was prepared for docking using receptor_setup and amber charges were employed. Finally, docking was performed with FRED51 using high-resolution and saving the top five poses.
Abbreviations
| ACN | Acetonitrilie |
| Ala | L-Alanine |
| D2O | Deuterium oxide |
| E. coli | Escherichia coli |
| EIC | Extracted ion chromatogram |
| EDTA | Ethylenediaminetetraacetic acid |
| ESI | Electrospray ionization |
| Fmoc | Fluorenylmethyloxycarbonyl |
| FPLC | Fast protein liquid chromatography |
| HEPES | 2-[4-(2-Hydroxyethyl)piperazin-1-yl]ethanesulfonic acid |
| HPLC | High performance liquid chromatography |
| HRMS | High-resolution mass spectrometry |
| LB | Lysogeny broth |
| IPTG | Isopropyl-β D-galactopyranoside |
| LCMS | Liquid chromatography-mass spectrometry |
| MFS | Major facilitator superfamily |
| MIC | Minimum inhibitory concentration |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| NMR | Nuclear magnetic resonance |
| PBP | Penicillin-binding protein |
| PG | Peptidoglycan |
| P. syringae | Pseudomonas syringae |
| Ser | L-Serine |
| SDS-PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| TβL | Tabtoxinine-β-lactam |
| TβL-COOH | Tabtoxinine |
| TβL-Thr | β-Tabtoxin |
| TβL-Thr-COOH | Tabtoxinine–threonine dipeptide |
| TδL | Tabtoxinine-δ-lactam |
| TδL-Thr | δ-Tabtoxin |
| TFA | Trifluoroacetic acid |
| Thr | L-Threonine |
| Tris | 2-Amino-2-hydroxymethyl-propane-1,3-diol |
| UPEC | Uropathogenic E. coli |
| UTI | Urinary tract infection |
Acknowledgements
We thank Mr. Garrett Patrick (Wencewicz Lab) for providing authentic samples of TβL-COOH and TβL-Thr-COOH and assisting with preparation of TβL-Thr and TβL samples. We thank Dr. Andre D'Avignon (formerly WUSTL, Dept. of Chemistry; currently Sanford Burnham Medical Research Institute, Orlando, FL) and Dr. Jeff Kao (WUSTL, Dept. of Chemistry) for assistance provided in the acquisition and processing of 2D NMR spectra. We thank Dr. Yu Chen (University of South Florida) for the gift of the plasmid encoding CTX-M-9. We thank the Proteomics & Mass Spectrometry Facility at the Danforth Plant Science Center for analysis of purified proteins and whose work is supported by the National Science Foundation under Grant No. DBI-0922879 for acquisition of the LTQ-Velos Pro Orbitrap LC-MS/MS. We thank Pfizer for providing the library of E. coli DH5α β-lactamase expressing bacteria. GRB holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund.References
- K. Schwab, in Global Risks 2014, Ninth Edition, Insight Report, World Economic Forum, 2014 Search PubMed.
- N. Gupta, B. M. Limbago, J. B. Patel and A. J. Kallen, Carbapenem-Resistant Enterobacteriaceae: Epidemiology and Prevention, Clin. Infect. Dis., 2011, 53, 60–67 CrossRef PubMed.
- L. E. Nicolle, Antimicrobial resistance: The never ending story, Can. J. Infect. Dis., 1991, 2, 47–48 CrossRef CAS PubMed.
- C. T. Walsh and T. A. Wencewicz, Prospects for new antibiotics: a molecule-centered perspective, J. Antibiot., 2014, 67, 7–22 CrossRef CAS PubMed.
- D. W. Woolley, R. B. Pringle and A. C. Braun, Isolation of the phytopathogenic toxin of Pseudomonas tabaci, an antagonist of methionine, J. Biol. Chem., 1952, 197, 409–417 CAS.
- W. W. Stewart, Isolation and proof of structure of wildfire toxin, Nature, 1971, 229, 174–178 CrossRef CAS PubMed.
- S. L. Sinden and R. D. Durbin, Glutamine synthetase inhibition: possible mode of action of wildfire toxin from Pseudomonas tabaci, Nature, 1968, 219, 379–380 CrossRef CAS PubMed.
