
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2-[18F]Fluoroethyl tosylate – a versatile tool for building 18F-based radiotracers for positron emission tomography
Torsten
Kniess
*,
Markus
Laube
,
Peter
Brust
and
Jörg
Steinbach
Helmholtz-Zentrum Dresden-Rossendorf, Institute of Radiopharmaceutical Cancer Research, Dresden, Germany. E-mail: t.kniess@hzdr.de
First published on 3rd September 2015
Abstract
Positron emission tomography (PET) is a modern in vivo imaging technique and an important diagnostic modality for clinical and pre-clinical research. The incorporation of a radionuclide like fluorine-18 into a target molecule to form PET radiopharmaceuticals is a repeated challenge for radiochemists. [18F]Fluoroethylation is a well-acknowledged method for 18F-radiolabeling and 2-[18F]fluoroethyl tosylate ([18F]FETs) is a preferred reagent because of its high reactivity to phenolic, thiophenolic, carboxylic and amide functionalities. This review will highlight the role of [18F]FETs in PET chemistry and summarize its applicability in radiotracer design. The radiolabeling conditions and the pros and cons of direct and indirect radiolabeling as well the aspects of the reactivity of [18F]FETs compared with other [18F]fluoroalkylating reagents will be discussed comprehensively.
 Torsten Kniess | Torsten Kniess graduated from Technical University Dresden and received his Ph.D. in Organic Chemistry in 1993. His career in radiopharmacy started in 1994 at Research Center Rossendorf (now Helmholtz-Zentrum Dresden-Rossendorf, HZDR) as a post-doc under the supervision of Prof. Bernd Johannsen and continued in 2001 at Instituto Tecnologico é Nuclear (ITN, Sacavém, Portugal) with Prof. Isabel Rego Dos Santos. He moved to the PET-Center in Dresden-Rossendorf with Prof. Frank Wüst in 2002 and dealt with palladium-mediated cross-coupling reactions in PET chemistry. Since 2008, he has been the head of the Organic and Radiochemistry group at HZDR. His actual research interests are radiolabeled inhibitors of tyrosine kinases (TKIs) and cyclooxygenase-2 (COX-2). |
 Markus Laube | Markus Laube studied food chemistry, graduated in 2009 at Technical University Dresden and received his Ph.D. in 2014 at Helmholtz-Zentrum Dresden-Rossendorf (HZDR) under the supervision of Professor Jörg Steinbach. His diploma and Ph.D. work dealt with the synthesis and radiolabeling of cyclooxygenase-2 inhibitors for imaging purposes with PET – his ongoing research interest. This and the development of novel carbon-11 labeled radiotracers are part of his continuing post-doc work at HZDR. Besides these, working in the project ForMaT HypoFood/HypoFeed (2009, Prof. T. Henle, TU Dresden) and a research stay at the Turku PET Centre, Finland (2014, Prof. O. Solin) enriched his academic career. |
 Peter Brust | Peter Brust received his M.S. in Immunology in 1981 and his Ph.D. in Neuroscience from Leipzig University in 1986. He worked as a postdoctoral fellow at Montreal Neurological Institute and Johns Hopkins University Baltimore from 1990 to 1991. He joined the Research Center Rossendorf (now known as Helmholtz-Zentrum Dresden-Rossendorf, HZDR) in 1992 and headed the Department of Biochemistry. Since 2002, he has worked in Leipzig first at the Institute of Interdisciplinary Isotope Research and since 2010, after operational transfer, again at HZDR where he leads the Department of Radiopharmaceutical Cancer Research. Since 2010, he has been an adjunct professor of Radiopharmacy at Leipzig University. |
 Jörg Steinbach | Jörg Steinbach is the Director of the Institute of Radiopharmaceutical Cancer Research, Helmholtz-Zentrum Dresden-Rossendorf and has been a Professor of Bioinorganic and Radiopharmaceutical Chemistry at Technische Universität Dresden, Germany since 2005. • 2001–2009: Full Professor of Isotope Research, Universität Leipzig, Germany, Director, Institute of Interdisciplinary Isotope Research • 1992–2001:Head of the PET Department, Institute of Bioinorganic and Radiopharmaceutical Chemistry, Forschungszentrum Dresden-Rossendorf • 1982–1991: Scientist, Central Institute of Nuclear Research Rossendorf, Department of Radioactive Isotopes • 1982: Ph.D. Inorganic Chemistry, Technical University Dresden • Research stays at Åbo Akademi, Turku, Finland (1986–1988), Research Fields • Radiochemistry and Radiopharmacy • Radiotracers and targeted radionuclide therapy • Radionuclide production and targetry. |
Introduction
Positron emission tomography (PET) is a modern in vivo imaging technique and has become an important diagnostic modality for clinical and pre-clinical research. By incorporation of a radionuclide like fluorine-18 (t1/2 = 109.8 min, β+ = 97%) into a target molecule, PET radiopharmaceuticals are formed, enabling the non-invasive visualization of biochemical processes and the imaging and quantification of pathological situations in vivo. The most prominent example, 2-[18F]fluoro-2-deoxy-D-glucose ([18F]FDG), a radiopharmaceutical for glucose metabolism, has been widely used for more than 25 years as a diagnostic tool for neuropsychiatric1,2 and cardiovascular2,3 disorders and is the working horse for the diagnosis, monitoring and staging of cancerous diseases.4Radiochemists have developed a number of methods of [18F]radiofluorination via electrophilic and nucleophilic pathways for the incorporation of fluorine-18 into molecules of interest. Due to its ease of handling, direct nucleophilic substitution using [18F]fluoride has been established as the preferred approach so far. However, the weak nucleophilic character of the fluoride ion often displays a major hindrance. Aprotic conditions, highly reactive leaving groups, and in the case of aromatic nucleophilic substitution, an electron-deficient aromatic system are the basic requirements for successful incorporation of [18F]fluoride into an organic molecule.5 Alternative labeling strategies have been developed, making use of so-called prosthetic groups by first attaching the radionuclide to a small bifunctional reactive unit and their subsequent coupling to the target compound. An overview on prosthetic groups for 18F-radiolabeling comprising activated esters, aldehydes and amines as well as halogenides, azide- and alkyne-based small molecules can be found in a number of excellent reviews.6–8 Beyond this, [18F]fluoroalkylating agents like [18F]fluoromethyl bromide, 2-[18F]fluoroethyl bromide ([18F]FEBr), 2-[18F]fluoroethyl triflate or 2-[18F]fluoroethyl tosylate ([18F]FETs) or even [18F]fluorocyclobutyl tosylate9 have gained increased interest because they provide high reactivity to phenolic, thiophenolic, carboxylic and amide functionalities.10 In general, the fluoroethyl group holds similar steric properties like ethyl and methyl groups and is expected to maintain the pharmacological properties of a lead compound. Compared to the methyl group, the fluoroethyl moiety is more lipophilic, so the shift in lipophilicity by introduction of a fluoroethyl moiety (Δlog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 0.70) is stronger than that when a methyl substituent is used (ΔlogP = 0.54).11 However, the increased lipophilicity originating from the fluoroethyl group may be beneficial for addressing specific targets or organs, e.g. the brain. From a radiochemist's point of view, the [18F]fluoroethyl group may act as a surrogate for a [11C]methyl radiolabel because it can be coupled to the same functional groups. Precursors already available for methylation with [11C]methyl iodide may be used without modification for [18F]fluoroethylation. This is advantageous for the transfer of a promising 11C-labeled radiotracer into its 18F-labeled counterpart by exploitation of the longer half-life of fluorine-18. Besides that, [18F]fluoroethylation can be performed with unprotected compounds and the reaction conditions are less restricting in comparison to those for direct nucleophilic substitution.
P = 0.70) is stronger than that when a methyl substituent is used (ΔlogP = 0.54).11 However, the increased lipophilicity originating from the fluoroethyl group may be beneficial for addressing specific targets or organs, e.g. the brain. From a radiochemist's point of view, the [18F]fluoroethyl group may act as a surrogate for a [11C]methyl radiolabel because it can be coupled to the same functional groups. Precursors already available for methylation with [11C]methyl iodide may be used without modification for [18F]fluoroethylation. This is advantageous for the transfer of a promising 11C-labeled radiotracer into its 18F-labeled counterpart by exploitation of the longer half-life of fluorine-18. Besides that, [18F]fluoroethylation can be performed with unprotected compounds and the reaction conditions are less restricting in comparison to those for direct nucleophilic substitution.
[18F]FETs is one of the mostly used reagents for [18F]fluoroethylation due to its beneficial properties; it is easy to prepare, possesses low volatility, is almost stable and shows high reactivity in correspondence with some chemoselectivity. On the other hand, some disadvantages are known: (1) [18F]FETs may be sensitive to distinct solvents, bases and high temperatures, (2) [18F]FETs may react unselectively, e.g. if multiple hydroxyl and amino groups are present in the target molecule, and (3) often, it shows inadequate chemical purity. To circumvent the latter drawback, intermediate purification steps for [18F]FETs have been developed comprising solid phase extraction (SPE) as well as HPLC-based procedures, partly remote-controlled, which as a consequence, afford the implementation of a two-pot radiolabeling procedure.12,13
This review will summarize the role of [18F]FETs as a labeling block in PET radiochemistry, its usage in the synthesis of radiopharmaceuticals with clinical relevance like [18F]fluoroethylcholine ([18F]FECh) or [18F]fluoroethyltyrosine ([18F]FET), and also its broad application in the radiolabeling of a multitude of radiotracers in pre-clinical research. The radiolabeling conditions for [18F]FETs and the aspects of reactivity towards functional groups will be reviewed for a number of O- and N-[18F]fluoroethylations. The pros and cons of direct ([18F]fluoride-based) and indirect ([18F]FETs-based) labeling approaches will be discussed as well as the different purification methods of [18F]FETs in accordance with the radiochemical yield.
Radiosynthesis, reactivity and purification
The first radiosynthesis of [18F]FETs and its methyl and propyl analogs was published in 1987 by the group of Stöcklin who described the reaction of the corresponding bis-tosylated, -mesylated and -brominated alkanes with [18F]fluoride, Kryptofix K222 and K2CO3 in acetonitrile.14 By this straightforward approach, [18F]FETs was obtained in 77–82% radiochemical yield (RCY) and this reaction is more or less still used without major modifications up to now (Fig. 1). Notably, dibromoalkane precursors gave lower yields of [18F]fluoroalkylated products than their bis-tosylated and bis-mesylated counterparts. The corresponding [18F]fluoromethylated compounds could be obtained in only 1% RCY. Furthermore, the impact of the solvent and reaction time on the synthesis of [18F]FETs was evaluated. Acetonitrile providing 82% RCY was superior to dichloromethane and acetone providing 52% and 28% RCY, respectively. A reaction time of 10–15 min was found to be optimal with a precursor concentration of ~0.015 mmol mL−1.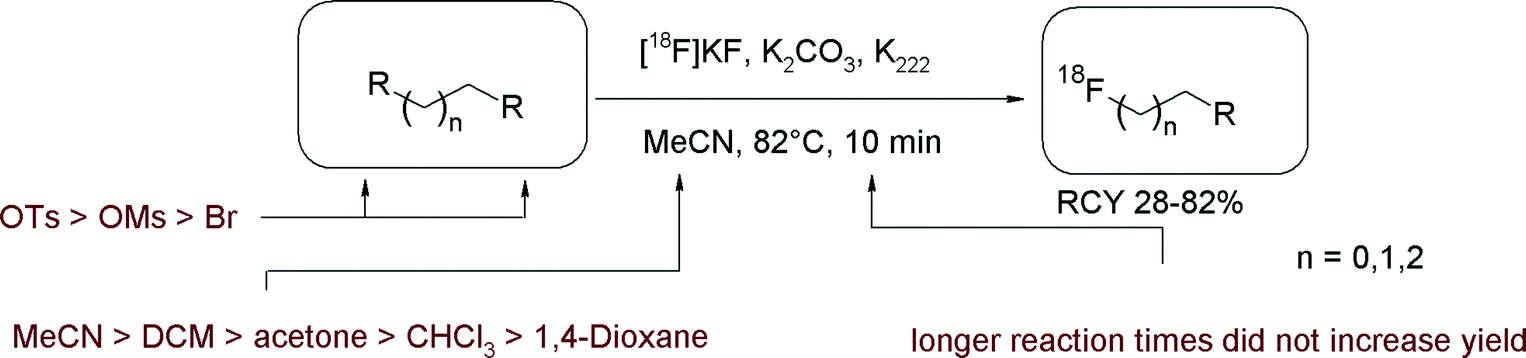 | ||
| Fig. 1 First [18F]FETs synthesis: influence of leaving group, solvent, chain length.14 | ||
Later on, Stöcklin et al. optimized the [18F]fluoroalkylation on simple H-acidic compounds with respect to leaving groups, reaction times, substrate concentration and basicity.15 Interestingly, the yield of the [18F]fluoroethylation of phenol in refluxing tetrahydrofuran or 1,4-dioxane (76–79%) was significantly higher than that in acetonitrile (50%). In a comparison of [18F]FETs with 2-[18F]fluoroethyl bromide and 2-[18F]fluoroethyl methanesulfonate in the reaction with phenol, [18F]FETs gave the highest RCY (51%) of [18F]fluoroethyl phenolate.15
In some cases, the reactivity of [18F]fluoroethylating reagents may be limited. Rösch et al. reported that the reaction of the weak nucleophilic amino group of p-anisidine with 2-[18F]fluoroethyl bromide or [18F]FETs using sodium hydride or lithium diisopropylamide (LDA) as a base, respectively, resulted in a low RCY of 15%.16 However, the authors found that the RCY was increased to 80% by adding 22–143 μmol of NaI, KI or LiI to the labeling mixture containing [18F]FEBr or [18F]FETs (Fig. 2). By application of these alkali iodides to [18F]fluoroethylation, the isolated radiochemical yield of clinically relevant radiopharmaceuticals, namely [18F]fluoroethyl-4-piperidyl benzilate and [18F]fluoroethylcholine, could almost be doubled. This most probably originates from the in situ formation of 2-[18F]fluoroethyl iodide which is a stronger fluoroethylating agent.16 From the series of alkali iodides examined, LiI was the most potent one and the more polar solvents DMF and DMSO were superior to acetonitrile.
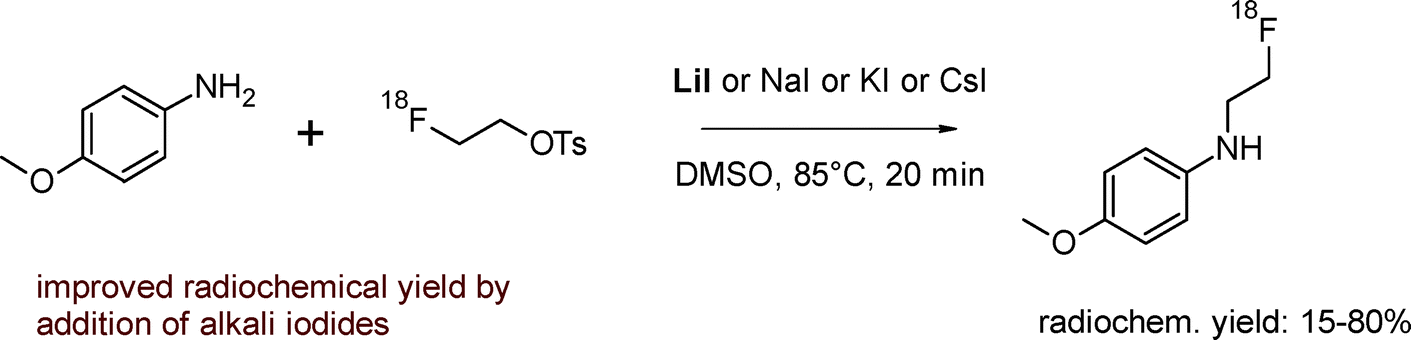 | ||
| Fig. 2 [18F]Fluoroethylation of model compounds.16 | ||
In recent years, microwave dielectric heating has brought a high impact on organic chemistry and radiochemistry.17 Accordingly, Lu et al. reported how to improve the RCY in the reaction of [18F]FETs with model compounds having carboxylic, secondary amine and phenolic functionalities.18 Under microwave-enhanced conditions (acetonitrile, 2–10 min, 300 W), the RCY was increased by ~20% compared to that obtained by conventional heating (Fig. 3). Notably, under microwave conditions, [18F]FETs reacts with DL-pipecolinic acid, having both carboxylic and amine functionalities, exclusively with the amino group.
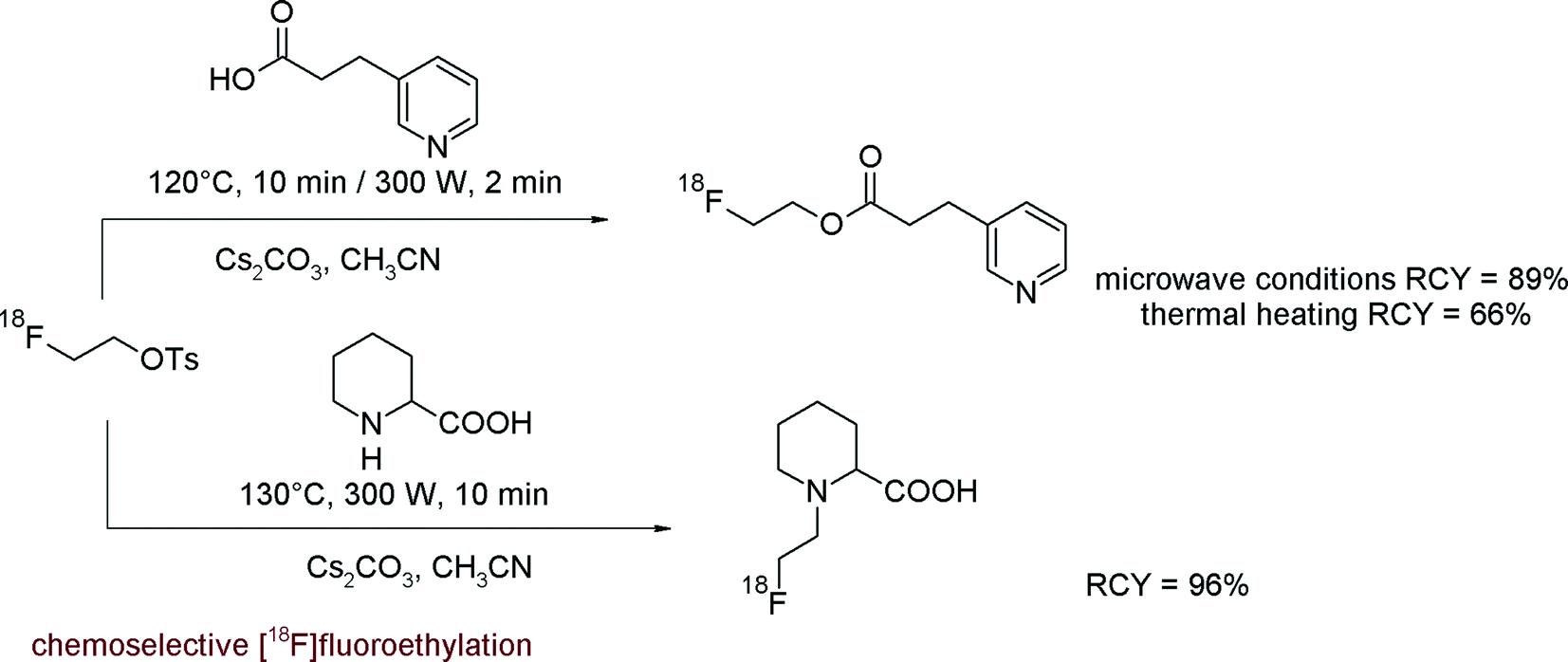 | ||
| Fig. 3 [18F]Fluorethylation of model compounds under microwave enhanced conditions.18 | ||
In 2005, the group of Pike reported the synthesis of modified 2-[18F]fluoroethyl arylsulfonates bearing a less electron-rich aryl group offering enhanced reactivity.19 A series of novel 2-[18F]fluoroethylated benzenesulfonyl ([18F]FEBs), 4-bromobenzenesulfonyl ([18F]FEBrBs), 4-nitrobenzenesulfonyl ([18F]FENs) and 3,4-dibromobenzenesulfonyl ([18F]FEBr2Bs) derivatives were synthesized and compared with [18F]FETs in terms of handling and reactivity (Fig. 4). The novel 2-[18F]fluoroethyl arylsulfonates were obtained in yields of 47–53%, suggesting that the substitution pattern on the phenyl ring has minimal effects on the yields of [18F]fluorine incorporation. However, in the subsequent [18F]fluoroethylation reaction with the target molecule 2β-carboxymethoxy-3β-(4-chlorophenyl)nortropane (CNT), [18F]FEBr2Bs provided a considerably higher RCY (87%) than [18F]FETs (64%). A similar tendency was observed with two other model compounds, an amyloid probe and p-nitrophenol where [18F]fluoroethylation with [18F]FEBr2Bs also provided a higher RCY compared with that using [18F]FETs. According to these results and the easy synthesis of [18F]FEBr2Bs, this reagent may be considered as a powerful alternative to [18F]FETs.
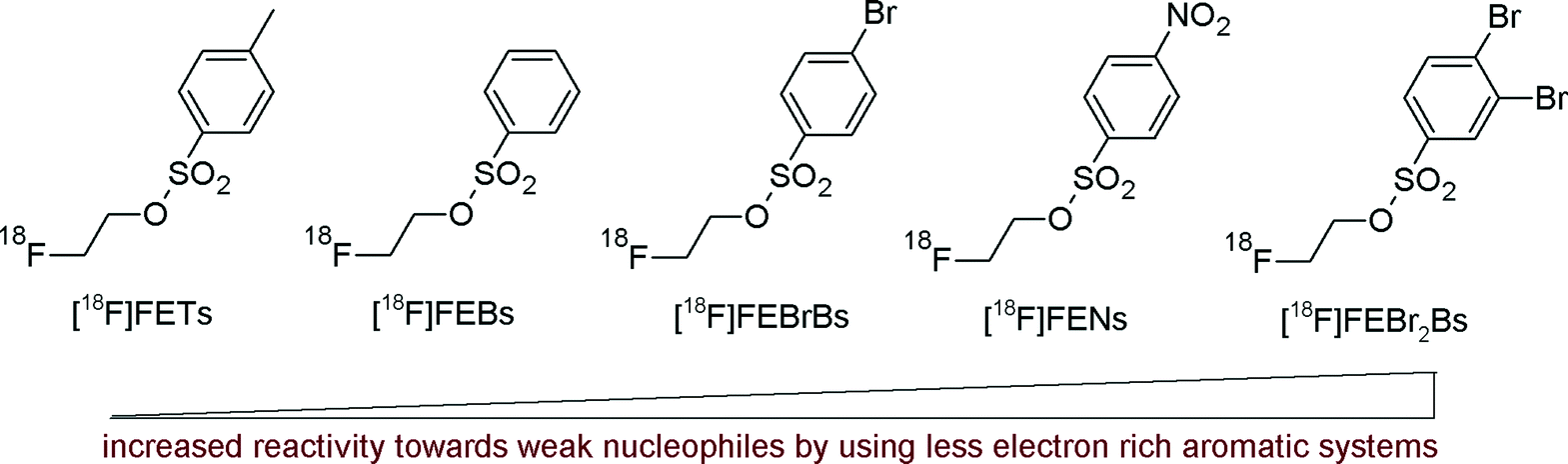 | ||
| Fig. 4 Graduation of reactivity of [18F]fluoroethylating reagents.19 | ||
In a recent study, the comparison of the reactivity of [18F]FETs, [18F]FENs and [18F]FEBr2Bs for the [18F]fluoroethylation of an azadipeptide nitrile-based inhibitor of cathepsin revealed that [18F]FENs provided a higher RCY (74%) compared with [18F]FETs and [18F]FEBr2Bs (Fig. 5).20
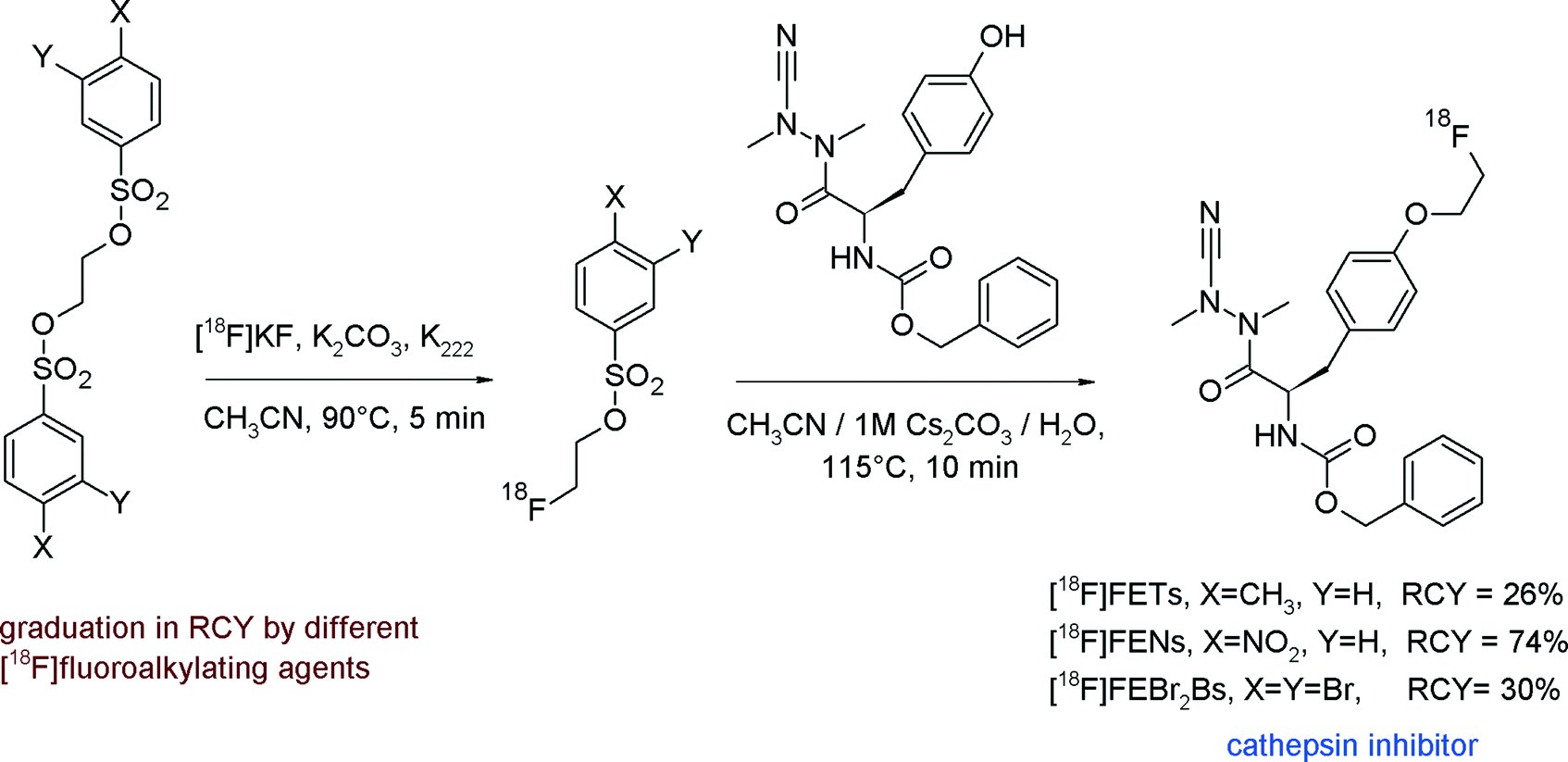 | ||
| Fig. 5 [18F]Fluoroethylation of a cathepsin inhibitor.20 | ||
A new approach for the synthesis of [18F]FETs was presented by Lemaire et al. using a technique circumventing the time-consuming azeotropic drying step of the [18F]fluoride/K222/K2CO3 complex, which is often characterized by loss of radioactivity.21 It is characterized by the elution of [18F]fluoride from an anion exchange cartridge (SAX) with acetonitrile containing water (6300 ppm) and the strong organic base tert-butyl-tetramethylguanidine (BTMG). The direct reaction of this mixture with 1,2-ethylene glycol-bis-ditosylate provided [18F]FETs in 83% RCY and 85% purity. This innovative technique using short reaction times of ~5 min and small solvent volumes of 0.5–1.0 mL offers advantages for the development of microreactors or microfluidic-based devices.
As another example for miniaturization, Pascali et al. reported the production of [18F]FETs in a microfluidic system, providing the [18F]fluoroethylating agent in 67% RCY within a reaction time of 90 s.22 As the [18F]fluoride drying procedure was performed on macro-scale and apart from the microfluidic system, the authors were able to divide the dry [18F]fluoride/K222/K2CO3 complex into several batches and perform a number of radiolabeling reactions from a single stock solution. This protocol may be useful to obtain individual doses of the labeling reagent permitting the dose-on-demand production of radiopharmaceuticals.
One major challenge in the synthesis and application of [18F]FETs is the chemical purity because the 1,2-ethylene glycol-bis-tosylate precursor, if not removed from the [18F]fluoroalkylating mixture, will act as a competitive reagent to the targeted functional groups resulting in the formation of tosyl-ethylated, hydroxyl-ethylated or cross-linked by-products (Fig. 6). The first two often show a similar chromatographic behavior like the [18F]fluoroethylated main product, making their purification by semi-preparative HPLC difficult and finally resulting in products with a similar binding behavior preventing specific binding of the radiotracer.
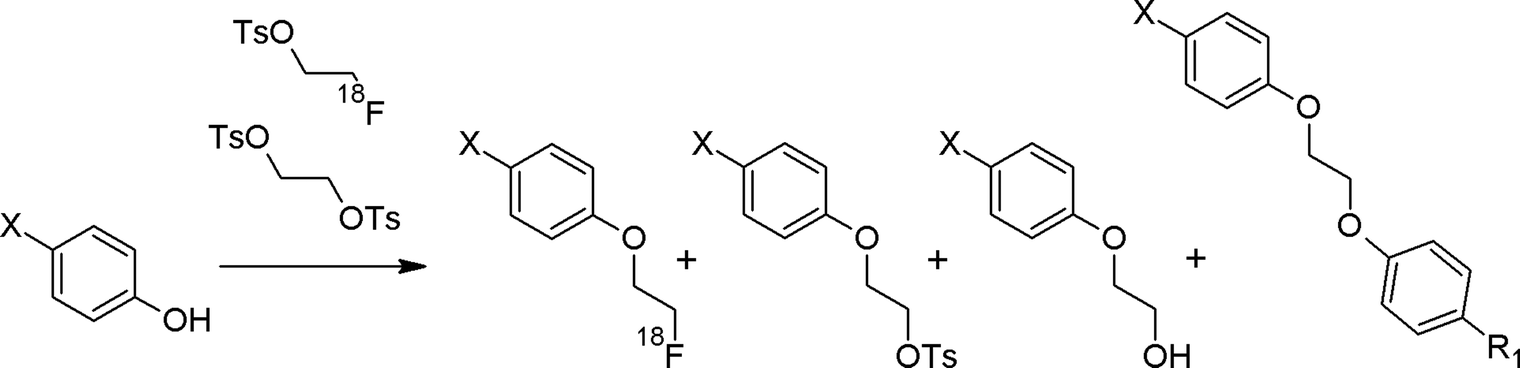 | ||
| Fig. 6 Expectable by-products in radiolabeling with [18F]FETs. | ||
To circumvent this disadvantage, a minimum amount of 1–2 mg of 1,2-ethylene glycol-bis-tosylate may be used as the precursor but this often results in a decreased RCY of [18F]FETs. Another and more sophisticated alternative is the implementation of a purification step for [18F]FETs by either solid phase extraction or semi-preparative HPLC. Both approaches result in enhanced technical expense, e.g. a two-pot radiolabeling device, additional valves and at least two HPLC steps. However, these expanded efforts are justified by the enhanced purity and reactivity of [18F]FETs resulting in higher RCY and improved specific activity of the final radiotracer.
The first purification of [18F]FETs was reported in 2000 by the group of Wester for the radiosynthesis and biological evaluation of 6-O-(2-[18F]fluoroethyl)-6-O-desmethyldiprenorphine ([18F]FDPN), a ligand for the opioid receptor system (Fig. 7).23 In brief, [18F]FETs was purified by RP18-based HPLC. After adsorption on a C18 cartridge and drying with a nitrogen stream, [18F]FETs was finally eluted with acetonitrile to react with the corresponding hydroxyl precursor. In this manner, [18F]FETs was obtained as an intermediate in 65% RCY. The two-step radiosynthesis provided [18F]FDPN within 100 min with an overall RCY of 22% and a specific activity of 37 GBq μmol−1 at the end of synthesis (EOS). The [18F]fluoroethylation was reproduced later by Mueller et al. under almost identical conditions providing [18F]FDPN in 19% RCY.24
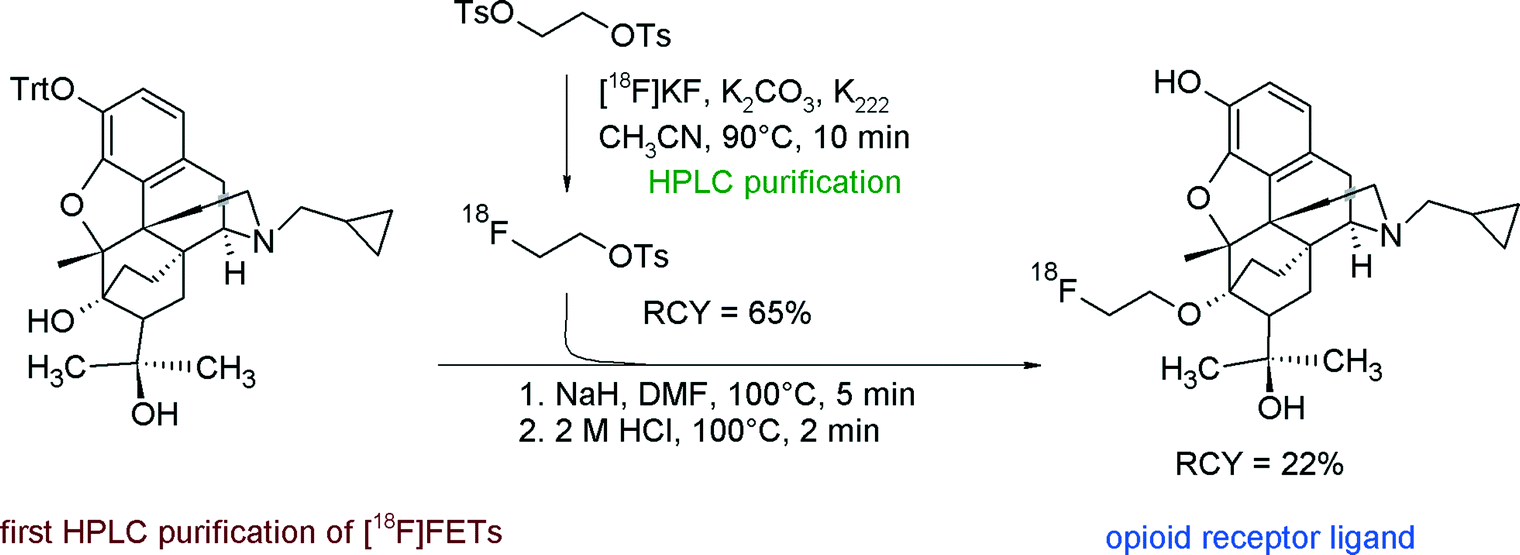 | ||
| Fig. 7 Radiosynthesis of [18F]FDPN.23 | ||
In two recent papers, Schoultz et al. published the full radiosynthesis of the opioid receptor ligand [18F]FDPN together with the partial agonist [18F]fluoroethyl-buprenorphine ([18F]FBPN) and the agonist [18F]fluoroethyl-phenethyl-orvinol ([18F]FBEO).12,25 The method is based on innovative automated production and purification of [18F]FETs where the excess of the 1,2-ethylene glycol-bis-tosylate precursor is removed by precipitation and subsequent filtration, and [18F]FETs is purified by gradient elution from the SPE cartridges. After elution of [18F]FETs with DMF, it was reacted with the FDPN precursor and NaH.12 This new two-pot two-cartridge-based system avoids HPLC purification of [18F]FETs and provides [18F]FDPN within 100 min in 25% overall RCY and high chemical and radiochemical purity. The other 18F-fluoroethylated orvinol-based PET tracers [18F]FBPN and [18F]FBEO were obtained by the same method in similar RCY from their O-desmethyl precursors, demonstrating the reliability of the method.
With the aim to translate 11C-labeled radiopharmaceuticals into their respective [18F]fluoroethylated analogs, Wadsak and Mitterhauser investigated several methods for the purification of [18F]FEBr and [18F]FETs in view of their implementation in commercially available synthesizers.13 In this study, the reactivity of [18F]FETs purified by SPE and HPLC-based methods was compared with that of [18F]FEBr and the results were transferred to the synthesis of five radiotracers of clinical interest. Several SPE materials like Silica SepPak®, C18plus SepPak® and AluminaN® have been evaluated for the purification of [18F]FETs. Finally, it was demonstrated that e.g. for the synthesis of 2-[18F]fluoroethyl-(R)-1-(1-phenylethyl)-1H-imidazole-5-carboxylate ([18F]FETO), HPLC-purified [18F]FETs provided the best results with regard to RCY and chemical and radiochemical purity. Additional [18F]FETs was synthesized in 55% RCY which was substantially higher than that for distilled [18F]FEBr (34%). However, this benefit is accompanied by an enhanced need for hardware components like additional HPLC and SPE units. However, for the highest demands on chemical purity due to the low receptor densities of the target, HPLC-purified [18F]FETs is the best choice.
[18F]FETs is a radiolabeling agent widely used to form a broad variety of radiotracers as will be demonstrated in the following sections. In the majority of cases, the resulting radiolabeled probe was comprehensively investigated regarding its in vivo behavior displayed in parameters such as biodistribution, stability and metabolism. However, to the best of our knowledge, no such studies were made concerning the radiopharmacology of [18F]FETs itself, thus its properties in vivo have still to be explored.
O-[18F]Fluoroethylation
O-[18F]Fluoroethyl phenols
The phenol group is a recurrent functionality in a variety of biomolecules, and is ideally suitable for the introduction of radionuclides such as 11C as [11C]methyl via [11C]CH3I or 18F as [18F]fluoroethyl via [18F]FETs. The following examples may demonstrate the successful application of [18F]FETs in the syntheses of a multitude of radiotracers by O-[18F]fluoroethylation.Radiolabeled amino acids have raised clinical interest in neurology and oncology because they are the biological building blocks for the synthesis of neurotransmitters and proteins. Especially for diagnosis of human brain tumors, radiolabeled amino acids are advantageous since they accumulate in tumor tissue because of upregulated transporters and higher protein synthesis rates.26,27
A number of radiolabeled amino acids are used for that purpose, such as [11C]L-methionine, 2-[18F]fluoro-L-tyrosine, 6-[18F]fluoro-L-3,4-dihydroxyphenylalanine ([18F]FDOPA) and 6-[18F]fluoro-L-3-methoxy-4-hydroxyphenylalanine ([18F]OMFD). After its introduction, O-(2-[18F]fluoroethyl)tyrosine ([18F]FET) has become a very useful PET tracer since it shows high brain uptake, has sufficient metabolic stability, is not involved in protein synthesis, and its accumulation in tumors is solely based on amino acid transporters.
The first radiosynthesis of [18F]FET by direct 18F-alkylation of tyrosine with [18F]FETs was published in 1999 by Wester et al. (Fig. 8).28 The disodium salts of unprotected L-tyrosine or D-tyrosine were reacted with HPLC-purified [18F]FETs in DMSO at 90 °C and the corresponding [18F]FET was provided within 60 min in 40% overall RCY.
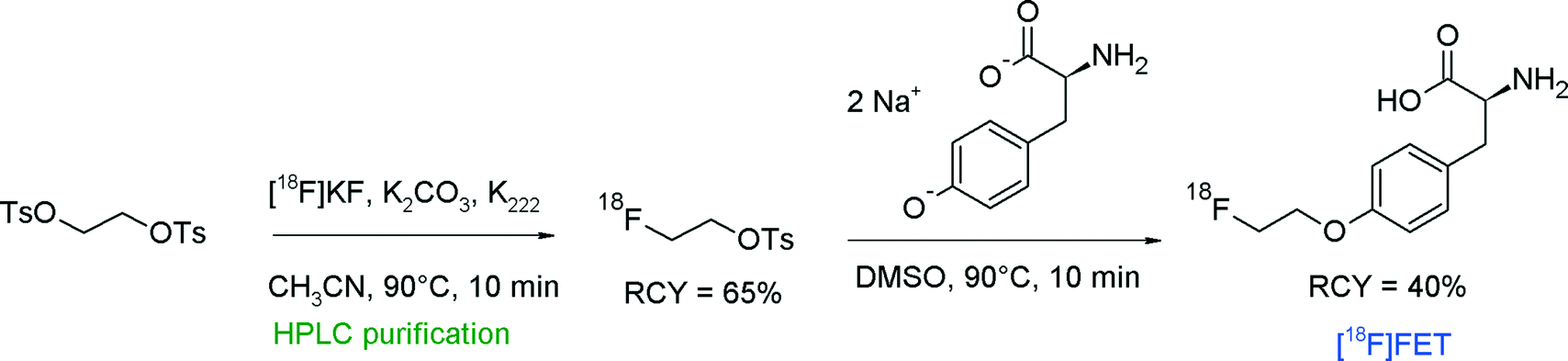 | ||
| Fig. 8 First radiosynthesis of O-(2-[18F]fluoroethyl)tyrosine.28 | ||
In a separate study, Tsukada et al. synthesized a series of radiolabeled unnatural amino acids, including D-[18F]FET to compare the properties with the L-isomer.29 Radiosynthesis of D-[18F]FET was performed by heating SPE-purified [18F]FETs with D-tyrosine in DMSO for 15 min and subsequent HPLC purification; no RCY was provided.