- M. D. Thomas, P. J. Langston-Unkefer, T. F. Uchytil and R. D. Durbin, Inhibition of Glutamine Synthetase from Pea by Tabtoxinine-β-lactam, Plant Physiol., 1983, 71, 912–915 CrossRef CAS PubMed.
- P. L. Langston-Unkefer, P. A. Macy and R. D. Durbin, Inactivation of Glutamine Synthetase by Tabtoxinine-β-lactam : Effects of Substrates and pH, Plant Physiol., 1984, 76, 71–74 CrossRef CAS PubMed.
- M. A. Fischbach and C. T. Walsh, Antibiotics for emerging pathogens, Science, 2009, 325, 1089–1093 CrossRef CAS PubMed.
- K. Bush and G. A. Jacoby, Updated functional classification of β-lactamases, Antimicrob. Agents Chemother., 2010, 54, 969–976 CrossRef CAS PubMed.
- D. R. Bush and P. J. Langston-Unkefer, Tabtoxinine-β-Lactam Transport into Cultured Corn Cells : Uptake via an Amino Acid Transport System, Plant Physiol., 1987, 85, 845–849 CrossRef CAS PubMed.
- T. A. Wencewicz and C. T. Walsh, Pseudomonas syringae self-protection from tabtoxinine-β-lactam by ligase TblF and acetylase Ttr, Biochemistry, 2012, 51, 7712–7725 CrossRef CAS PubMed.
- C. T. Knight, R. D. Durbin and P. J. Langston-Unkefer, Role of glutamine synthetase adenylylation in the self-protection of Pseudomonas syringae subsp. “tabaci” from its toxin, tabtoxinine-β-lactam, J. Bacteriol., 1986, 166, 224–229 CrossRef PubMed.
- P. J. Kinscherf and D. K. Willis, The biosynthetic gene cluster for the β-lactam antibiotic tabtoxin in Pseudomonas syringae, J. Antibiot., 2005, 58, 817–821 CrossRef PubMed.
- R. H. Coleman, J. Shaffer and H. True, Properties of β-lactamase from Pseudomonas syringae, Curr. Microbiol., 1996, 32, 147–150 CrossRef CAS PubMed.
- E. Arrebola, F. M. Cazorla, A. Perez-Garcia and A. Vicente, Genes Involved in the Production of Antimetabolite Toxins by Pseudomonas syringae Pathovars, Genes, 2011, 2, 640–660 CrossRef CAS PubMed.
- P. Y. Savard and S. M. Gagne, Backbone dynamics of TEM-1 determined by NMR: evidence for a highly ordered protein, Biochemistry, 2006, 45, 11414–11424 CrossRef CAS PubMed.
- M. Sabate, R. Tarrago, F. Navarro, E. Miro, C. Verges, J. Barbe and G. Prats, Cloning and sequence of the gene encoding a novel cefotaxime-hydrolyzing β-lactamase (CTX-M-9) from Escherichia coli in Spain, Antimicrob. Agents Chemother., 2000, 44, 1970–1973 CrossRef CAS PubMed.
- X. Wang, G. Minasov and B. K. Shoichet, Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs, J. Mol. Biol., 2002, 320, 85–95 CrossRef CAS PubMed.
- M. L. Salverda, J. A. De Visser and M. Barlow, Natural evolution of TEM-1 β-lactamase: experimental reconstruction and clinical relevance, FEMS Microbiol. Rev., 2010, 34, 1015–1036 CrossRef CAS PubMed.
- J. Starr, M. F. Brown, L. Aschenbrenner, N. Caspers, Y. Che, B. S. Gerstenberger, M. Huband, J. D. Knafels, M. M. Lemmon, C. Li, S. P. McCurdy, E. McElroy, M. R. Rauckhorst, A. P. Tomaras, J. A. Young, R. P. Zaniewski, V. Shanmugasundaram and S. Han, Siderophore receptor-mediated uptake of lactivicin analogues in gram-negative bacteria, J. Med. Chem., 2014, 57, 3845–3855 CrossRef CAS PubMed.
- H. Jeong, V. Barbe, C. H. Lee, D. Vallenet, D. S. Yu, S. H. Choi, A. Couloux, S. W. Lee, S. H. Yoon, L. Cattolico, C. G. Hur, H. S. Park, B. Segurens, S. C. Kim, T. K. Oh, R. E. Lenski, F. W. Studier, P. Daegelen and J. F. Kim, Genome sequences of Escherichia coli B strains REL606 and BL21(DE3), J. Mol. Biol., 2009, 394, 644–652 CrossRef CAS PubMed.
- C. Levi and R. D. Durbin, The isolation and properties of a tabtoxin-hydrolysing aminopeptidase from the periplasm of Pseudomonas syringae pv. tabaci, Physiol. Mol. Plant Pathol., 1986, 28, 345–352 CrossRef CAS.
- P. Swaren, L. Maveyraud, V. Guillet, J. M. Masson, L. Mourey and J. P. Samama, (1995) Electrostatic analysis of TEM1 β-lactamase: effect of substrate binding, steep potential gradients and consequences of site-directed mutations, Structure, 1993, 3, 603–613 CrossRef.
- D. C. Marciano, N. G. Brown and T. Palzkill, Analysis of the plasticity of location of the Arg244 positive charge within the active site of the TEM-1 β-lactamase, Protein Sci., 2009, 18, 2080–2089 CrossRef CAS PubMed.
- B. P. Atanasov, D. Mustafi and M. W. Makinen, Protonation of the β-lactam nitrogen is the trigger event in the catalytic action of class A β-lactamases, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3160–3165 CAS.
- A. Matagne, J. Lamotte-Brasseur and J. M. Frere, Interactions between active-site serine β-lactamases and so-called β-lactamase-stable antibiotics. Kinetic and molecular modelling studies, Eur. J. Biochem., 1993, 217, 61–67 CrossRef CAS PubMed.
- D. J. Waxman and J. L. Strominger, Penicillin-binding proteins and the mechanism of action of β-lactam antibiotics, Annu. Rev. Biochem., 1983, 52, 825–869 CrossRef CAS PubMed.
- J. A. Kelly, O. Dideberg, P. Charlier, J. P. Wery, M. Libert, P. C. Moews, J. R. Knox, C. Duez, C. Fraipont and B. Joris, and et al. On the origin of bacterial resistance to penicillin: comparison of a β-lactamase and a penicillin target, Science, 1986, 231, 1429–1431 CrossRef CAS PubMed.
- L. Varetto, F. De Meester, D. Monnaie, J. Marchand-Brynaert, G. Dive, F. Jacob and J. M. Frere, The importance of the negative charge of β-lactam compounds in the interactions with active-site serine DD-peptidases and β-lactamases, Biochem. J., 1991, 278(Pt 3), 801–807 CrossRef CAS PubMed.
- J. Fishovitz, J. A. Hermoso, M. Chang and S. Mobashery, Penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus, IUBMB Life, 2014, 66, 572–577 CrossRef CAS PubMed.
- M. W. Staude, T. E. Frederick, S. V. Natarajan, B. D. Wilson, C. E. Tanner, S. T. Ruggiero, S. Mobashery and J. W. Peng, Investigation of signal transduction routes within the sensor/transducer protein BlaR1 of Staphylococcus aureus, Biochemistry, 2015, 54, 1600–1610 CrossRef CAS PubMed.
- C. Jacobs, J. M. Frere and S. Normark, Cytosolic intermediates for cell wall biosynthesis and degradation control inducible β-lactam resistance in gram-negative bacteria, Cell, 1997, 88, 823–832 CrossRef CAS PubMed.
- K. Dave, T. Palzkill and R. F. Pratt, Neutral β-Lactams Inactivate High Molecular Mass Penicillin-Binding Proteins of Class B1, Including PBP2a of MRSA, ACS Med. Chem. Lett., 2014, 5, 154–157 CrossRef CAS PubMed.
- O. Kocaoglu, H. C. Tsui, M. E. Winkler and E. E. Carlson, Profiling of β-lactam selectivity for penicillin-binding proteins in Streptococcus pneumoniae D39, Antimicrob. Agents Chemother., 2015, 59, 3548–3555 CrossRef CAS PubMed.
- Z. Yao, D. Kahne and R. Kishony, Distinct single-cell morphological dynamics under β-lactam antibiotics, Mol. Cell, 2012, 48, 705–712 CrossRef CAS PubMed.