The first automated synthesis of [18F]FET using a commercial synthesizer was reported in 2003 by Tang et al.30 In this case, the [18F]fluoroalkylating agent [18F]FETs was synthesized starting from [18F]fluoride that was previously adsorbed on a quaternary 4-(4-methylpiperidinyl)pyridinium-functionalized anion exchange resin (SAX). The reaction was performed directly on the resin by passing 1,2-ethylene glycol-bis-tosylate in acetonitrile through the heated cartridge. [18F]FETs was eluted from the resin and added to a mixture of L-tyrosine and NaOH. After evaporation of the solvent, DMSO was added and the mixture was heated to form [18F]FET. This protocol avoids any HPLC-purification step and provided [18F]FET within 52 min in 8–10% overall RCY.
A further automated, high-yielding radiosynthesis of [18F]FET was presented by Mueller et al. using a modified synthesizer designed for 11C-radiolabeling chemistry consisting of two reaction vessels.31 [18F]FETs was formed in the first reactor by conversion of 1,2-ethylene glycol-bis-tosylate with the dried [18F]KF/K222/K2CO3 complex. After addition of water, [18F]FETs was collected by SPE and eluted with hot DMSO into the second reactor containing the L-tyrosine disodium salt. The formed [18F]FET was purified by SAX and SCX-based SPE systems which provided the radiotracer in typically 41% RCY with chemical and radiochemical purity that meets the current Pharmacopeia requirements. Nowadays, the preferred synthesis of [18F]FET is based on direct nucleophilic substitution of a protected para-(2-tosylethoxy)-tyrosine with [18F]fluoride yielding the radiotracer in about 60% RCY.32 However, the abovementioned automatic [18F]FETs-based radiolabeling method may serve as a very useful radiolabeling protocol for further [18F]fluoroethylated radiotracers.
A convenient remote-controlled radiosynthesis of a new tryptophan analog L-5-(2-[18F]fluoroethoxy)-tryptophan (5-[18F]FETP) by [18F]fluoroethylation of the 5-hydroxy-L-tryptophan disodium salt was reported in 2010 (Fig. 9).33 The radiolabeling took place in a two-pot synthesizer, where [18F]FETs was formed in the first reactor in 45–60% RCY and transferred into the second vessel for [18F]fluoroethylation in DMSO. After purification of 5-[18F]FETP by silica- and RP18-based SPE, the radiolabeled amino acid was isolated within 65 min in 12–16% overall RCY.
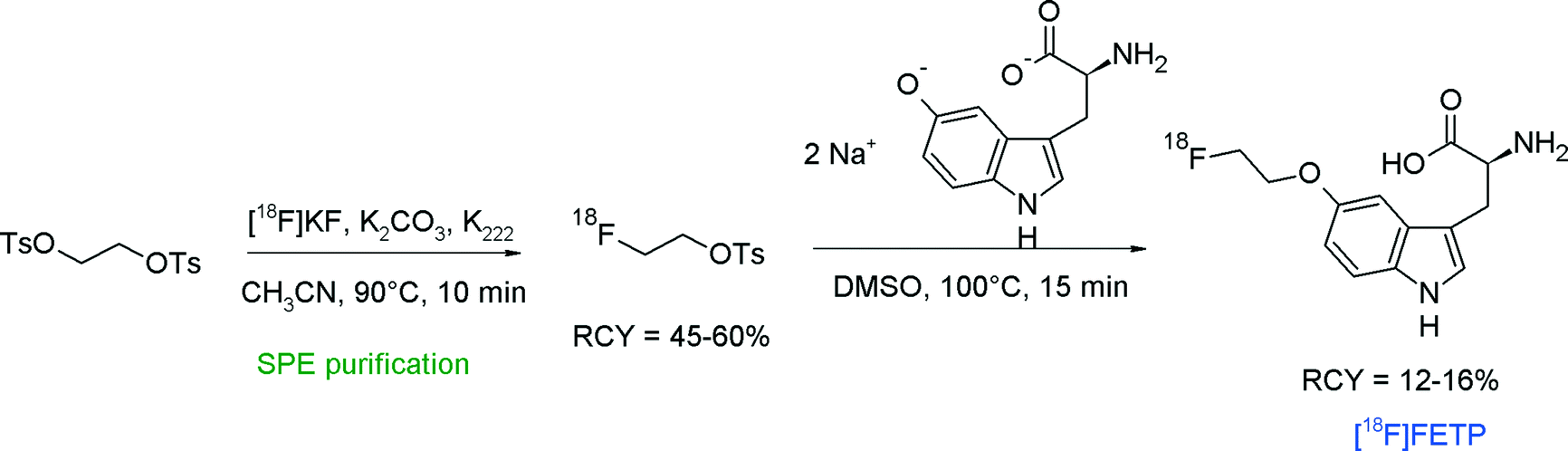 | ||
| Fig. 9 Radiosynthesis of L-5-(2-[18F]fluoroethoxy)tryptophan.33 | ||
4-Phenylpiperazines have attracted radiopharmaceutical interest since their derivatives such as FAUC346 have proven to be highly selective and affine dopamine D3 receptor antagonists. Aiming at the development of dopamine D3 radioligands, Hocke et al. synthesized 18F-labeled compounds derived from FAUC346 with the methoxy functionality being replaced with a fluoroethoxy bioisostere (Fig. 10).34O-[18F]Fluoroethylation with [18F]FETs was the method of choice to obtain four different radioligands for the D3 receptor. HPLC-purified [18F]FETs was reacted with the phenol precursor and a base in DMF at 120 °C; under these conditions, 24–65% RCYs were observed. Interestingly, the best RCY was achieved by using N(Bu)4OH as a base, whereas no radiotracers were formed with NaH and NaOCH3.
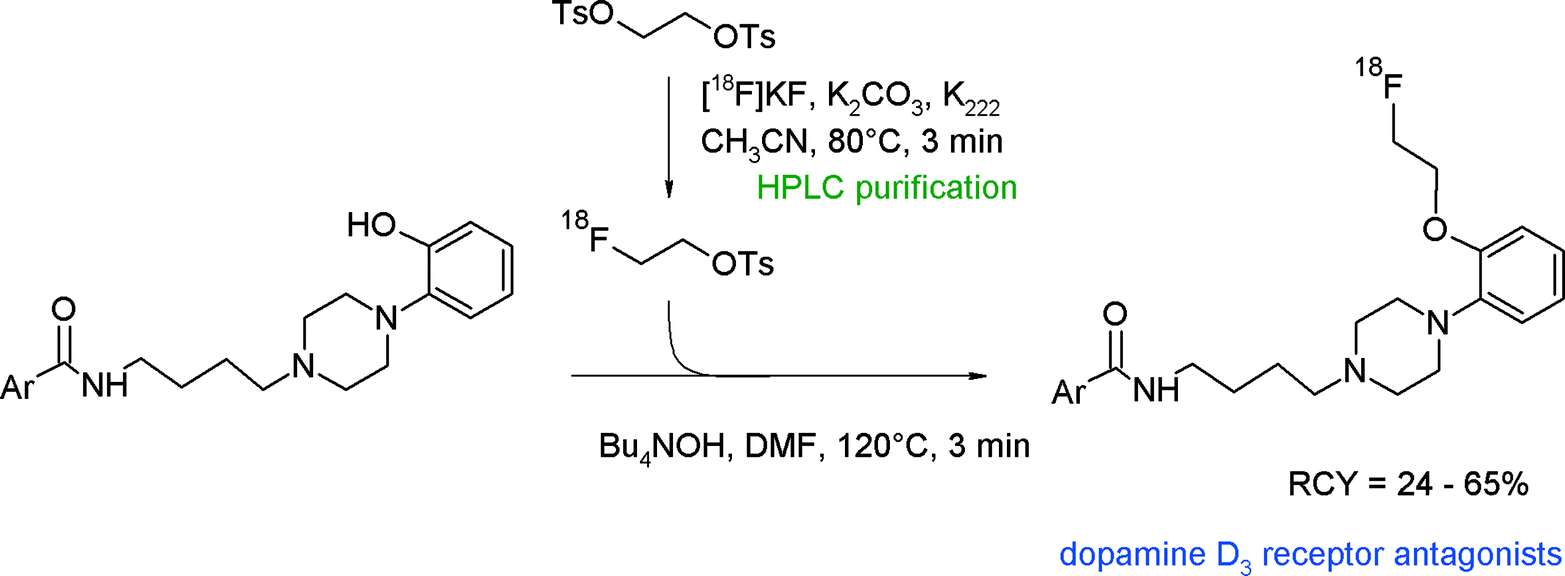 | ||
| Fig. 10 Synthesis of O-[18F]fluoroethylated 4-phenylpiperazine.34 | ||
Another series of radiolabeled dopamine D2/3 receptor agonists, [18F]fluoroalkylated derivatives of 2-aminomethylchromane, has been presented by van Wieringen et al. (Fig. 11).35 In this study, [18F]FETs and 4-[18F]fluorobutyl tosylate were synthesized from the corresponding bis-tosylated alkyl precursors and the intermediates were purified by semi-preparative HPLC. The O-[18F]fluoroethylation was performed by addition of the methoxymethyl-protected phenolic precursor, sodium hydride and DMF followed by a deprotection step. The protective group was required to prevent [18F]fluoroethylation at the second hydroxyl group of the target molecule. The overall RCY of this three-step radiosynthesis involving two HPLC runs was up to 11%. Notably, the reaction with 4-[18F]fluorobutyl tosylate provided a higher RCY up to 22%.
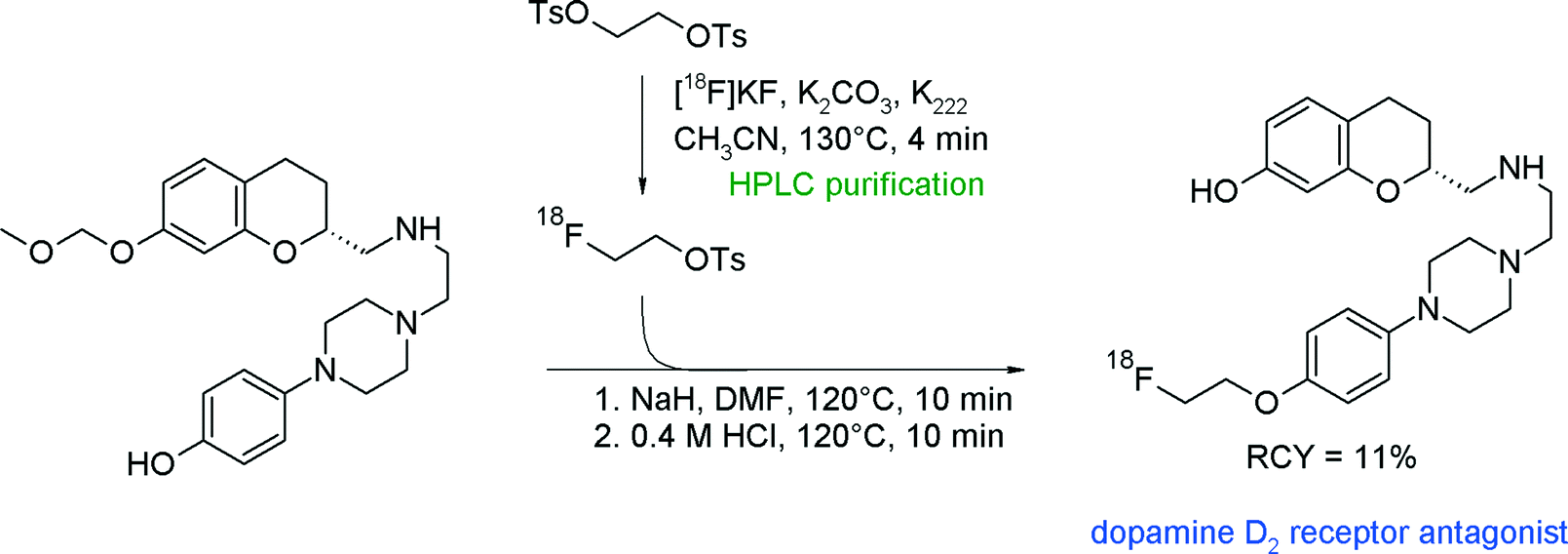 | ||
| Fig. 11 Synthesis of an O-[18F]fluoroethylated 2-aminomethylchromane.35 | ||
To provide potential PET imaging probes for the dopamine D4 receptor, Tietze et al. developed [18F]fluoroethylated 5-cyano-indole derivatives and reported optimized radiolabeling conditions with [18F]FETs.36 [18F]FETs was synthesized within a reaction time of 3 min at 90 °C, purified by gradient reversed-phase HPLC, fixed on a SPE unit and eluted with dry DMF into a reaction vessel containing the hydroxyl precursor in DMSO and a base (Fig. 12). Surprisingly, the optimized reaction conditions for the ortho-fluoroethylation could not be transferred to the para-derivative. As an alternative to the reagent system DMSO/NaOMe, which was efficient for the ortho- but failed for the para-hydroxyl group, the mixture DMF/N(Bu)4OH finally led to effective para-[18F]fluoroethylation. The RCY of the O-[18F]fluoroethylation step was 81% for the 2-hydroxyphenyl and 47% for the 4-hydroxyphenyl precursor. As part of these investigations, the authors compared the solvents DMF and DMSO and pointed out that the use of DMSO resulted in accelerated product formation (80% in DMSO versus 43% in DMF) under identical reaction conditions. By optimizing the reaction temperature, a significant drop of the RCY was observed at 140 °C due to decomposition of [18F]FETs and oxidation to 2-[18F]fluoroacetaldehyde according to Kornblum conditions (see Fig. 70).
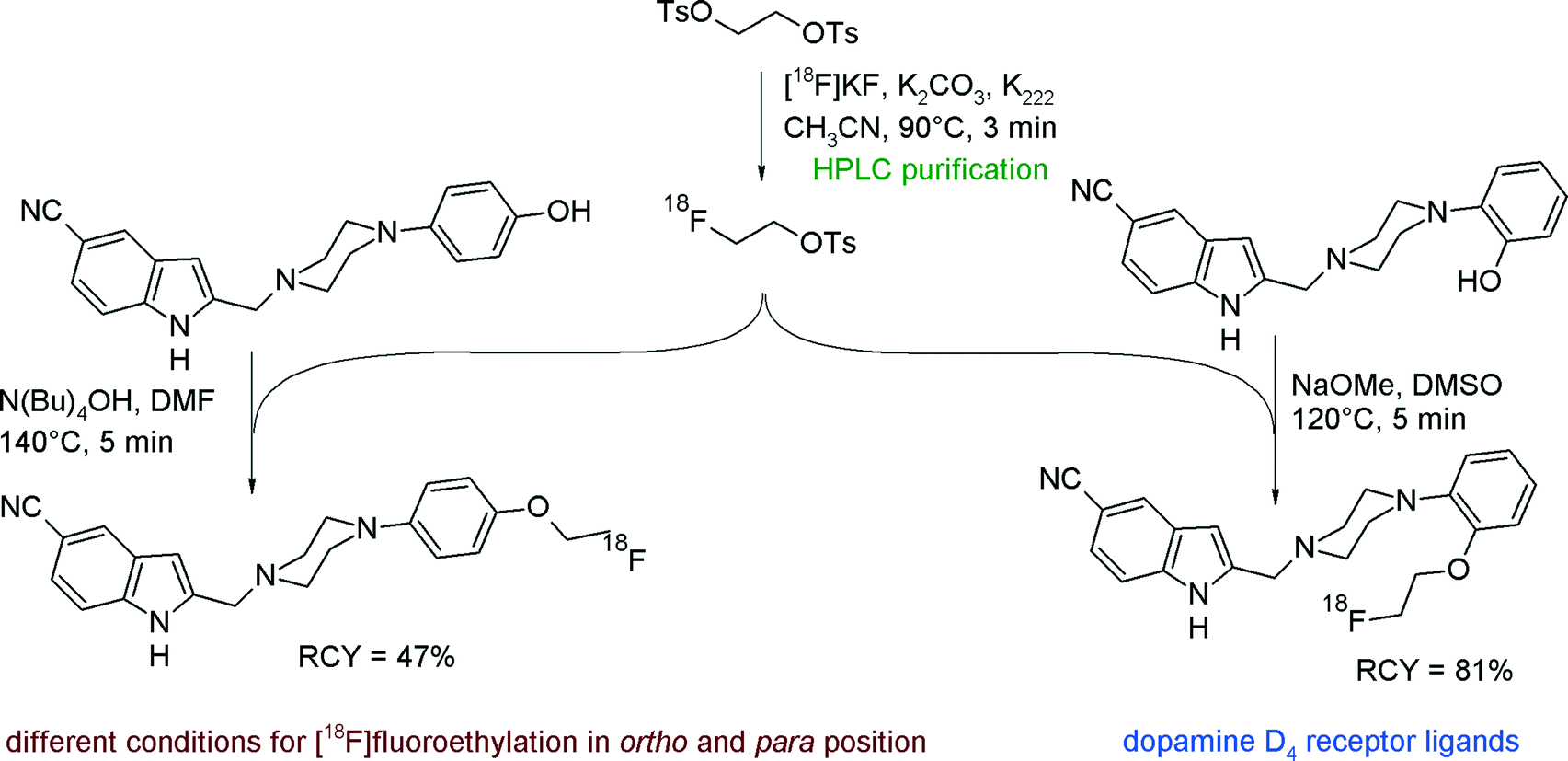 | ||
| Fig. 12 Synthesis of O-[18F]fluoroethylated 5-cyano-indoles.36 | ||
A further radioligand targeting the dopamine D4 receptor was prepared by O-[18F]fluoroethylation of a dimethoxy-substituted benzylpiperazine derivative.37 Employing HPLC-purified [18F]FETs, the hydroxyl precursor was reacted in DMF and N(Bu)4OH for 3 min to provide the [18F]fluoroethylated product in 92% RCY. The desired radiotracer was isolated after semi-preparative HPLC purification in high purity (Fig. 13).
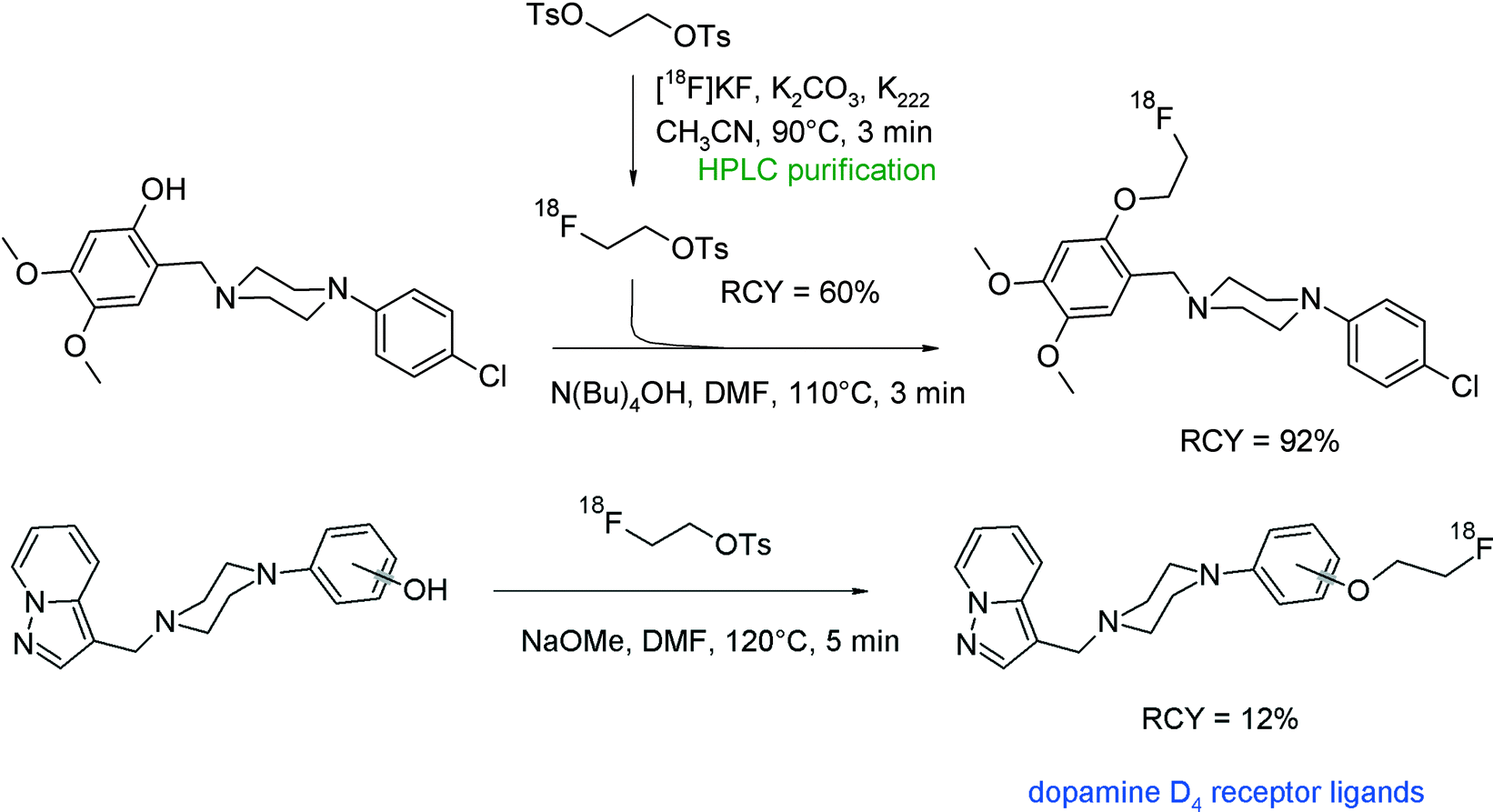 | ||
| Fig. 13 Synthesis of O-[18F]fluoroethylated piperazines.37,38 | ||
Prante et al. published a series of piperazine-based radioligands with high affinity towards the dopamine D4 receptor, including 2- and 3-substituted pyrazolo[1,5-a]pyridines.38 Two compounds were selected for [18F]fluoroethylation. For that purpose, the corresponding hydroxyl precursors were allowed to react with [18F]FETs in DMF and NaOMe (Fig. 13). After 3 min and 5 min, respectively, at 120 °C, 87–92% of the [18F]fluoroethylated products were detected. The overall RCY of the dopamine D4 receptor ligands after purification and formulation for in vivo experiments was 12–13% within a total synthesis time of 120 min. In this case and opposite to previous findings,36 no difference in reactivity towards [18F]FETs between the 4-hydroxyl- and 2-hydroxyl-phenyl derivatives was observed. Interestingly, the authors synthesized in a parallel approach one of the radioligands by direct kryptate-assisted nucleophilic [18F]fluorination using a tosylate precursor and standard conditions. By this direct method, the rate of 18F-incorporation was 26% but after workup, purification and formulation, the radioligand was obtained in less than 10% overall RCY. Both approaches provide more or less a similar RCY. This is not always the case as will be shown later on in a separate chapter of this review comparing [18F]fluoroethylation with [18F]FETs with direct [18F]fluoride substitution to generate the same radiolabeled species.
The compound class of imidazobenzodiazepines is known to be antagonists for the γ-aminobutyric acid (GABAA) receptor. A prominent example is flumazenil which has been radiolabeled as well with carbon-11 and fluorine-18.39 With the aim to find alternative radiolabeling routes, Jackson et al. synthesized a [18F]fluoroethylated derivative which was obtained by the reaction of a hydroxyl precursor with [18F]FETs (Fig. 14).39 The overall RCY for the three-step synthesis of [18F]fluoroethylated flumazenil was 18–20%.
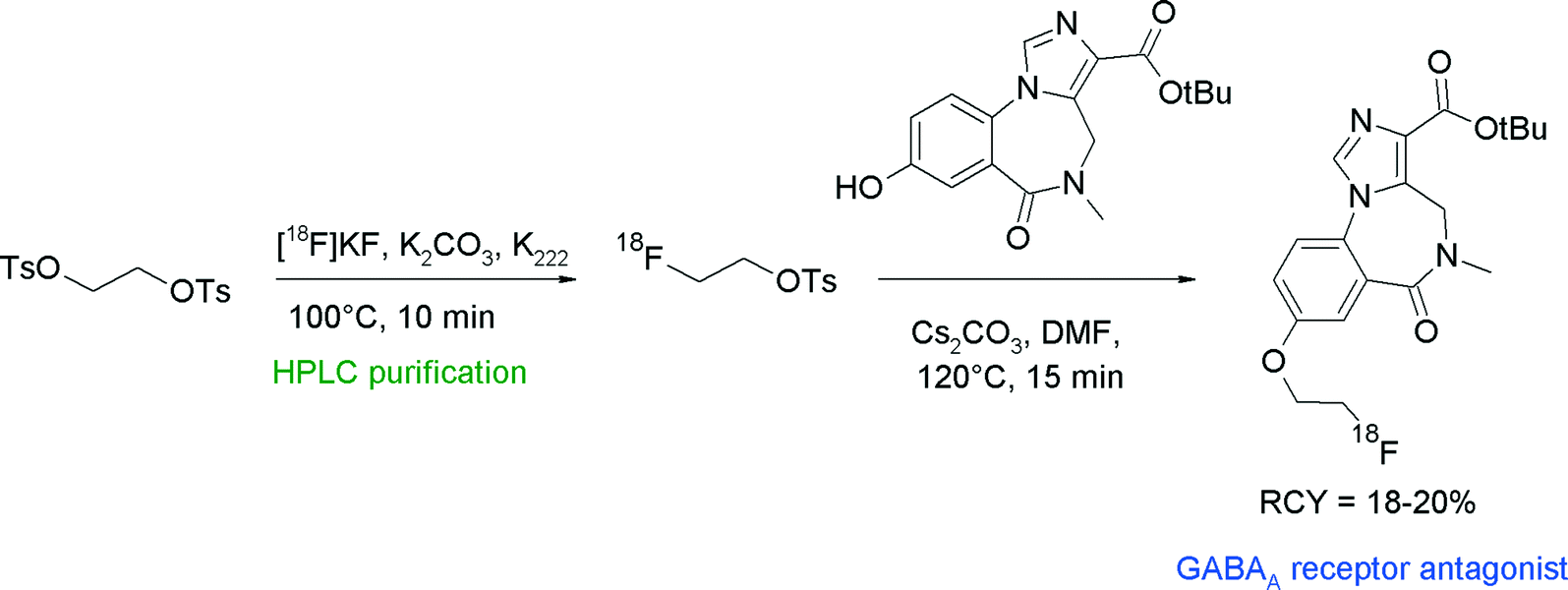 | ||
| Fig. 14 Radiosynthesis of O-[18F]fluoroethylated flumazenil.39 | ||
Since serotonin (5-hydroxytryptamine, 5-HT) is involved in important brain functions, radiolabeled serotonin receptor antagonists are highly desired compounds for the investigation of human brain disorders. Herth et al. reported one radioligand in a series of piperazine-substituted azatricyclo-dec-8-ene-3,5-diones targeting the serotonin 5-HT1A receptor, which was synthesized via [18F]fluoroethylation with [18F]FETs.40 HPLC-purified [18F]FETs was reacted for 20 min at 120 °C with the corresponding hydroxyl precursor (Fig. 15). The [18F]fluoroethylated 5-HT1A receptor ligand was provided as an injectable solution within 130 min in 25% overall RCY.
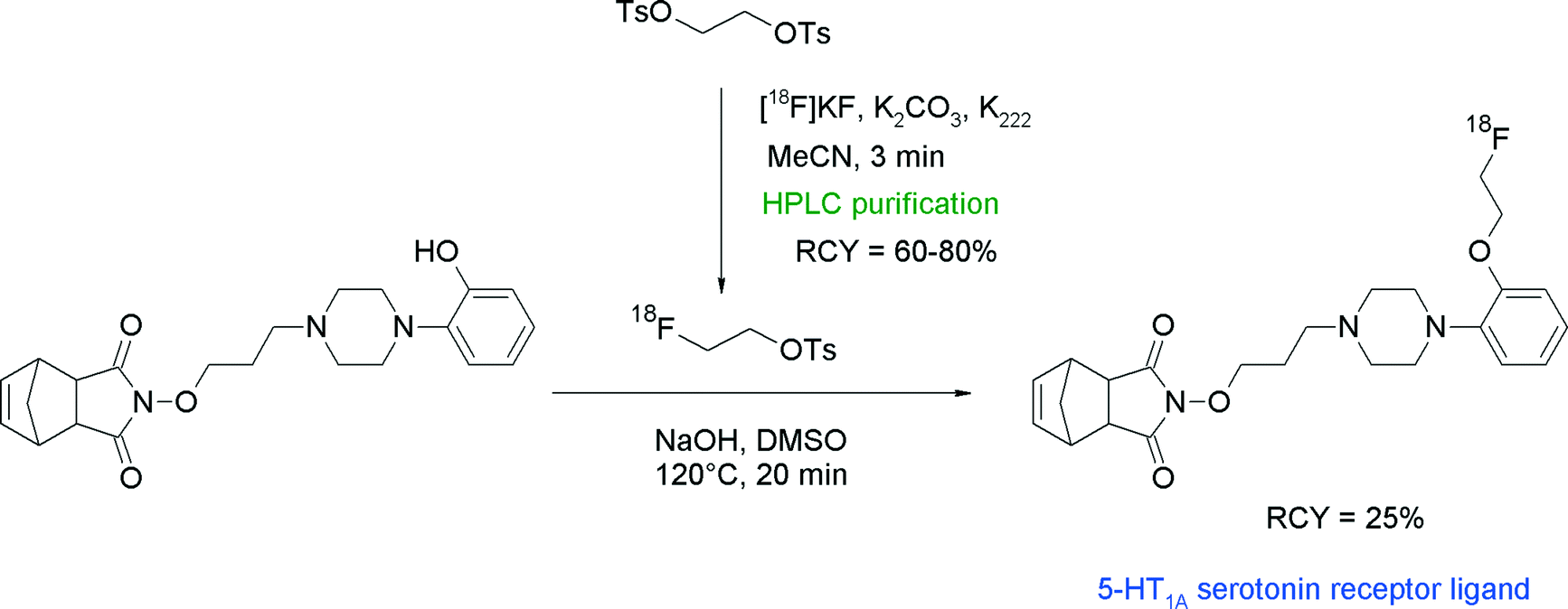 | ||
| Fig. 15 Radiosynthesis of an O-[18F]fluoroethylated azatricyclo-dec-8-ene-3,5-dione.40 | ||
Funke et al. developed 18F-labeled aminocyclohexyl-substituted indole carbonitriles having nanomolar affinities to the serotonin transporter (SERT) as important targets for PET.41 The [18F]fluoroethylation was performed as a one-pot two-step procedure. [18F]FETs was added to the dried hydroxyl precursor which was beforehand activated with K2CO3 and K222 in refluxing methanol (Fig. 16). The HPLC-purified SERT radioligands were obtained within 150 min in 11–22% overall RCY.
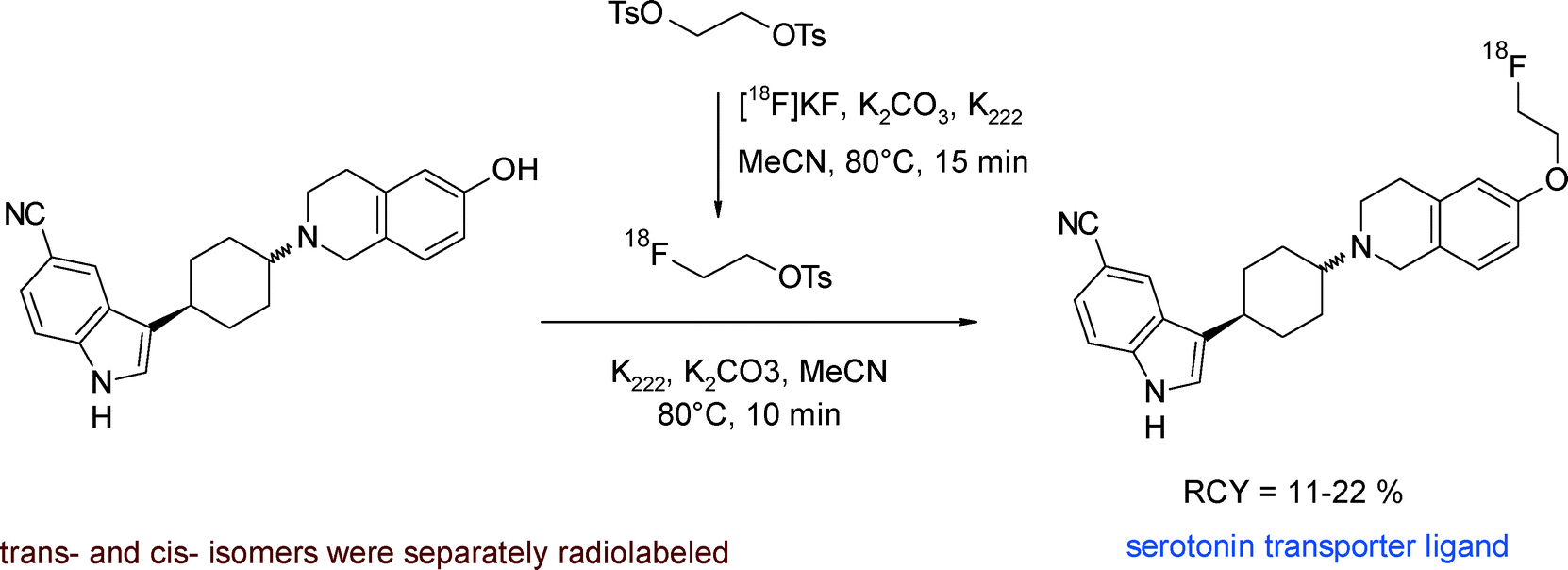 | ||
| Fig. 16 Radiosynthesis of O-[18F]fluoroethylated aminocyclohexyl indole carbonitriles.41 | ||
N-Alkylated benzoylpiperidines (e.g. MDL 105725, MH.MZ) represent a compound class targeting the 5-HT2A receptor and have been successfully radiolabeled with [18F]FETs (Fig. 17).42,43 [18F]FETs was purified by SPE prior to the reaction with the hydroxyl precursor which was activated by NaOH in DMSO. Two radiotracers were obtained within 100 min in about 40% overall RCY. Notably, one of the precursors bearing an aliphatic hydroxyl group did not undergo [18F]fluoroalkylation on the alcohol, indicating that under the standard basic conditions used, the phenolic functionality is more reactive towards [18F]FETs.
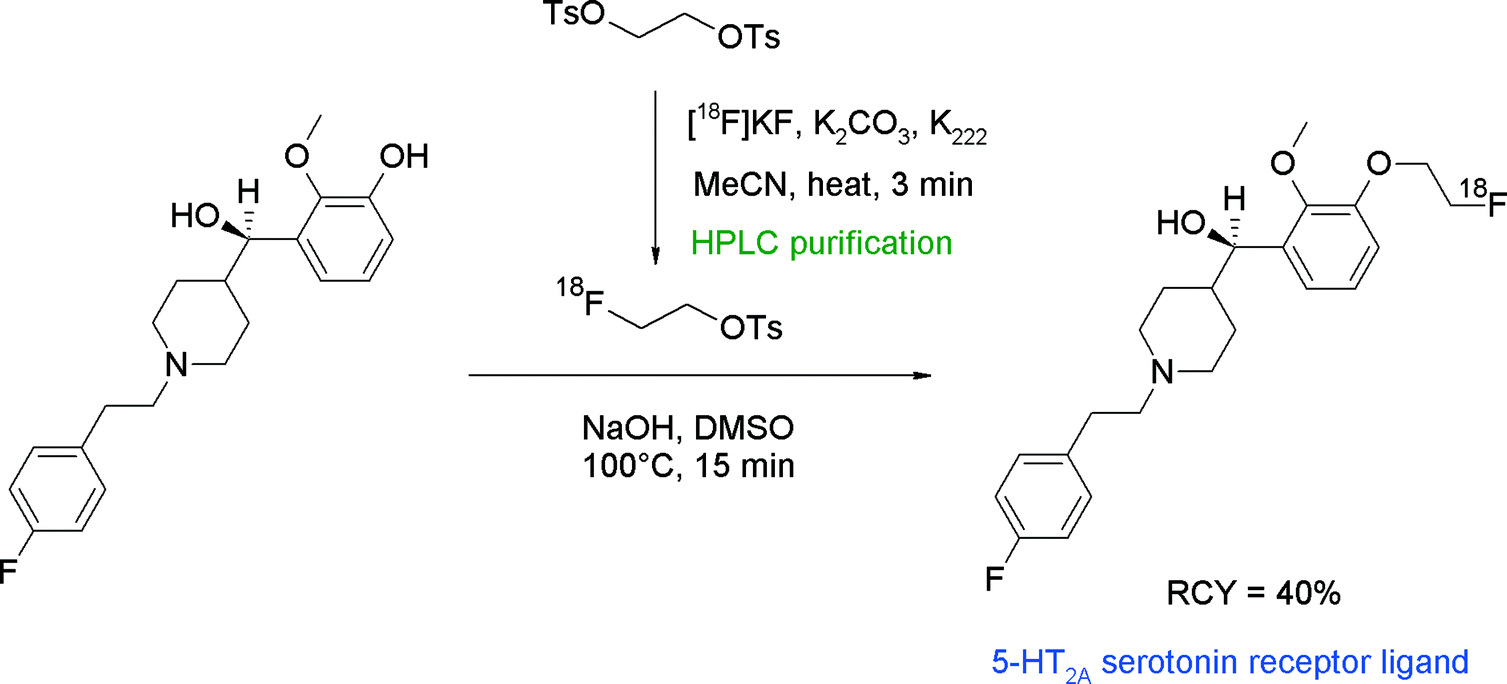 | ||
| Fig. 17 Radiosynthesis of an O-[18F]fluoroethylated benzoylpiperidine.42,43 | ||
Majo et al. developed phenylpiperazine-substituted 6-aza-uracil derivatives as promising antagonists of the 5-HT1A serotonin receptor.44 Since the corresponding carbon-11 labeled radiotracer showed a promising in vivo behavior but suffered from the usual limitations (short half-life), the authors decided to develop an O-[18F]fluoroethylated analogue. Radiosynthesis was performed by two-step fluoroalkylation of the phenolate. HPLC-purified [18F]FETs was reacted with phenol in the presence of K2CO3 in DMSO at 110 °C to provide the [18F]fluoroethylated 5-HT1A receptor antagonist after 60 min in 45% overall RCY (Fig. 18). When aqueous NaOH was used as a base, decomposition of [18F]FETs was observed at temperatures of about 125 °C and the RCY dropped to 25%. The authors developed a one-pot modification of their radiosynthesis in which the HPLC purification of [18F]FETs was replaced with SPE-based purification. After drying and elution of [18F]FETs with diethyl ether, the ether layer was removed and [18F]FETs was reacted with the phenol precursor. However, by application of this protocol, the radiotracer was obtained in only 20–25% overall RCY, suggesting that HPLC-purified [18F]FETs is more reactive compared to the SPE-purified batch.
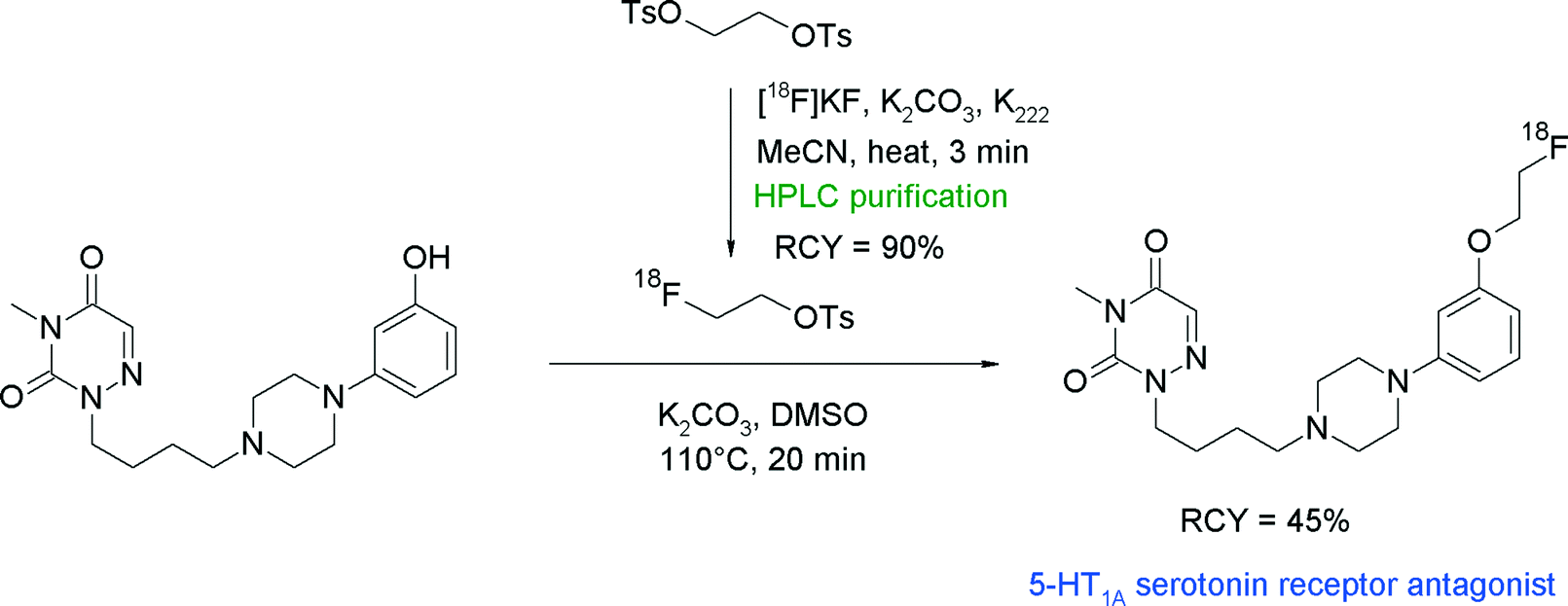 | ||
| Fig. 18 Radiosynthesis of an O-[18F]fluoroethylated phenylpiperazine-substituted 6-aza-uracil.44 | ||
N-Methyl-D-aspartate (NMDA) receptor represents another important target in neurological and psychiatric disorders and hydantoin-substituted indole carboxylic acids are lead compounds for the development of radiolabeled NMDA receptor ligands. The group of Rösch developed a series of ortho-, meta- and para-(2-fluoroethoxy)phenyl-substituted derivatives of this class and performed O-[18F]fluoroethylation at different positions (Fig. 19).45 The initial reaction of the free carboxylic acid with HPLC-purified [18F]FETs in aprotic solvents and NaOH as a base resulted in [18F]fluoroethylation RCY <4% connected with the formation of two unknown by-products. Hence, the authors decided to start from the corresponding ethyl esters. Under optimized conditions using the ethyl ester precursor in DMSO and NaOH at 100 °C, 25–40% [18F]fluoroethylation was achieved within 5 min. Prolongation of the reaction time resulted in a drop of the RCY caused by cleavage of the ester moiety and thermal decomposition. Although different base systems had been tested, the subsequent cleavage of the ethyl ester group with 1 M NaOH was incomplete. Finally, further HPLC purification was performed providing the [18F]fluoroethylated NMDA receptor ligand in 5–7% overall RCY.
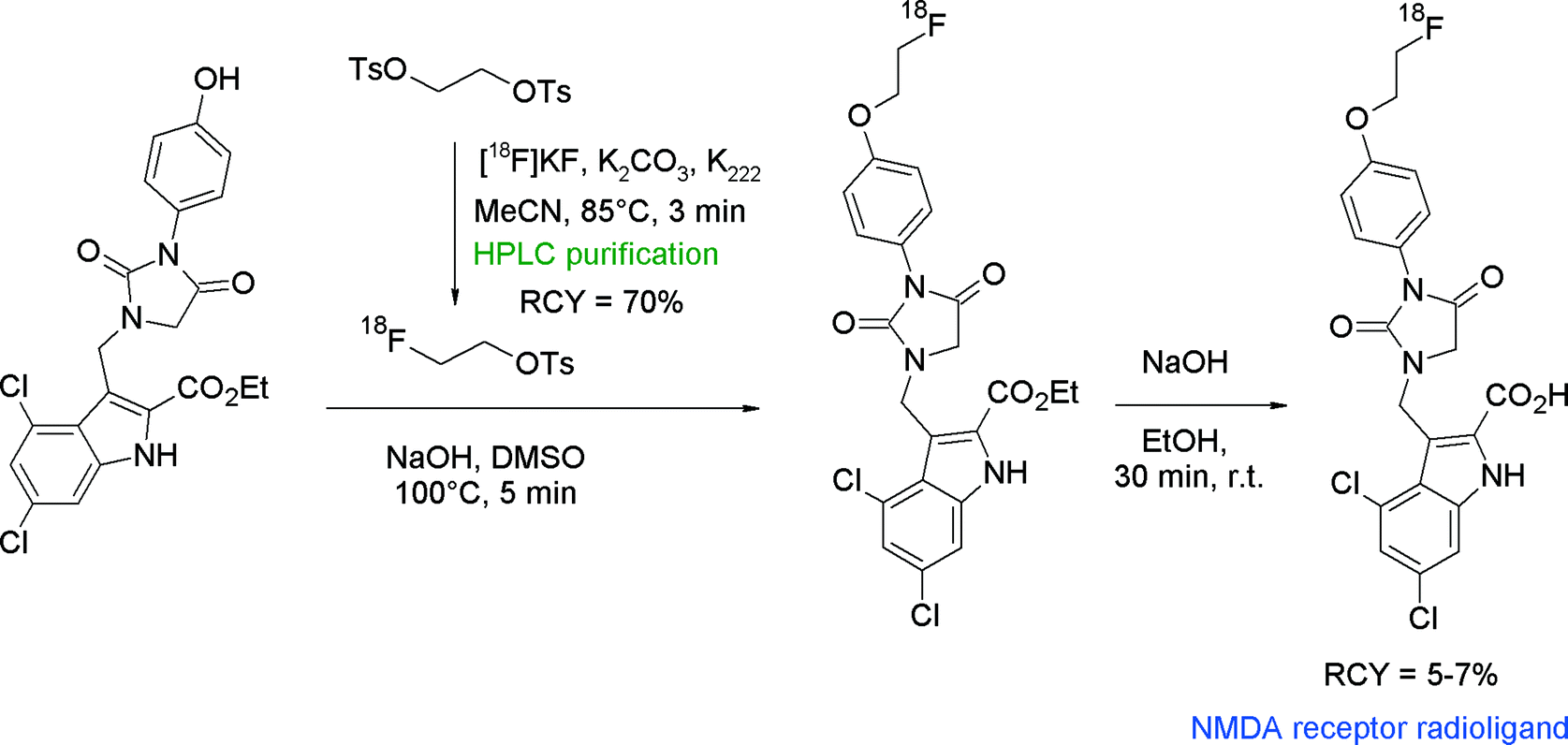 | ||
| Fig. 19 Synthesis of an O-[18F]fluoroethylated hydantoin derivative.45 | ||
Non-invasive detection of β-amyloid plaques and neurofibrillary tangles in the human brain would help in the early identification of patients potentially having Alzheimer's disease. With regard to β-amyloid plaques, 2-(4′-(methylamino)phenyl)-6-hydroxybenzothiazole (PIB) is an intensively examined compound due to its significant accumulation in tissue of high plaque density in the human brain. The corresponding carbon-11 labeled radiotracer [11C]PIB was the first prominent PET radiotracer in this context that had achieved clinical relevance. To obtain a [18F]fluoroethylated PIB analogue, the free hydroxyl group of PIB was reacted with [18F]FETs in DMSO in the presence of K2CO3 for 20 min at 100 °C, followed by HPLC purification (Fig. 20).46 The high chemoselectivity of [18F]FETs allowed selective O-alkylation without the need of a protective group on the nitrogen. Although the labeling yields were high for the single steps (90 and 85%, respectively), the overall RCY of [18F]FE-PIB was only 5–10% attributed to a considerable loss of [18F]FETs during the SPE-based purification process.
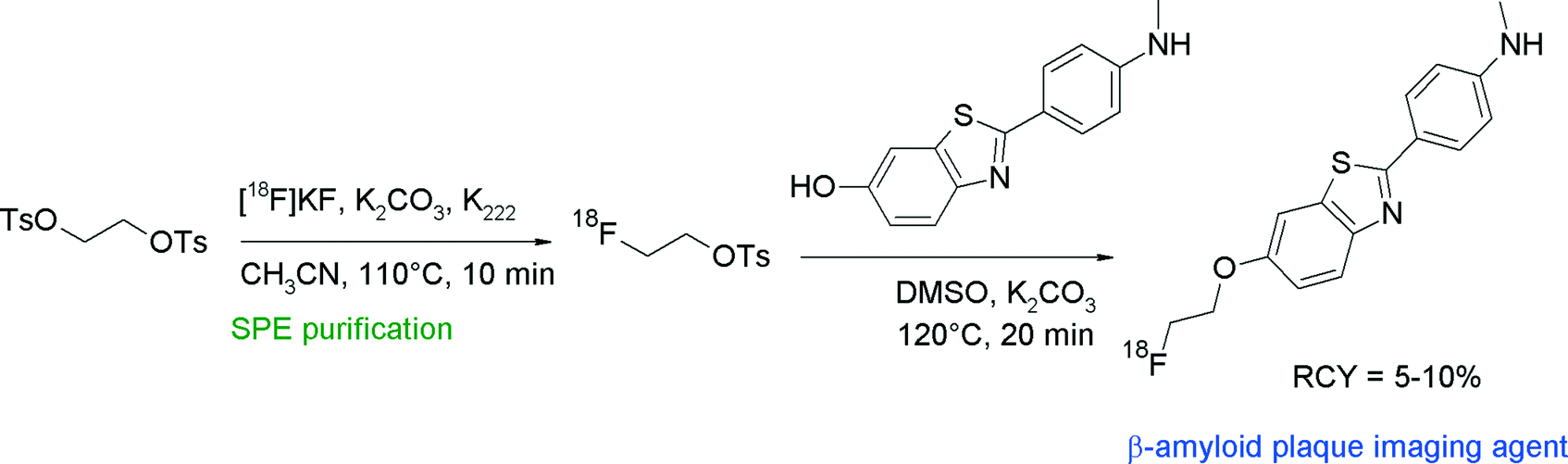 | ||
| Fig. 20 Radiosynthesis of [18F]fluoroethylated benzothiazole.46 | ||
Recently, the class of imidazo[2,1-b]benzothiazoles (IBTs) has been developed as a new compound class having high affinity to β-amyloid plaques. An O-[18F]fluoroethylated IBT derivative was synthesized and evaluated as a PET radiotracer.47 In that study, direct one-step [18F]fluorination of the corresponding tosylated precursor of IBT as well as two-step [18F]fluoroethylation at the phenolic hydroxyl group with [18F]FETs was performed (Fig. 21). The one-step procedure provided [18F]FE-IBT in 24% RCY. For the two-step radiofluorination, [18F]FETs was purified by HPLC, providing the intermediate adsorbed on the SPE cartridge in 45% yield. After drying, [18F]FETs was eluted with DMF to the hydroxyl precursor in DMF, which was deprotonated with sodium hydride beforehand. [18F]Fluoroethylation was performed for 5 min at 90 °C and provided the desired [18F]fluoroethylated IBT in 58% RCY.
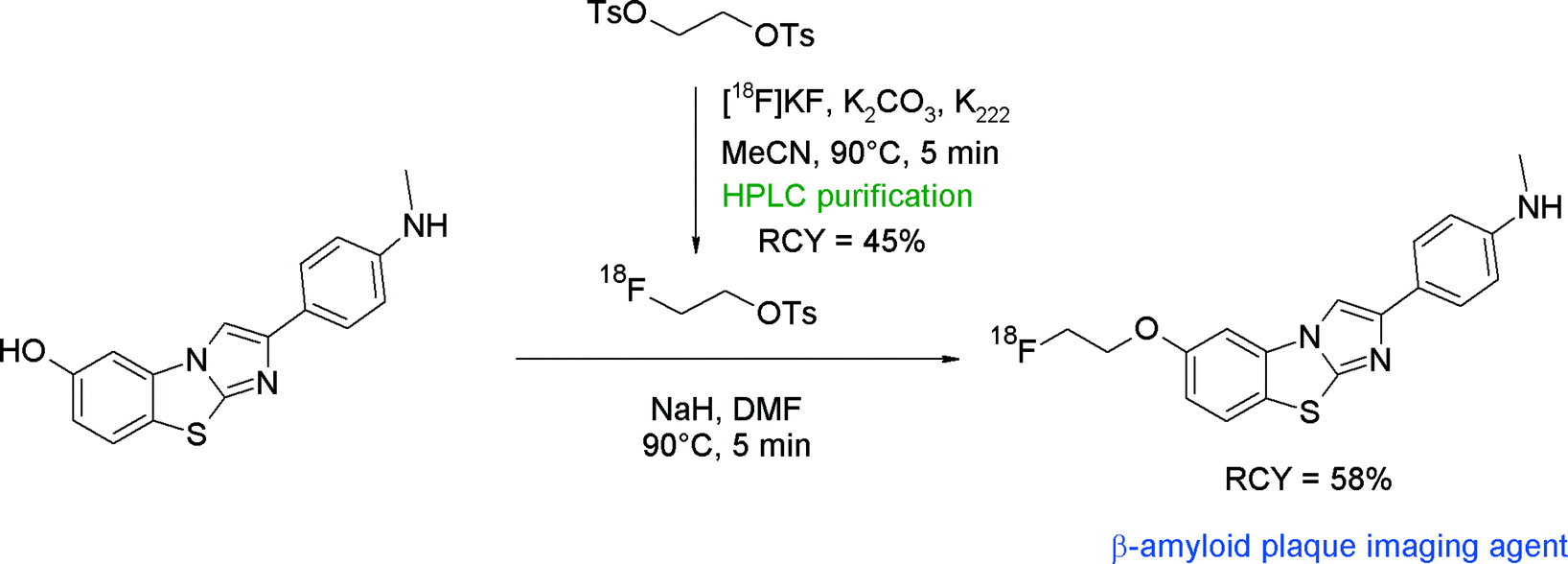 | ||
| Fig. 21 Radiosynthesis of O-[18F]fluoroethylated imidazo[2,1-b]benzothiazole.47 | ||
Since sigma receptors are involved in diseases of the central nervous system and have been found to be expressed in human tumor entities, radiolabeled sigma receptor ligands are of high scientific interest and their role has to be studied in vivo. Among the diversity of structures, the piperazine derivative SA4503 is a promising ligand and has been radiolabeled with carbon-11 for imaging sigma-1 receptors with PET. Consequently, the corresponding O-[18F]fluoroethylated radiotracer was synthesized to study its pharmacokinetics in vivo (Fig. 22).48,49 Elsinga et al. used SPE-purified [18F]FETs to perform [18F]fluoroethylation of the hydroxyl precursor in the presence of NaH in DMF. After reaction for 5 min at 125 °C, [18F]FE-SA4503 was isolated after HPLC purification in 4–7% overall RCY. The low RCY was explained by the authors with the relatively large dilution of [18F]FETs in the DMF solution (1 mL) in relation to the small amount of the hydroxyl precursor (0.2 mg).
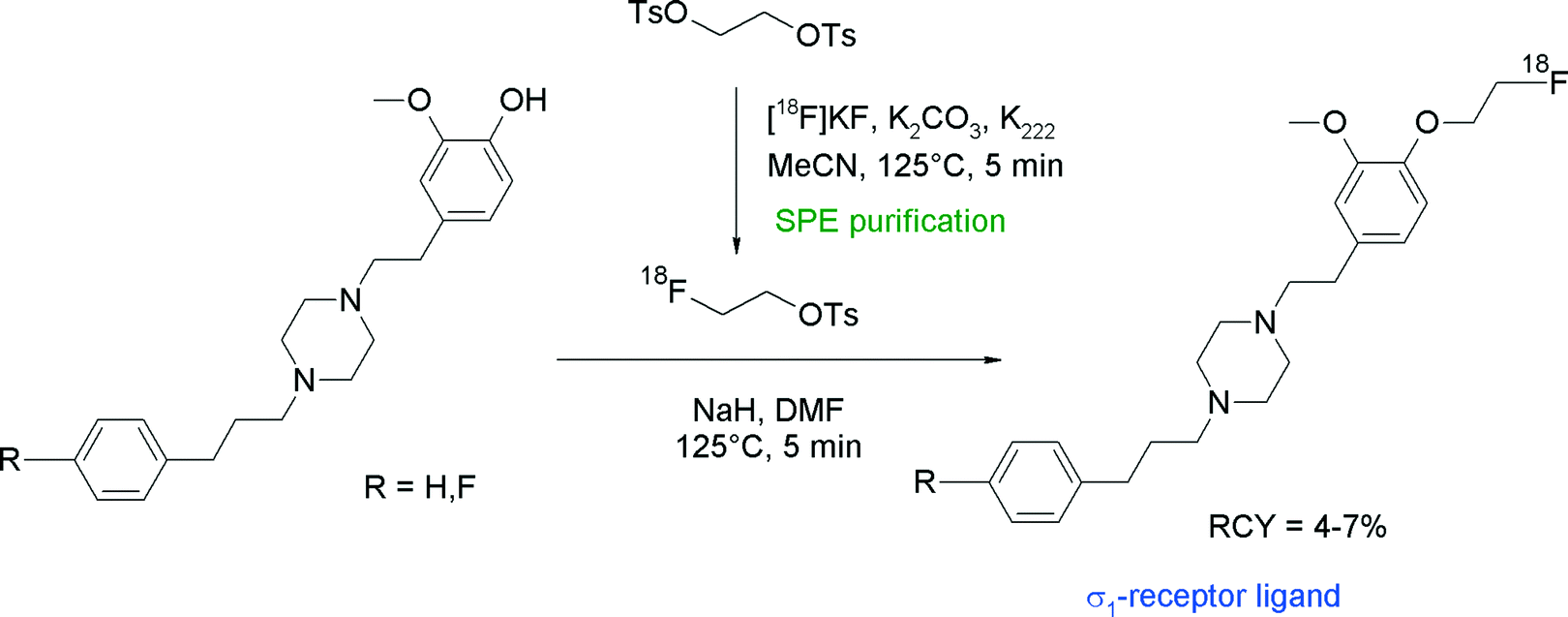 | ||
| Fig. 22 Synthesis of O-[18F]fluoroethylated SA4503.48,49 | ||
Recently, spirocyclic piperidine derivatives were found to possess nanomolar affinities for the sigma-1 receptor subtype and two compounds have been selected for radiotracer development via [18F]fluoroethylation with [18F]FETs (Fig. 23).50,51 Radiolabeling of the spirocyclic piperidine was performed in an automated synthesizer using a one-pot two-step procedure without purification of [18F]FETs. After the preparation of [18F]FETs, the corresponding hydroxyl precursor with sodium hydroxide in DMSO50 or with Cs2CO3 in DMF51 was added and [18F]fluoroethylation was performed by heating at 110 °C for 10–20 min. Final HPLC purification provided the O-[18F]fluoroethylated spirocyclic sigma-1 ligands within 100 min in 15–20% overall RCY.
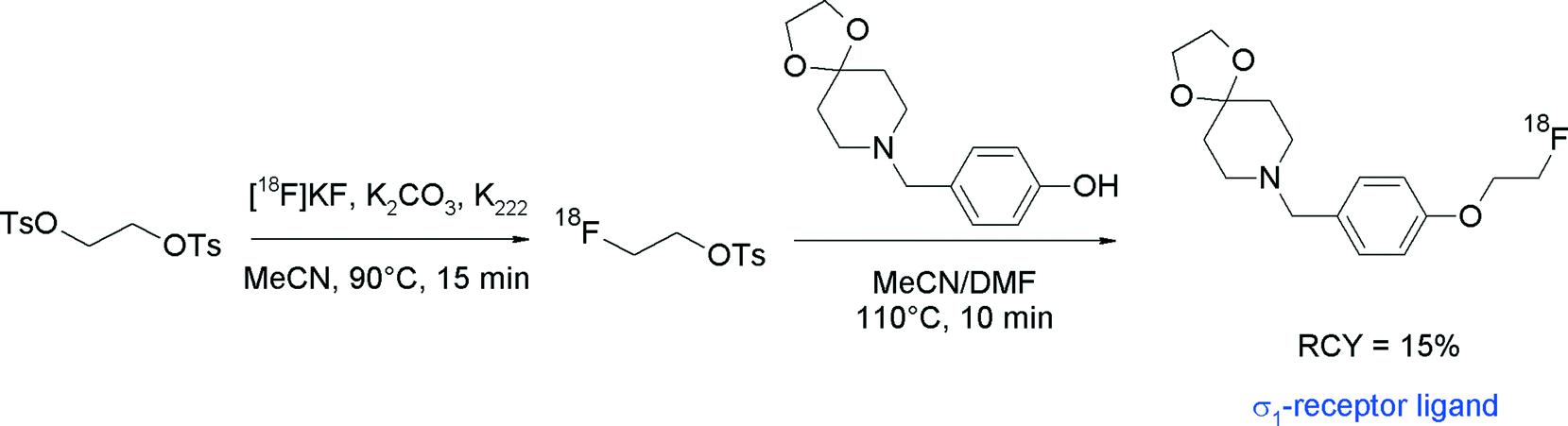 | ||
| Fig. 23 Synthesis of an O-[18F]fluoroethylated spirocyclic piperidine.51 | ||
The translocator protein (TSPO) is an established target for the imaging of neuroinflammation with PET. To develop radiolabeled TSPO ligands, Wadsworth et al. performed [18F]fluoroethylation of diaryl-substituted acetanilides (Fig. 24).52 For the radiolabeling reaction, HPLC-purified [18F]FETs was used which was heated with the phenol precursor in acetonitrile in the presence of Cs2CO3 at 120 °C for 15 min. After HPLC purification, the 18F-fluoroethylated TSPO ligand was isolated in 16% overall RCY within a synthesis time of 120 min.
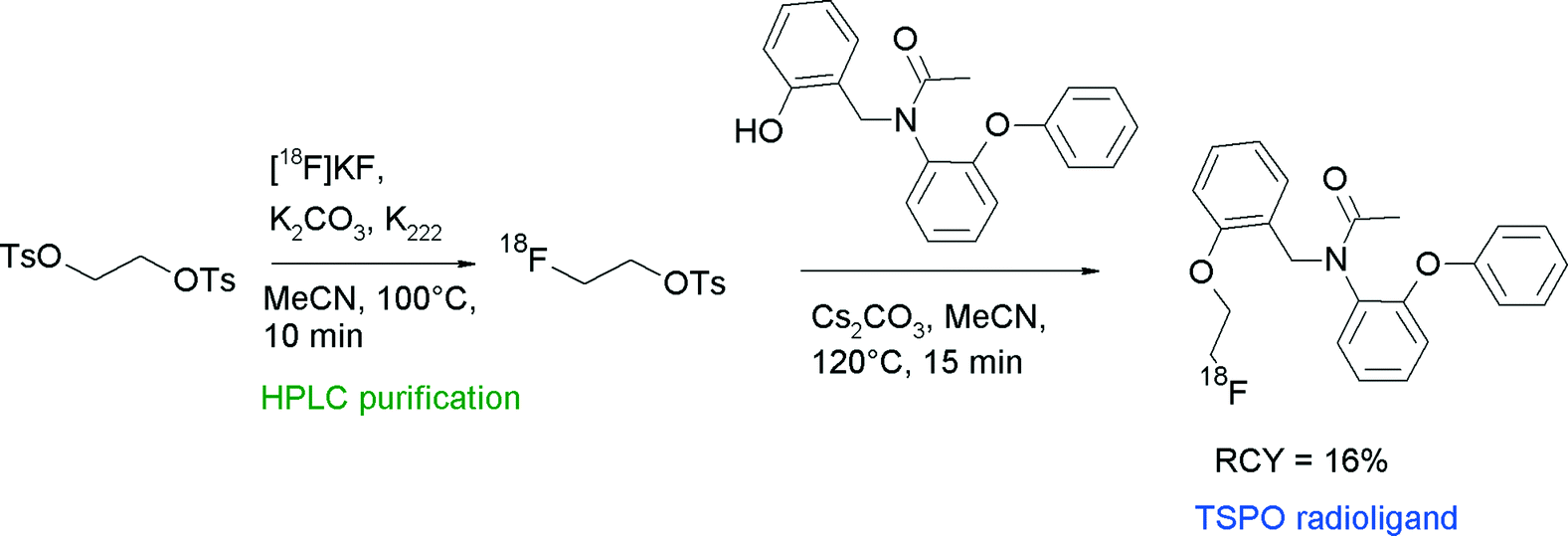 | ||
| Fig. 24 Radiosynthesis of O-[18F]fluoroethylated diaryl-substituted acetanilide.52 | ||
Adenosine A2 receptors are further attractive therapeutic targets for the treatment of neurodegenerative disorders and radiolabeled AA2 ligands could be used to assess changes in receptor density. SCH442416 is a furanyl-substituted pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine compound with high affinity to the AA2A receptor. Radiotracer design aimed at the replacement of a methoxy group of SCH442416 with a 18F-fluoroethoxy moiety (Fig. 25).53 [18F]FETs was purified by SPE and reacted with the phenol precursor in acetonitrile and 10 μL of 40% aqueous N(Bu)4OH solution at 115 °C for 15 min. The O-[18F]fluoroethylated AA2A receptor ligand was obtained within a synthesis time of 114 min in 7% overall RCY.
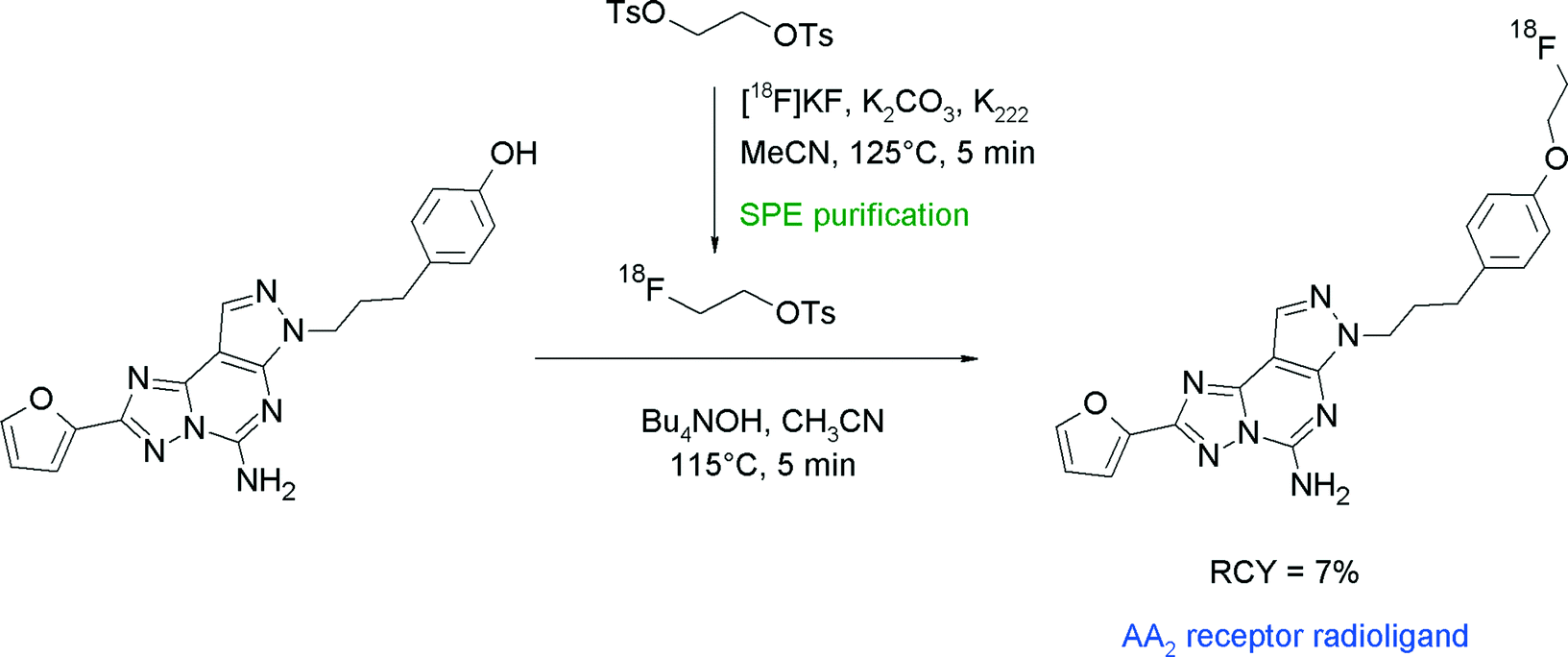 | ||
| Fig. 25 Radiosynthesis of O-[18F]fluoroethylated SCH442416.53 | ||
As the enzyme monoamine oxidase (MAO) is involved in psychiatric and neurodegenerative disorders, treatment with MAO inhibitors is a common therapeutic approach. For the in vivo determination of MAO levels in the brain, radiotracers have been developed, most of them radiolabeled with carbon-11, e.g. [11C]harmine. In an effort to produce a 18F-radiolabeled harmine derivative, Schieferstein et al. performed O-[18F]fluoroethylation of harmine with [18F]FETs (Fig. 26).54 [18F]FETs was synthesized using a homemade automated synthesis unit and reacted with harmol and NaOH after SPE purification by heating for 10 min at 130 °C. Final HPLC purification delivered the 2-[18F]fluoroethyl harmol in 47% overall RCY within 100 min.
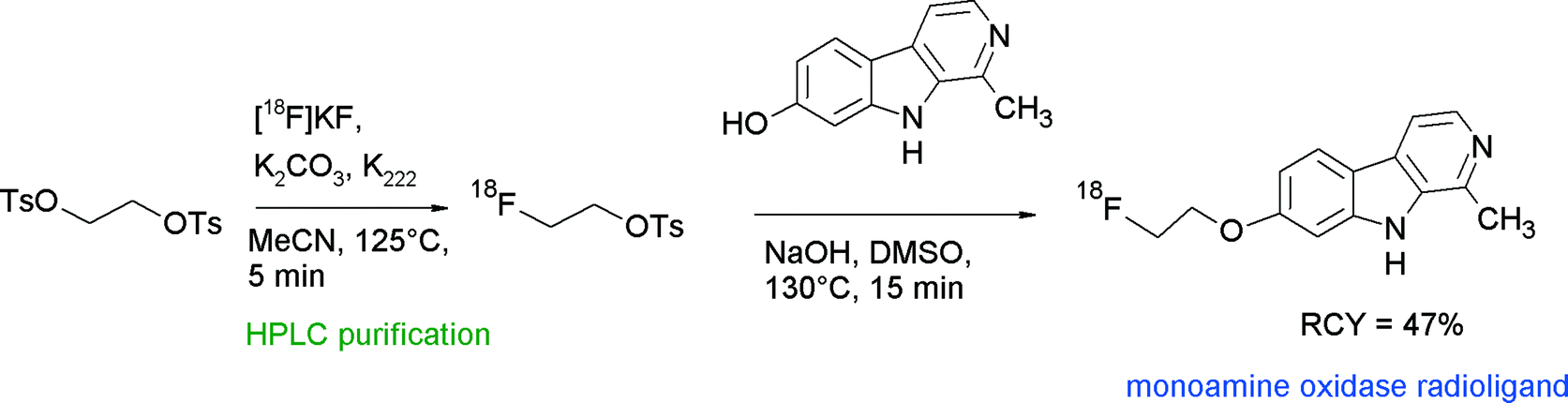 | ||
| Fig. 26 Radiosynthesis of O-[18F]fluorethylated harmine.54 | ||
The non-invasive imaging of the endocrine pancreas with suitable radiotracers might be a useful tool in the diagnostics of e.g. diabetes mellitus. Sulfonylureas which can block pancreatic ATP-sensitive potassium channels, located at the β-cells of the islets of Langerhans, have been found to act as antidiabetic compounds, therefore fluorine-18 labeled sulfonylureas such as glyburide may serve as β-cell imaging agents. Shiue et al. developed 2-[18F]fluoroethoxy glyburide as a potential radiotracer.55 The corresponding phenol served as a precursor for the O-[18F]fluoroethylation to form the 2-[18F]fluoroethoxy glyburide by a one-pot two-step reaction (Fig. 27). [18F]FETs was reacted without purification with hydroxyl glyburide in DMSO and NaOH for 30 min at 90 °C. 2-[18F]Fluoroethoxy glyburide was isolated in 5–10% overall RCY after HPLC purification within a synthesis time of 100 min. The corresponding 2-[18F]fluoroethoxy-5-iodoglyburide was later synthesized by the same group in the same manner and in comparable RCY.56
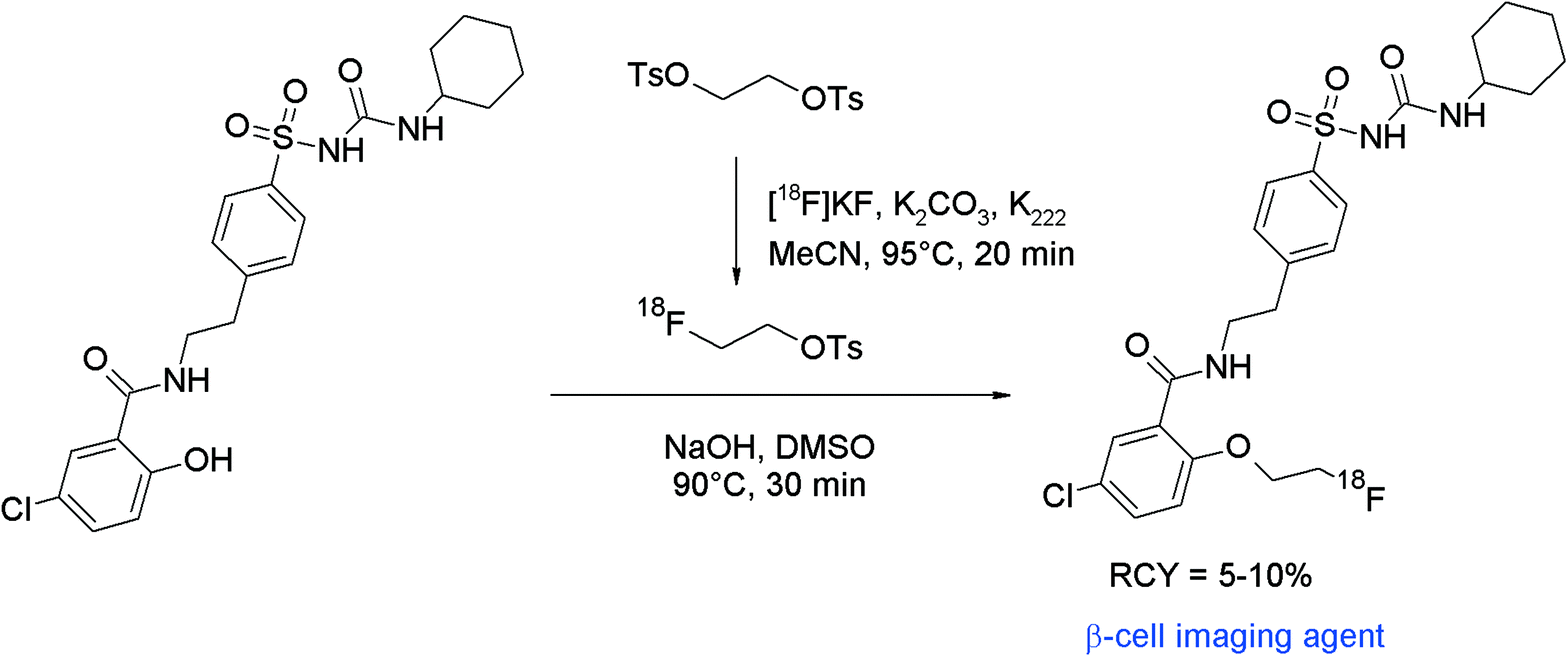 | ||
| Fig. 27 Radiosynthesis of 2-O-[18F]fluoroethoxy glyburide.55 | ||
Repaglinide is a non-sulfonylurea-based glucose regulator and, in clinical use, is employed to block ATP-sensitive potassium channels to regulate the secretion of insulin. An 18F-labeled repaglinide derivative was developed by Wängler et al. for the in vivo visualization and quantification of human pancreatic β-cells.57 Since the chemical structure of repaglinide is characterized by the presence of an ethoxy group, a radiolabeling strategy comprising [18F]fluoroethylation was suggested. Radiolabeling was performed as a two-pot procedure with HPLC-purified [18F]FETs which, after a drying step, was dissolved in DMSO and added to the hydroxyl precursor in the presence of NaOH solution (Fig. 28). The carboxylic functionality of des-ethoxy-repaglinide was protected beforehand by a methyl ester group to avoid the formation of a [18F]fluoroethyl ester. O-[18F]Fluoroethylation was performed for 10 min at 150 °C, the intermediate product was purified by HPLC and deprotected with 1 M NaOH at 80 °C for 35 min. The O-[18F]fluoroethyl repaglinide derivative was isolated in an overall RCY of 20%. The fact that the [18F]fluoroethyl moiety was unharmed during the relatively harsh deprotection step gives evidence for the stability of this labeling group.
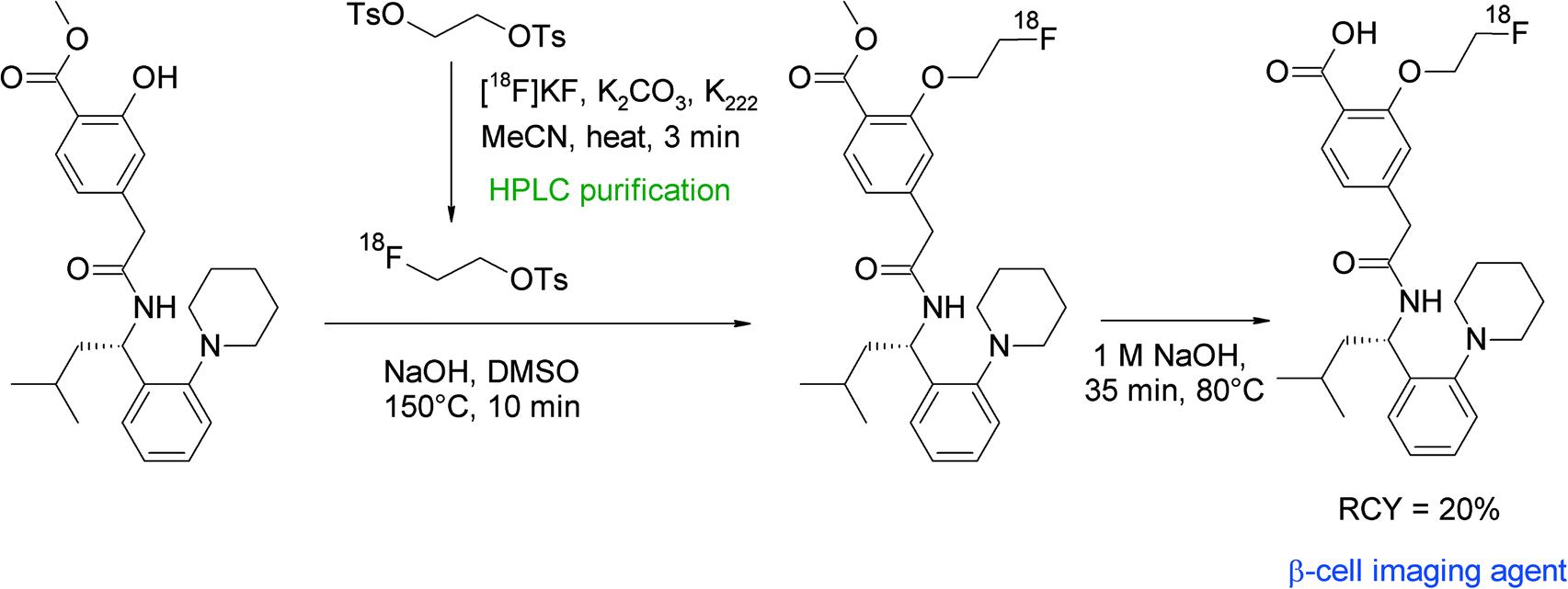 | ||
| Fig. 28 Radiosynthesis of O-[18F]fluoroethylated repaglinide.57 | ||
In humans, the peripheral beta-adrenoceptors are involved in the neuronal regulation of the epithelial, glandular and muscular lung function. Fenoterol represents a highly affine subtype-specific β2 receptor antagonist showing three phenolic groups, hence Schirrmacher et al. developed a fluorine-18 labeled derivative of fenoterol via O-[18F]fluoroethylation.58 The radiolabeling was achieved using (R,R)(S,S)fenoterol and [18F]FETs by heating in DMF at 130 °C for 8 min in the presence of 1 M NaOH (Fig. 29). The RCY of [18F]FE-fenoterol was 60% based on [18F]FETs. As a side reaction, the formation of the corresponding N-[18F]fluoroethylated compound was observed in 40% yield. Taking into account the difference in acidity of the hydroxyl groups, it turned out that the hydroxyl functionality on the phenol moiety was more easily deprotonated than that of the resorcinol; hence, selective [18F]fluoroethylation was achieved using 0.9 equivalents of NaOH. The use of an additional equivalent of the base resulted in the formation of an [18F]fluoroethylated resorcinol derivative as a side product. The purification of the radiotracer was performed by HPLC whereby the O-[18F]fluoroalkylated radiotracer [18F]FE-(R,R)(S,S)fenoterol was delivered in 20% overall RCY. In conclusion, this reaction gives a good illustration of the chemoselectivity of [18F]FETs by selective labeling of different acidic phenols avoiding the need of protective groups.
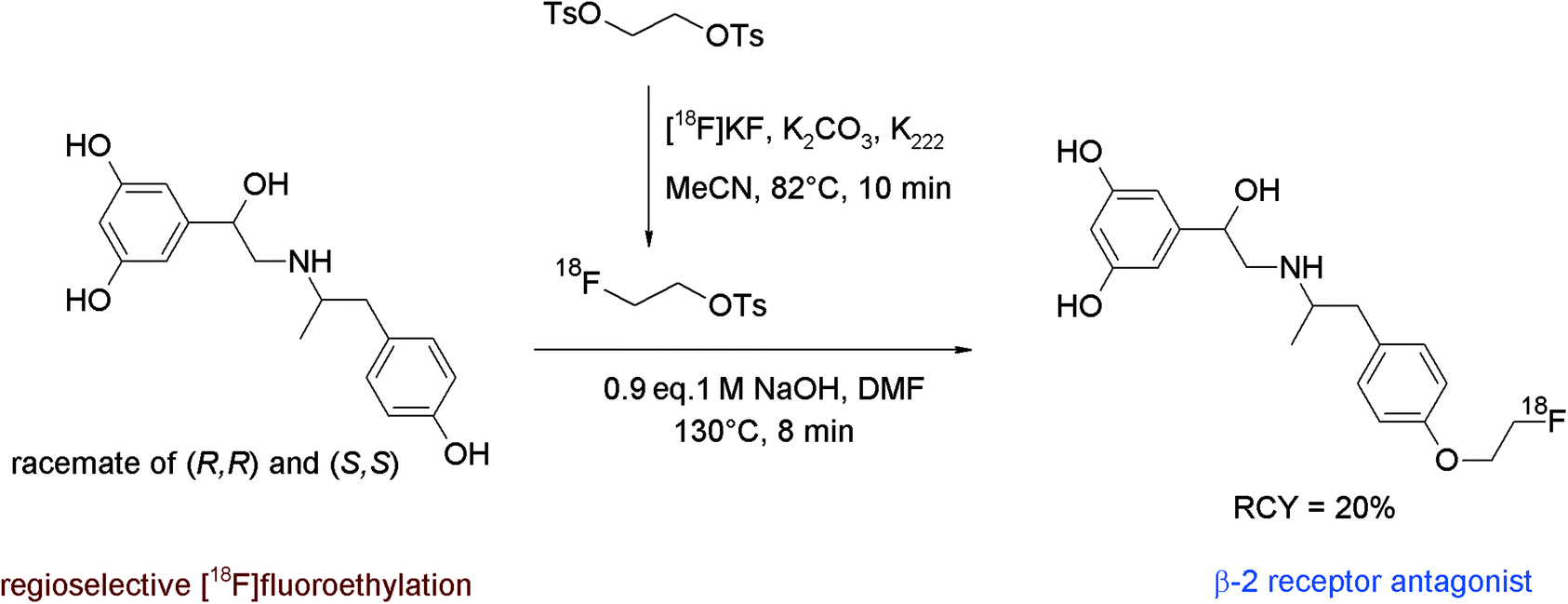 | ||
| Fig. 29 Radiosynthesis of O-[18F]fluoroethylated fenoterol.58 | ||
Tyrosine kinases play pivotal roles in the pathogenesis of cancer and tyrosine kinase inhibitors (TKIs) represent promising candidates for therapeutic intervention. Accordingly, radiolabeled TKIs would be useful tools to assess the status of cancer disease and help to determine the optimal course of treatment. The class of aminoquinazolines is part of many drugs with antibacterial, antiviral and anticancer activity, so approved TKIs like gefitinib, lapatinib and erlotinib are composed of this important pharmacophore. Chen et al. developed a series of 18F-radiolabeled aminoquinazoline derivatives as potential tumor imaging tracers by radiolabeling the appropriate phenol precursors with [18F]FETs.59 The radiotracers were formed via a one-pot two-step procedure without isolation of the intermediate labeling agent. The final O-[18F]fluoroethylated aminoquinazolines were provided in 21–24% overall RCY (not decay corrected) (Fig. 30).