- K. Poole, Resistance to β-lactam antibiotics, Cell. Mol. Life Sci., 2004, 61, 2200–2223 CrossRef CAS PubMed.
- S. S. Pao, I. T. Paulsen and M. H. Saier, Major Facilitator Superfamily, Microbiol. Mol. Biol. Rev., 1998, 62, 1–34 CAS.
- V. S. Reddy, M. A. Shlykov, R. Castillo, E. I. Sun and M. H. Saier Jr., The major facilitator superfamily (MFS) revisited, FEBS J., 2012, 279, 2022–2035 CrossRef CAS PubMed.
- A. L. Flores-Mireles, J. N. Walker, M. Caparon and S. J. Hultgren, Urinary tract infections: epidemiology, mechanisms of infection and treatment options, Nat. Rev. Microbiol., 2015, 13, 269–284 CrossRef CAS PubMed.
- G. Harth and M. A. Horwitz, An inhibitor of exported Mycobacterium tuberculosis glutamine synthetase selectively blocks the growth of pathogenic mycobacteria in axenic culture and in human monocytes: extracellular proteins as potential novel drug targets, J. Exp. Med., 1999, 189, 1425–1436 CrossRef CAS PubMed.
- D. Eisenberg, H. S. Gill, G. M. Pfluegl and S. H. Rotstein, Structure-function relationships of glutamine synthetases, Biochim. Biophys. Acta, 2000, 1477, 122–145 CrossRef CAS.
- S. L. Mowbray, M. K. Kathiravan, A. A. Pandey and L. R. Odell, Inhibition of glutamine synthetase: a potential drug target in Mycobacterium tuberculosis, Molecules, 2014, 19, 13161–13176 CrossRef PubMed.
- C. Couturier, S. Silve, R. Morales, B. Pessegue, S. Llopart, A. Nair, A. Bauer, B. Scheiper, C. Pöverlein, A. Ganzhorn, S. Lagrange and E. Bacqué, Nanomolar inhibitors of Mycobacterium tuberculosis glutamine synthetase 1: Synthesis, biological evaluation and X-ray crystallographic studies, Bioorg. Med. Chem. Lett., 2015, 25, 1455–1459 CrossRef CAS PubMed.
- J. Ghrayeb, H. Kimura, M. Takahara, H. Hsiung, Y. Masui and M. Inouye, Secretion cloning vectors in Escherichia coli, EMBO J., 1984, 3, 2437–2442 CAS.
- H. Edelhoch, Spectroscopic determination of tryptophan and tyrosine in proteins, Biochemistry, 1967, 6, 1948–1954 CrossRef CAS PubMed.
- CLSI, in Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard-Ninth Edition, CLSI document M07-A9, Clinical and Laboratory Standards Institute, Wayne, PA, 2012 Search PubMed.
- C. H. O'Callaghan, A. Morris, S. M. Kirby and A. H. Shingler, Novel method for detection of β-lactamases by using a chromogenic cephalosporin substrate, Antimicrob. Agents Chemother., 1972, 1, 283–288 CrossRef.
- P. C. Hawkins, A. G. Skillman, G. L. Warren, B. A. Ellingson and M. T. Stahl, Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database, J. Chem. Inf. Model., 2010, 50, 572–584 CrossRef CAS PubMed.
- M. McGann, FRED pose prediction and virtual screening accuracy, J. Chem. Inf. Model., 2011, 51, 578–596 CrossRef CAS PubMed.
- G. A. Jacoby and L. S. Munoz-Price, The new β-lactamases, N. Engl. J. Med., 2005, 352, 380–391 CrossRef CAS PubMed.
Footnotes |
| † Research was supported by funds from Washington University in St. Louis. |
| ‡ The authors declare no competing financial interest. |
| § Electronic supplementary information (ESI) available: SDS-PAGE and electrospray mass spectrometry analysis of purified TEM-1 and CTX-M-9 enzymes. Procedures for synthesis of TβL-Thr-COOH, and TβL-COOH analytical standards. NMR spectra of purified TβL-Thr, TβL-Thr-COOH, and TβL-COOH. HPLC and LCMS traces for β-lactamase activity assays. Additional TEM-1 docking poses for TβL-Thr and TβL. See DOI: 10.1039/c5md00325c |
| This journal is © The Royal Society of Chemistry 2016 |