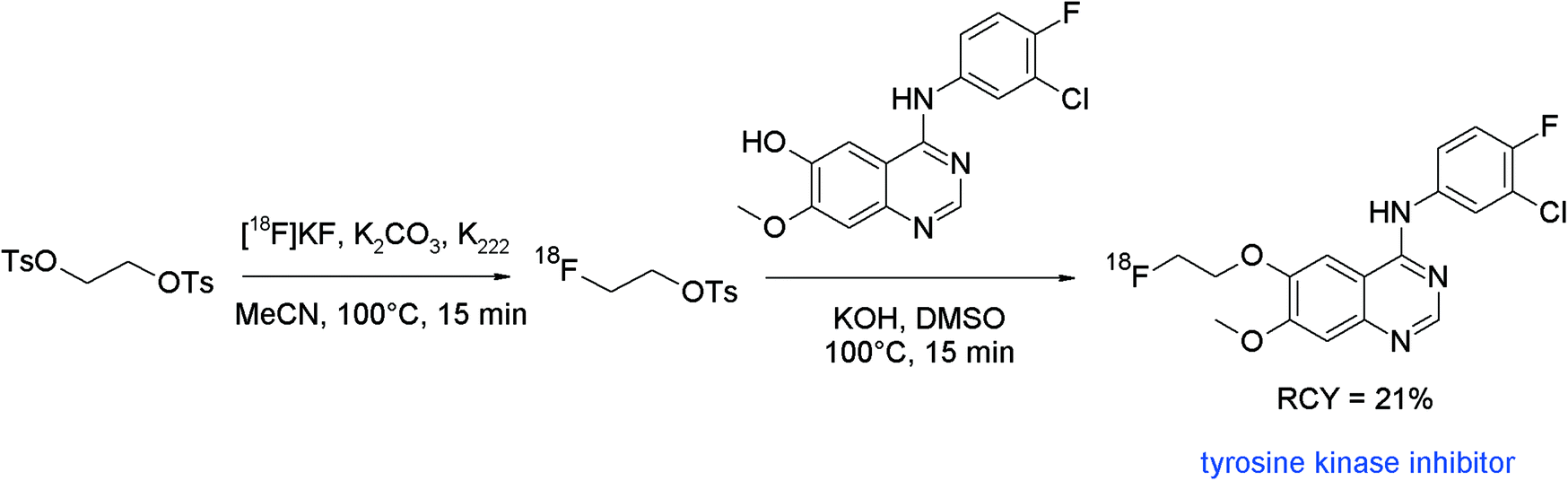 | ||
| Fig. 30 Radiosynthesis of an O-[18F]fluoroethylated aminoquinazoline.59 | ||
Poly-N-(2-hydroxypropyl)methacrylamide (HPMA) is a promising nontoxic, non-immunogenic and biocompatible drug for anticancer treatment and recently lauryl methacrylate segments were added to HPMAs to improve the delivery of these drugs through the blood brain barrier.60 To track the distribution and accumulation of such lauryl methacrylate-substituted HPMAs in vivo, these drugs have been converted into radiotracers by 18F-radiolabeling with [18F]FETs.60,61 Radiolabeling was accomplished in two steps comprising the synthesis and HPLC purification of [18F]FETs and subsequent attachment to the hydroxyl group of the linker tyramine in DMSO in the presence of NaOH (Fig. 31). The radiolabeled polymeric substances were purified by size exclusion chromatography. Altogether, the yield of 18F-incorporation did not exceed 4% and the labeling efficiency decreased with higher molecular weight of the polymers, most likely caused by the decreased accessibility of the tyramine linkers.
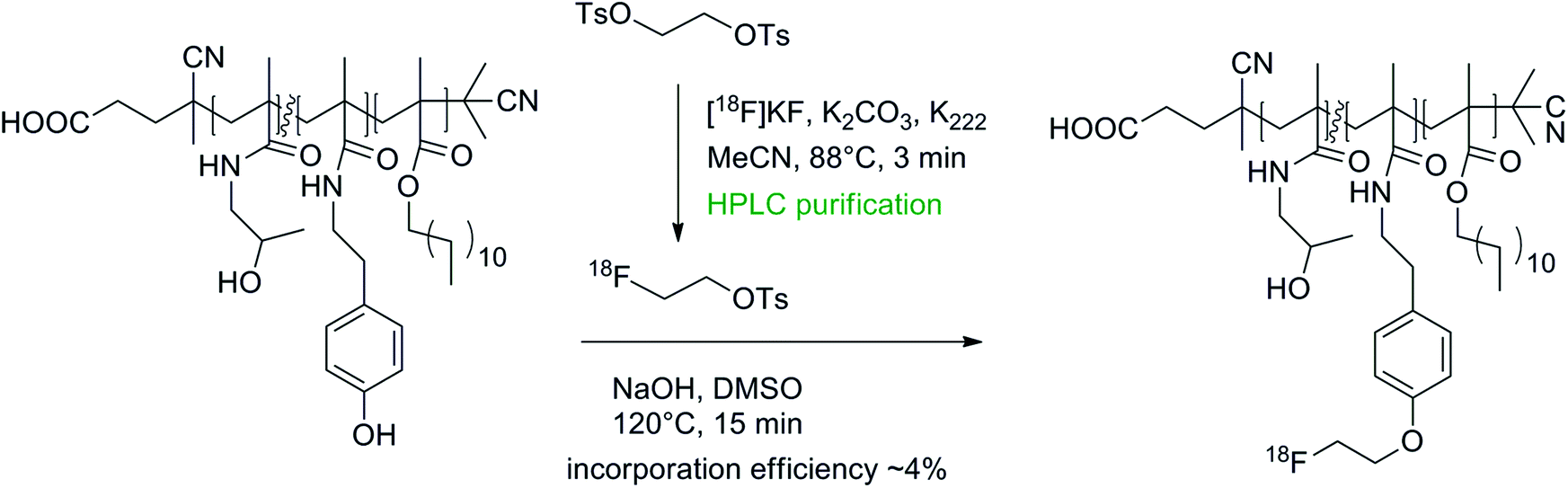 | ||
| Fig. 31 O-[18F]Fluoroethylation of poly-N-(2-hydroxypropyl)methacrylamide.60,61 | ||
A relatively new approach for the radiolabeling of bio(macro)molecules is the application of bioorthogonal coupling reactions because they are fast and highly selective. Staudinger ligation is a powerful conjugation reaction in this field using functionalized phosphane analogs to be coupled with an azide-substituted biomolecule. To prepare a prosthetic group suitable as a radiolabeling agent for the Staudinger ligation, Mamat et al. designed a [18F]fluoroethoxy-benzoate-substituted phosphane.62 This new bifunctional labeling agent was prepared using a one-pot method by generation of [18F]FETs and subsequent reaction with the phenol group of 2-(diphenylphosphano)phenyl 4-hydroxybenzoate (Fig. 32). As a third step, azide-functionalized model compounds were added. In this manner, the 18F-radiolabeled Staudinger coupling products were formed in 12–17% overall RCY.
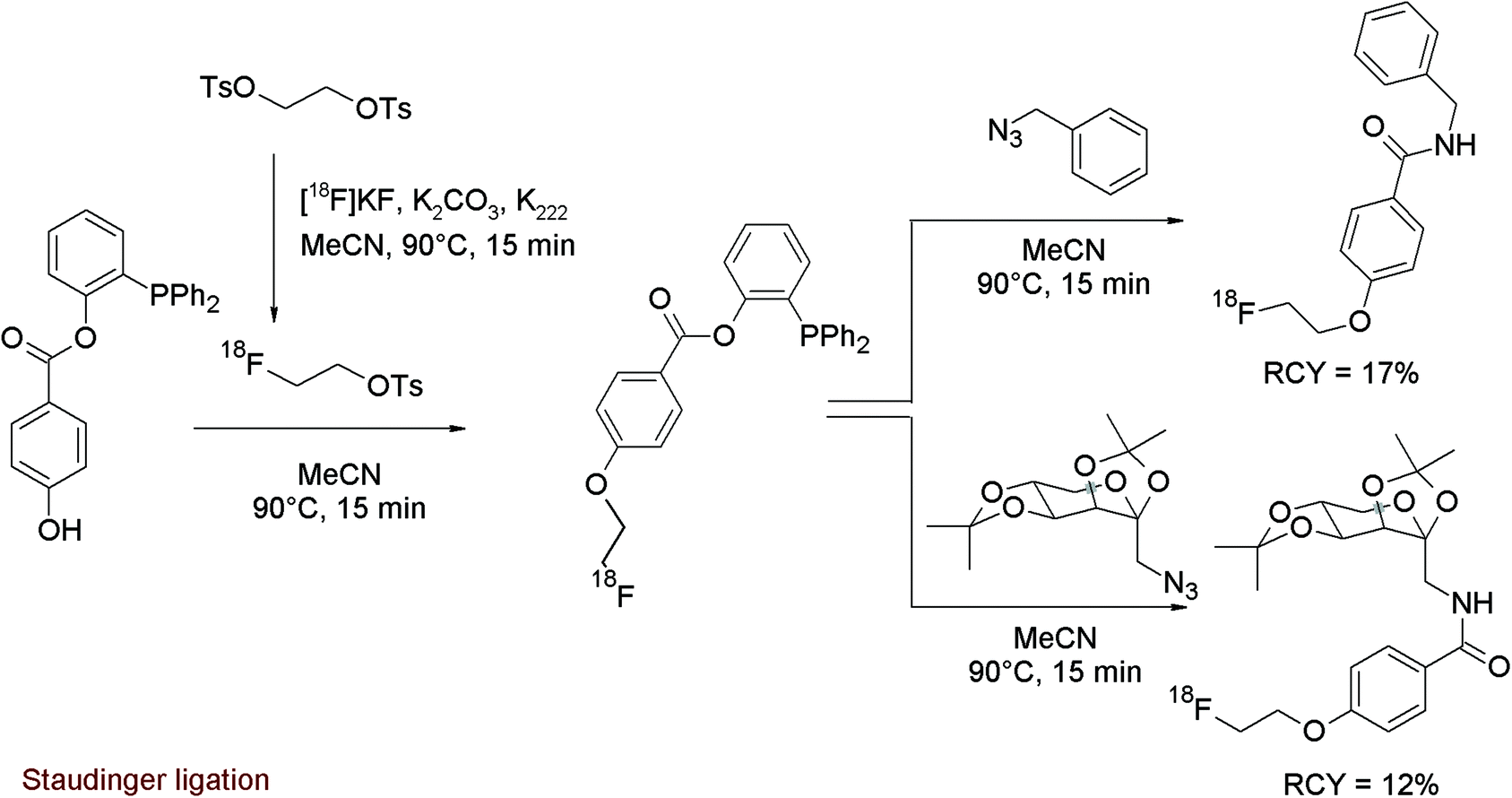 | ||
| Fig. 32 Radiosynthesis of an O-[18F]fluoroethoxy-benzoate-substituted phosphane.62 | ||
[18F]Fluoroethyl esters
[18F]FETs can also be used as a labeling reagent to form 2-[18F]fluoroethyl esters when a carboxylic acid is provided as the precursor as demonstrated in the following examples.Carfentanil, an anilinopiperidine derivative, is a potent agonist of the μ-opioid receptor. A number of 18F-labeled carfentanil derivatives have been evaluated intensively by Henriksen et al. including the 2-[18F]fluoroethyl ester, obtained by the reaction of the carboxylic acid sodium salt with [18F]FETs (Fig. 33).63,64 The carfentanil-2-[18F]fluoroethyl ester was prepared within 100 min in 36% overall RCY. The authors performed a series of optimization experiments to determine the relation of the RCY on temperature, solvent and addition of NaI. When the [18F]fluoroethylation was performed in DMF at 100 °C, only 7% of the radiolabeled product was obtained. Increasing the temperature to 120 °C yielded about 20% of the product. Compared to DMF, DMSO resulted in lower yields under otherwise identical conditions. Addition of NaI to the reaction mixture led to 25–55% overall RCY, so the application of 20 mM NaI was optimal.
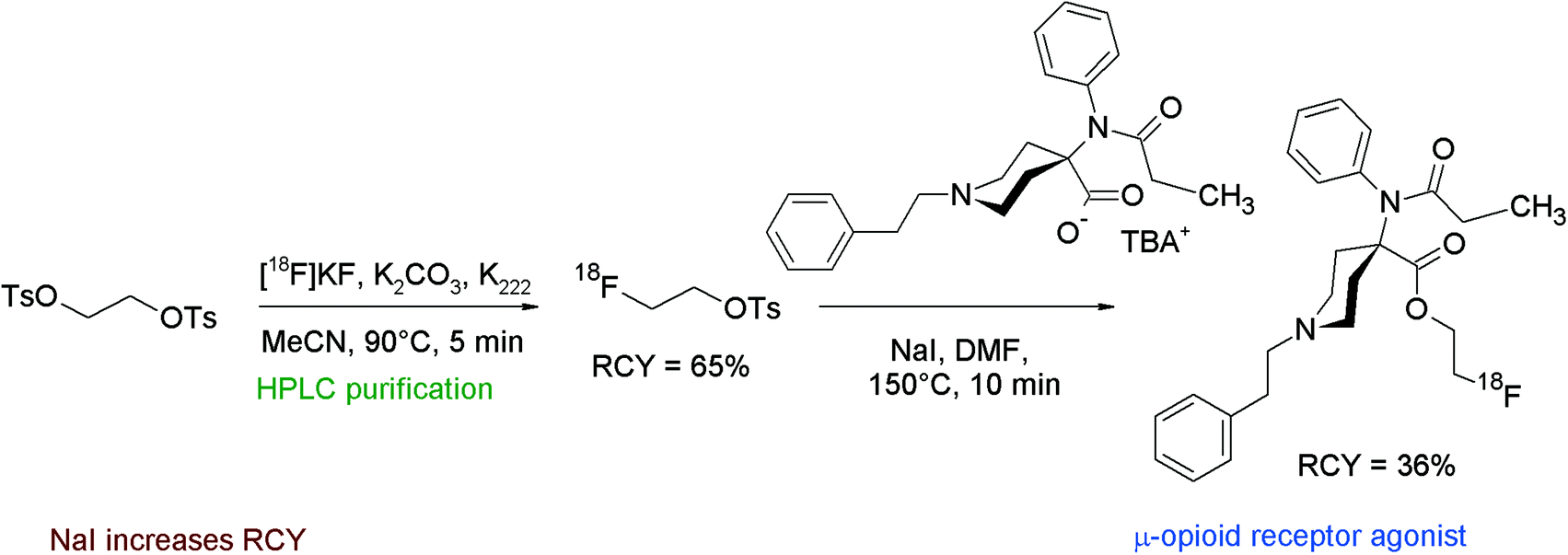 | ||
| Fig. 33 Radiosynthesis of carfentanil-2-[18F]fluoroethyl ester.63,64 | ||
Another example of [18F]fluoroethyl ester formation was described by Philippe et al. in the synthesis of a new PET ligand for the melanin-concentrating hormone receptor-1 (MCHR1), which plays a key role in energy homeostasis and is involved in anxiety, diabetes and adiposity.65 The [11C]methyl ester of the MCHR1 ligand served as a lead for the development of the corresponding 2-[18F]fluoroethyl ester. For radiolabeling, the carboxylic acid precursor of SNAP-7941, activated by addition of tetrabutylammonium hydroxide, was reacted with [18F]fluoroethylating reagents [18F]FEBr, [18F]FETs and 2-[18F]fluoroethyl triflate ([18F]FETf) in a vessel-based approach (Fig. 34). Surprisingly, the authors did not observe any 18F-incorporation into SNAP-7941 for all of these reagents; even the direct [18F]fluorination of a tosylated precursor failed. To overcome this situation, a microfluidic approach was attempted that was finally successful and delivered the 2-[18F]fluoroethyl ester of SNAP-7941 in 44% RCY starting from the tosylated precursor. However, the [18F]FETs-based method did not work in the microfluidic device either.
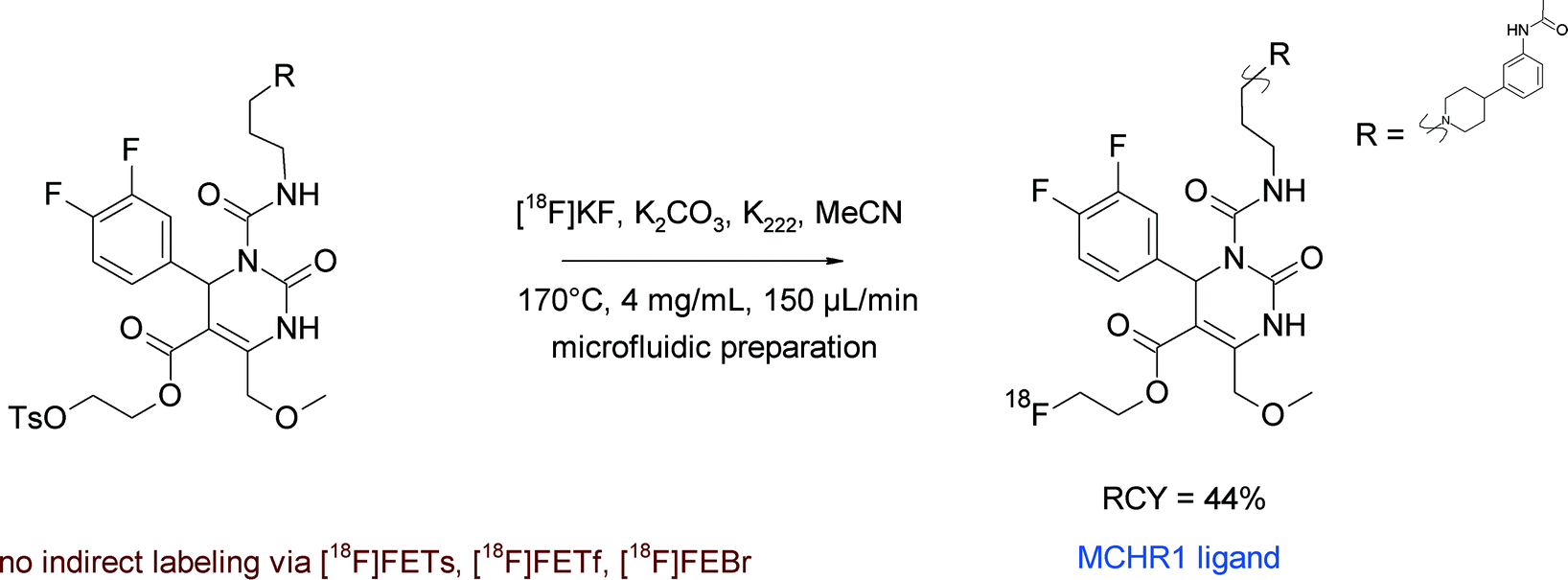 | ||
| Fig. 34 Radiosynthesis of 2-[18F]fluoroethyl ester of SNAP-7941 (direct 18F-fluorination).65 | ||
A third example of [18F]fluoroethyl ester formation with [18F]FETs was presented by Heinrich et al. who developed the 2-[18F]fluoroethyl ester of rhodamine B for imaging myocardial perfusion with PET.66 Rhodamines are lipophilic cations which are known to accumulate in heart tissue and bind at the mitochondrial membrane. The lactone of rhodamine B served as a precursor for [18F]fluoroethyl ester formation, which was heated with unpurified [18F]FETs in acetonitrile at 160 °C in the presence of diisopropylethylamine, a base required for inducing the ring opening of the lactone (Fig. 35). The authors found that optimal yields were obtained by slowly evaporating the solvent during the reaction and isolated the rhodamine B 2-[18F]fluoroethyl ester after HPLC purification in 35% overall RCY within a total synthesis time of 90 min.
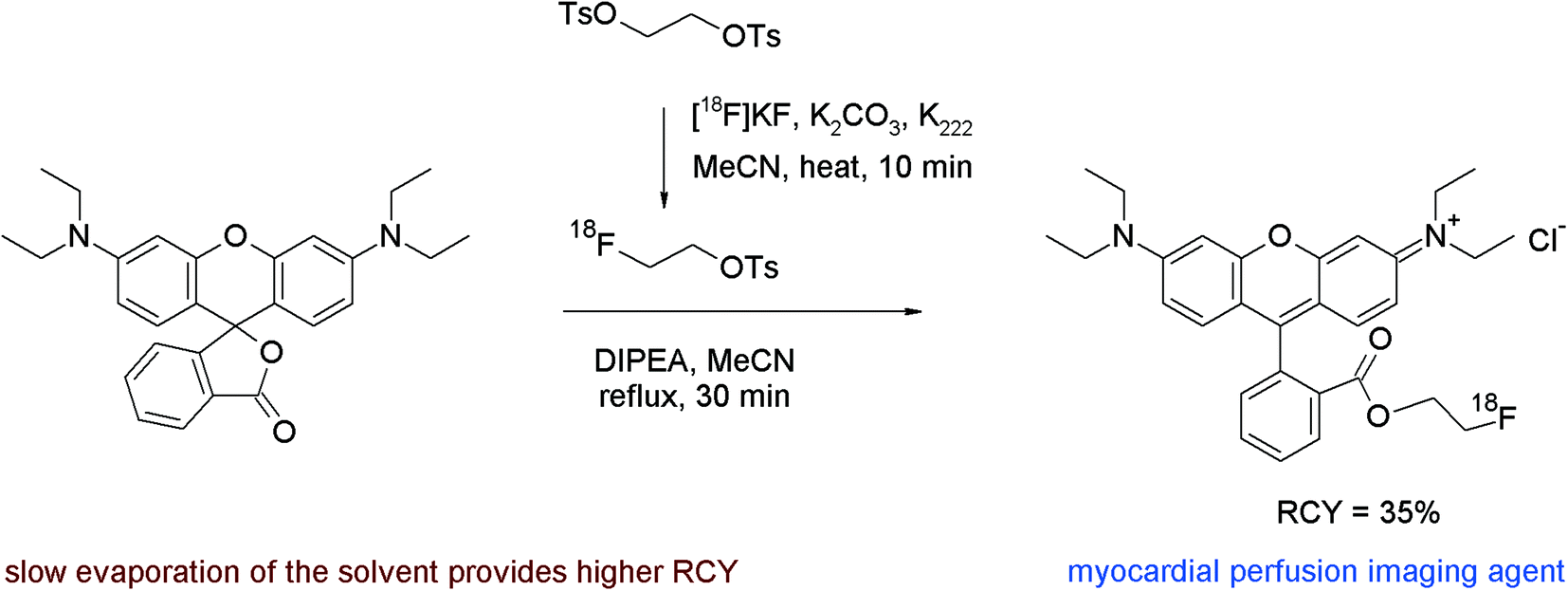 | ||
| Fig. 35 Radiosynthesis of a 2-[18F]fluoroethyl ester of rhodamine B.66 | ||
N-[18F]Fluoroethylation
2-[18F]Fluoroethylcholine
The amine group is an important functionality in numerous biologically active compounds and alkylation of primary and secondary amines by treatment with alkyl halides or sulfonates is a common and straightforward route in organic chemistry. N-[18F]Fluoroethylation using [18F]FETs or [18F]FEBr stands for a simple and efficient radiolabeling strategy, however this method is sometimes limited due to the high basicity of the amine, needing strict aprotic conditions and high temperatures. The following examples may illustrate the potential of [18F]FETs in the radiolabeling of amines.The finding that radiolabeled choline accumulates in certain tumor cells, e.g. of prostate cancer, because of elevated uptake of choline as the precursor of phosphatidylcholine in tumors promoted the use of carbon-11 and fluorine-18 labeled choline as preferred PET tracers for prostate cancer imaging. Hara et al. first described the PET imaging of prostate cancer using [11C]choline and performed in 1997 the first radiosynthesis of 2-[18F]fluoroethylcholine ([18F]FEC) by reacting [18F]FETs with neat N,N-dimethylethanolamine at 80 °C for 20 min.67 [18F]FEC was obtained using a one-pot procedure after ion exchange-based SPE purification in 50% overall RCY. Later, an automated synthesis of [18F]FEC was presented including the one-pot two-step radiolabeling starting from 1,2-ethylene glycol-bis-tosylate providing the intermediate [18F]FETs, its reaction with N,N-dimethylethanolamine and subsequent purification by semi-preparative HPLC, and the final trapping of [18F]FEC on a SCX cartridge (Fig. 36).68 By following this protocol, [18F]FEC was obtained after 65 min as a hydrochloride in 40% overall RCY with a specific activity of 74 GBq μmol−1 (EOS).
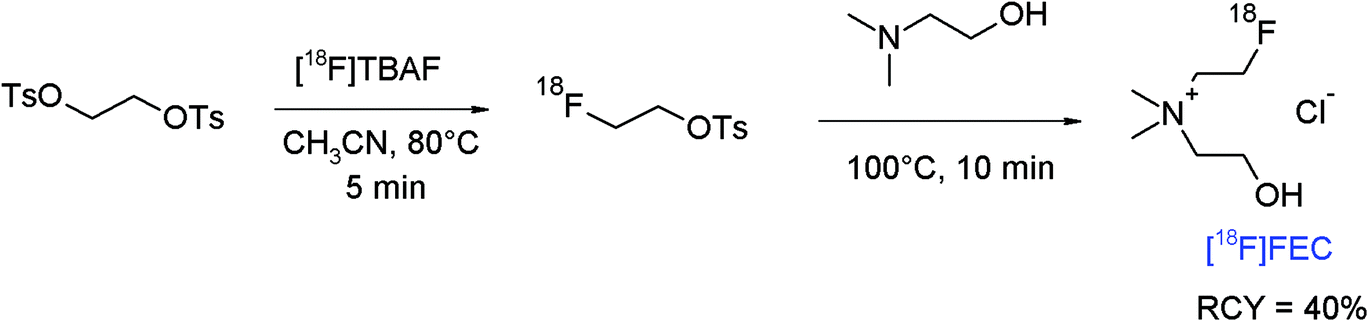 | ||
| Fig. 36 Radiosynthesis of 2-[18F]fluoroethylcholine.68 | ||
In 2004, Yu et al. published a study comparing the radiosynthesis of 2-[18F]fluoroethyl-, 3-[18F]fluoropropyl- and 4-[18F]fluorobutyl-choline from the corresponding bis-tosylated and bis-brominated alkane precursors.69 The reactions were performed as a one-pot procedure; the radiolabeled choline derivatives were purified by SCX SPE which provided the radiotracers in 18–24% overall RCY. The authors found that the dibromo-substituted alkane precursors provided a considerably lower yield of 18F-incorporation compared to the corresponding bis-tosylated counterparts. Furthermore, one-step radiofluorination of [18F]FEC was attempted by starting from e.g. the N-2-acetoxyethyl-N,N-dimethyl-N-2-p-toluenesulfonyloxyethyl ammonium salt precursors, however this approach was unsuccessful due to supposed ion pair formation between the ammonium cation and the [18F]fluoride anion.
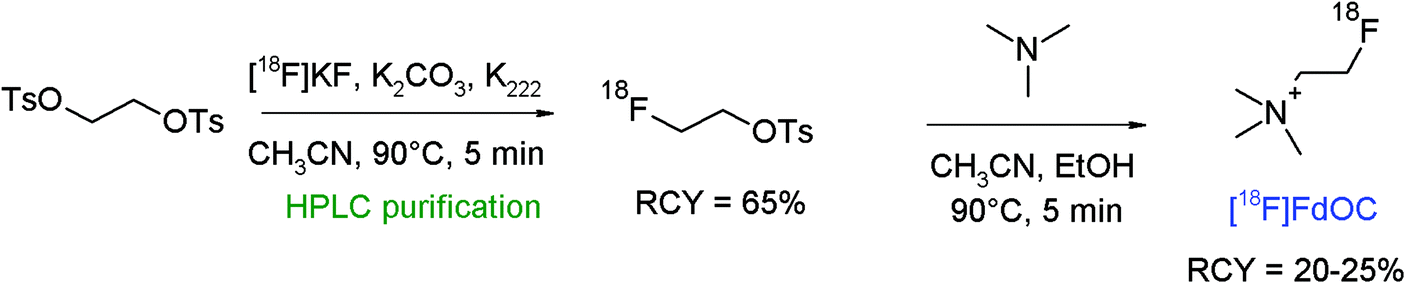
As a further radiotracer for choline transport, deshydroxy-[18F]fluorocholine ([18F]FdOC) was developed by Henriksen et al.70 [18F]FdOC was synthesized by a two-step procedure consisting of the [18F]-fluorination of 1,2-ethylene glycol-bis-tosylate and [18F]fluoroethylation of N,N,N-trimethylamine (Fig. 37). The intermediate [18F]FETs was obtained after reversed-phase HPLC purification in 65% RCY. The final [18F]FdOC was isolated after SCX-based purification within a synthesis time of 70 min in 20–25% overall RCY.
 | ||
| Fig. 37 Radiosynthesis of deshydroxy-[18F]fluorocholine.70 | ||
A reliable, fully automated synthesis of [18F]FEC using a commercially available synthesis unit was published in 2007 by the group of Rösch.71 Since the authors could show that addition of alkali iodides, especially LiI, to the fluoroalkylating agents [18F]FETs or [18F]FEBr significantly improved the RCY,16 they transferred this method successfully to the synthesis of [18F]FEC. Due to the toxicity of lithium, 6.5 mg of NaI was used instead; as a result, higher reaction temperatures (120 °C) were needed to obtain [18F]fluoroethylation yields of 85–90%. For the automated radiosynthesis using a two-pot synthesizer, the intermediate [18F]FETs was purified by C18-based SPE while [18F]FEC purification was performed by semi-preparative HPLC. This iodine-promoted protocol provided [18F]FEC after a synthesis time of 50 min in 30% overall RCY with a specific activity of about 55 GBq μmol−1 (EOS).
The radiosynthesis of [18F]fluoromethyl-, [18F]fluoroethyl- and [18F]fluoropropyl-choline by a microfluidic approach was reported by Pascali et al. with a view on dose-on-demand production of radiopharmaceuticals.72 The aim of the study was to obtain a single dose of these radiotracers in a short time. To enable the two-step 18F-labeling procedure in the microfluidic system, additional storage loops and a second microreactor were implemented. The production of [18F]FETs was performed e.g. by delivering 10 μL of a [18F]KF/K222 complex (50–300 MBq) into a 15.6 μL volume microreactor together with 10 μL of a 1,2-ethylene glycol-bis-tosylate solution (33 mg mL−1) and reacting it for 23 s at 150 °C. Then, 20 μL of the [18F]FETs solution was applied into the second microreactor and mixed with N,N-dimethylethanolamine (4–100 μL) at 80 °C for 62 s and the 18F-fluoroalkylated choline derivatives were purified by SPE. [18F]FEC was obtained after 15 min in 36% overall RCY with a specific activity of 149 MBq μmol−1. Interestingly, [18F]fluoropropyl-choline provided better RCY (49%) than the [18F]fluoroethylated (36%) and [18F]fluoromethylated (20%) analogs.
N-[18F]Fluoroethylamines
An N-[18F]fluoroethylated L-tryptophan analog was obtained by the reaction of N-Boc-L-tryptophan ethyl ester with [18F]FETs and subsequent removal of the BOC and ester protecting groups (Fig. 38).73 Briefly SPE-purified [18F]FETs was reacted with the protected tryptophan precursor in DMSO/NaOH followed by hydrolysis in hydrochloric acid. The overall RCY of N-[18F]fluoroethyl-L-tryptophan was only 0.9%, which was explained by a troublesome purification procedure of the final product.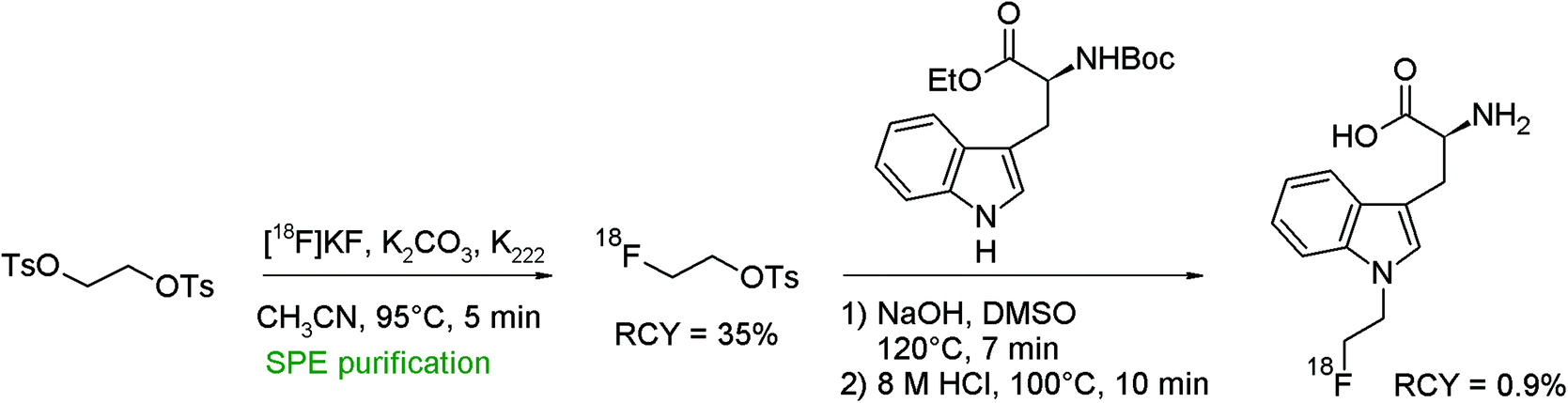 | ||
| Fig. 38 Radiosynthesis of N-[18F]fluoroethylated L-tryptophan.73 | ||
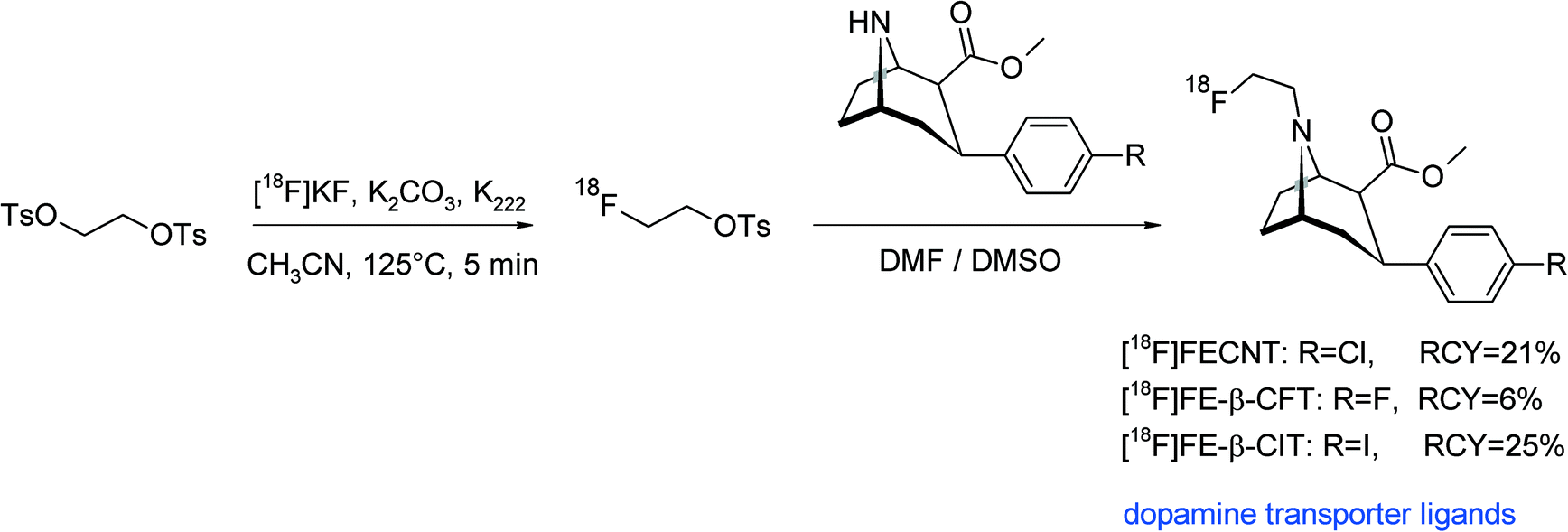
The dopamine transporter is a key protein in the pharmacology and pathophysiology of the brain. Accordingly, a number of radiolabeled dopamine transporter ligands have been developed, among them 2β-carbomethoxy-3β-aryl-substituted tropanes. An N-[18F]fluoroethylated tropane derivative ([18F]FECNT) was used as an imaging ligand for the dopamine transporter with PET and synthesized by N-[18F]fluoroethylation of nortropane.74 [18F]FETs, purified via SiO2-based SPE, was reacted at 135 °C for 45 min with the nortropane precursor in DMF without addition of a base (Fig. 39). HPLC purification provided [18F]FECNT in 21% overall RCY.
 | ||
| Fig. 39 Synthesis of N-[18F]fluoroethylated tropane derivatives.74–76 | ||
A similar tropane-based dopamine transporter ligand was presented in 2004, the labeling approach was likewise [18F]fluoroethylation on a secondary amine (Fig. 39).75 In contrast to the abovementioned publication, the use of 5 mg of the precursor resulted in only 2.7% RCY. By using 10 mg of the nortropane precursor for [18F]fluoroethylation, the final radiotracer [18F]FE-β-CFT was formed in 6% overall RCY. Riss et al. developed the two-step radiosynthesis of [18F]FE-β-CIT, another dopamine transporter imaging agent based on 3-phenyltropane.76 [18F]FETs was produced using an automated synthesis module with integrated semi-preparative HPLC purification. Subsequent reaction with the nortropane derivative in DMSO at 120 °C for 20 min resulted in a [18F]fluoroethylation yield of 64%, whereby the purified [18F]FE-β-CIT was obtained after 100 min in 25% overall RCY (Fig. 39). The authors studied the impact of reaction time and temperature and found that 120 °C resulted in higher RCY than 105 °C and that 20 min was superior to 10 min. Notably, the addition of LiI did not lead to improved RCY because in that case, the reaction temperature had to be kept at 80 °C to avoid evaporation of the volatile intermediate 2-[18F]fluoroethyl iodide.
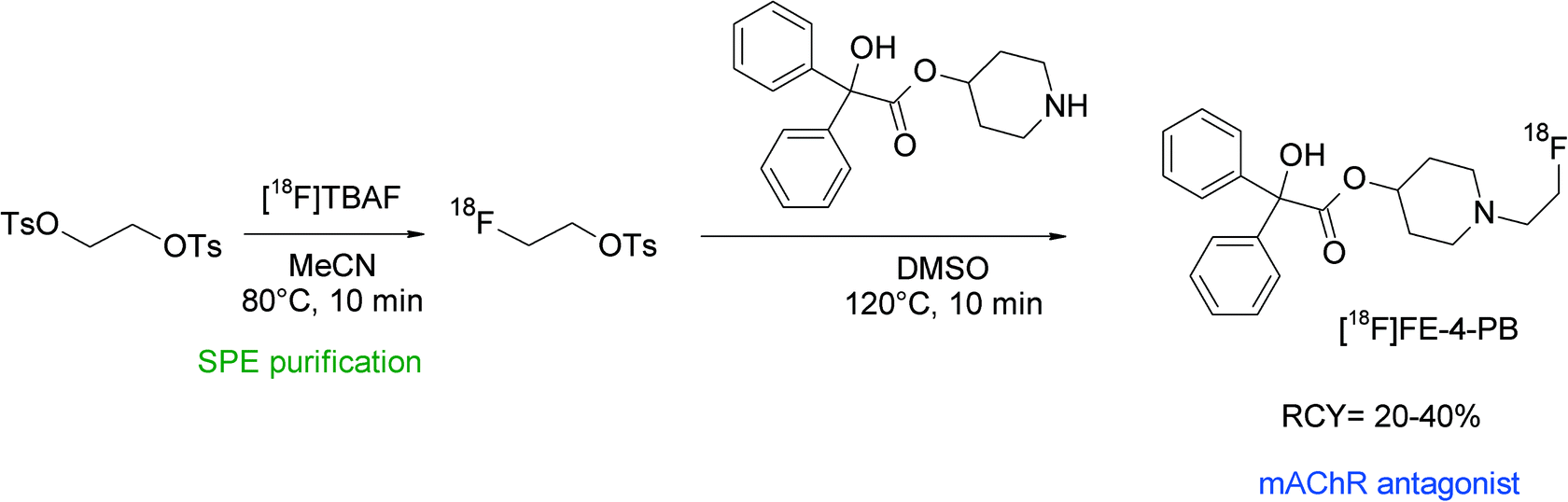
[18F]Fluoroethyl-4-piperidyl benzilate ([18F]FE-4-PB) was reported to be a suitable antagonist of the muscarinic acetylcholine receptor (mAChR) and its radiosynthesis was performed by N-[18F]fluoroethylation of the corresponding piperidyl precursor (Fig. 40).77 [18F]FETs was formed by the reaction of 1,2-ethylene glycol-bis-tosylate with tetrabutylammonium [18F]fluoride in acetonitrile at 80 °C for 10 min and was purified using silica-based SPE. N-Alkylation was performed in DMSO at 120 °C with the piperidyl precursor within a total synthesis time of 90 min and provided HPLC-purified [18F]FE-4-PB in 20–40% overall RCY. The [18F]FE-4-PB synthesis has been re-evaluated by Skaddan et al. in 2001. In addition, [18F]fluoroethyl-3-piperidyl benzilate and [18F]fluoroethyl-3-pyrrolidyl benzilate have been synthesized as possible radioligands for the mAChR.78 The 18F-fluoroethylation was performed as a two-step procedure in one pot just by adding the amine precursor dissolved in DMSO to [18F]FETs. By this method, the HPLC-purified mACh receptor ligands were isolated in 14–24% overall RCY.
 | ||
| Fig. 40 Synthesis of N-[18F]fluoroethylated 4-piperidyl benzilate.77 | ||
Caillé et al. reported the synthesis and 18F-radiolabeling of two benzodioxane derivatives as new serotonin 5-HT4 receptor antagonists.79 [18F]Fluoroethylation was achieved at the secondary amine, i.e. [18F]FETs was reacted with a piperidine derivative in DMF in the presence of different bases (Cs2CO3, K2CO3, NaOH, and Bu4NOH) at 130 °C (Fig. 41). Unfortunately, the N-[18F]fluoroalkylation of piperidine was difficult and only the application of DMF/Cs2CO3 led to the radiolabeled 5-HT4 receptor antagonists, although in only 1.0% overall RCY. The authors could not achieve any improvements neither by using HPLC-purified [18F]FETs nor by application of a one-pot labeling procedure or by using NaI in DMSO as recommended by the literature.16 Despite the low yield, the radiosynthesis provided sufficient amounts of the radiotracers to permit PET imaging studies. Noteworthy, [18F]fluoroethylation was the only successful labeling approach since direct [18F]fluorination was impossible due to the failure in the synthesis of the corresponding ethyl-tosylated precursor.
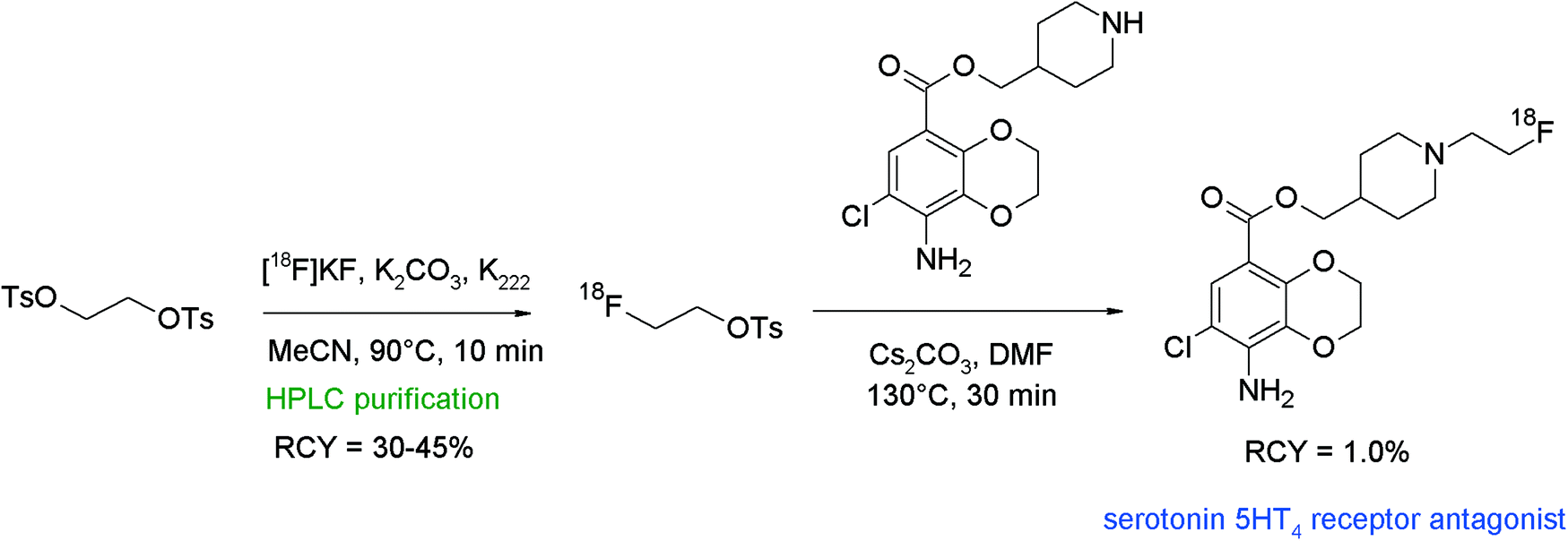 | ||
| Fig. 41 Radiosynthesis of an N-[18F]fluoroethylated benzodioxane.79 | ||
Brain cholinesterase activity has successfully been determined with radiolabeled N-[18F]fluoroalkylated piperidine and pyrrolidine esters.80 The [18F]fluoroethylation was performed as a one-pot reaction without purification of [18F]FETs. The free bases were prepared beforehand from the hydrochloride salts of the piperidine or pyrrolidine precursors by treatment with aqueous NaHCO3 in diethyl ether, drying of the organic phase with sodium sulfate, filtration, addition of DMF, and ether removal. The activated precursors dissolved in DMF were added to the reaction mixture containing the [18F]FETs and radiolabeling was performed at 120 °C for 15 min (Fig. 42). After workup and HPLC purification, the corresponding radiolabeled substrates of cholinesterase were obtained within 90 min in 8–11% overall RCY.
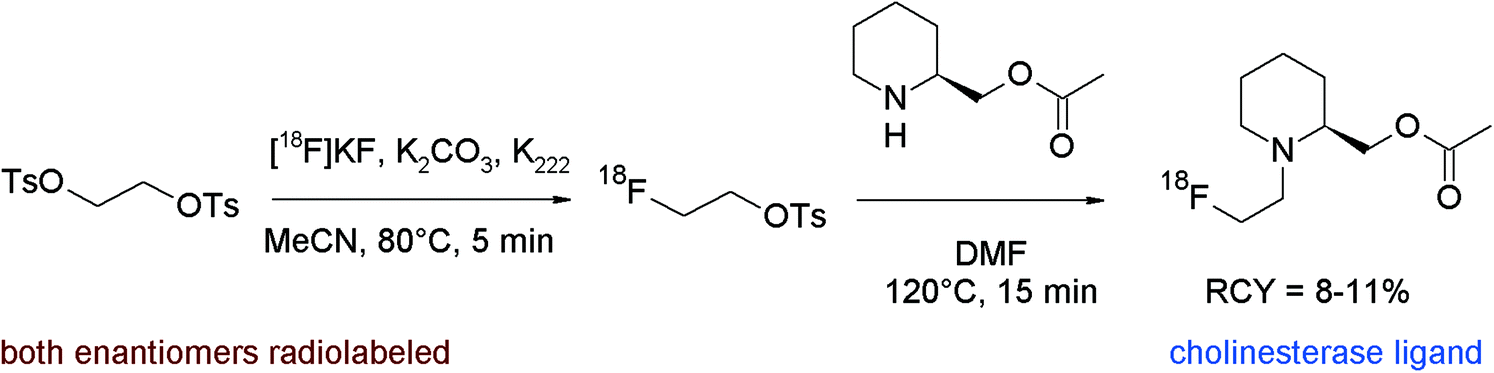 | ||
| Fig. 42 Synthesis of N-[18F]fluoroethylated piperidine esters.80 | ||
N-Methyl-D-aspartate (NMDA) receptors represent an important target in neurological and psychiatric disorders. The discovery of compounds providing nanomolar affinities to NMDA receptors, among them trans-5,7-dichloro 4-substituted 2-carboxytetrahydroquinolines, has triggered the development of adequate radiotracers. One example is the N-[18F]fluoroethylation of a piperazine derivative as shown in Fig. 43.81 The radiolabeling was realised by using HPLC-purified [18F]FETs which was added to the piperazine precursor in DMSO in the absence of a base. The mixture was stirred at 140 °C for 25 min and subsequently purified by HPLC. For removal of a methyl ester protecting group, the radiotracer was reacted with LiOH in methanol, followed by SPE-based purification of the final product. Despite the complex 3-step radiosynthesis involving two HPLC runs, the NMDA receptor ligand was isolated within 110 min in 35% overall RCY.
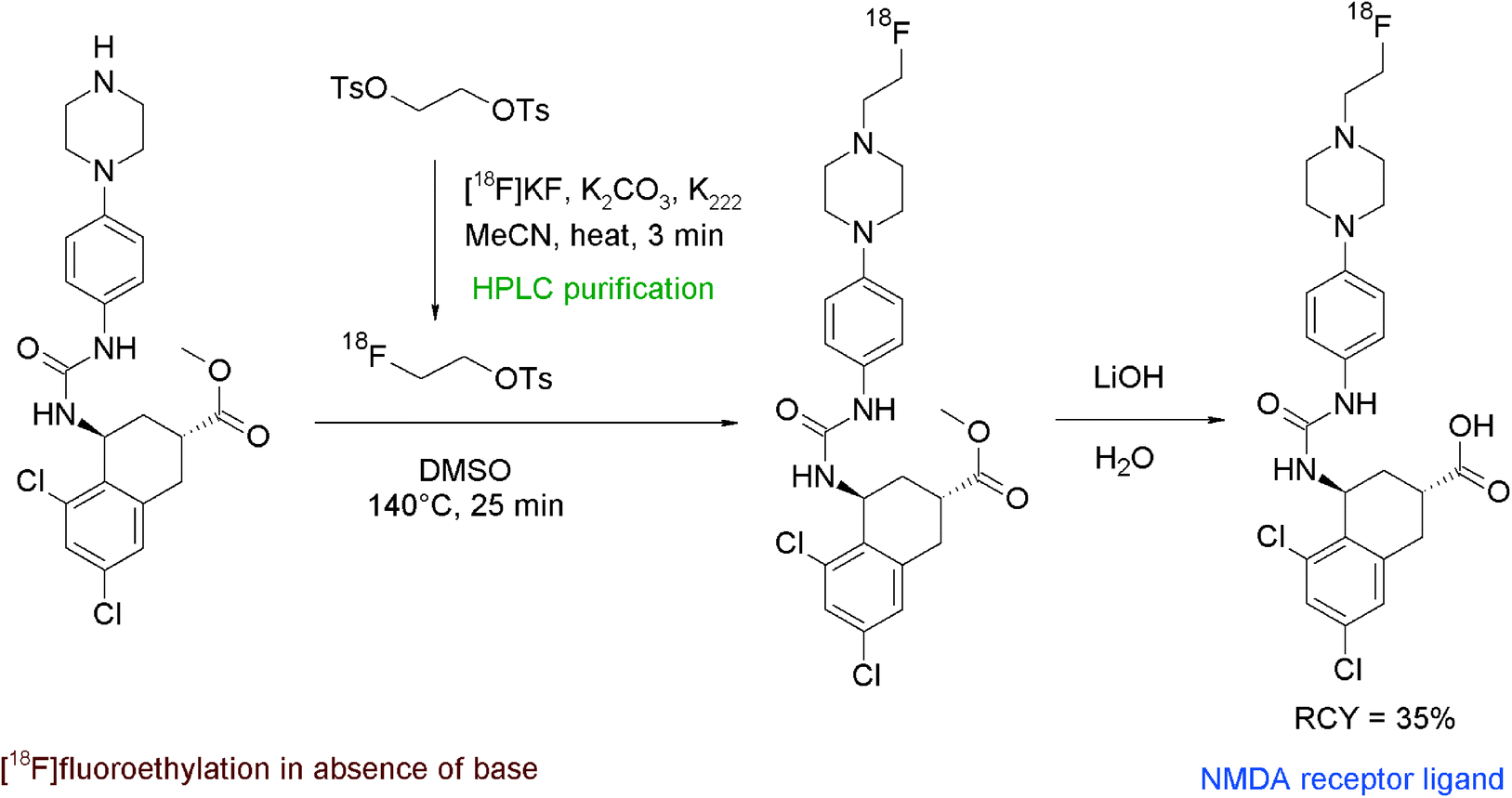 | ||
| Fig. 43 Synthesis of an N-[18F]fluoroethylated piperazine.81 | ||
For non-invasive detection of β-amyloid plaques and neurofibrillary tangles in the human brain, a number of radiolabeled biomarkers have been developed during the past decade. The SPECT tracer 6-[123I]iodo-2-(4′-N,N-dimethylamino)phenylimidazo[1,2-a]pyridine ([123I]IMPY) has been identified to have high uptake in mouse brain. Therefore, the N-[18F]fluoroethylated analogue was developed for PET.82 One-pot radiosynthesis was performed based on the generation of [18F]FETs, followed by [18F]fluoroethylation of the corresponding N-methylaniline precursor at 135 °C for 45 min without addition of a base (Fig. 44). HPLC purification and formulation in saline delivered [18F]fluoroethylated IMPY in 26–51% overall RCY. Notably, also the D4-[18F]fluoroethylated analog was prepared to provide higher metabolic stability. However, no improvement was found in comparison with the non-deuterated radiotracer.
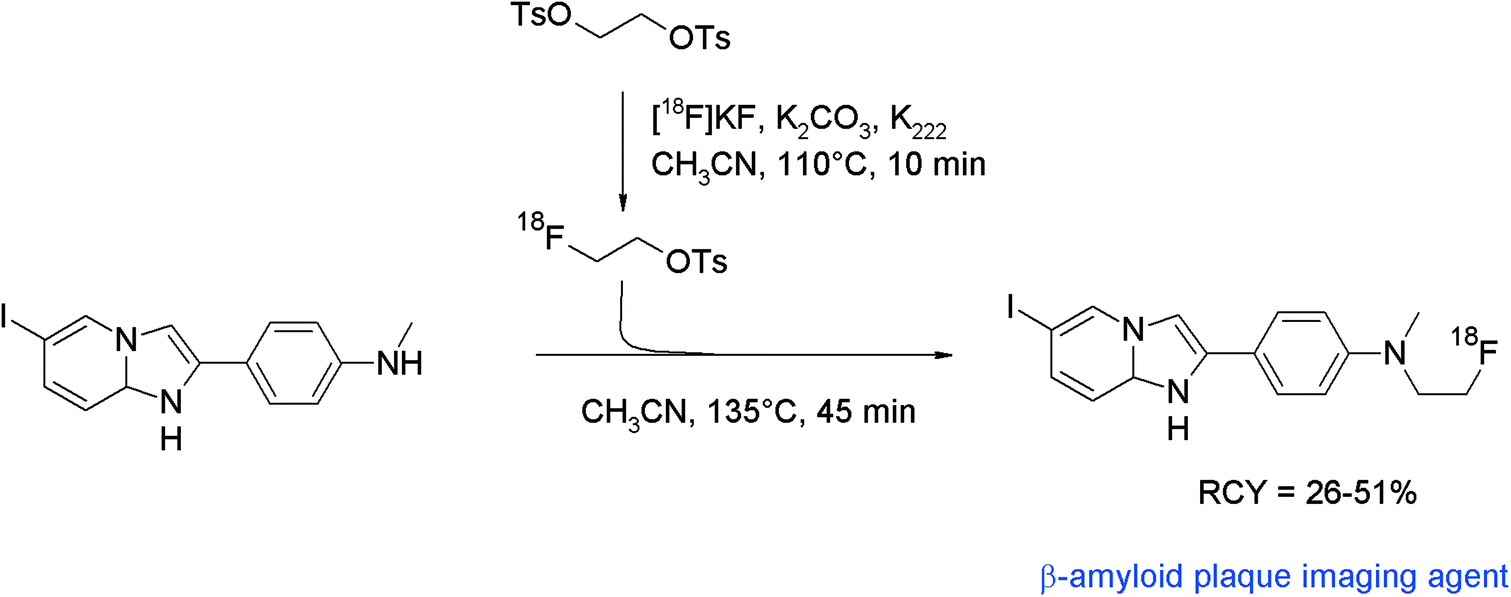 | ||
| Fig. 44 Radiosynthesis of N-[18F]fluoroethylated IMPY.82 | ||
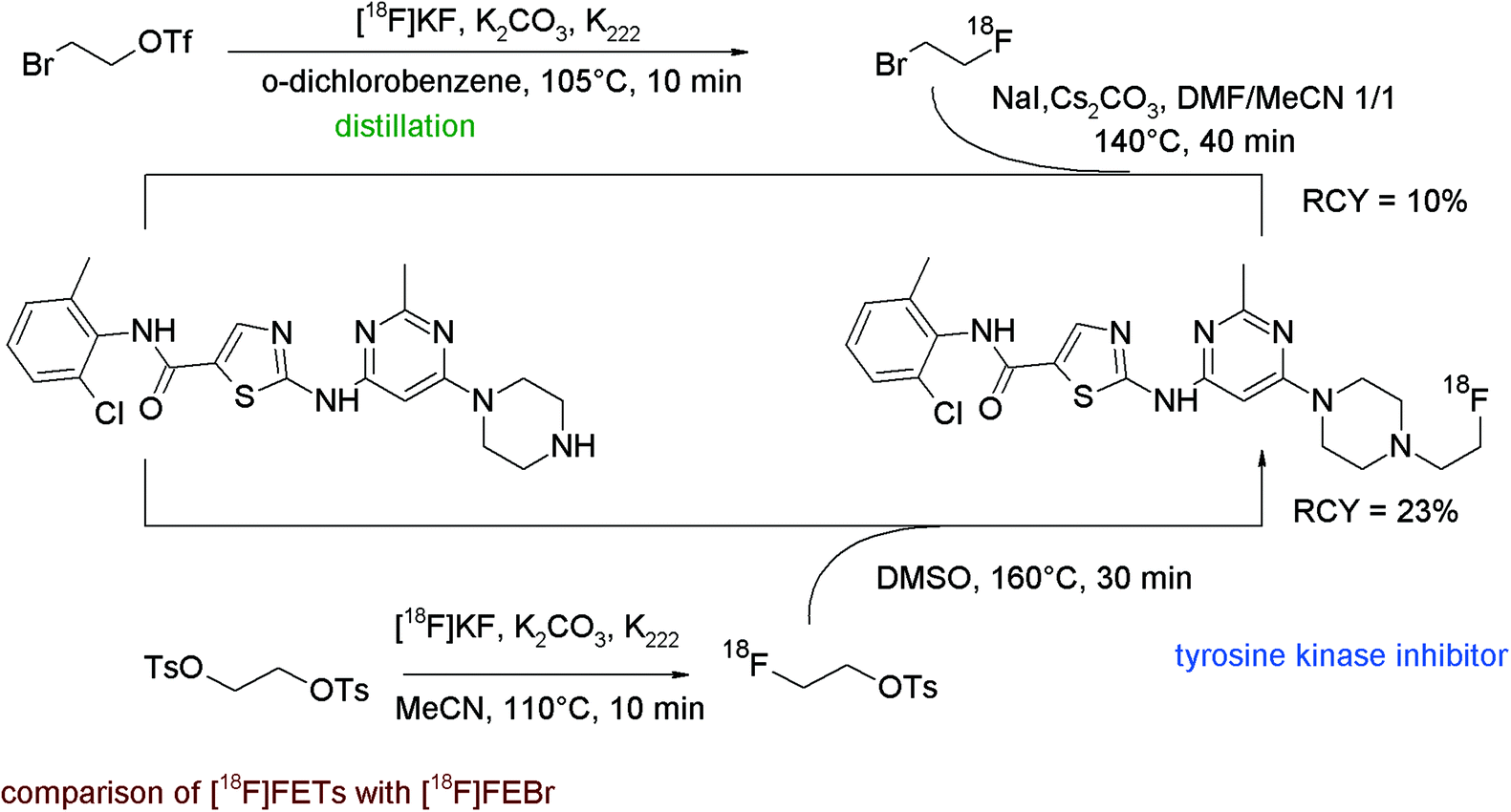
The tyrosine kinase inhibitor dasatinib is known to be a multi-kinase inhibitor with anti-proliferative and cytostatic activity; its structure comprises a hydroxyethyl-substituted piperazine representing a suitable position for N-[18F]fluoroethylation. To obtain [18F]fluoroethylated dasatinib as a radiolabeled probe, Veach et al. compared N-[18F]fluoroethylation with [18F]FEBr and with [18F]FETs in parallel.83 For [18F]FEBr, the 2-bromoethyl triflate precursor was reacted with [18F]fluoride and the labeling agent was purified by distillation. Subsequent N-alkylation of the piperazine precursor in the presence of NaI and Cs2CO3 resulted in 10% overall RCY of 18F-labeled dasatinib (Fig. 45). In contrast, the [18F]fluoroethylation with [18F]FETs was performed as a one-pot method and the piperazine in DMSO was added to the labeling agent. By using this approach, the overall RCY could be enhanced to 23% but surprisingly, as a drawback, the specific activity dropped from 92 to 0.11–0.22 GBq μmol−1 (EOS) in comparison to the [18F]FEBr method. Hence, in this example, [18F]FETs has gained advantage over [18F]FEBr in terms of reactivity but not regarding the specific activity.
 | ||
| Fig. 45 Radiosynthesis of N-[18F]fluoroethylated dasatinib.83 | ||
A further comparison of [18F]FEBr and [18F]FETs was made for the radiolabeling of a 4-benzylpiperidine derivative as a model compound.84 In this work, 80% product formation was achieved by using [18F]FEBr in contrast to 10% in the case of [18F]FETs.
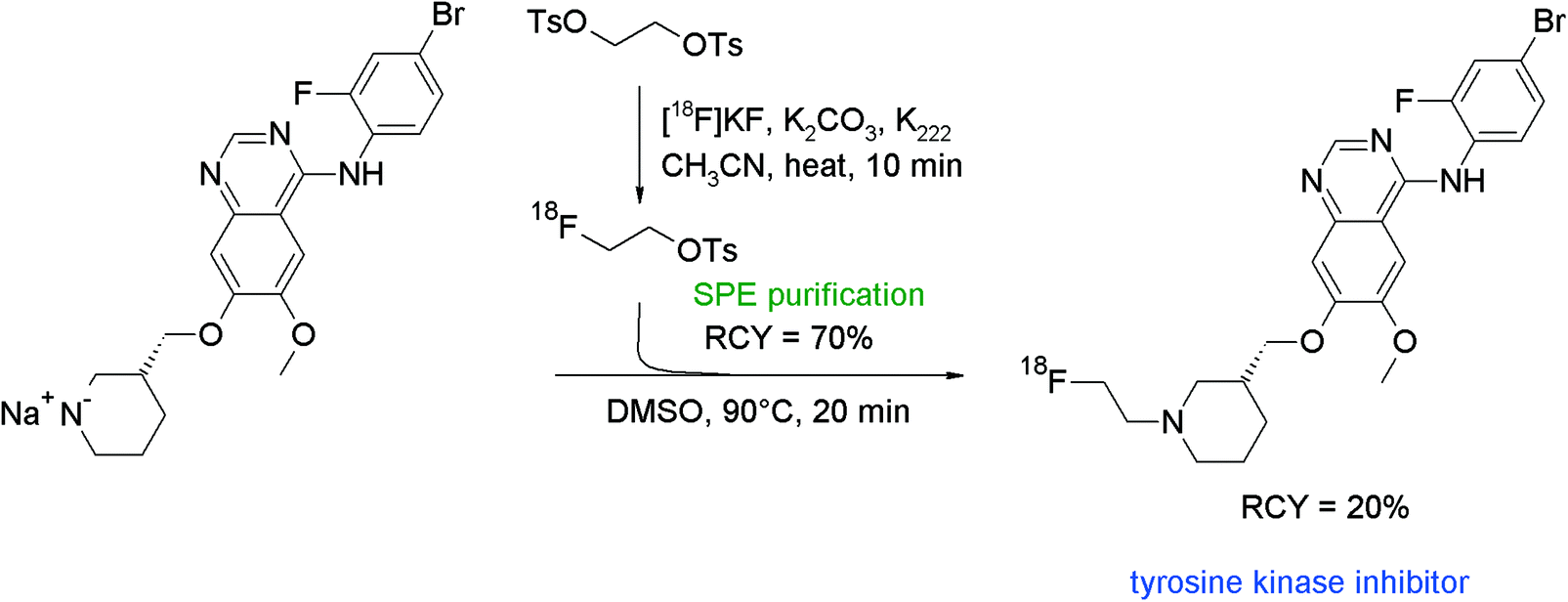
Vandetanib is an antiangiogenic TKI also showing a methylpiperidine moiety and served as a lead for the development of an N-[18F]fluoroethylated radiotracer which was built from a piperidine-substituted quinazoline and [18F]FETs (Fig. 46).85 Briefly, the labeling agent was purified by SPE and subsequently reacted with the freshly prepared sodium salt of the piperidine in DMSO. The radiotracer was isolated in 20% overall RCY after HPLC purification within a synthesis time of 75 min with a specific activity about 37–74 GBq μmol−1 (EOS). Here, it could be demonstrated that by using SPE-purified [18F]FETs, the [18F]fluoroethylation gave an adequate RCY and a satisfying specific activity.
 | ||
| Fig. 46 Radiosynthesis of N-[18F]fluoroethylated vandetanib.85 | ||
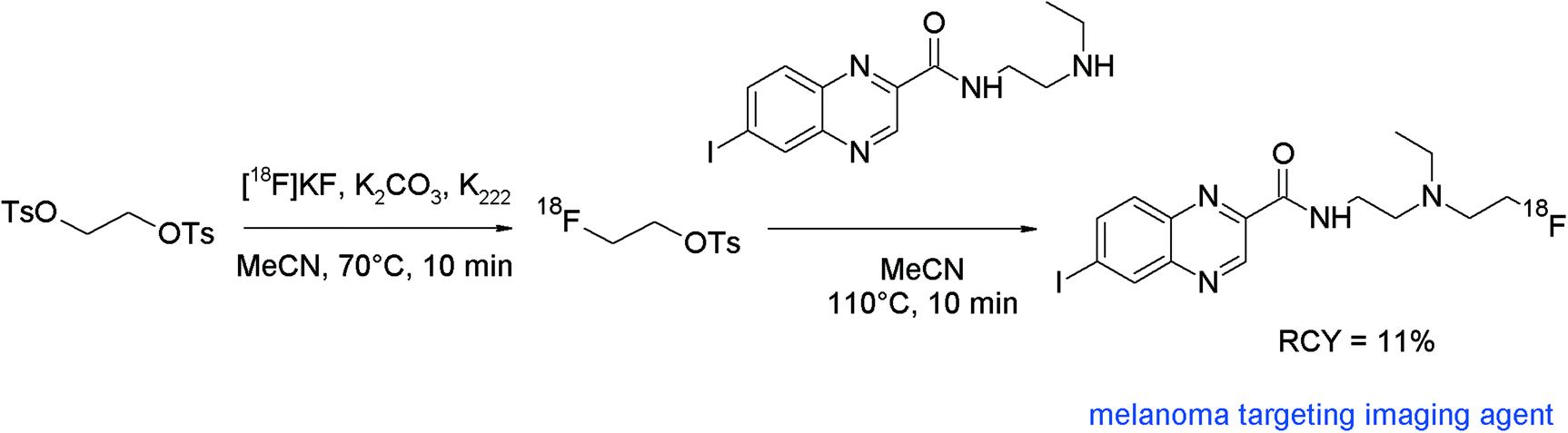
Malignant melanoma affects an increasing number of patients worldwide and early diagnosis of melanoma is the patient's best opportunity to be cured. In this context, Billaud et al. developed 18F-radiolabeled quinoxaline carboxamide derivatives targeting melanin-containing cells as imaging probes for PET and radioiodinated (iodine-131) derivatives for targeted radiotherapy.86 In several examples, [18F]-radiofluorination was performed directly by nucleophilic substitution of the mesylate precursors with [18F]fluoride. However, in the case of 2-hydroxyethylamine, the synthesis of the mesylate precursor failed, thus the authors decided to perform N-[18F]fluoroalkylation with [18F]FETs (Fig. 47). [18F]FETs was synthesized in one pot and used unpurified. The secondary amine precursor was added in acetonitrile without a base and the mixture was heated at 110 °C for 10 min. The [18F]fluoroethylated quinoxaline carboxamide was provided in 11% overall RCY after HPLC separation. This is lower in comparison with the RCY of the other derivatives obtained by direct [18F]fluorination (16–54%) but the [18F]FETs reaction represents in that example the only access to the desired radiotracer. Notably, the amide group of the quinoxaline carboxamide precursor was unaffected by [18F]FETs due to its lower basic character.
 | ||
| Fig. 47 Radiosynthesis of an N-[18F]fluoroethylated quinoxaline carboxamide.86 | ||
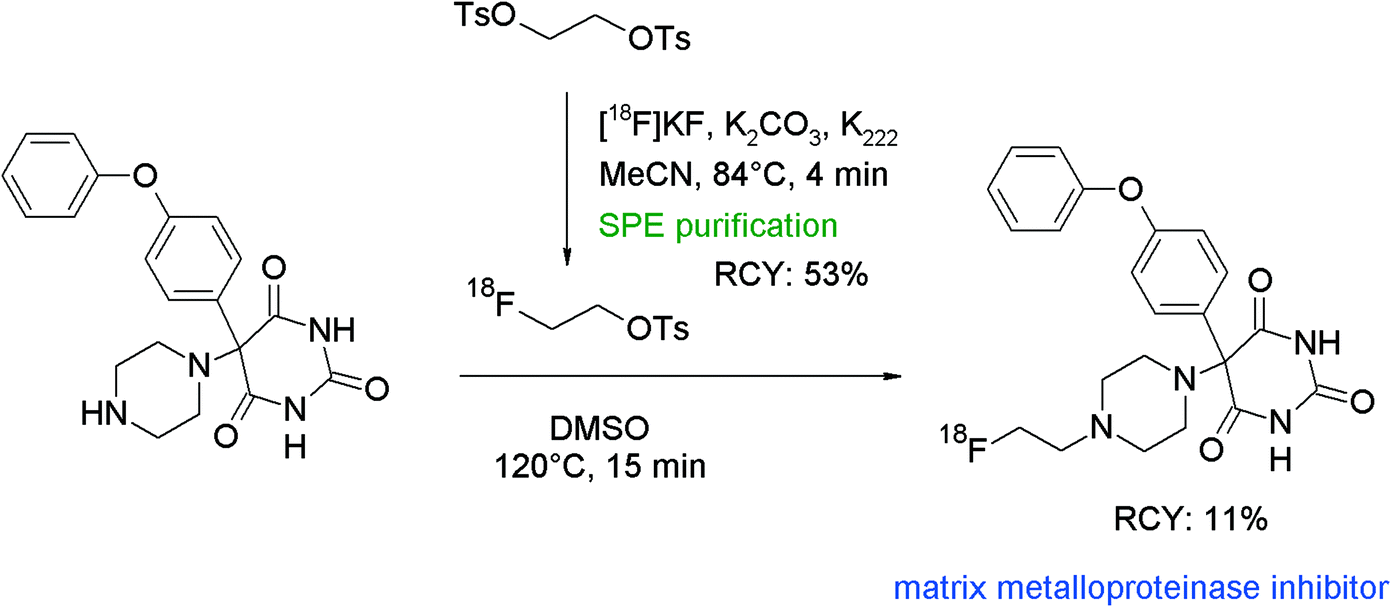
An interesting contribution to N-[18F]fluoroalkylation with [18F]FETs was made by Kopka et al. who developed radiolabeled matrix metalloproteinase inhibitors (MMPIs) as molecular probes for imaging of upregulated levels of MMPs in living organisms.87 The chemical structure of the pharmacophore is based on phenylpiperazine-substituted pyrimidine-2,4,6-triones and [18F]fluoroethylation at the piperazine was the preferred route to obtain the radiotracer (Fig. 48). [18F]FETs was purified by a SPE-based method and subsequently reacted with the pyrimidine-2,4,6-trione precursor at 120 °C for 15 min. After HPLC purification, the [18F]fluoroethylated MMPI was obtained in 11% overall RCY in a synthesis time of 142 min. Remarkably, the pyrimidine-2,4,6-trione precursor displayed both amine and amide functionalities, and again the [18F]fluoroalkylation occurred at the amine.
 | ||
| Fig. 48 Radiosynthesis of an N-[18F]fluoroethylated pyrimidine-2,4,6-trione.87 | ||
Phosphatidylserine (PS), an anionic phospholipid in the plasma membrane, may be used as a target for imaging of metastatic infectious diseases and apoptosis. Li et al. developed an 18F-radiolabeled dipicolylamine derivative showing high affinity to PS as a promising radiotracer.88 In order to find the ideal prosthetic group for the amine functionality, the authors used three 18F-bearing labeling synthons in parallel: 4-nitrophenyl 2-[18F]fluoropropionate ([18F]NFP), N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) and [18F]FETs (Fig. 49). Each [18F]fluorine-bearing labeling reagent was produced in a previous reaction and purified prior to usage. [18F]Fluoroethylation was performed by reacting a solution of [18F]FETs in DMSO with the amine precursor and diisopropylethylamine for 10 min at 100 °C. The [18F]-fluoroethylated dipicolylamine was isolated after HPLC purification in 76% overall RCY within 65 min. By using the other prosthetic groups [18F]NFP and [18F]SFB, overall RCYs of 68–71% were achieved and the total synthesis time was 105 and 75 min, respectively. Comparing all three labeling agents in terms of labeling efficiency, there was no clear advantage between them. However, [18F]FETs turned out to be the one that could be prepared the easiest, affording a one-step reaction only.
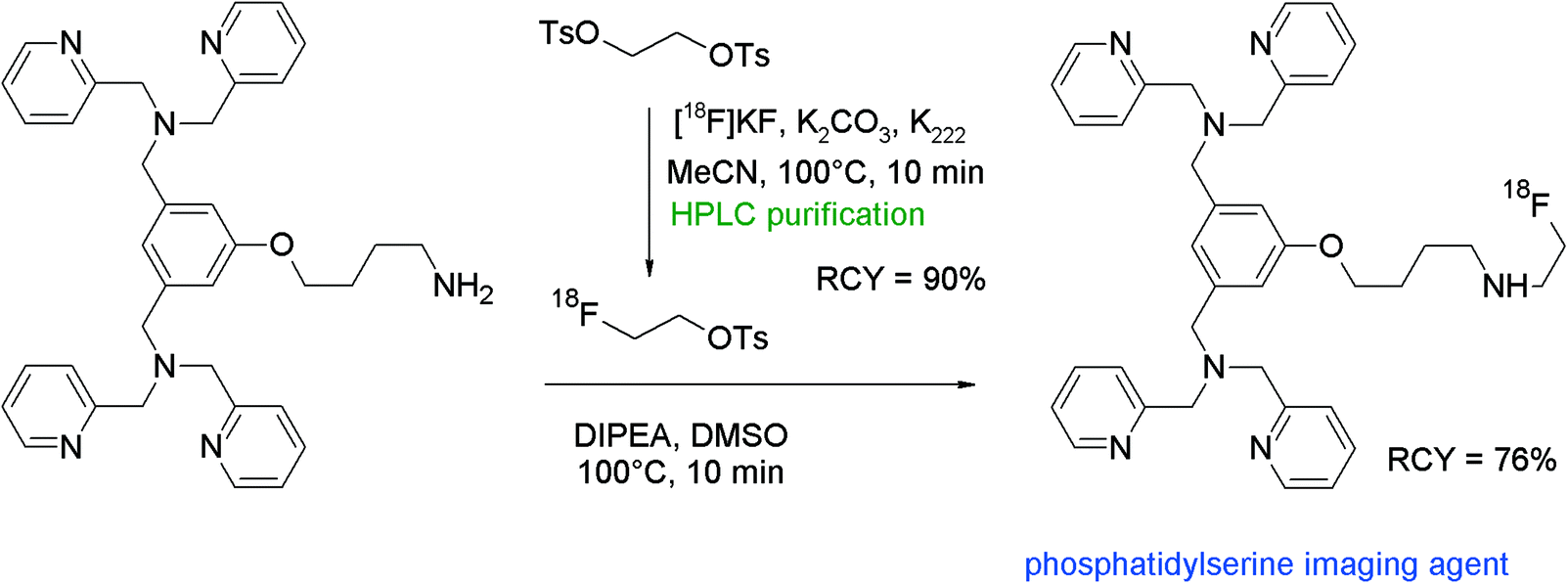 | ||
| Fig. 49 Radiosynthesis of an N-[18F]fluoroethylated dipicolylamine.88 | ||
N-[18F]Fluoroethyl amides
The only example of N-[18F]fluoroethylation on an amide functionality was reported by Rösch et al. who developed two radiotracers based on pirenzepine, a well-established muscarinergic M1 antagonist that is clinically used in gastric ulcer therapy and as a selective proton-pump inhibitor.89 [18F]FETs prepared by automated synthesis with integrated HPLC was dissolved in DMSO and added to one of the selected pirenzepine precursors and a base (Fig. 50). [18F]Fluoroethylation was performed for 15 and 20 min at 120 °C and the corresponding radiotracers were provided after HPLC purification in 30% and 15% overall RCY, respectively. It is worth remarking that in the case of the diazepine-6-one precursor, showing both amine and amide functionalities, the amide is preferred, which means that the N-[18F]fluoroethylation occurs exclusively on that position.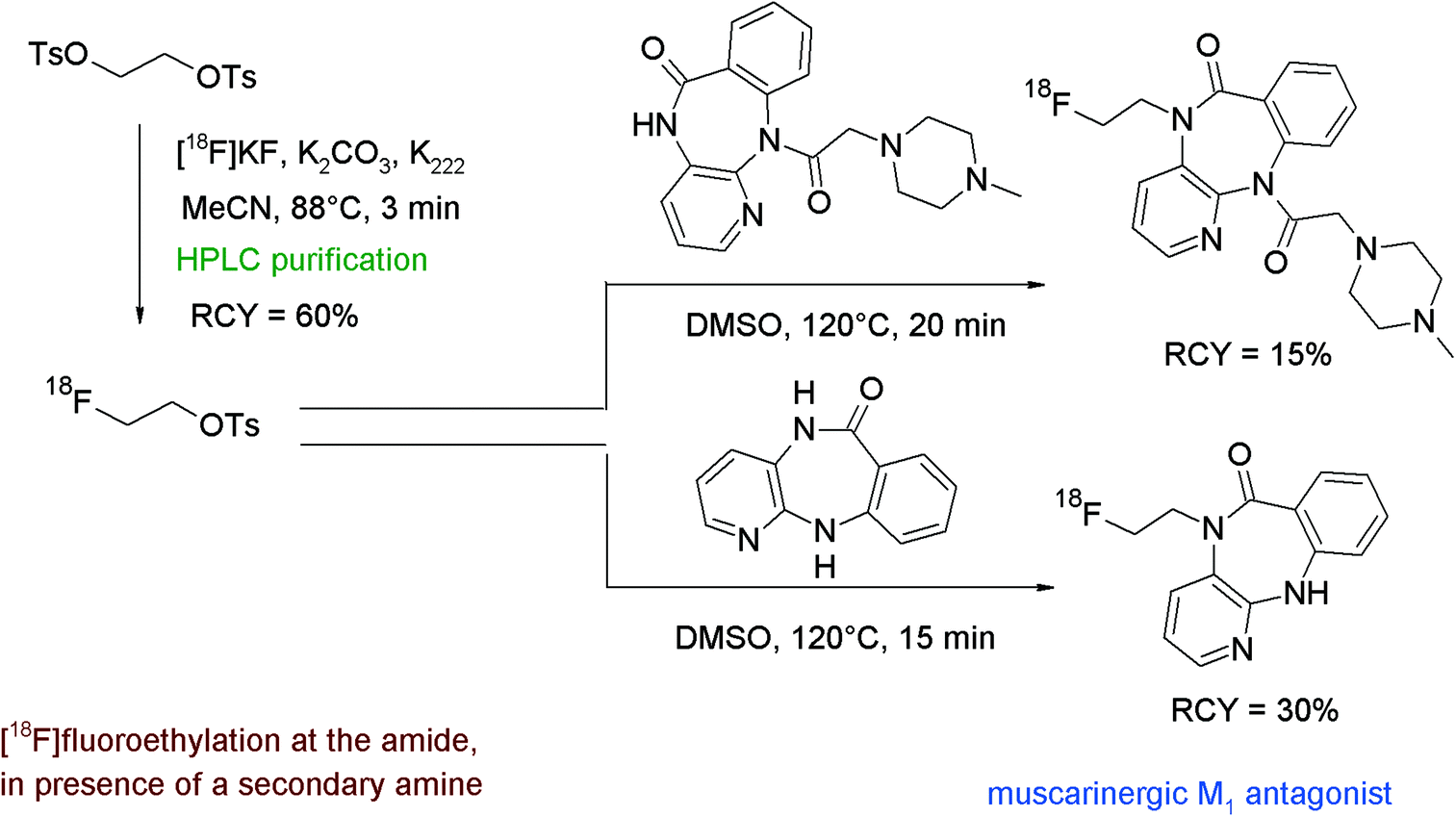 | ||
| Fig. 50 Radiosynthesis of two N-[18F]fluoroethylated pirenzepine derivatives.89 | ||
Direct versus indirect radiolabeling
A leading point in radiotracer design is the radiolabeling approach, which means finding the optimal step in the synthesis route for introducing the radiolabel. The most preferred method, by doing the radiolabeling in the last step of the synthesis, is described as the so-called direct radiolabeling. In contrast, the application of a prosthetic group or a pre-labeled reagent such as [18F]FETs is called as indirect radiolabeling, and often it is difficult to decide what is the method of choice. In this review and with reference to [18F]fluoroethylation with [18F]FETs, we found that for a number of radiotracers, both strategies were pursued and we took the opportunity of comparing these two approaches.As we have shown before, the N-[18F]fluoroethylated dopamine transporter ligand ([18F]FECNT) was synthesized by N-[18F]fluoroethylation of nortropane with [18F]FETs in 21% overall RCY.74 To simplify and improve the radiosynthesis, Chen et al. and Pijarowska-Kruszyna et al. developed an automated one-step procedure for [18F]FECNT synthesis starting from the mesylate precursor and [18F]KF/K222 providing the radiotracer in 33 ± 9% RCY which suggests an advantage of the direct radiolabeling (Fig. 51).90
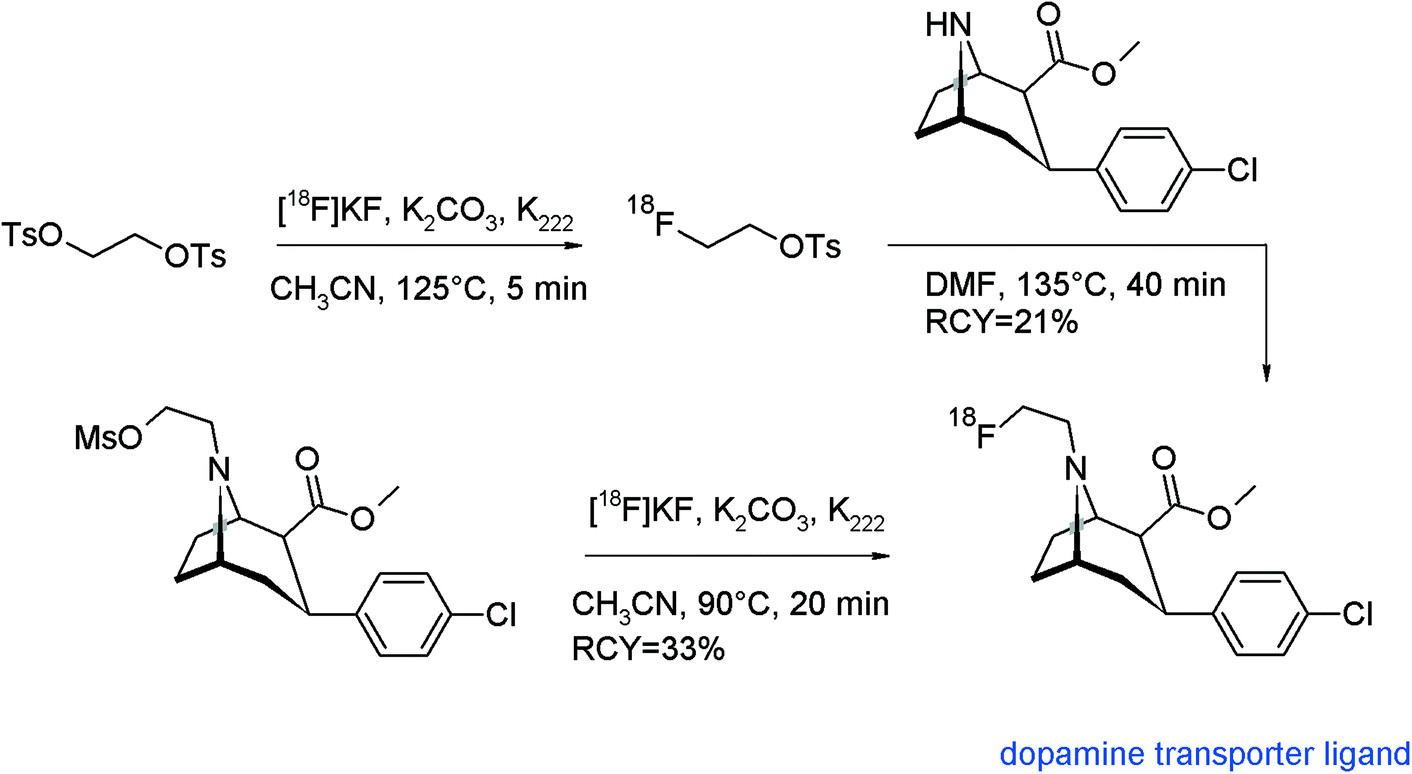 | ||
| Fig. 51 Radiosynthesis of [18F]FECNT.74,90 | ||
To study the γ-aminobutyric acid (GABA)-based neurotransmission, radiolabeled GABA transporter ligands have been developed. Two radiotracers were reported, available by 11C-methylation and O-[18F]fluoroethylation of a triphenyl-substituted piperidine-3-carboxylic acid derivative.91 The initial reaction of the carboxymethyl ester-substituted desmethyl precursor with [18F]FETs in the presence of NaOH resulted in 80% [18F]fluoroethylation. However, a subsequent ester cleavage reaction was required to obtain the final radiotracer. To circumvent this deprotection step, [18F]fluoroethylation with the free carboxylic acid moiety was performed by using a variety of bases and solvents. It turned out that in the presence of NaH in DMSO, the highest RCY of about 90% was achieved, whereas in the presence of NaOH and K2CO3 under identical conditions, only 65% and 40% RCYs were obtained.
In a subsequent paper, the authors compared the [18F]fluoroethylation with the direct [18F]fluorination of the ethyl-tosylated precursor with [18F]fluoride. By [18F]fluoroethylation, the radiotracer was formed within 5 min in 37–40% RCY, whereas direct nucleophilic fluorination with [18F]fluoride provided the radiotracer in only 19% RCY within 40 min (Fig. 52).92 In summary, the direct radiolabeling approach is less efficient. Additionally, it should be considered that the ethyl-tosylated precursor was difficult to synthesize with a yield of only 2.6%.
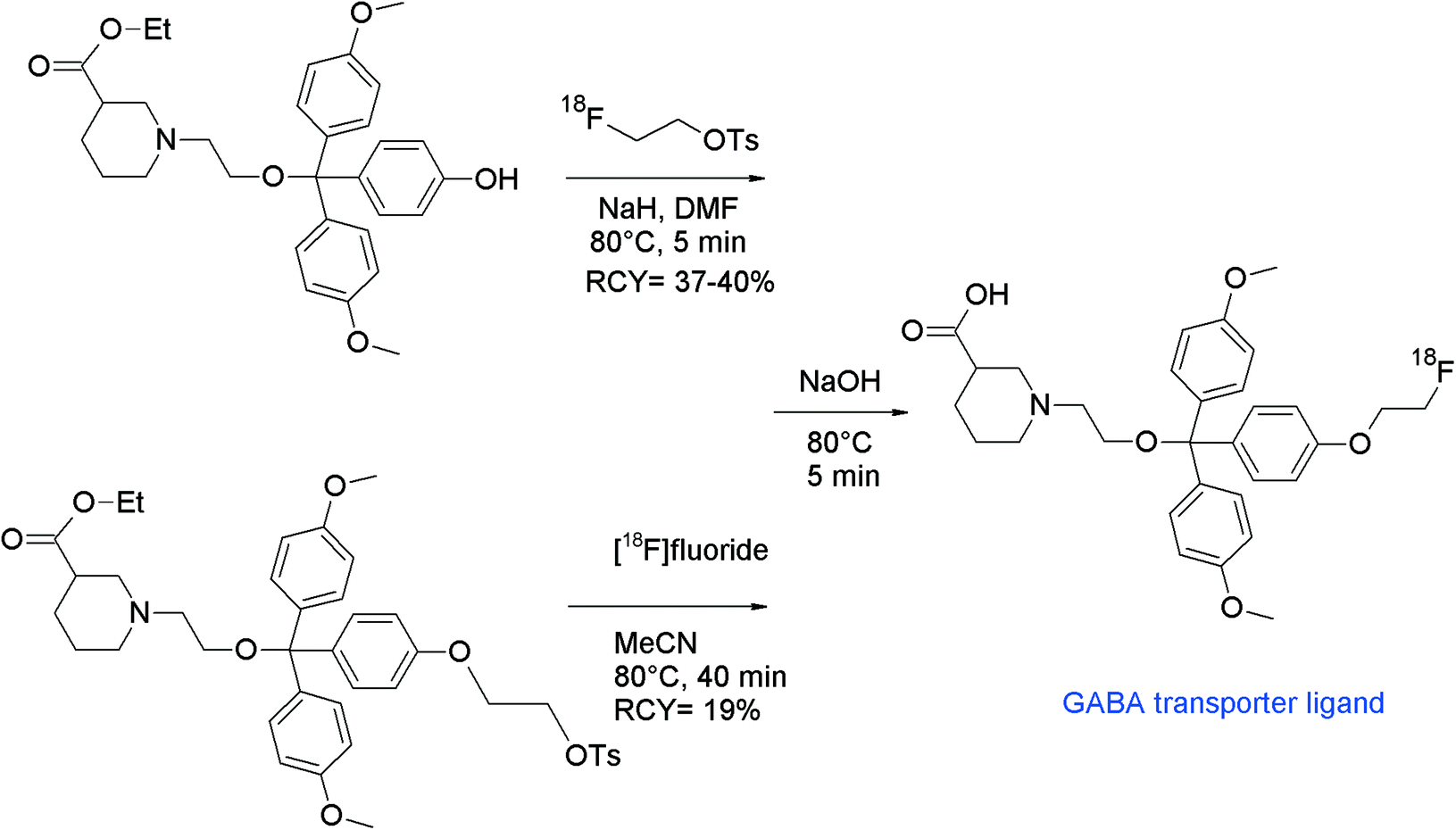 | ||
| Fig. 52 Synthesis of an O-[18F]fluoroethylated triphenyl-substituted piperidine-3-carboxylic acid.91,92 | ||
Radiolabeled analogs of spiperone and raclopride were the first radiopharmaceuticals targeted towards the dopaminergic D2 receptor. The synthesis of a series of O-ω-fluoroalkylated derivatives of raclopride was published describing the radiosynthesis of a [18F]fluoroethylated raclopride which was realized via two different approaches,93 firstly, by using [18F]FETs and secondly, by direct radiofluorination starting from an appropriate ethyl-tosylated raclopride precursor (Fig. 53). In the indirect one-pot process, [18F]FETs was not purified and reacted with the hydroxyl precursor by adding the latter into the reaction vessel. The overall RCY of the HPLC-purified [18F]fluoroethylated raclopride was 12%. In the direct procedure, the tosylate precursor of raclopride was reacted with the [18F]KF/K2CO3/K222 complex to provide the radiotracer in 35% RCY. By comparison of both radiolabeling protocols, it turned out that the direct approach is superior to the [18F]fluoroethylation with [18F]FETs due to a shorter reaction time and a higher RCY. However, it was pointed out that HPLC purification of the [18F]FE-raclopride synthesized by the one-step procedure was difficult because the hydrolyzed precursor and the final radiotracer show nearly identical retention. Furthermore, the synthetic efforts for the preparation of the tosylated raclopride precursor are challenging.
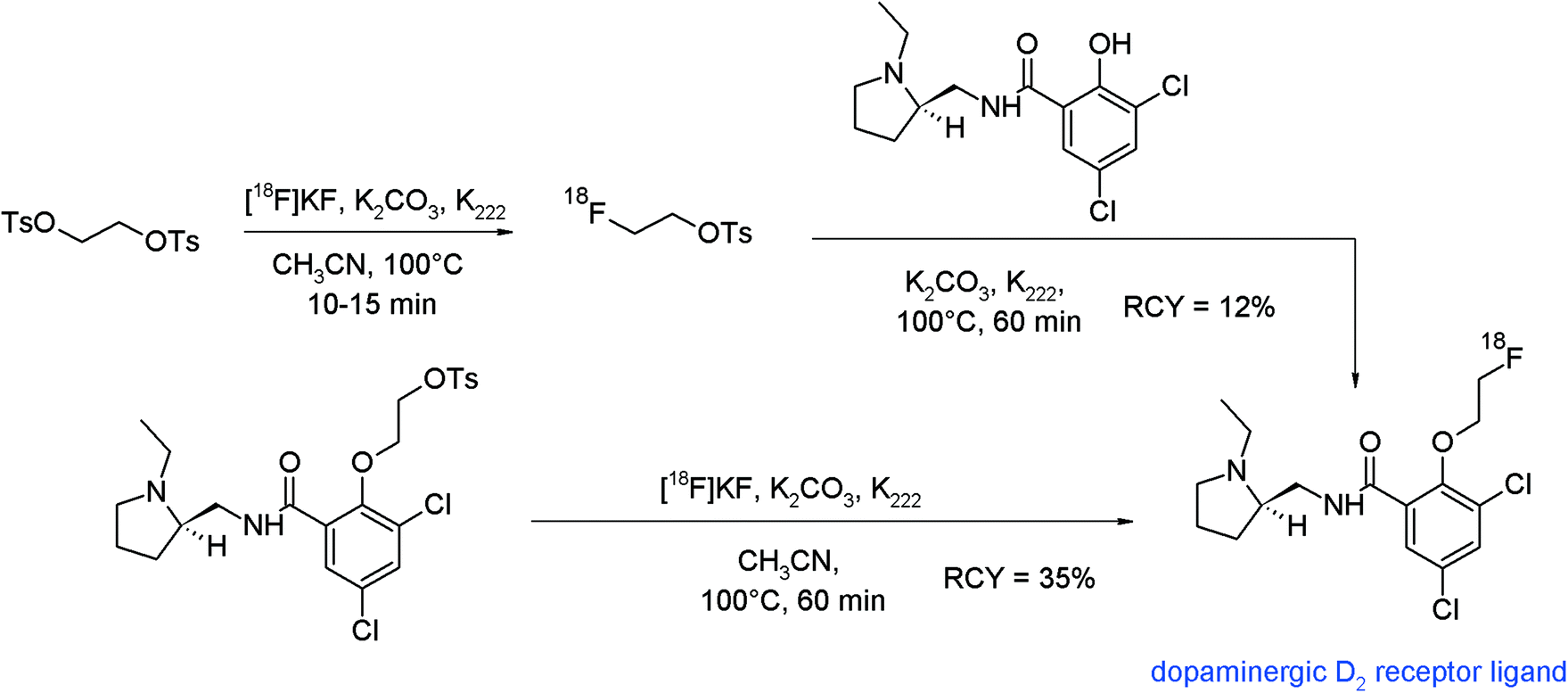 | ||
| Fig. 53 Synthesis of O-[18F]fluoroethylated raclopride.93 | ||
The development of selective PET radiotracers for imaging of neurofibrillary tangles (NFTs) is expected to reinforce insights into Alzheimer's disease. Astemizole, having the basic structure of a phenyl piperidyl-substituted benzimidazole, was found to bind to NFTs with low nanomolar affinity. For development of a [18F]fluoroethylated astemizole, the direct radiofluorination with [18F]fluoride and the indirect O-[18F]fluoroethylation with [18F]FETs were investigated (Fig. 54).94 The direct nucleophilic substitution of the tosylated precursor in acetonitrile at 100 °C provided the radiotracer in 29% RCY. The indirect radiolabeling of [18F]fluoroethyl astemizole was performed as a one-pot two-step procedure using an automated synthesizer. [18F]FETs was reacted with the hydroxyl precursor and aqueous NaOH in DMF by heating for 20 min at 120 °C. Following this route, [18F]fluoroethyl astemizole was isolated in 5–10% overall RCY. Accordingly, for this radiotracer, the direct radiofluorination was found to be superior.
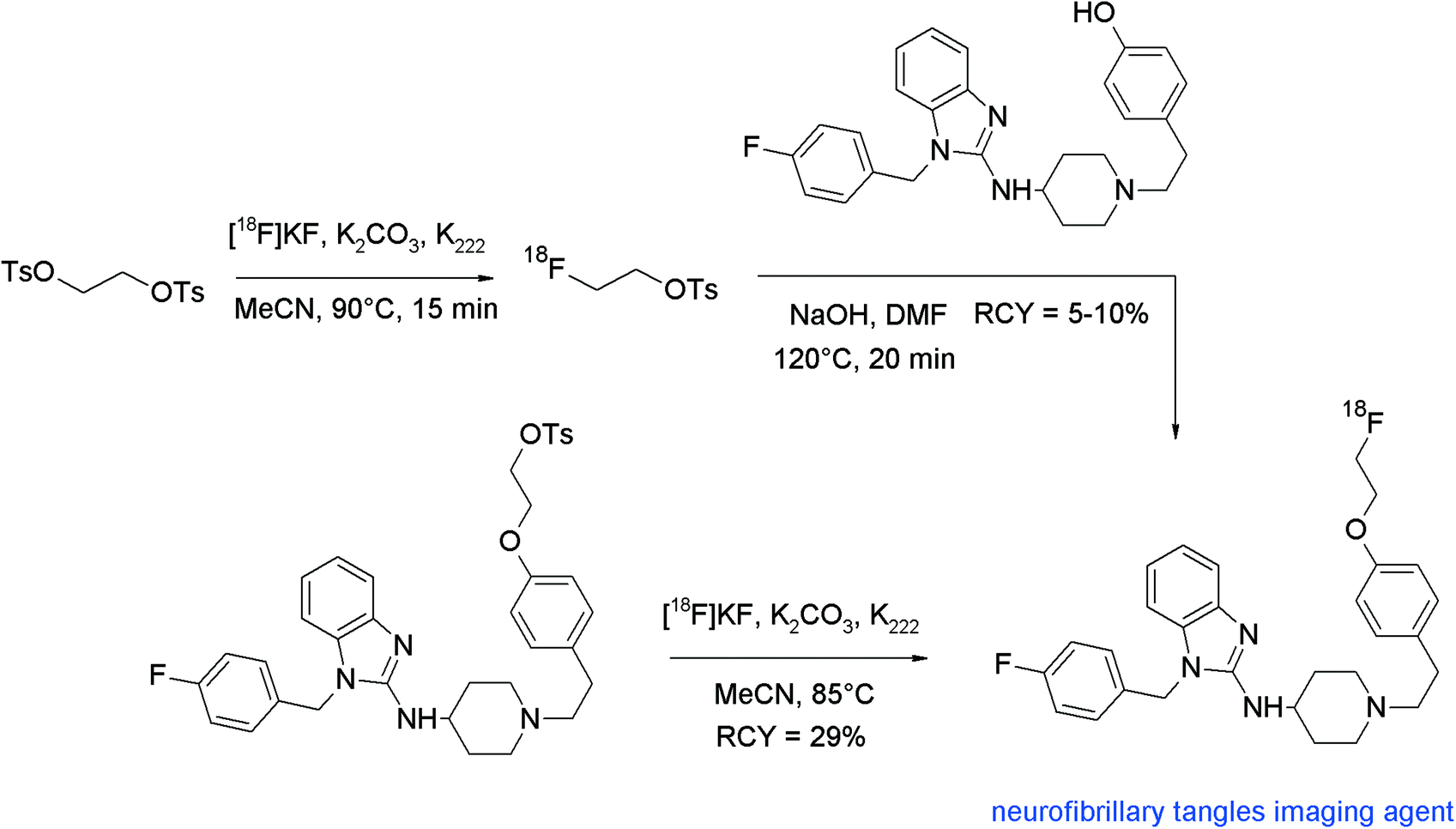 | ||
| Fig. 54 Radiosynthesis of O-[18F]fluoroethylated astemizole.94 | ||
A further contribution in regard to direct and indirect [18F]radiofluorination was made by Skaddan et al. who had developed a PET ligand for imaging of fatty acid amide hydrolase (FAAH) in the brain.95 The target molecule PF-9811 contains a 2-fluoroethoxypyridine moiety and the initially chosen route was the nucleophilic substitution of a tosylated precursor with [18F]fluoride (Fig. 55). However, all efforts to produce [18F]PF-9811 by this route proved to be unsuccessful, so the authors decided to perform [18F]fluoroethylation by a two-step one-pot process. The desired radiotracer was formed but could not be separated from other chemical impurities by HPLC. To solve this problem, [18F]FETs was purified beforehand by HPLC and reacted with the hydroxyl precursor and K2CO3 in DMA but the chemical purity of the radiotracer was not significantly improved. The finally successful route was a three-step one-pot reaction starting with direct [18F]fluorination of a BOC-protected tosylated amine, followed by deprotection and subsequent coupling with phenyl pyrazin-3-ylcarbamate providing [18F]PF-9811 in 11% overall RCY.
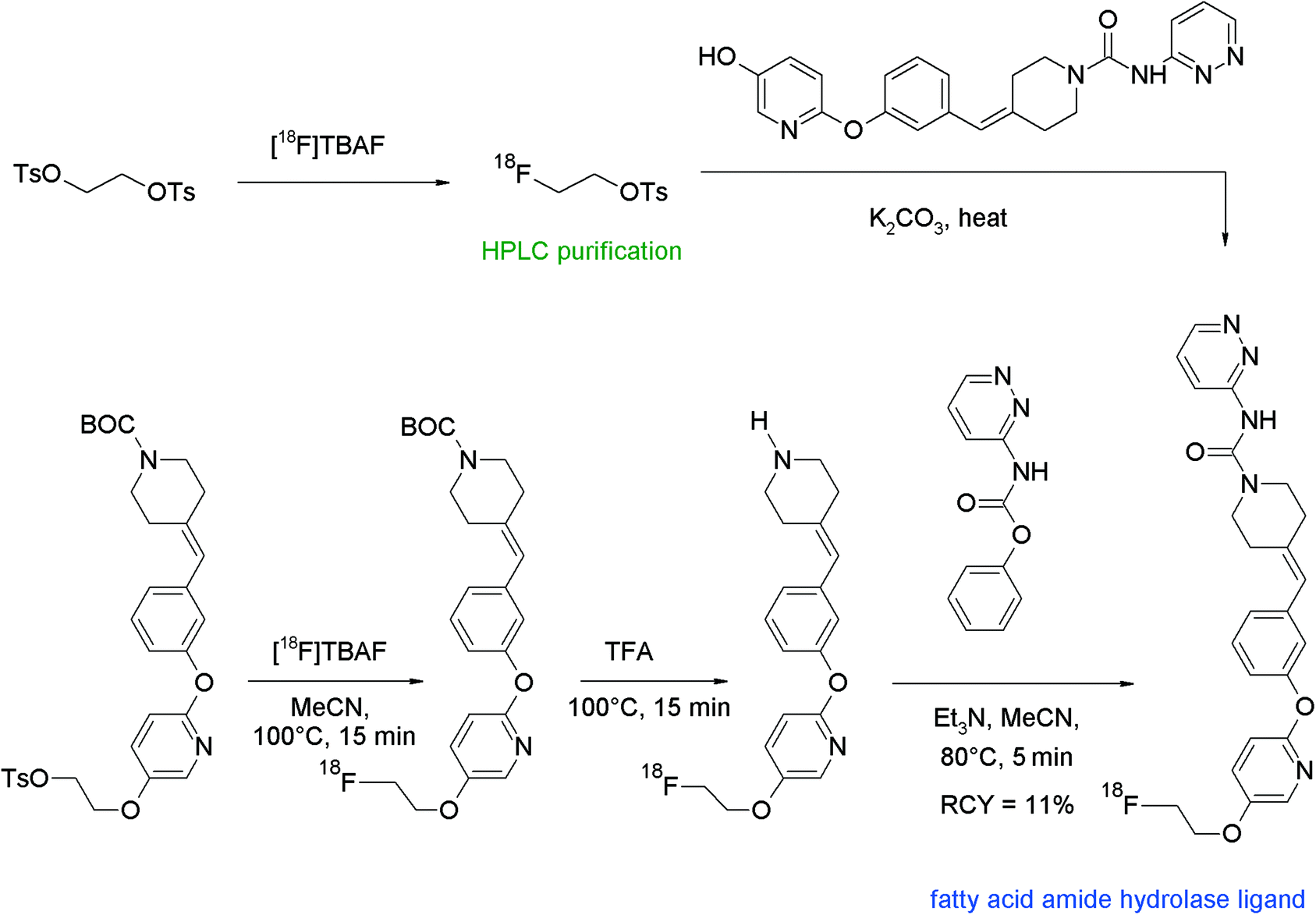 | ||
| Fig. 55 Synthesis of an O-[18F]fluoroethylated pyridine.95 | ||
A 18F-labeled phosphodiesterase 10A (PDE10A) ligand based on 6,7-dimethoxy-4-pyrrolidinylquinazoline was developed by Funke et al.96 Also in this work, the direct and indirect radiolabeling approach was compared. For O-[18F]fluoroethylation with [18F]FETs, the labeling reagent was added to the phenolate-containing solution after activating the hydroxyl precursor with K2CO3/K222 at 60 °C (Fig. 56). Interestingly, the solvent showed a significant effect on [18F]fluoroethylation. By using DMF at 110 °C, only 5% RCY of [18F]FE-PDE10A was observed but upon shifting the solvent to acetonitrile, the labeling efficiency was improved up to 30–40%. Purification of the final product was difficult due to lipophilic non-radioactive by-products and because of this, the authors decided to develop a direct radiofluorination route starting from the corresponding tosylated precursor. By using this approach, [18F]FE-PDE10A was formed with a smaller amount of by-products and could be isolated in 17–40% RCY with high chemical and radiochemical purity.
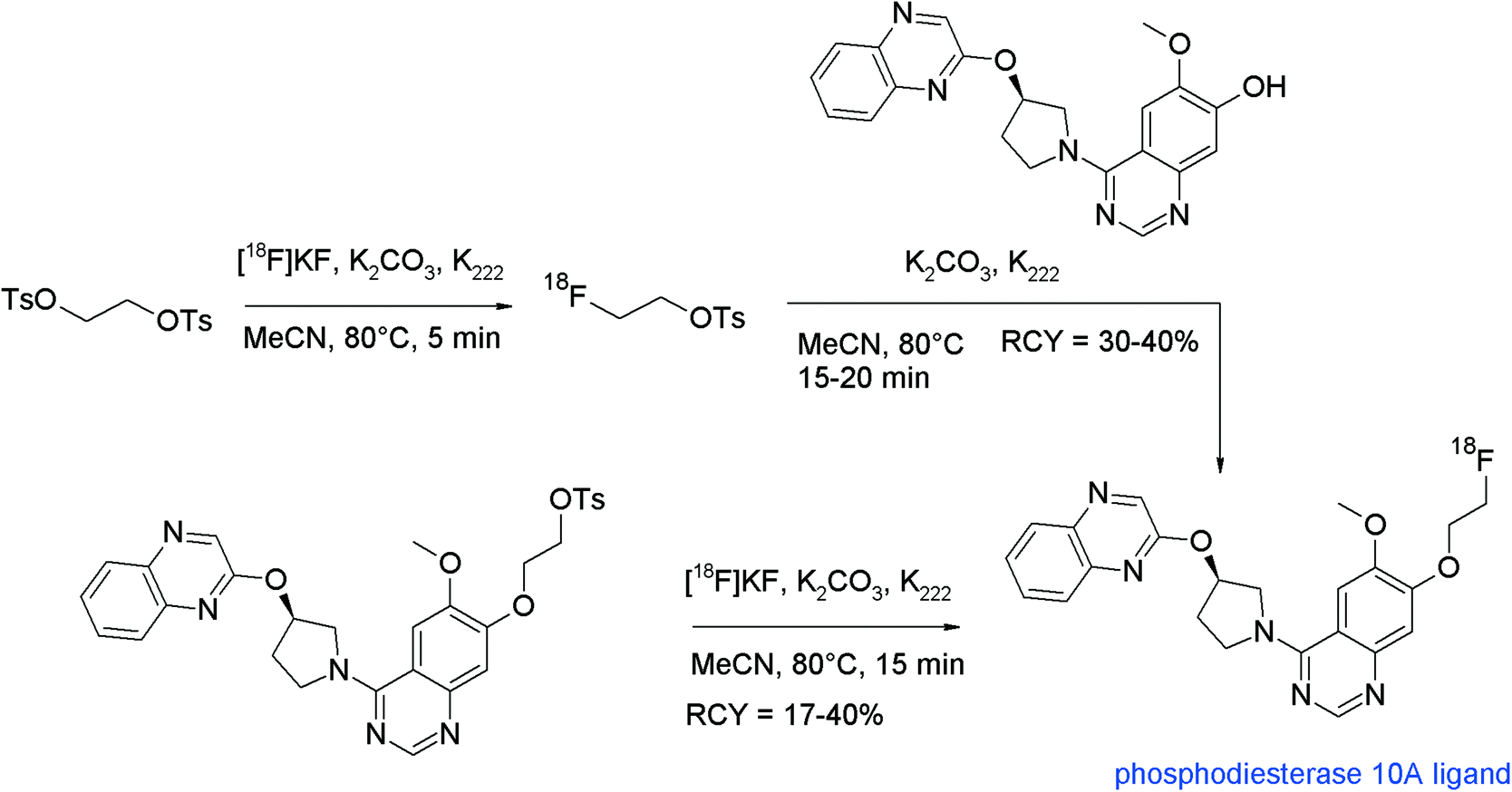 | ||
| Fig. 56 Synthesis of an O-[18F]fluoroethylated pyrrolidinylquinazoline.96 | ||
[18F]NS14490 was synthesized and evaluated recently by Rötering et al. as a potential radiotracer for neuroimaging of α7 nicotinic acetylcholine receptors.97 NS14490 contains 1,4-diazabicyclo[3.2.2]nonane, oxadiazole and indole moieties, and the radiolabeling was performed by N-[18F]fluoroethylation with [18F]FETs at the indole nitrogen (Fig. 57). By using non-purified [18F]FETs as the labeling reagent and reacting it with the precursor in acetonitrile at 82 °C in a one-pot reaction, [18F]NS14490 was formed in 7% overall RCY. In contrast, the direct nucleophilic substitution of a tosylated precursor of NS14490 with [18F]fluoride under equal conditions gave the [18F]NS14490 within 2 hours in 36% isolated RCY. The authors did not explore the cause of the low yield of N-[18F]fluoroethylation, but one reason may be the need for strong deprotonation of the indole hampering sufficient alkylation with [18F]FETs.
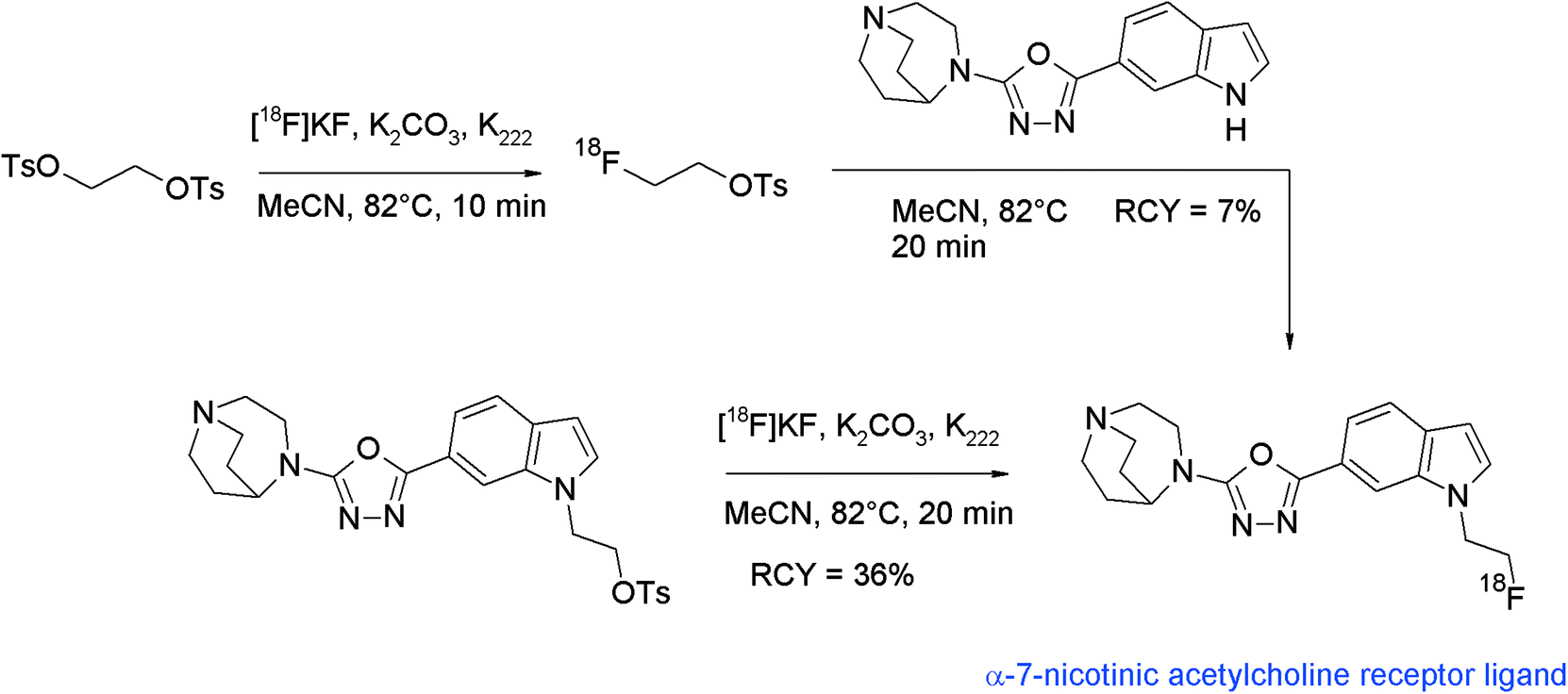 | ||
| Fig. 57 Synthesis of an N-[18F]fluoroethylated indole.97 | ||
Another example of direct and indirect radiofluorination of an indole was described by O'Shea et al. with the synthesis of a radioligand for the translocator protein (TSPO), which is an established target for the imaging of neuroinflammation with PET.98 For the indirect pathway, [18F]FETs was reacted with the indole in DMF in the presence of sodium hydride for 10 min at 100 °C (Fig. 58). After workup and semi-preparative HPLC purification, the [18F]fluoroethylated TSPO ligand was isolated in 4% overall RCY. In contrast, when the radiosynthesis was performed via direct nucleophilic substitution of the corresponding mesylate precursor for 10 min at 100 °C, the RCY of the final radiotracer was 18%.
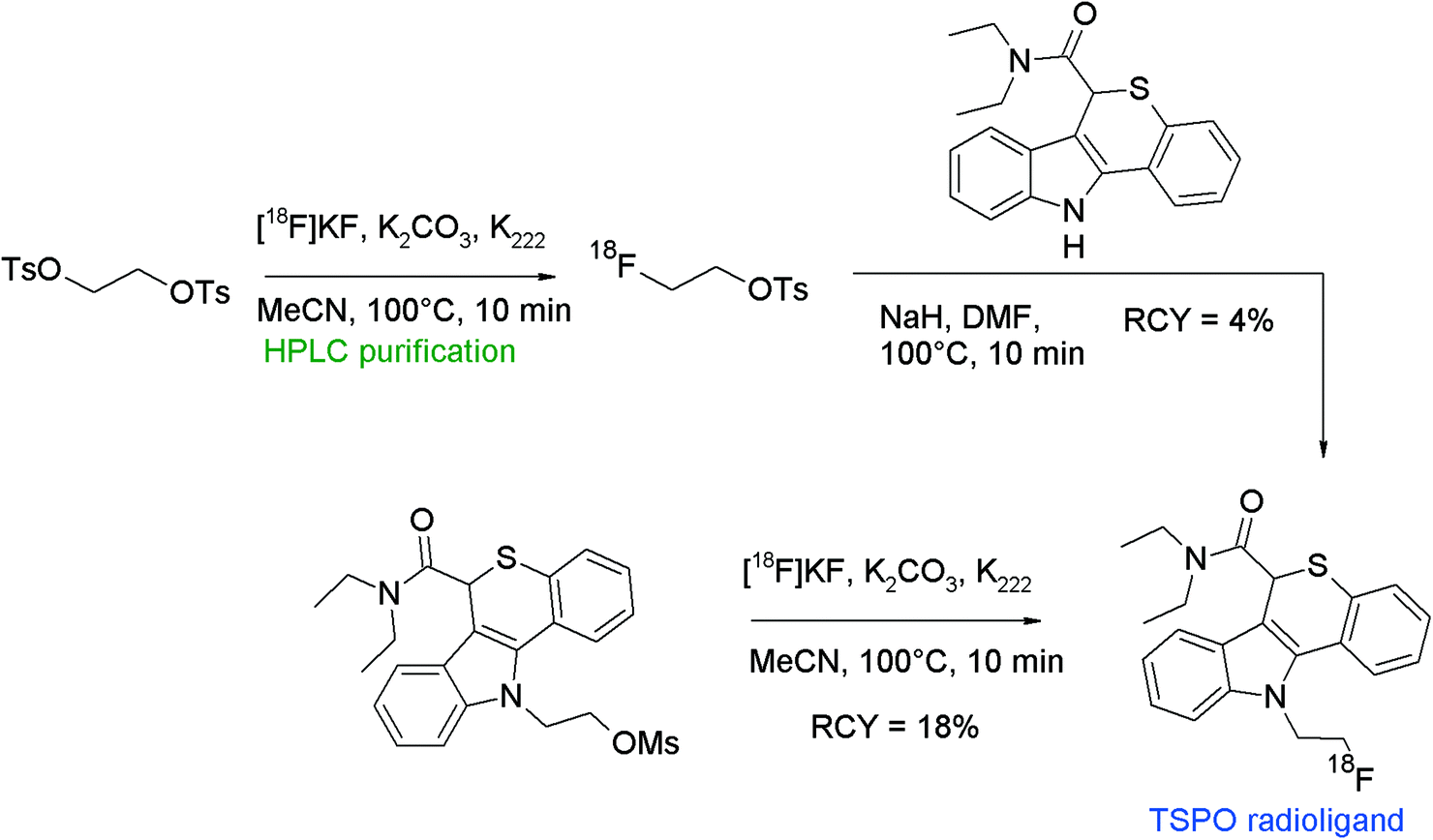 | ||
| Fig. 58 Synthesis of an N-[18F]fluoroethylated indole.98 | ||
For development of radiolabeled tolbutamide to visualize and quantify β-cell concentrations in the pancreas, the methyl group of tolbutamide was replaced with a hydroxyl functionality and O-[18F]fluoroethylation and direct [18F]fluorination were applied (Fig. 59).99 HPLC-purified [18F]FETs was used to react with the phenol precursor which was deprotonated beforehand with NaOH in DMF at 80 °C. [18F]Fluoroethylation was performed for 10 min at 120 °C and [18F]FE-tolbutamide was isolated after HPLC purification in 45% RCY, which represents an excellent overall yield for this indirect labeling procedure. Notably, the direct nucleophilic substitution by reacting a nitro precursor of tolbutamide with [18F]fluoride to produce [18F]fluoro-tolbutamide gave only a very low yield with a mixture of by-products.55 This is caused by the acidic character of the sulfonamide proton due to the unprotected precursor, hampering the nucleophilic substitution with [18F]fluoride.
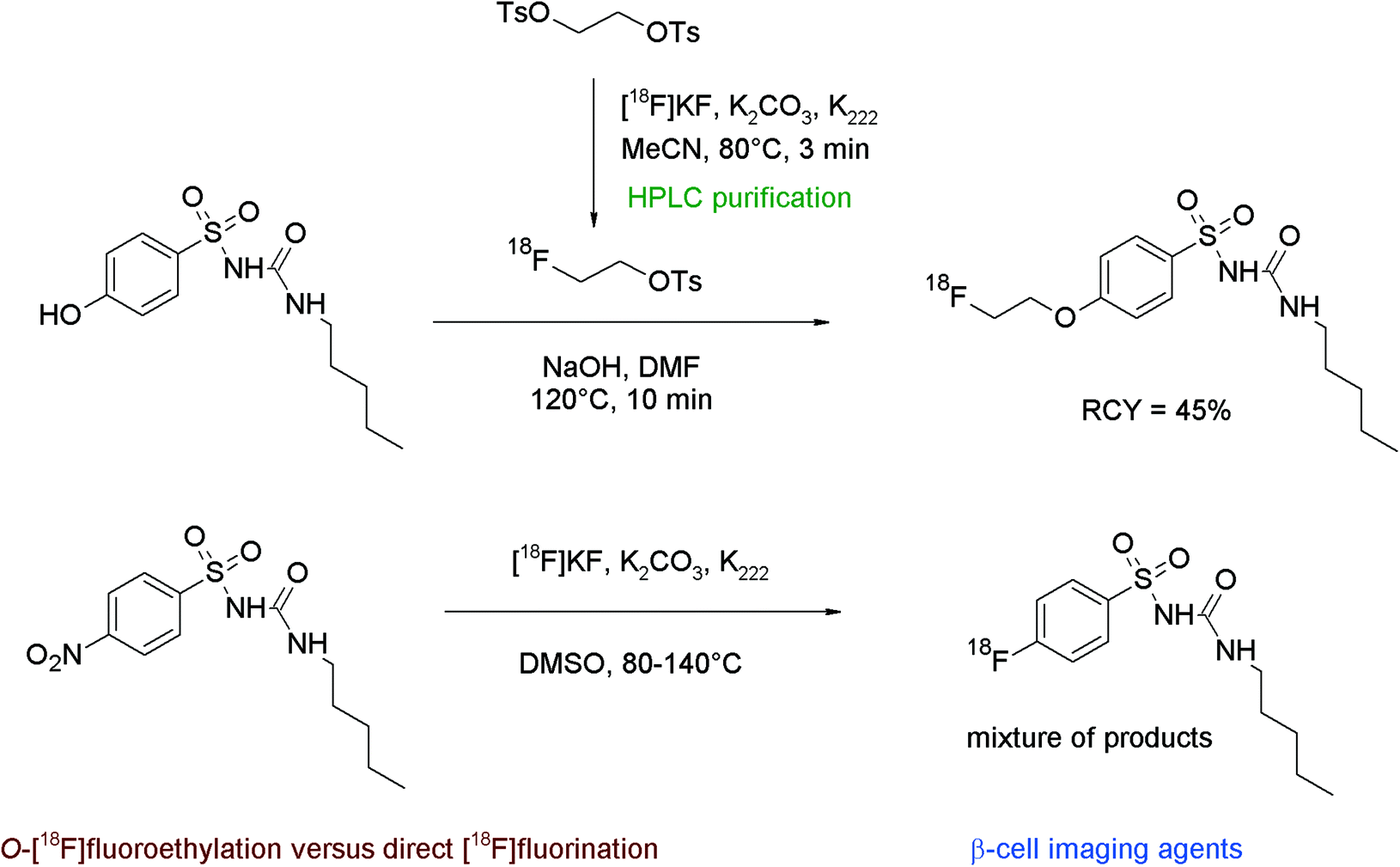 | ||
| Fig. 59 Radiosynthesis of O-[18F]fluoroethylated and [18F]labeled tolbutamide.55,99 | ||
ICI 89406 has been reported to be a β1-selective adenoreceptor antagonist and was chosen for the development of new radiotracers. Firstly, iodine-123 and iodine-125 radiolabeled derivatives of ICI 89406 were synthesized followed by a carbon-11 labeled radiotracer. However, due to the supposed in vivo de-iodination and the superior physical decay characteristics of 18F compared to 11C, Kopka et al. developed a [18F]fluoroethylated ICI 89406 derivative.100 Also here, two synthetic routes for 18F-radiolabeling, the direct and the indirect, via [18F]FETs were explored (Fig. 60). The first one started with the corresponding tosylate precursor of ICI 89406 and [18F]fluoride, but by this route, the radiotracer was obtained in poor RCY (3%) with inadequate specific activity caused by incomplete HPLC purification of the product. The indirect labeling approach based on the synthesis and purification of [18F]FETs followed by [18F]fluoroethylation which was performed by the reaction of [18F]FETs with the phenol precursor and NaOH in DMF and resulted in 38% product formation with a specific activity of 40 GBq μmol−1 (EOS). Finally, an overall RCY of 16% for [18F]FE-ICI 89406 was provided. These results have been reproduced later on by Radeke et al. who had performed [18F]fluoroethylation and [18F]fluoropropylation using 3- and 4-hydroxyl precursors of ICI 89406.101 In that publication, the average overall RCY for [18F]fluoroalkylation using the indirect method was 30%. Also, in this example, the direct labeling process using the corresponding tosylate precursors demonstrated a rather poor chemical efficiency (<2% radiochemical yield). For this compound class, the superiority of [18F]fluoroethylation with [18F]FETs towards direct [18F]fluorination was demonstrated.
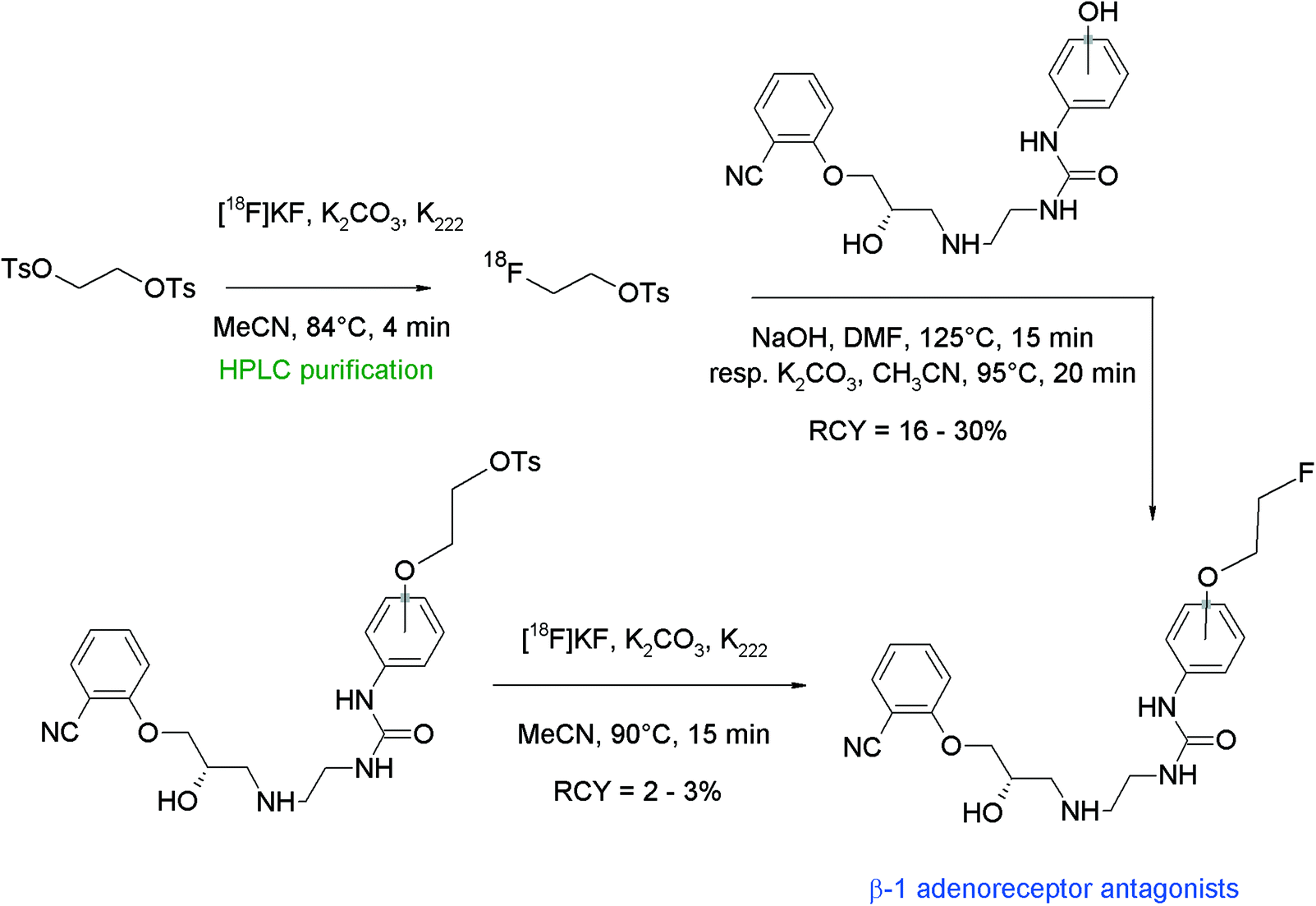 | ||
| Fig. 60 Radiosynthesis of O-[18F]fluoroethylated ICI 89406 derivative.100,101 | ||
Metomidate is a drug interacting selectively with the mitochondrial cytochrome P450 species in the adrenal cortex and has been radiolabeled with carbon-11 for adrenocortical imaging. Also, the 18F-labeled tracer analog 2-[18F]fluoroethyl-(R)-1-(1-phenylethyl)-1H-imidazole-5-carboxylate ([18F]FETO) has entered clinical studies. Accordingly, Långström et al. developed further 18F-labeled derivatives of metomidate, some of them via [18F]fluoroethylation with [18F]FETs.102 The 18F-radiolabeling was performed via two routes, the direct nucleophilic substitution with [18F]fluoride and the indirect approach via [18F]FETs. Here, we find another example for O-[18F]fluoroethyl ester formation starting from the carboxylic acid precursor. [18F]FETs was reacted without isolation with the appropriate ammonium salt precursor of the carboxylic acid in DMF at 150 °C for 15 min (Fig. 61). The corresponding 2-[18F]fluoroethyl esters of metomidate were provided in 18–29% overall RCY. In comparison, if the same tracer was produced by the direct [18F]-radiofluorination, a slightly higher RCY was obtained (27–37%). However, this benefit was diminished by the fact that the synthesis of the tosylated precursors afforded two additional steps with extremely low yields (2–3%) in some cases. In the end, it can be noted that [18F]fluoroethyl ester formation with [18F]FETs is a further effective approach for building [18F]FETO-derived radiotracers.
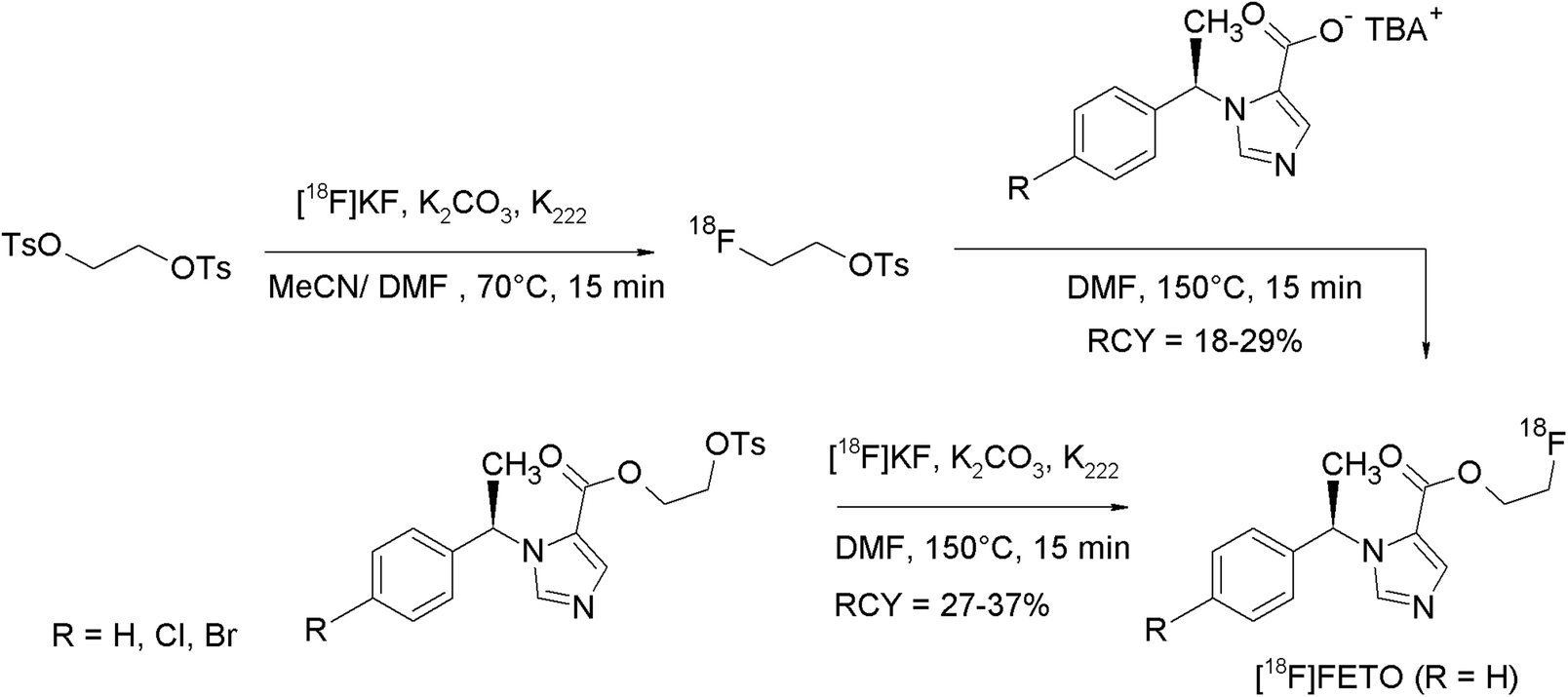 | ||
| Fig. 61 Radiosynthesis of O-[18F]fluoroethylated metomidate esters.102 | ||
N-[11C]Methylvorozole, a substituted benzotriazole, was found to be an interesting highly affine aromatase-binding radiotracer and, consistently, a corresponding [18F]fluoroethylated derivative was designed.103 Also, for this radiotracer, the 18F-radiolabeling was performed via direct and indirect approach (Fig. 62). For the latter, [18F]FETs and an additional ethoxy group-enlarged derivative 2-(2-[18F]fluoroethoxy)ethyl tosylate ([18F]FEETs) were used, both prepared from the appropriate bis-tosylated precursors by reaction with [18F]fluoride. Subsequently, the benzotriazole precursor in DMF was deprotonated with 1 M KOH, added to the previously formed [18F]FETs or [18F]FEETs, and heated at 150 °C for 15 min. The overall RCY of the [18F]fluoroalkylated vorozole derivatives was determined to be 11% for the reaction with [18F]FETs and 15% for that with [18F]FEETs. In contrast, the radiosynthesis via the direct labeling of the corresponding tosylated or brominated precursors provided significantly higher RCYs of 99% and 36%, respectively, but afforded two additional steps for the precursor synthesis with yields between 22–31%.
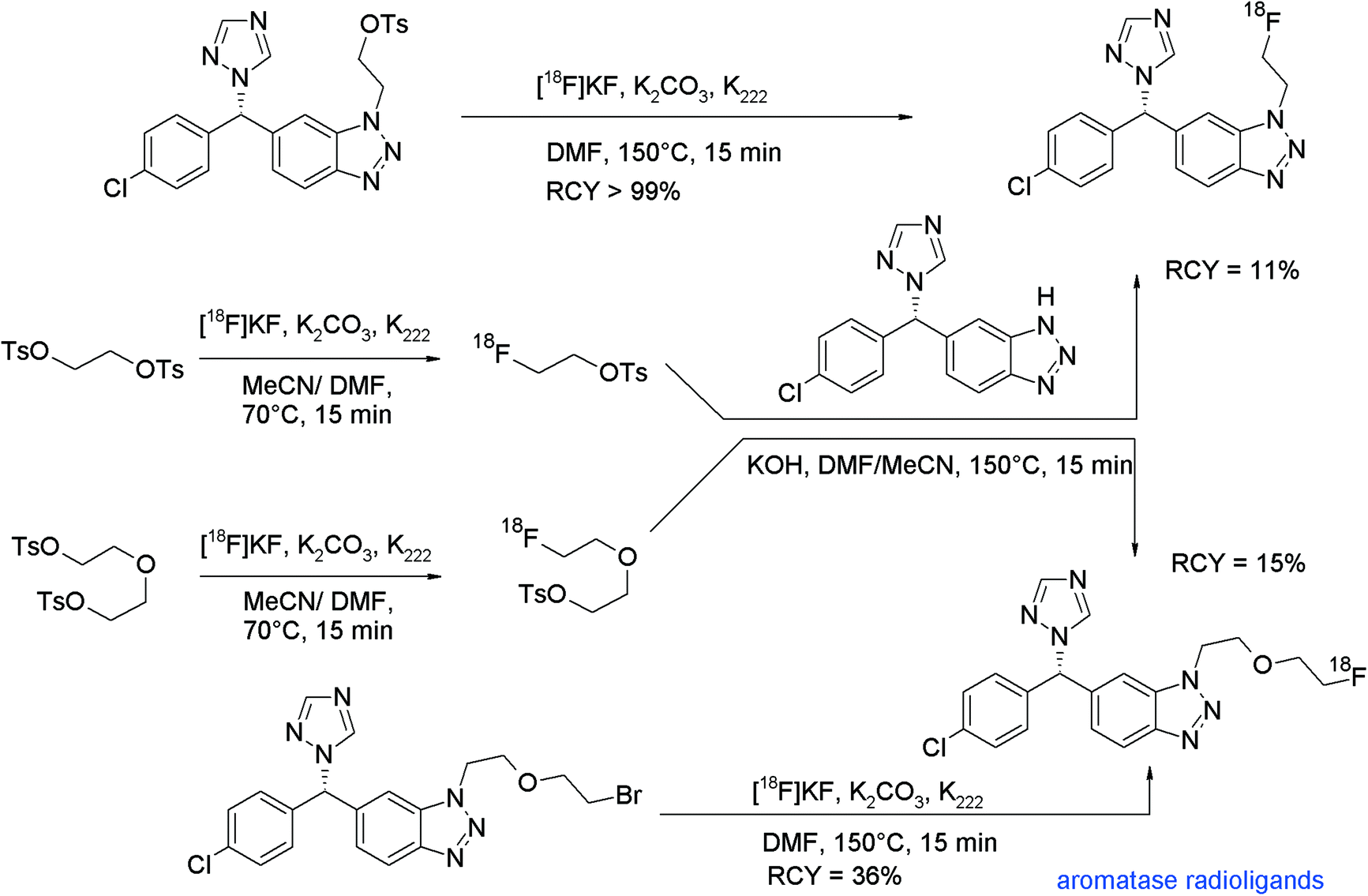 | ||
| Fig. 62 Radiosynthesis of O-[18F]fluoroalkylated vorozoles.103 | ||
The group of Ametamey evaluated the role of L-type amino acid transporters in tumor imaging with the tryptophan isomers L-5-[18F]FEHTP and DL-5-[18F]FEHTP.104 Both isomers were obtained by [18F]fluoroethylation of the corresponding tryptophan precursors with SPE-purified [18F]FETs in 15% overall RCY. Further evaluation of the tryptophan-based radiotracers was carried out recently by using a series of 18F-radiolabeled DL-tryptophan derivatives displaying the [18F]fluoroethylation either at position 4, 6, or 7 of the indole core (Fig. 63).105 [18F]FETs was used as the [18F]fluoroethylating agent, however only with 7-hydroxytryptophan a pure radiotracer was formed. The reaction of 4-hydroxy- and also 6-hydroxy-tryptophan with [18F]FETs resulted in the formation of a more polar radiolabeled by-product, most probably attributed to [18F]-fluoroalkylation at the N-1 position of the indole. To circumvent this problem, the authors performed the direct nucleophilic substitution of the appropriate BOC-protected mesylate precursors of 4- and 6- hydroxyethyl-substituted tryptophan with [18F]fluoride and obtained 4-[18F]FEHTP and 6-[18F]FEHTP in 13–18% RCY.
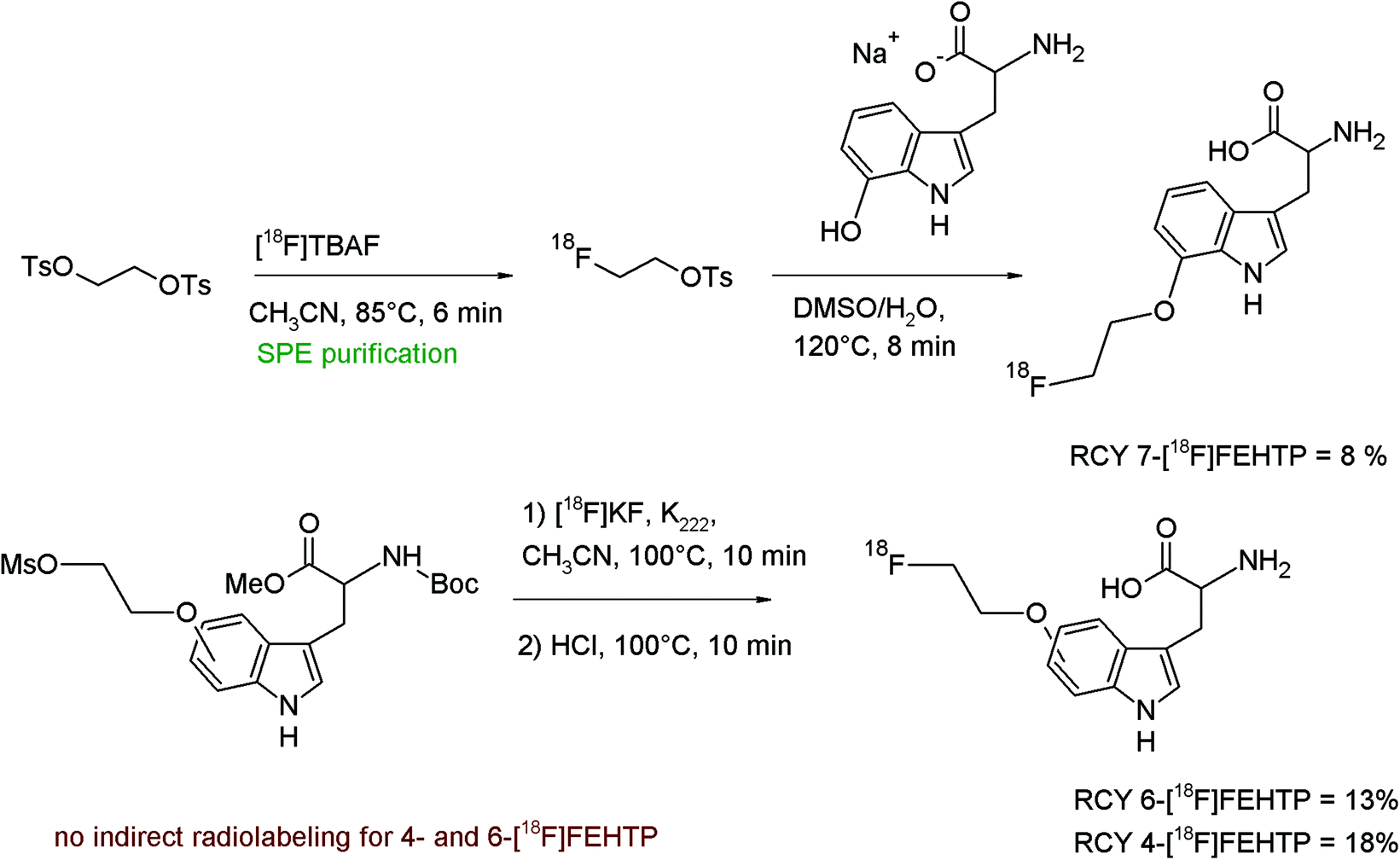 | ||
| Fig. 63 Radiosynthesis of 4-, 6- and 7-O-[18F]fluoroethylated tryptophans.105 | ||
Sulfur- and phosphoric acid-[18F]fluoroethylation and reactions with [18F]FETs in aqueous medium
In contrast to the often used labeling reaction on the oxygen and nitrogen atom, [18F]fluoroethylation is rarely applied to sulfur or phosphorus, probably due to the fact that compounds having free thiol and phosphane groups may represent less suitable precursors for radiolabeling. Nevertheless, there exist a number of these kinds of [18]fluoroethylations as will be demonstrated in the following examples.S-[18F]Fluoroethyl thioethers
Tang et al. introduced S-(2-[18F]fluoroethyl)-L-methionine ([18F]FEMET), a new amino acid analogue of L-[11C]methionine, to overcome the limitations of the short half-life of the carbon-11 labeled radiotracer.106 [18F]FETs was produced on a functionalized polystyrene SAX resin by direct nucleophilic substitution of [18F]fluoride with 1,2-ethylene glycol-bis-tosylate followed by the reaction of [18F]FETs with L-homocysteine thiolactone hydrochloride in the presence of sodium hydroxide and DMSO (Fig. 64). By using a fully automated synthesizing device, the new [18F]fluoroethylated amino acid [18F]FEMET was provided in 10% overall RCY.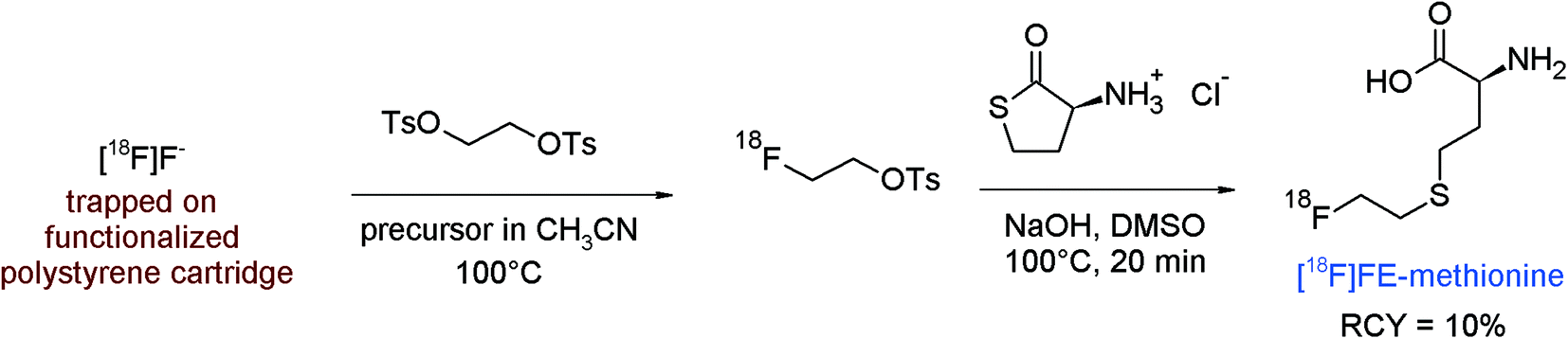 | ||
| Fig. 64 Radiosynthesis of S-(2-[18F]fluoroethyl)-L-methionine.106 | ||
Muscarinic receptors are interesting targets for PET because the receptor density is diminished in patients with Alzheimer's disease. In this context, radiolabeled selective muscarinic receptor ligands like [18F]fluoropropyl- and [18F]fluoroethyl-substituted 1,2,5-thiadiazol-4-yl derivatives such as [18F]FE-TZTP have been synthesized by Kiesewetter et al. by [18F]fluoroethylation of the free thiol group of a tetrahydro-1-methylpiperidine thiadiazole (Fig. 65).107 The radiosynthesis was performed as follows: after the preparation of [18F]FETs, it was transferred without purification to the thiol precursor in DMF, which was deprotonated with 8 M KOH; after heating at 100 °C for 5 min and semi-preparative HPLC purification, [18F]FE-TZTP was provided within a synthesis time of 80 min in 22% overall RCY.
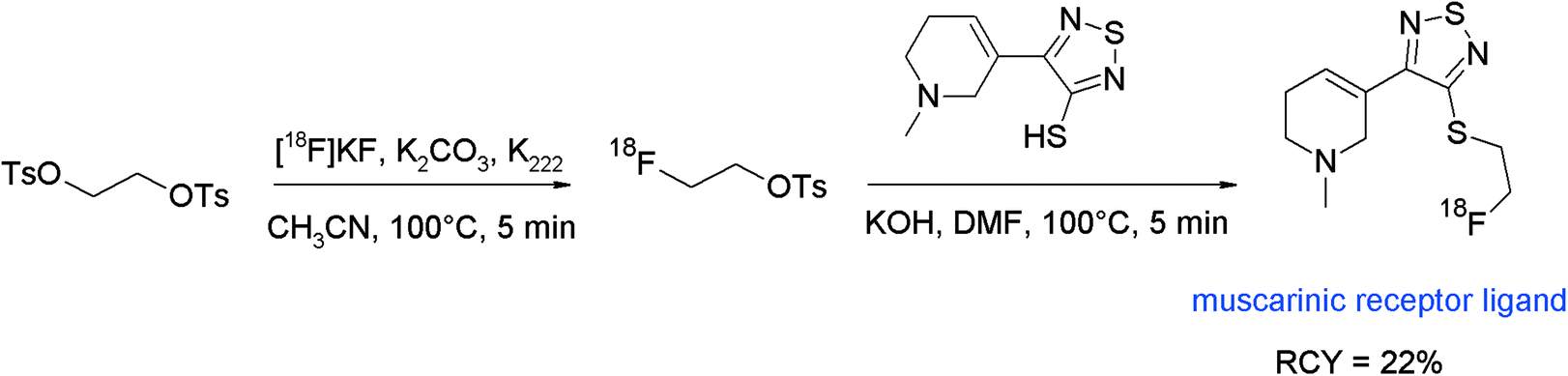 | ||
| Fig. 65 Synthesis of S-[18F]fluoroethylated 1,2,5-thiadiazol-4-yl derivative.107 | ||
Furthermore, diaryl methylguanidine derivatives were found to have high affinity and selectivity to the NMDA receptor and be suitable for radiolabeling through [18F]fluoroalkylation by Robins et al.108 [18F]FETs was used without purification to react with the thiol-substituted guanidine precursor in the presence of Cs2CO3 in acetonitrile at 110 °C (Fig. 66). The low overall yield of the S-[18F]fluoroethylated diaryl guanidine of only 4–9% RCY obtained after HPLC purification was primarily caused by the low yield of the intermediate [18F]FETs that was obtained, according to the authors, in no more than 10–17% RCY. Surprisingly, the overall procedure took the authors 3–4 hours to accomplish the reaction and purification of the radiotracer.
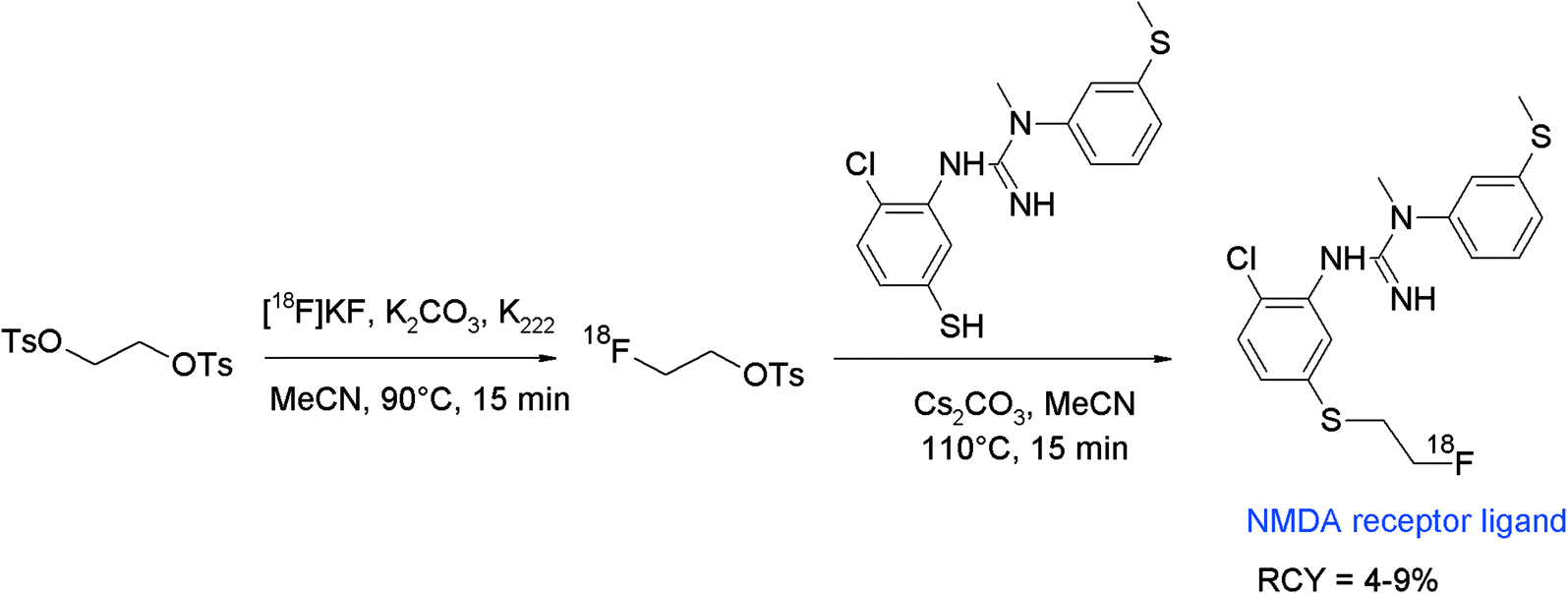 | ||
| Fig. 66 S-[18F]Fluoroethylation of a diaryl methylguanidine.108 | ||
[18F]Fluoroethylated phosphorus compounds
Antisense oligonucleotides (ODNs) are molecules which have typically 10–25 nucleotides and show potential therapeutic and diagnostic capabilities because hybridization of antisense ODNs with target mRNA can interrupt the translation process and inhibit the expression of a specific gene. Radiolabeled antisense ODNs would represent useful tools for imaging the status of hybridization; likewise, the biodistribution and pharmacokinetics of ODNs could be studied in vivo. De Vries et al. performed a comparative study for the radiolabeling of adenosine 5′-O-thiomonophosphate as a model compound using six 18F-labeled alkylating agents, among them N-(4-[18F]fluorobenzyl)-2-bromoacetamide, α-bromo-α′-[18F]fluoro-m-xylene and [18F]FETs (Fig. 67).109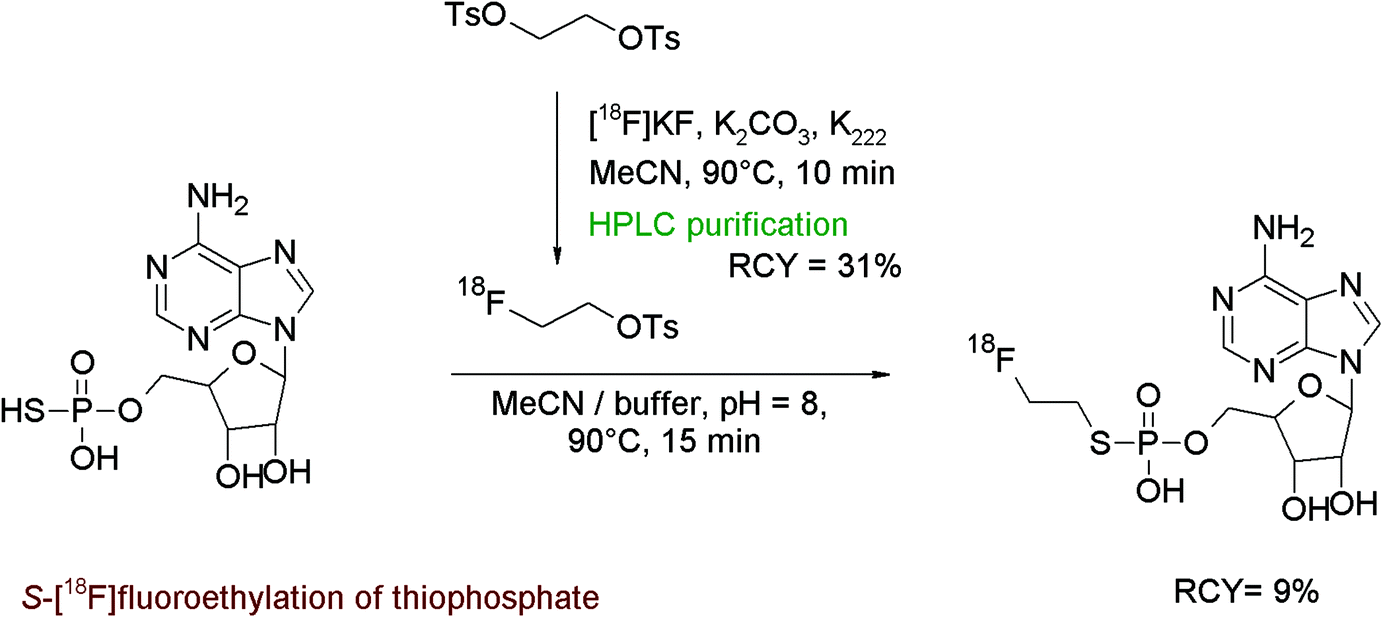 | ||
| Fig. 67 S-[18F]Fluoroethylation of adenosine 5′-O-thiomonophosphate.109 | ||
It should be noted that this is the only example of S-alkylation of a thiosphosphate moiety with [18F]FETs. The S-[18F]fluoroalkylation with [18F]FETs provided the radiolabeled nucleotide in 9% RCY. In contrast, the non-radioactive fluoroethylation for obtaining the reference compound yielded 44% of the product. To provide an explanation for the low RCY, the authors investigated the by-products during the radiofluorination. They observed an unknown volatile side product and postulated the formation of [18F]vinyl fluoride from [18F]FETs as a result of β-elimination.
Special organophosphorus esters are known to be highly toxic to the nervous system; their action is generally attributed to the inhibition of acetylcholinesterase (AChE) which is responsible for the hydrolysis of the neurotransmitter acetylcholine. To obtain radiolabeled inhibitors of AChE, studies were performed to radiolabel organic phosphorus compounds with fluorine-18. For this purpose, the 4-nitrophenyl ester of methylphosphonic acid was [18F]fluoroethylated with [18F]FETs (Fig. 68).110 In this sole example of [18F]fluoroethylation of a phosphorus-bound hydroxyl group, [18F]FETs was reacted with the organic phosphorus precursor in acetonitrile in the presence of Cs2CO3 and activated molsieves under microwave conditions to provide the radiotracer within 90 min in 6.5% overall RCY.
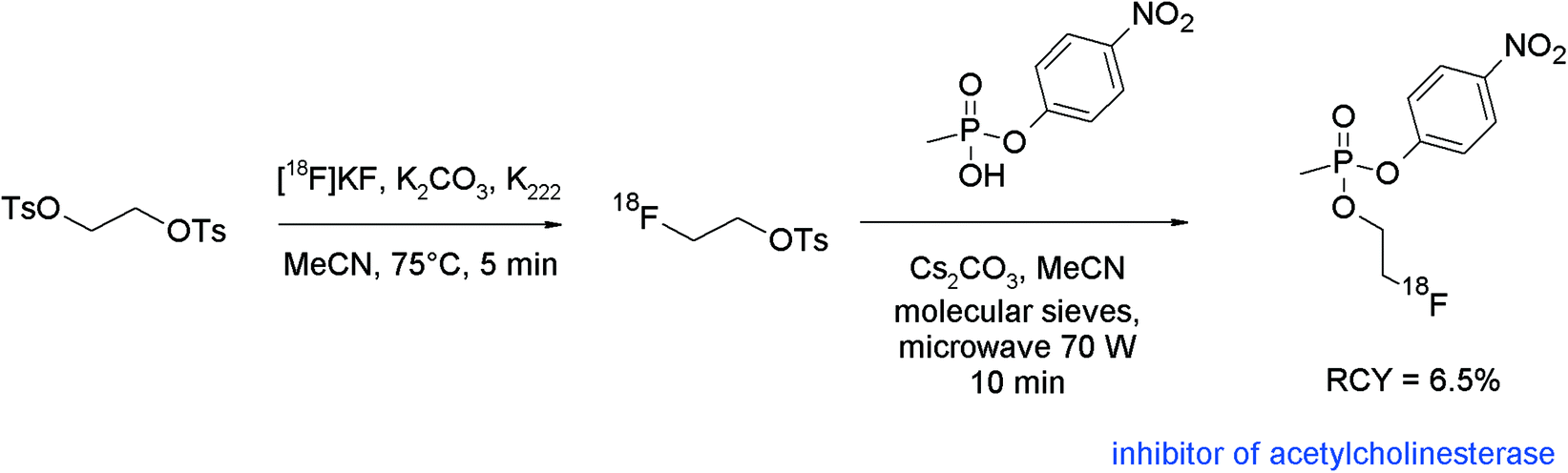 | ||
| Fig. 68 [18F]Fluoroethylation of methylphosphonic acid-4-nitrophenyl ester.110 | ||
[18F]Fluoroethylation in aqueous medium
Generally, all reactions with [18F]FETs were performed under non-aqueous conditions to avoid hydrolysis of alkylating agents, however two papers have been recently published demonstrating the practicality of [18F]fluoroethylation in aqueous medium.Quantum dots are semiconductor crystals consisting of a semiconductor core and an outer surface that can be easily functionalized to introduce functionalities having an affinity to specific biological targets. The application of quantum dot-based imaging agents in humans is hampered by missing data about their behavior in vivo, therefore they have been radiolabeled with carbon-11 and fluorine-18. The [18F]fluoroethylation of quantum dots was performed by Patt et al. in DMSO/borate buffer, and the reaction mixture was analyzed by size exclusion chromatography to determine the radiolabeling yield.111 Even though the DMSO/buffer system is not ideal for the radiolabeling agent [18F]FETs, [18F]fluoroethylated quantum dots could be obtained with up to 5% overall RCY. Notably, amino-functionalized quantum dots gave a slightly better yield compared to their carboxyl-functionalized counterparts. In comparison, 11C-radiolabeling of quantum dots with [11C]methyl iodide delivered 35–45% RCY.111
For diagnosis of prostate cancer, the most promising target is the prostate-specific membrane antigen (PSMA) that is highly expressed in this disease. Besides 68Ga-labeled peptides, other small amino acid conjugates based on the glutamate–urea–lysine moiety with high affinity to PSMA have been developed and radiolabeled with technetium-99m and fluorine-18.112 Recently, Al-Momani et al. synthesized an alternative small 18F-labeled peptide, [18F]FE-Tyr-urea-Glu ([18F]FETUG) as a PMSA-affine ligand using [18F]FETs as the labeling precursor (Fig. 69).113 This is an innovative approach since usually for labeling of peptides, prosthetic groups like 6-[18F]fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester ([18F]FPyTFP) or N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) are used. Briefly, HPLC-purified [18F]FETs was eluted from the C18 cartridge with 80% aqueous acetonitrile and added to the peptide TUG dissolved in 15 μL of an aqueous base, and the reaction was performed for 15 min at 80 °C. The authors found out that the labeling efficiency as well the selectivity of radiolabeling strongly depended on the choice and concentration of the supporting base. By using a peptide/NaHCO3 ratio of 1/20, a radiolabeling yield of 40% was achieved, reducing the amount of the base to 10 equivalents resulted in a RCY of only 7% of [18F]FETUG together with a radiolabeled by-product that was supposed to be a [18F]-fluoroethyl ester derivative. Finally, by application of 10 M NaOH in a 1/18 peptide/base ratio, the RCY of [18F]FETUG was increased to 77%. This efficient radiolabeling of TUG is remarkable since it stands for successful [18F]-fluoroethylation with [18F]FETs in aqueous milieu and is, to the best of our knowledge, the first example of peptide radiolabeling using this radiolabeling agent.
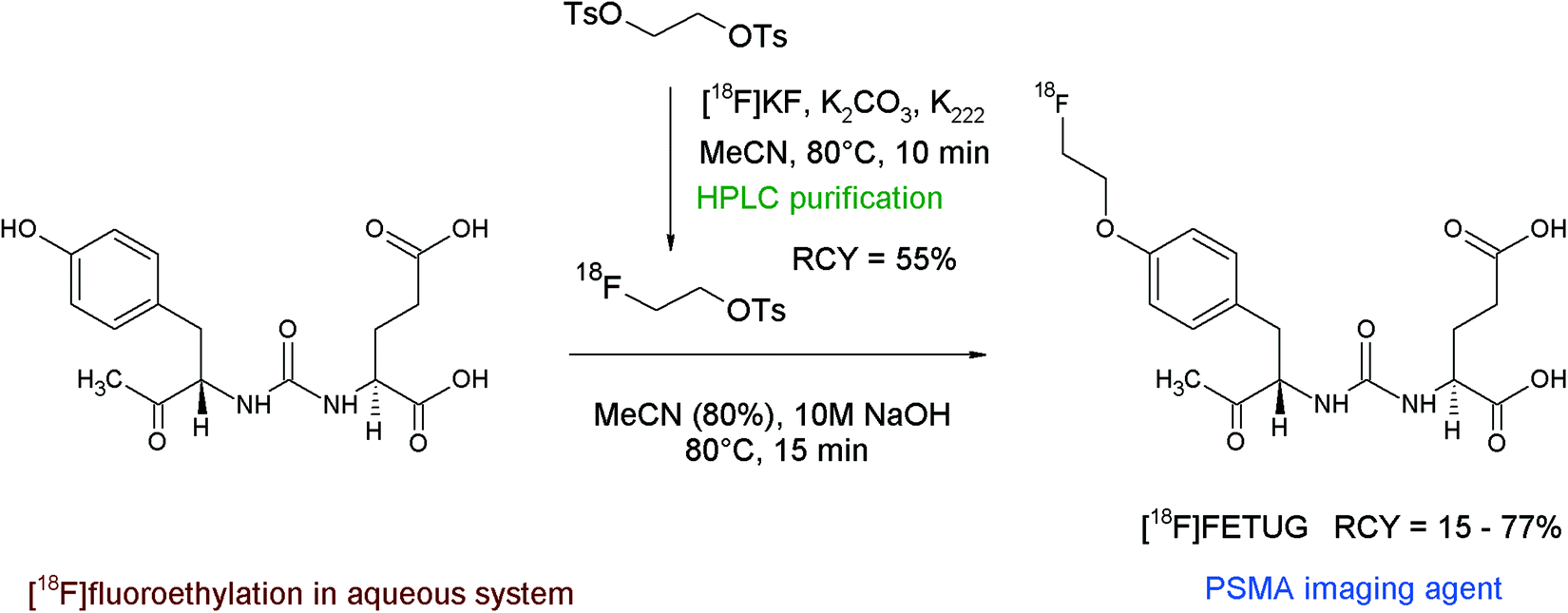 | ||
| Fig. 69 O-[18F]Fluoroethylation of a small peptide.113 | ||
[18F]FETs as an intermediate for building blocks
[18F]FETs is known as a [18F]fluoroalkylating reagent and is used for integration of [18F]fluoride into molecules of pharmaceutical interest. However, in a few cases, [18F]FETs was used as an intermediate to form other building blocks, for example, in the formation of [18F]fluoroacetaldehyde as a reductive 18F-alkylating agent.114 In selected cases, [18F]fluoroacetaldehyde is expected to be superior to [18F]FETs because it enables labeling in aqueous solution and thus should be advantageous for the radiolabeling of peptides, proteins and antibodies. Formation of [18F]fluoroacetaldehyde was achieved by oxidation of [18F]FETs in DMSO in the presence of K2CO3 according to Kornblum conditions (Fig. 70). Briefly, [18F]FETs was synthesized in acetonitrile, the solvent was removed in vacuum, and DMSO was added to the intermediate. [18F]Fluoroacetaldehyde was then generated by heating the mixture to 150 °C and separated by distillation under a stream of nitrogen into a vial containing water. The RCY of [18F]fluoroacetaldehyde from the starting compound [18F]FETs was 31–37%. As a proof-of-principle, the reaction of [18F]fluoroacetaldehyde with two model compounds was investigated; the formation of the corresponding 2,4-dinitrophenylhydrazone and the reductive alkylation with NaBH3CN to form N-benzyl-2-[18F]fluoroethanamine.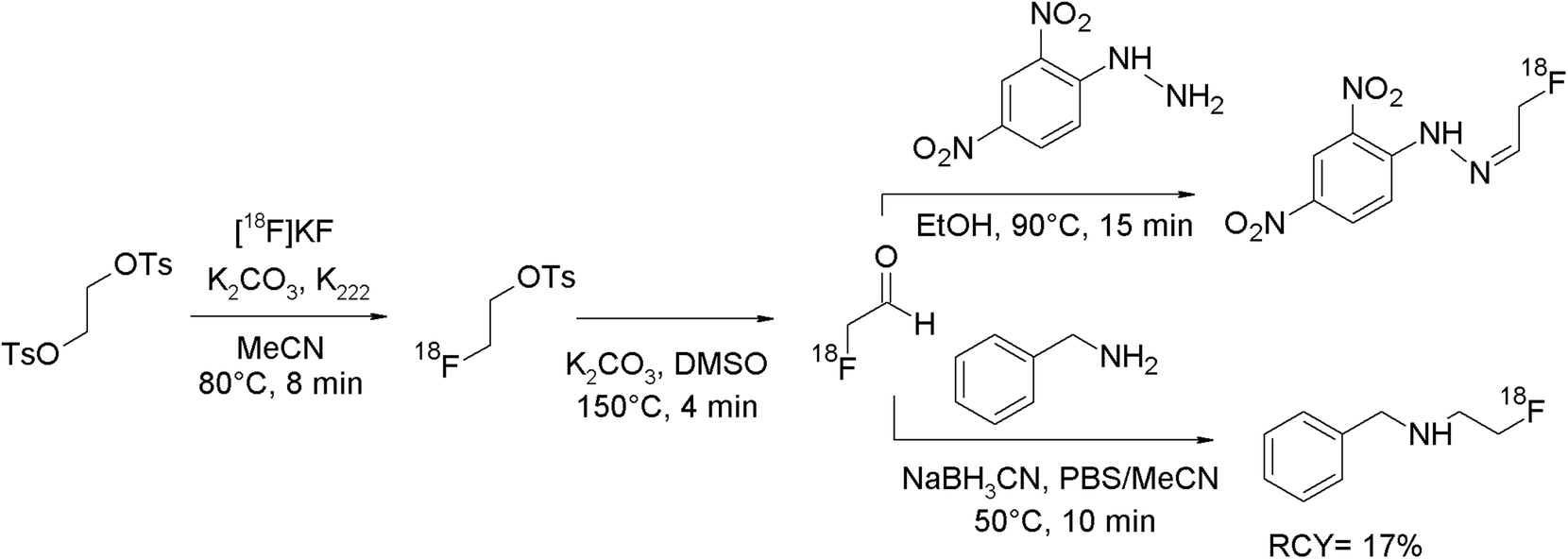 | ||
| Fig. 70 Radiosynthesis of [18F]fluoroacetaldehyde from [18F]FETs and reaction with model compounds.114 | ||
Summary
[18F]Fluoroalkylation holds growing interest in PET chemistry due to the easy introduction of a 18F-radiolabel into phenolic, thiophenolic, carboxylic, amine and amide functionalities and the supposed minimal modification of the pharmacological properties of the lead compound. [18F]FETs is one of the mostly used [18F]fluoroalkylating agents because it is simple to prepare, almost stable and provides high reactivity together with – to some extent – chemoselectivity.Since the introduction of [18F]FETs in 1987,14 much work was performed to optimize the manufacturing and purification and to improve the overall radiolabeling yields. An important step was the finding that by addition of alkali iodides, the yield of [18F]fluoroethylation could be significantly increased10 as well as the fact that the reactivity of [18F]FETs could be further improved by introduction of strong electron-withdrawing substituents such as e.g. in 2-[18F]fluoroethyl-3,4-dibromobenzenesulfonate ([18F]FEBr2Bs) and 2-[18F]fluoroethyl-4-nitrobenzenesulfonate ([18F]FENs).19,20 Acknowledging that application of HPLC-purified [18F]FETs had a significant impact on the labeling yield, chemical and radiochemical purity of the final radiotracer13,44 has triggered the development of innovative automated methods for production and purification of [18F]FETs and [18F]fluoroethylation.12,46,48,71
[18F]FETs has been approved as a building block in the synthesis of clinically relevant radiotracers such as 2-[18F]fluoroethylcholine ([18F]FEC)67,67a,b,68,69 and O-(2-[18F]fluoroethyl)tyrosine ([18F]FET)28–31 and equally in the [18F]fluoroethylation of other amino acids like methionine and tryptophan.33,73,104–106 In radiotracer building, the reaction with [18F]FETs is indispensable as has been demonstrated for a multitude of O- and N-[18F]fluoroethylations, the radiolabeling of thiols106,107,109 and the formation of [18F]fluoroethyl-carboxylic esters,63–66,102 -amides89 and -phosphorus esters.109,110
By comparison of [18F]FETs with other [18F]fluoroalkylating reagents such as 2-[18F]fluoroethyl bromide, in majority, [18F]FEBr showed higher RCY but this benefit is diminished by the need of additional efforts for purification (distillation) of [18F]FEBr.13,83,84 Generally, the application of HPLC-purified [18F]FETs was found to be advantageous44 but it was also demonstrated that by using SPE-purified [18F]FETs, high RCYs and sufficient specific activities could be achieved.12,25,27,74,77,85
In terms of chemoselectivity, phenolic groups are preferred to react with [18F]FETs, and in molecules having both amino and phenol functionalities, [18F]fluoroethylation will normally occur on the phenol.28,46,47,100 If the target compound shows several phenol groups, regioselective [18F]fluoroethylation can be achieved by the character of the added base,58 otherwise a selective protective group may be used.35 In existence of concurrent amino and carboxylic groups, [18F]fluoroethylation is preferred on the amine;18 when amide and amine groups are present, there is no clear chemoselectivity observed.86,87,89
In a number of protocols besides the [18F]fluoroethylation with [18F]FETs (indirect approach), the direct radiolabeling of a corresponding mesylated or tosylated precursor with [18F]fluoride has been performed in a parallel approach. By comparison of these two synthesis pathways, it is shown at first glance that in several protocols, direct [18F]fluorination with [18F]fluoride provided more than doubled overall yields compared to [18F]fluoroethylation93–95,97,98,103 suggesting the direct method as the superior one. However, in a closer look, it turned out that often the RCY of [18F]fluoroethylation was diminished by the use of e.g. non-purified [18F]FETs or the insufficient production of [18F]FETs. For two examples, the total RCY for direct and indirect approach was almost comparable,96,102 whereas in a notable set of protocols, indirect [18F]fluoroethylation with [18F]FETs gave a significantly higher RCY in comparison to direct radiofluorination.36,47,55,79,87,91,101 The reason is that the synthesis of the corresponding tosylated/mesylated precursors has failed79,86 or afforded extremely low yields (2–3%).92,102 Under these circumstances, [18F]fluoroethylation is the sole method to provide the desired radiotracer. Whether [18F]fluoroethylation may be performed either by direct labeling or by introduction of the radiolabeled precursor [18F]FETs (indirect approach) is dependent on the chemical behavior and reactivity of a particular precursor compound. A prognosis about the optimal reaction route may be very difficult in most cases. For preventing the use of protective groups and avoiding additional precursor synthesis, the indirect way may be favorable.
A further benefit of [18F]FETs as one of the preferred labeling agents is that it is ideal for the easy transfer of promising 11C-labeled radiotracers into their 18F-labeled counterparts as shown for several examples.13,34,39,44,46,48,49,53,54,82,100,102 One main concern by replacing the [11C]methyl group with a [18F]fluoroethyl labeling unit is the preservation of the physical and chemical properties of the radiotracer, i.e. will the affinity to the designated target be substantially altered? To answer this question, we have extracted, for a series of radiotracers, the in vitro data assessed for their methyl and the corresponding fluoroethyl-substituted derivatives. The results are displayed in Table 1. In most cases, there is only moderate alteration in the in vitro behavior by substitution of methyl with fluoroethyl; there are even examples where the receptor affinity is improved by the introduction of a fluoroethyl group.
| –CH3 substituent | –CH2CH2F substituent | |
|---|---|---|
| GABAA ligand102 | MTO: IC50 = 9 μM115 | FETO: IC50 = 4.8 μM115 |
| GABAA ligand39 | K i = 0.29 nM116 | K i = 0.52 nM39 |
| Dopamine D3 antagonist34 | K i = 0.32 nM34 | K i = 0.12 nM34 |
| 5-HT1A SR antagonist44 | K i = 1.1 nM117 | K i = 0.1 nM44 |
| Sigma ligand48 | IC50 σ1 = 17.4 nM48 | IC50 σ1 = 6.48 nM48 |
| IC50 σ2 = 1784 nM | IC50 σ2 = 2.11 nM | |
| Sigma ligand49 | IC50 σ1 = 33 nM49 | IC50 σ1 = 3.1 nM49 |
| IC50 σ2 = 9.5 nM | IC50 σ2 = 6.8 nM | |
| MAO inhibitor54 | K i = 2.5 nM54 | K i = 0.54 nM54 |
| AA2 receptor ligand53 | K i = 4.1 nM118 | K i = 12.4 nM118 |
| β1 AR antagonist100 | K i = 0.067/0.288 nM119 | K i = 0.049/0.297 nM100 |
| β-Amyloid plaque imaging46 | K i = 4.9 nM120 | K i = 0.17 nM46 |
| β-Amyloid plaque imaging82 | K i = 15 nM82 | K i = 27 nM82 |
In summary, with respect to its broad application in radiolabeling molecules with pharmaceutical relevance, [18F]FETs is an absolutely essential building block in 18F-radiochemistry. It enables rapid radiolabeling and investigation of a potential radiotracer without increased organic synthetic effort for precursor synthesis. That [18F]FETs can also be used for peptide 18F-radiolabeling radiolabeling in aqueous medium was recently successfully demonstrated, a result which opens the door to additional applications of this compound.113 In the future and due to the fact that a number of reliable protocols for the – preferably automated – synthesis and purification of [18F]FETs nowadays are available, [18F]fluoroethylation with [18F]FETs will stay as a versatile tool for building 18F-based radiotracers.
References
- K. Ishii, Ann. Nucl. Med., 2002, 16, 515–525 CrossRef.
- A. Alavi, J. W. Kung and H. Zhuang, Semin. Nucl. Med., 2002, 32, 2–5 CrossRef PubMed.
- J. M. Tarkin, F. R. Joshi and J. H. Rudd, Nat. Rev. Cardiol., 2014, 11, 443–457 CrossRef CAS PubMed.
- A. Gallamini, C. Zwarthoed and A. Borra, Cancers, 2014, 6, 1821–1889 CrossRef PubMed.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- S. Banister, D. Roeda, F. Dollè and M. Kassiou, Curr. Radiopharm., 2010, 3, 68–80 CrossRef CAS.
- M. Pretze, D. Pietzsch and C. Mamat, Molecules, 2013, 18, 8618–8665 CrossRef CAS PubMed.
- C. Wängler, R. Schirrmacher, P. Bartenstein and B. Wängler, Curr. Med. Chem., 2010, 17, 1092–1116 CrossRef.
- D. Franck, T. Kniess, J. Steinbach, S. Zitzmann-Kolbe, M. Friebe, L. M. Dinkelborg and K. Graham, Bioorg. Med. Chem., 2013, 21, 643–652 CrossRef CAS PubMed.
- M. R. Zhang and K. Suzuki, Curr. Top. Med. Chem., 2007, 7, 1817–1828 CrossRef CAS.
- D. Block, H. H. Coenen and G. Stöcklin, J. Labelled Compd. Radiopharm., 1987, 25, 201–216 CrossRef PubMed.
- B. W. Schoultz, B. J. Reed, J. Marton, F. Willoch and G. Henriksen, Molecules, 2013, 18, 7271–7278 CrossRef CAS PubMed.
- W. Wadsak, L. K. Mien, D. Ettlinger, H. Eidherr, D. Haeusler, K. M. Sindelar, B. K. Keppler, R. Dudczak, K. Kletter and M. Mitterhauser, Nucl. Med. Biol., 2007, 34, 1019–1028 CrossRef CAS PubMed.
- D. Block, H. H. Coenen and G. Stöcklin, J. Labelled Compd. Radiopharm., 1987, 24, 1029–1042 CrossRef CAS PubMed.
- D. Block, H. H. Coenen and G. Stöcklin, J. Labelled Compd. Radiopharm., 1988, 25, 201–216 CrossRef CAS PubMed.
- A. Bauman, M. Piel, R. Schirrmacher and F. Rösch, Tetrahedron Lett., 2003, 44, 9165–9167 CrossRef CAS PubMed.
- S. Stone-Elander, N. Elander, J. O. Thorell and A. Fredrikson, PET Chemistry, The Driving Force in Molecular Imaging, Ernst Schering Research Foundation, Springer, 2007, pp. 243–269 Search PubMed.
- S. Y. Lu, F. T. Chin, J. A. McCarron and V. W. Pike, J. Labelled Compd. Radiopharm., 2004, 47, 289–297 CrossRef CAS PubMed.
- J. L. Musachio, J. Shah and V. W. Pike, J. Labelled Compd. Radiopharm., 2005, 48, 735–747 CrossRef CAS PubMed.
- R. Löser, R. Bergmann, M. Fritzler, B. Mosch, L. Dombrowski, M. Kuchar, J. Steinbach, M. Gütschow and J. Pietzsch, ChemMedChem, 2013, 8, 1330–1344 CrossRef PubMed.
- C. F. Lemaire, J. J. Aerts, S. Voccia, L. C. Libert, F. Mercier, D. Goblet, A. R. Plenevaux and A. J. Luxen, Angew. Chem., 2010, 122, 3229–3232 CrossRef PubMed.
- G. Pascali, G. Mazzone, G. Saccomanni, C. Manera and P. A. Salvadori, Nucl. Med. Biol., 2010, 37, 547–555 CrossRef CAS PubMed.
- H. J. Wester, F. Willoch, T. R. Tölle, F. Munz, M. Herz, I. Øye, J. Schadrack, M. Schwaiger and P. Bartenstein, J. Nucl. Med., 2000, 41, 1279–1286 CAS.
- C. Mueller, A. Klega, H. G. Buchholz, R. Rolke, W. Magerl, R. Schirrmacher, E. Schirrmacher, F. Birklein, R. D. Treede and M. Schreckenberger, NeuroImage, 2010, 49, 731–737 CrossRef PubMed.
- B. W. Schoultz, T. Hjørnevik, B. J. Reed, J. Marton, C. S. Coello, F. Willoch and G. Henriksen, J. Med. Chem., 2014, 57, 5464–5469 CrossRef CAS PubMed.
- M. Piel, I. Vernaleken and F. Rösch, J. Med. Chem., 2014, 57, 9232–9258 CrossRef CAS PubMed.
- C. Juhász, S. Dwivedi, D. O. Kamson, S. K. Michelhaugh and S. Mittal, Mol. Imaging, 2014, 13, 1535–3508 Search PubMed.
- H. J. Wester, M. Herz, W. Weber, P. Heiss, R. Senekowitsch-Scmidtke, M. Schwaiger and G. Stöcklin, J. Nucl. Med., 1999, 40, 205–212 CAS.
- H. Tsukada, K. Sato, D. Fukumoto and T. Kakiuchi, Eur. J. Nucl. Med. Mol. Imaging, 2006, 33, 1017–1024 CrossRef CAS PubMed.
- G. Tang, X. Tang, M. Wang, L. Luo, M. Gan and Z. Huan, J. Labelled Compd. Radiopharm., 2003, 46, 661–668 CrossRef CAS PubMed.
- D. Mueller, I. Klette, F. Kalb and R. P. Baum, Nucl. Med. Biol., 2011, 38, 653–658 CrossRef CAS PubMed.
- K. Hamacher and H. H. Coenen, Appl. Radiat. Isot., 2002, 57, 853–856 CrossRef CAS.
- R. Li, S. C. Wu, S. C. Wang, Z. Fu, Y. Dang and L. Huo, Appl. Radiat. Isot., 2010, 68, 303–308 CrossRef CAS PubMed.
- C. Hocke, O. Prante, I. Salama, H. Hübner, S. Löber, T. Kuwert and P. Gmeiner, ChemMedChem, 2008, 3, 788–793 CrossRef CAS PubMed.
- J. P. van Wieringen, V. Shalgunov, H. M. Janssen, P. M. Fransen, A. G. M. Janssen, M. C. Michel, J. Booij and P. H. Elsinga, J. Med. Chem., 2014, 57, 391–410 CrossRef CAS PubMed.
- R. Tietze, C. Hocke, S. Löber, H. Hübner, T. Kuwert, P. Gmeiner and O. Prante, J. Labelled Compd. Radiopharm., 2006, 49, 55–70 CrossRef CAS PubMed.
- R. Tietze, S. Löber, H. Hübner, P. Gmeiner, T. Kuwert and O. Prante, Bioorg. Med. Chem. Lett., 2008, 18, 983–988 CrossRef CAS PubMed.
- O. Prante, R. Tietze, C. Hocke, S. Löber, H. Hübner, T. Kuwert and P. Gmeiner, J. Med. Chem., 2008, 51, 1800–1810 CrossRef CAS PubMed.
- A. Jackson, B. B. Guilbert, S. D. Plant, J. Goggi, M. R. Battle, J. L. Woodcraft, A. Gaeta, C. L. Jones, D. R. Bouvet, P. A. Jones, D. M. O. Shea, P. H. Zheng, S. L. Brown, A. L. Ewan and W. Trigg, Bioorg. Med. Chem. Lett., 2013, 23, 821–826 CrossRef CAS PubMed.
- M. M. Herth, V. Kramer and F. Rösch, J. Labelled Compd. Radiopharm., 2009, 52, 201–207 CrossRef CAS PubMed.
- U. Funke, S. Fischer, A. Hiller, M. Scheunemann, W. Deuther-Conrad, P. Brust and J. Steinbach, Bioorg. Med. Chem. Lett., 2008, 18, 4727–4730 CrossRef CAS PubMed.
- M. M. Herth, M. Piel, F. Debus, U. Schmitt, H. Lüddens and F. Rösch, Nucl. Med. Biol., 2009, 36, 447–454 CrossRef CAS PubMed.
- F. Debus, M. M. Herth, M. Piel, H. G. Buchholz, N. Bausbacher, V. Kramer, H. Lüddens and F. Rösch, Nucl. Med. Biol., 2010, 37, 487–495 CrossRef CAS PubMed.
- V. J. Majo, M. S. Milak, J. Prabhakaran, P. Mali, L. Savenkova, N. R. Simpson, J. J. Mann, R. V. Parsey and J. S. D. Kumar, Bioorg. Med. Chem., 2013, 21, 5598–5604 CrossRef CAS PubMed.
- A. Bauman, M. Piel, S. Höhnemann, A. Krauss, M. Jansen, C. Solbach, G. Dannhardt and F. Rösch, J. Labelled Compd. Radiopharm., 2011, 54, 645–656 CrossRef CAS PubMed.
- M. Q. Zheng, D. Z. Yin, J. P. Qiao, L. Zhang and Y. X. Wang, J. Fluorine Chem., 2008, 129, 210–216 CrossRef CAS PubMed.
- B. H. Yousefi, A. Drzezga, B. von Reutern, A. Manook, M. Schwaiger, H. J. Wester and G. Henriksen, ACS Med. Chem. Lett., 2011, 2, 673–677 CrossRef CAS PubMed.
- P. H. Elsinga, K. Kawamura, T. Kobashi, H. Tsukanda, M. Senda, W. Vaalburg and K. Ishiwata, Synapse, 2002, 43, 259–267 CrossRef CAS PubMed.
- K. Kawamura, P. H. Elsinga, T. Kobayashi, S. I. Ishii, W. F. Wang, K. Matsuno, W. Vaalburg and K. Ishiwata, Nucl. Med. Biol., 2003, 30, 273–284 CrossRef CAS.
- Y. Y. Chen, X. Wang, J. M. Zhang, W. Deuther-Conrad, X. J. Zhang, Y. Huang, Y. Li, J. J. Ye, M. C. Cui, J. Steinbach, P. Brust, B. L. Liu and H. M. Jia, Bioorg. Med. Chem., 2014, 22, 5270–5278 CrossRef CAS PubMed.
- F. Xie, R. Bergmann, T. Kniess, W. Deuther-Conrad, C. Mamat, C. Neuber, B. Liu, J. Steinbach, P. Brust, J. Pietzsch and H. Jia, J. Med. Chem., 2015, 58, 5395–5407 CrossRef CAS PubMed.
- H. Wadsworth, P. A. Jones, W. F. Chau, C. Durrant, V. Morisson-Iveson, J. Passmore, D. O'Shea, D. Wynn, I. Khan, A. Black, M. Avory and W. Trigg, Bioorg. Med. Chem. Lett., 2012, 22, 5795–5800 CrossRef CAS PubMed.
- S. Khanapur, S. Paul, A. Shah, S. Vatakuti, M. J. B. Koole, R. Zijlma, R. A. J. O. Dierckx, G. Luurtsema, P. Garg, A. van Waarde and P. H. Elsinga, J. Med. Chem., 2014, 57, 6765–6780 CrossRef CAS PubMed.
- H. Schieferstein, M. Piel, F. Beyerlein, H. Lüddens, N. Bausbacher, H. G. Buchholz, T. L. Ross and F. Rösch, Bioorg. Med. Chem., 2015, 23, 612–623 CrossRef CAS PubMed.
- G. G. Shiue, R. Schirrmacher, C. Y. Shiue and A. A. Alavi, J. Labelled Compd. Radiopharm., 2001, 44, 127–139 CrossRef CAS PubMed.
- A. Schmitz, C. Y. Shiue, Q. Feng, G. G. Shiue, S. Deng, M. T. Pourdehnad, R. Schirrmacher, M. Vatamaniuk, N. Doliba, F. Matschinsky, B. Wolf, F. Rösch, A. Naji and A. A. Alavai, Nucl. Med. Biol., 2004, 31, 483–491 CrossRef CAS PubMed.
- B. Wängler, S. Schneider, O. Thews, E. Schirrmacher, S. Comagic, P. Feilen, C. Schwanstecher, M. Schwanstecher, C. Y. Shiue, A. Alavi, S. Höhnemann, M. Piel, F. Rösch and R. Schirrmacher, Nucl. Med. Biol., 2004, 31, 639–647 CrossRef PubMed.
- E. Schirrmacher, R. Schirrmacher, O. Thews, W. Dillenburg, A. Helisch, I. Wessel, R. Buhl, S. Höhnemann, H. G. Buchholz, P. Bartenstein, H. J. Machulla and F. Rösch, Bioorg. Med. Chem. Lett., 2003, 13, 2687–2692 CrossRef CAS.
- Y. Chen, M. Feng, S. Li, J. Xu, H. Ning, Y. He, X. Wang, R. Ding and C. Qi, Bioorg. Med. Chem. Lett., 2012, 22, 4745–4749 CrossRef CAS PubMed.
- M. Allmeroth, D. Moderegger, D. Gründel, K. Koynov, H. G. Buchholz, K. Mohr, F. Rösch, R. Zentel and O. Thews, Biomacromolecules, 2013, 14, 3091–3101 CrossRef CAS PubMed.
- M. M. Herth, M. Barz, D. Moderegger, M. Allmeroth, M. Jahn, O. Thews, R. Zentel and F. Rösch, Biomacromolecules, 2009, 10, 1697–1703 CrossRef CAS PubMed.
- C. Mamat, M. Franke, T. Peppel, M. Köckerling and J. Steinbach, Tetrahedron, 2011, 67, 4521–4529 CrossRef CAS PubMed.
- G. Henriksen, M. Herz, M. Schwaiger and H. J. Wester, J. Labelled Compd. Radiopharm., 2005, 48, 771–779 CrossRef CAS PubMed.
- G. Henriksen, S. Platzer, J. Marton, A. Hauser, A. Bethele, M. Schwaiger, L. Marinelli, A. Lavecchina, E. Novellino and H. J. Wester, J. Med. Chem., 2005, 48, 7720–7732 CrossRef CAS PubMed.
- C. Philippe, J. Ungersboeck, E. Schirmer, M. Zdravkovic, L. Nics, M. Zeilinger, K. Shanab, R. Lanzenbeger, G. Karanikas, H. Speitzer, H. Viernstein, M. Mitterhauser and W. Wadsak, Bioorg. Med. Chem., 2012, 20, 5936–5940 CrossRef CAS PubMed.
- T. K. Heinrich, V. Gottumukkala, E. Snay, P. Dunning, F. H. Fahey, S. T. Treves and A. B. Packard, Appl. Radiat. Isot., 2010, 68, 96–100 CrossRef CAS PubMed.
- (a) T. Hara, N. Kosaka and H. Kishi, J. Nucl. Med., 1998, 39, 990–995 CAS; (b) T. Hara, N. Kosaka, M. Yuasa and H. Yoshida, J. Labelled Compd. Radiopharm., 1997, 40, 670 CrossRef PubMed.
- T. Hara, N. Kosaka and H. Kishi, J. Nucl. Med., 2002, 43, 187–199 CAS.
- K. H. Yu, J. H. Park and S. D. Yang, Bull. Korean Chem. Soc., 2004, 25, 506–510 CrossRef CAS.
- G. Henriksen, M. Herz, A. Hauser, M. Schwaiger and H. J. Wester, Nucl. Med. Biol., 2004, 31, 851–858 CrossRef CAS PubMed.
- M. Piel, A. Bauman, R. P. Baum, S. Höhnemann, I. Klette, R. Wortmann and F. Rösch, Bioorg. Med. Chem., 2007, 15, 3171–3175 CrossRef CAS PubMed.
- G. Pascali, G. Nannavecchia, S. Pitzianti and P. A. Salvadori, Nucl. Med. Biol., 2011, 38, 637–644 CrossRef CAS PubMed.
- T. Sun, G. Tang, H. Tian, X. Wang, X. Chen, Z. Chen and S. C. Wang, Appl. Radiat. Isot., 2012, 70, 676–680 CrossRef CAS PubMed.
- M. M. Goodman, C. D. Kilts, R. Keil, B. Shi, L. Martarello, D. Xing, J. Votaw, T. D. Ely, P. Lambert, M. J. Owens, V. M. Camp, E. Malveaux and J. M. Hoffman, Nucl. Med. Biol., 2000, 27, 1–12 CrossRef CAS.
- N. Harada, H. Ohba, D. Fukumoto, T. Kakiuchi and H. Tsukada, Synapse, 2004, 54, 37–45 CrossRef CAS PubMed.
- P. J. Riss, S. Hoehnemann, M. Piel and F. Rösch, J. Labelled Compd. Radiopharm., 2013, 56, 356–359 CrossRef CAS PubMed.
- K. Takahashi, S. Miura, J. Hatazawa, E. Shimosegawa, T. Kinoshita, H. Ito, H. Tamura and I. Kanno, J. Labelled Compd. Radiopharm., 1999, 42, S1–S497 CrossRef.
- M. B. Skaddan, P. S. Sherman and M. R. Kilbourn, Nucl. Med. Biol., 2001, 28, 735–759 CrossRef.
- F. Caillé, T. J. Morley, A. A. S. Tavares, C. Papin, N. M. Twardy, D. Alagille, H. S. Lee, R. M. Baldwin, J. P. Seibyl, O. Barret and G. D. Tamagnan, Bioorg. Med. Chem. Lett., 2013, 23, 6243–6247 CrossRef PubMed.
- X. Shao, E. R. Butch, M. R. Kilbourn and S. E. Snyder, Nucl. Med. Biol., 2003, 30, 491–500 CrossRef CAS.
- M. Piel, R. Schirrmacher, S. Höhnemann, W. Hamkens, B. Kohl, M. Jansen, U. Schmitt, H. Lüddens, G. Dannhardt and F. Rösch, J. Labelled Compd. Radiopharm., 2006, 46, 645–659 CrossRef PubMed.
- L. Cai, F. T. Chin, V. W. Pike, H. Toyama, J. S. Liow, S. S. Zoghbi, K. Modell, E. Briard, H. U. Shetty, K. Sinclair, S. Donohue, D. Tipre, M. P. Kung, C. Dagostin, D. A. Widdowson, M. Green, W. Gao, M. M. Herman, M. Ichise and R. B. Innis, J. Med. Chem., 2004, 47, 2208–2218 CrossRef CAS PubMed.
- D. R. Veach, M. Namavari, N. Pillarsetty, E. B. Santos, T. Beresten-Kochetov, C. Lambeck, B. J. Punzalan, C. Antczak, P. M. Smith-Jones, H. Djaballah, B. Clarkson and S. M. Larson, J. Med. Chem., 2007, 50, 5853–5857 CrossRef CAS PubMed.
- S. Comagic, M. Piel, R. Schirrmacher, S. Höhnemann and F. Rösch, Appl. Radiat. Isot., 2002, 56, 847–851 CrossRef CAS.
- J. Prabhakaran, V. Arango, V. J. Majo, N. R. Simpson, S. A. Kassir, M. D. Underwood, H. Polavarapu, J. N. Bruce, P. Canoll, J. J. Mann and J. S. D. Kumar, Bioorg. Med. Chem. Lett., 2012, 22, 5104–5107 CrossRef CAS PubMed.
- E. M. F. Billaud, L. Rbah-Vidal, A. Vidal, S. Besse, S. Tarrit, S. Askienazy, A. Maisonial, N. Moins, J. C. Madelmont, E. Miot-Noirault, J. M. Chezal and P. Auzeloux, J. Med. Chem., 2013, 56, 8455–8467 CrossRef CAS PubMed.
- H. J. Breyholz, S. Wagner, A. Faust, B. Riemann, C. Höltke, S. Hermann, O. Schober, M. Schäfers and K. Kopka, ChemMedChem, 2010, 5, 777–789 CrossRef CAS PubMed.
- J. Li, B. D. Gray, K. Y. Pak and C. K. Ng, J. Labelled Compd. Radiopharm., 2012, 55, 149–154 CrossRef CAS PubMed.
- P. J. Riss, V. Soskic, A. Schrattenholz and F. Rösch, J. Labelled Compd. Radiopharm., 2009, 52, 576–579 CAS.
- (a) Z. P. Chen, S. P. Wang, X. M. Li, C. Y. Liu, J. Tang, G. X. Cao, S. N. Luo, L. F. Zhang and J. Jin, Appl. Radiat. Isot., 2008, 66, 1818–1885 CrossRef PubMed; (b) J. Pijarowska-Kruszyna, A. W. Jaron, A. Kachniarz, K. Kasprzak, A. Kowalska, B. Malkowski, S. Demphel, F. Dollè and R. Mikolajczak, J. Labelled Compd. Radiopharm., 2013, 57, 148–157 CrossRef PubMed.
- R. Schirrmacher, W. Hamkens, M. Piel, J. Brockmann and F. Rösch, J. Labelled Compd. Radiopharm., 1999, 42(S1), S339 Search PubMed.
- R. Schirrmacher, W. Hamken, M. Piel, U. Schmitt, H. Lüddens, C. Hiemke and F. Rösch, J. Labelled Compd. Radiopharm., 2001, 44, 627–642 CrossRef CAS PubMed.
- W. R. Banks, S. M. Moerlein, D. Parkinson and M. J. Welch, Med. Chem. Res., 1994, 5, 150–173 Search PubMed.
- P. J. Riss, L. Brichard, V. Ferrari, D. J. Williamson, T. D. Fryer, Y. T. Hong, J. C. Baron and F. I. Aigbirhio, Med. Chem. Commun., 2013, 4, 852–855 RSC.
- M. B. Skaddan, L. Zhang, D. S. Johnson, A. Zhu, K. R. Zasadny, R. V. Coelho, K. Kuszpit, G. Currier, K. H. Fan, E. M. Beck, L. Chen, S. E. Drozda, G. Balan, M. Niphakis, B. F. Cravatt, K. Ahn, T. Bocan and A. Villalobos, Nucl. Med. Biol., 2012, 29, 1058–1067 CrossRef PubMed.
- U. Funke, W. Deuther-Conrad, G. Schwan, A. Maisonial, M. Scheunemann, S. Fischer, A. Hiller, D. Briel and P. Brust, Pharmaceuticals, 2012, 5, 169–188 CrossRef CAS PubMed.
- S. Rötering, M. Scheunemann, S. Fischer, A. Hiller, D. Peters, W. Deuther-Conrad and P. Brust, Bioorg. Med. Chem., 2013, 21, 2635–2642 CrossRef PubMed.
- D. O'Shea, R. Ahmad, E. Årstad, M. Avory, W. F. Chau, C. Durrant, E. Hirani, P. A. Jones, I. Khan, S. K. Luthra, D. Mantzilas, V. Morisson-Iveson, J. Passmore, E. G. Robins, B. Shan, H. Wadsworth, S. Walton, Y. Zhao and W. Trigg, Bioorg. Med. Chem. Lett., 2013, 23, 2368–2373 CrossRef PubMed.
- R. Schirrmacher, M. Weber, A. Schmitz, C. Y. Shiue, A. A. Alavi, P. Feilen, S. Schneider, P. Kann and F. Rösch, J. Labelled Compd. Radiopharm., 2002, 45, 763–774 CrossRef CAS PubMed.
- S. Wagner, M. P. Law, B. Riemann, V. W. Pike, H. J. Breyholz, C. Höltke, A. Faust, C. Renner, O. Schober, M. Schäfers and K. Kopka, J. Labelled Compd. Radiopharm., 2006, 49, 177–195 CrossRef CAS PubMed.
- H. S. Radeke, A. Purohit, T. D. Harris, K. Hanson, R. Jones, C. Hu, P. Yalamanchili, M. Hayes, M. Yu, M. Guaraldini, M. Kagan, M. Azure, M. Cdebaca, S. Robinson and D. Casebier, ACS Med. Chem. Lett., 2011, 2, 650–655 CrossRef CAS PubMed.
- M. Erlandson, F. Karimi, Ö. Lindhe and B. Långström, Nucl. Med. Biol., 2009, 36, 435–445 CrossRef PubMed.
- M. Erlansson, F. Karimi and K. Takahashi, J. Labelled Compd. Radiopharm., 2008, 51, 207–212 CrossRef PubMed.
- S. D. Krämer, L. Mu, A. Müller, C. Keller, O. F. Kuznetsova, C. Schweinsberg, D. Franck, C. Müller, T. L. Ross, R. Schibli and S. M. Ametamey, J. Nucl. Med., 2012, 53, 434–442 CrossRef PubMed.
- A. Chiotellis, A. Müller, L. Mu, C. Keller, R. Schibli, S. D. Krämer and S. M. Ametamey, Mol. Pharmaceutics, 2014, 11, 3839–3851 CrossRef CAS PubMed.
- G. Tang, M. Wang, X. Tang, L. Luo and M. Gan, Nucl. Med. Biol., 2003, 30, 509–512 CrossRef CAS.
- D. O. Kiesewetter, R. E. Carson, E. M. Jagoda, P. Herscovitch and W. E. Eckelman, Synapse, 1999, 31, 29–40 CrossRef CAS.
- E. G. Robins, Y. Zhao, I. Khan, A. Wilson, S. K. Luthra and E. Arstad, Bioorg. Med. Chem. Lett., 2010, 20, 1749–1751 CrossRef CAS PubMed.
- E. F. J. de Vries, J. Vroegh, P. H. Elsinga and W. Vaalburg, Appl. Radiat. Isot., 2003, 58, 469–476 CrossRef CAS.
- S. L. James, S. K. Ahmed, S. Murphy, M. R. Braden, Y. Belabassi, H. F. VanBrocklin, C. M. Thompson and J. M. Gerdes, ACS Chem. Neurosci., 2014, 5, 519–524 CrossRef CAS PubMed.
- M. Patt, A. Schildan, B. Habermann, O. Mishchenko, J. T. Patt and O. Sabri, J. Radioanal. Nucl. Chem., 2010, 283, 487–491 CrossRef CAS.
- A. Afshar-Oromieh, A. Malcher, M. Eder, M. Eisenghut, H. G. Linhart, B. A. Hadaschik, T. Holland-Letz, F. L. Giesel, C. Kratochwil, S. Haufe, U. Haberkorn and C. M. Zechmann, Eur. J. Nucl. Med. Mol. Imaging, 2013, 40, 486–495 CrossRef CAS PubMed.
- E. Al-Momani, N. Malik, H. J. Machulla, S. N. Reske and C. Solbach, J. Radioanal. Nucl. Chem., 2013, 295, 2289–2294 CrossRef CAS.
- C. Prenant, J. Gillies, J. Bailey, G. Chimon, N. Smith, G. C. Jayson and J. Zweit, J. Labelled Compd. Radiopharm., 2008, 51, 262–267 CrossRef CAS PubMed.
- E. Atucha, F. Hammerschmidt, I. Zolle, W. Sieghart and M. L. Berger, Bioorg. Med. Chem. Lett., 2009, 19, 4284–4287 CrossRef CAS PubMed.
- X. Li, C. Ma, X. He, J. Yu, D. Han, C. Zhang, J. R. Atack and C. M. Cook, Med. Chem. Res., 2002, 11, 504–537 CAS.
- J. Prabhakaran, R. V. Parsey, V. J. Majo, S. C. Hsiung, M. S. Milak, H. Tamir, N. R. Sinpson, R. L. van Heertum, J. J. Mann and J. S. D. Kumar, Bioorg. Med. Chem. Lett., 2006, 16, 2101–2104 CrossRef CAS PubMed.
- B. A. Shinkre, T. S. Kumar, Z. G. Gao, F. Deflorian, K. A. Jacobson and W. C. Trenkle, Bioorg. Med. Chem. Lett., 2010, 20, 5690–5694 CrossRef CAS PubMed.
- S. Wagner, M. P. Law, B. Riemann, V. W. Pike, H. J. Breyholz, C. Höltke, A. Faust, O. Schober, M. Schäfers and K. Kopka, J. Labelled Compd. Radiopharm., 2005, 48, 721–733 CrossRef CAS PubMed.
- C. A. Matis, Y. Wang, D. P. Holt, G. F. Huang, M. L. Debnath and W. E. Klunk, J. Med. Chem., 2003, 46, 2740–2754 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |