
Towards understanding cell penetration by stapled peptides†
Qian
Chu‡
ab,
Raymond E.
Moellering‡
ab,
Gerard J.
Hilinski
ab,
Young-Woo
Kim
ab,
Tom N.
Grossmann
ab,
Johannes T.-H.
Yeh
ab and
Gregory L.
Verdine
*abcd
aDepartment of Stem Cell & Regenerative Biology, Harvard University, 12 Oxford Street, Cambridge, Massachusetts 02138, USA. E-mail: gregory_verdine@harvard.edu; Tel: +1-617-495-5323
bChemistry & Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, Massachusetts 02138, USA
cMolecular & Cellular Biology, Harvard University, 12 Oxford Street, Cambridge, Massachusetts 02138, USA
dProgram in Cancer Chemical Biology, Dana-Farber Cancer Institute, Boston, Massachusetts 02115, USA
First published on 14th October 2014
Abstract
Hydrocarbon-stapled α-helical peptides are a new class of targeting molecules capable of penetrating cells and engaging intracellular targets formerly considered intractable. This technology has been applied to the development of cell-permeable ligands targeting key intracellular protein–protein interactions. However, the properties governing cell penetration of hydrocarbon-stapled peptides have not yet been rigorously investigated. Herein we report our studies to systematically probe cellular uptake of stapled peptides. We developed a high-throughput epifluorescence microscopy assay to quantitatively measure stapled peptide intracellular accumulation and demonstrated that this assay yielded highly reproducible results. Using this assay, we analyzed more than 200 peptides with various sequences, staple positions and types, and found that cell penetration ability is strongly related to staple type and formal charge, whereas other physicochemical parameters do not appear to have a significant effect. We next investigated the mechanism(s) involved in stapled peptide internalization and have demonstrated that stapled peptides penetrate cells through a clathrin- and caveolin-independent endocytosis pathway that involves, in part, sulfated cell surface proteoglycans, but that also seems to exploit a novel, uncharacterized pathway. Taken together, staple type and charge are the key physical properties in determining the cell penetration ability of stapled peptides, and anionic cell surface proteoglycans might serve as receptors to mediate stapled peptide internalization. These findings improve our understanding of stapled peptides as chemical probes and potential targeted therapeutics, and provide useful guidelines for the design of next-generation stapled peptides with enhanced cell permeability.
Introduction
Hydrocarbon stapled α-helical peptides are an exciting new class of investigational agents capable of targeting and interfering with intracellular protein–protein interactions.1,2 (For reviews on hydrocarbon stapled peptides, see ref. 3 and 4, and for reviews on synthetic α-helix stabilization in general, see ref. 5 and 6.) These peptides contain a synthetic brace, referred to as a staple, introduced across one face of an α-helix (Fig. 1), that in favorable cases can increase α-helical content and protease resistance, enhance target binding affinity, promote cell membrane penetration, and suppress clearance in vivo.7–10 Stapled peptides are synthesized via incorporation of two α-methyl, α-alkenyl amino acids at defined positions in a synthetic peptide, followed by ring-closing olefin metathesis to close the helix-spanning hydrocarbon bridge (Fig. 1a).11,12 The two components of the staple, namely the hydrocarbon bridge and terminal methyl groups, are both important to obtain maximal effectiveness of the conformationally constrained peptide products. This technology has been successfully utilized to target several classes of proteins formerly considered intractable, including multi-component transcription factor complexes and protein–protein interactions having extended interfaces, such as the NOTCH transcription factor complex,13 the β-catenin–TCF interaction in the oncogenic Wnt signaling pathway,14 and the epigenetic modulator PRC2 complex.15 Given the difficulties of developing traditional small molecule drugs that can successfully target intracellular protein–protein interactions, hydrocarbon stapling technology is widely considered to represent a promising avenue of research for the development of chemical probes and potential targeted therapeutics.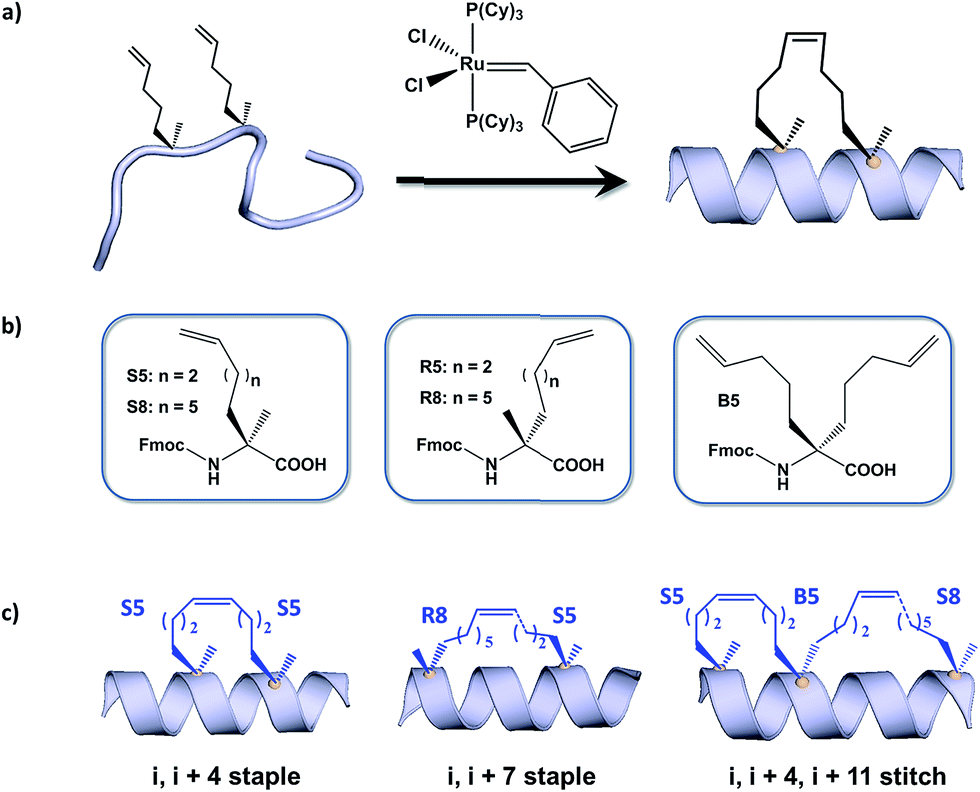 | ||
| Fig. 1 All-hydrocarbon stapled peptide technology. (a) Schematic illustration of peptide stapling. Two alpha-methylated, alkenyl-bearing non-natural amino acids are incorporated at two or more positions in the peptide chain and then cross-linked by ruthenium-catalyzed ring-closing olefin metathesis. (b) Different types of alkenyl-containing non-natural amino acids with distinct stereochemistry at the α-carbon and varied lengths of alkenyl side chains. (c) Three types of stapled peptides used in this study with optimized combinations of non-natural amino acids. | ||
Multiple types of hydrocarbon staples have been obtained by varying the relative placement of the cross-linking α,α-disubstituted amino acids, as well as the stereochemistry at the α-carbon and the lengths of the alkenyl substituents (Fig. 1b).16,17 These staple types were optimized to provide robust α-helical stabilization and confer the potential for in vitro and in vivo activity. As a result of the combinatorial search process used to identify helix-stabilizing hydrocarbon staples, the diversity of the resulting macrocyclic bridges has revealed stapled peptides with different physicochemical properties. Recently, a new hyperstable version of stapled peptide with tandem crosslinks, referred to as a stitched peptide, was generated by introduction of S5 at the i position, B5 at the i + 4 position, and S8 at the i + 11 position (Fig. 1c) (Y.-W. Kim and G. L. Verdine, to be published).
Of the physicochemical properties demonstrated by peptide bearing hydrocarbon staples, the capacity to promote cellular membrane penetration is perhaps the most significant and yet remains the most poorly understood. Independent of hydrocarbon-stapled peptides, several classes of cell penetrating peptides (CPPs) have been discovered, including naturally occuring transcription factor domains such as pennetratin18 and HIV-Tat19 and synthetic cationic peptides such as poly-Arginine peptides.20 Notably, despite extensive exploration during the past two decades, the mechanism(s) by which CPPs enter cells remain unclear.21–23 In contrast to CPPs, in which cell penetration appears to be sequence-dependent, numerous cell permeable stapled peptides have been discovered for peptide scaffolds with little sequence homology. These divergent observations regarding cell penetration is proposed to result from several features of stapled peptides that differentiate them from typical CPPs. For example, the introduction of an all-hydrocarbon cross-link results in a constrained α-helical conformation, which embeds the hydrophilic amide backbone in the core of the folded structure. Furthermore, the hydrocarbon brace itself introduces a significantly hydrophobic patch to one face of the peptide. The exposure of the hydrophobic moiety as well as the masking of the hydrophilic peptide backbone may facilitate the interaction of stapled peptides with the hydrophobic interior of the cell membrane and thereby enhance the cellular uptake. As cell penetration is a critical property of stapled peptides, we sought to develop quantitative methods to correlate a battery of stapled peptide properties with the capacity for cellular uptake. A direct comparison with several well-known CPPs has revealed that stapled peptides, including some stapled versions of the CPPs, exhibit more robust cell penetration. Lastly, we have demonstrated that stapled peptides penetrate cells through a clathrin- and caveolin-independent endocytosis pathway that involves, in part, sulfated cell surface proteoglycans. These findings significantly expand our current understanding of cell penetration by stapled peptides and provide useful information for the future rational design of cell penetrating stapled peptides with novel applications.
Results and discussion
Development of a high-throughput assay to quantitatively measure cellular uptake of peptides
Understanding the internalization process of cell penetrating peptides (CPPs), especially stapled peptides, has been a subject of great interest. The majority of previous studies have been performed by either using high-resolution microscopy to show the existence of fluorophore-labeled CPPs inside cells, or by quantitatively measuring intracellular fluorescence by flow cytometry.24,25 Although these two methods can provide important information regarding cell penetration, their respective limitations prompted us to adopt an assay that combines high-resolution imaging with reliable quantitation of intracellular accumulation to better analyze and understand the cell penetration of stapled peptides. In recent years, high-throughput cell-based imaging platforms have become increasingly popular to screen for small molecule modulators of various biological processes.26,27 Taking advantage of one of these platforms, high-content epifluorescence microscopy, we developed a high-throughput quantitative assay to measure stapled peptide intracellular access.Proof-of-principle experiments were performed to determine whether epifluorescence microscopy could be used to quantitatively compare stapled peptide intracellular access. Human U2OS osteosarcoma cells were seeded in black, clear-bottom 384-well plates and then incubated in serum-containing media supplemented with fluorescein-labeled peptides or DMSO vehicle for 12 hours. After the treatment, cells were washed thoroughly with PBS to remove excess peptide, fixed with 4% formaldehyde, and stained with Hoechst dye to visualize nuclei. Once prepared, the plates were imaged and quantified by epifluorescence microscopy according to a protocol developed and discussed in detail in Experimental methods. An initial z-scan was performed using the Hoechst channel to locate the cells, and the microscope parameters were subsequently adjusted to optimize the cell size and fluorescence intensity. The parameters from this acquisition were then applied to the FITC channel, and the microscope scanned and recorded images of the FITC-labeled peptides within the z-plane of the cell. This assay was performed in a high-throughput manner, resulting in a panel of Hoechst/FITC images from individual wells (Fig. S1†). The raw image data was then analyzed using MetaXpress® software (Fig. 2a). Cells were identified based on the Hoechst stain of nuclei, with the requirement that they were a contiguous fluorescent region having a specific intensity above local background as well as having a diameter between defined minimum and maximum to be designated as “positive” cells. The cytoplasm of each cell was then identified according to the spatial location of FITC signal in relation to the nuclei as well as empiric parameters (details in Experimental methods). The FITC intensities in the cytoplasm and nuclei were then quantified separately, and the sum of these two values yielded the FITC signal for the whole cell, which can be considered the relative intracellular peptide intensity. In addition, FITC negative cells were identified on the basis of a positive Hoechst stain, which was accompanied by an absence of appreciable signal in the FITC channel.
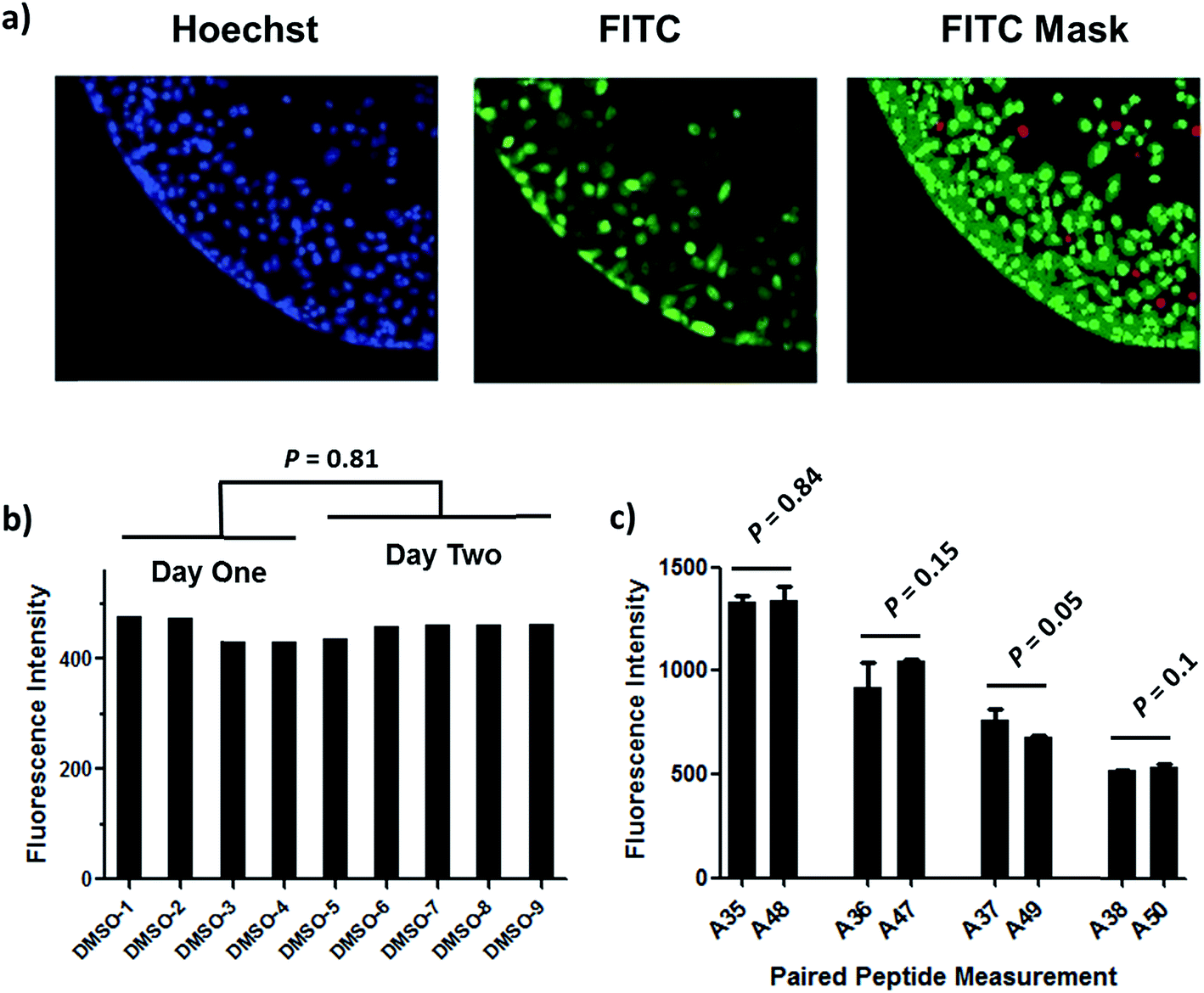 | ||
| Fig. 2 Quantitative measurement of cellular peptide intensity. (a) Hoechst channel (left) showing the location and size of nuclei, FITC channel (middle) showing the fluorescence intensity of the same cells. Information about cell size and fluorescence intensity was integrated to identify the FITC positive (green mask) and negative (red mask) cell (right). For positive cells, additional parameters allowed determination of the fluorescence intensity in the nucleus (inner intense green) and the cytoplasm (outer dim green). (b) The background fluorescence the DMSO vehicle was almost identical among different experiments. (c) Four stapled peptides from different batches of synthesis generated similar intracellular fluorescence intensity in different tests. Error bars represent the S.D. of two measurements. | ||
We found that this system generated highly reproducible and reliable results from assay-to-assay and with different stocks of the same stapled peptides. As shown in Fig. 2b, there were negligible fluorescence differences among experiments for cells treated with DMSO vehicle, which could be used as a fluorescence background for all subsequent experiments. In addition, the same stapled peptides from different batches of synthesis and stocks featured almost identical intracellular fluorescence signals in different tests (Fig. 2c), indicating that the assay developed in this study produces repeatable and reliable results that could be directly combined and compared from a large set of experiments. Furthermore, to determine how this assay performs as a screening tool, we have calculated Z′ factor of 0.54 by using the most penetrant A6 peptide as a positive control and DMSO background as negative control, which also indicates a statistically good assay quality.
Analysis of cell penetration by stapled peptides
The development of this quantitative high-throughput assay enabled a broad investigation of the physicochemical properties governing the cell uptake of a diverse set of hydrocarbon-stapled peptides synthesized in our laboratory. We postulated that any correlation between cellular uptake and physicochemical properties would illuminate characteristics associated with productive cellular uptake and inform the future design of stapled peptides with improved cell penetration.28 To this end, we screened and analyzed more than 200 discrete FITC-labeled peptides belonging to three different classes: wild-type (unmodified), stapled and stitched peptides. All peptides were converted to two-dimensional structures and analyzed for theoretical physicochemical properties with the publicly available MarvinView software package from ChemAxon. Properties including the molecular weight, theoretical pI, calculated 2D polar surface area (PSA), theoretical log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P and formal charge at pH 7.5 were calculated for each peptide (Table S1†). In general, the unmodified, stapled and stitched peptide libraries present in this screen had relatively similar physicochemical characteristics (Fig. S2†). The mean molecular weight and calculated PSA values were nearly identical among the three peptide classes. A notable difference was observed among theoretical logP values, which were significantly higher for the stapled and stitched peptides relative to the unmodified peptides, which is not surprising as these modified peptides contain a solvent-exposed hydrocarbon crosslink. Additionally, the stapled peptide class had a mean formal charge of approximately zero while the stitched and unmodified peptide classes exhibited a positive mean charge. Overall, the calculated physicochemical properties indicated that the peptide classes were quite similar in terms of their mean properties, which is useful when making comparisons among their cell penetration properties.
P and formal charge at pH 7.5 were calculated for each peptide (Table S1†). In general, the unmodified, stapled and stitched peptide libraries present in this screen had relatively similar physicochemical characteristics (Fig. S2†). The mean molecular weight and calculated PSA values were nearly identical among the three peptide classes. A notable difference was observed among theoretical logP values, which were significantly higher for the stapled and stitched peptides relative to the unmodified peptides, which is not surprising as these modified peptides contain a solvent-exposed hydrocarbon crosslink. Additionally, the stapled peptide class had a mean formal charge of approximately zero while the stitched and unmodified peptide classes exhibited a positive mean charge. Overall, the calculated physicochemical properties indicated that the peptide classes were quite similar in terms of their mean properties, which is useful when making comparisons among their cell penetration properties.
We next performed an intracellular access screen by treating U2OS cells with 1 μM of FITC-labeled peptide for 12 hours in duplicate. All assays contained control DMSO wells and positive control peptides, which were compared among assays to ensure plate-to-plate reproducibility (Fig. 2b and c). The primary readout of the screen was mean cellular fluorescence intensity. As the DMSO background was highly consistent between wells and experiments, a mean background value was subtracted from all data. The results of the screen were used to generate plots comparing cell penetration with peptide physicochemical parameters. Interestingly, as a class, stapled and stitched peptides exhibited significantly higher cell penetration compared with wild-type unmodified peptides, which contained several established cell penetrating peptides (CPPs; Fig. 3a). Given that all three peptide classes have similar physicochemical properties in general, the benefit in cell penetration can be largely attributed to the synthetic stabilization of the α-helical peptides with all-hydrocarbon peptide stapling technology. Furthermore, we found that peptide charge near physiologic pH exhibited a strong correlation with intracellular access and could be fitted into a Gaussian distribution with a population centroid at a formal charge of +4 (Fig. 3b). In particular, peptides exhibiting a net negative charge (−7 to −1) exhibited little cellular uptake, whereas peptides of approximately neutral charge (−1 to +1) displayed moderate cell penetration above background. Interestingly, peptides with a net positive charge (+1 to +7) showed significantly higher cell penetration as a group. Cellular uptake did not appear to increase linearly with charge, as the cell penetration decreases dramatically for the peptides in this study with charge greater than +7. The same trend between formal charge and cellular uptake were observed for individual stapled and stitched peptide classes as well (Fig. S3†). This observation is not consistent with previously reported models that indicate that peptides/mini-proteins with more positive charge have better penetration properties due to tighter electrostatic interactions with the negatively charged phospholipid membrane.29,30 The lower penetration for highly charged peptides in this study could result from any one of many factors including, for example, peptide aggregation in solution, the disruption of peptide packing during internalization or difficulty in dissociation from cell membrane. Additional tests with a larger number of peptides could further our understanding of this phenomenon. In addition, there was no discernible correlation between cell penetration and peptide molecular weight, logP, pI value or PSA (Fig. S4†). Taken together, these data demonstrated that the staple type and peptide charge are key physical properties correlated with peptide cell penetration ability, whereas the other parameters do not appear to be significantly associated.
 | ||
| Fig. 3 Relationship between mean cellular fluorescence intensity and (a) staple type, and (b) formal charge at pH 7.5, showing that stapled and stitched peptides exhibited a significant increase in cell penetration compared with wild-type unmodified peptides and that the intracellular access was correlated to peptide charge, which could be fitted to a Gaussian distribution with a population centroid at approximated +4 charge. The mean DMSO background has been subtracted from the fluorescence intensity for each peptide before plotting. Error bars represent the S.E.M. of mean cellular intensity of indicated peptide classes. *P < 0.05, ***P < 0.001. | ||
In order to further investigate the cell penetration properties for stapled peptides and to systematically analyze the similarities and differences in cellular uptake between stapled peptides and other wild-type cell penetrating peptides, we compared cell penetration of several stapled peptides to that of three well-known wild-type CPPs: Tat (48–60), penetratin (Antennapedia 43–58) and poly-Arg8 (Table S2†). First, we investigated the cellular uptake at varied peptide concentrations. As shown in Fig. 4a and b, both wild-type CPPs and stapled peptides showed dose-dependent increases in cell penetration. Strong intracellular fluorescence was detected in the low micromolar range, and although the levels of accumulation were different for distinct peptides, stapled peptides featured more robust dose-dependent cell penetration at lower concentrations relative to wild-type CPPs, in general. It is also interesting to note that while significant increases in intracellular fluorescence were mostly evident in the 1–10 μM range for stapled peptides, distinct profiles were observed for specific peptides. For example, TNG147 showed little cell penetration at 1 μM but showed a dramatic increase at 5 μM, which might suggest that concentration-dependent peptide packing or a receptor-mediated mechanism may facilitate the cell penetration process, and these processes may be triggered at different concentrations for distinct peptides. Furthermore, it is worth noting that the stapled peptides studied here were more cell permeable than wild-type CPPs at most concentrations tested, exhibiting nearly an order of magnitude higher intracellular fluorescence at the same treatment concentrations.
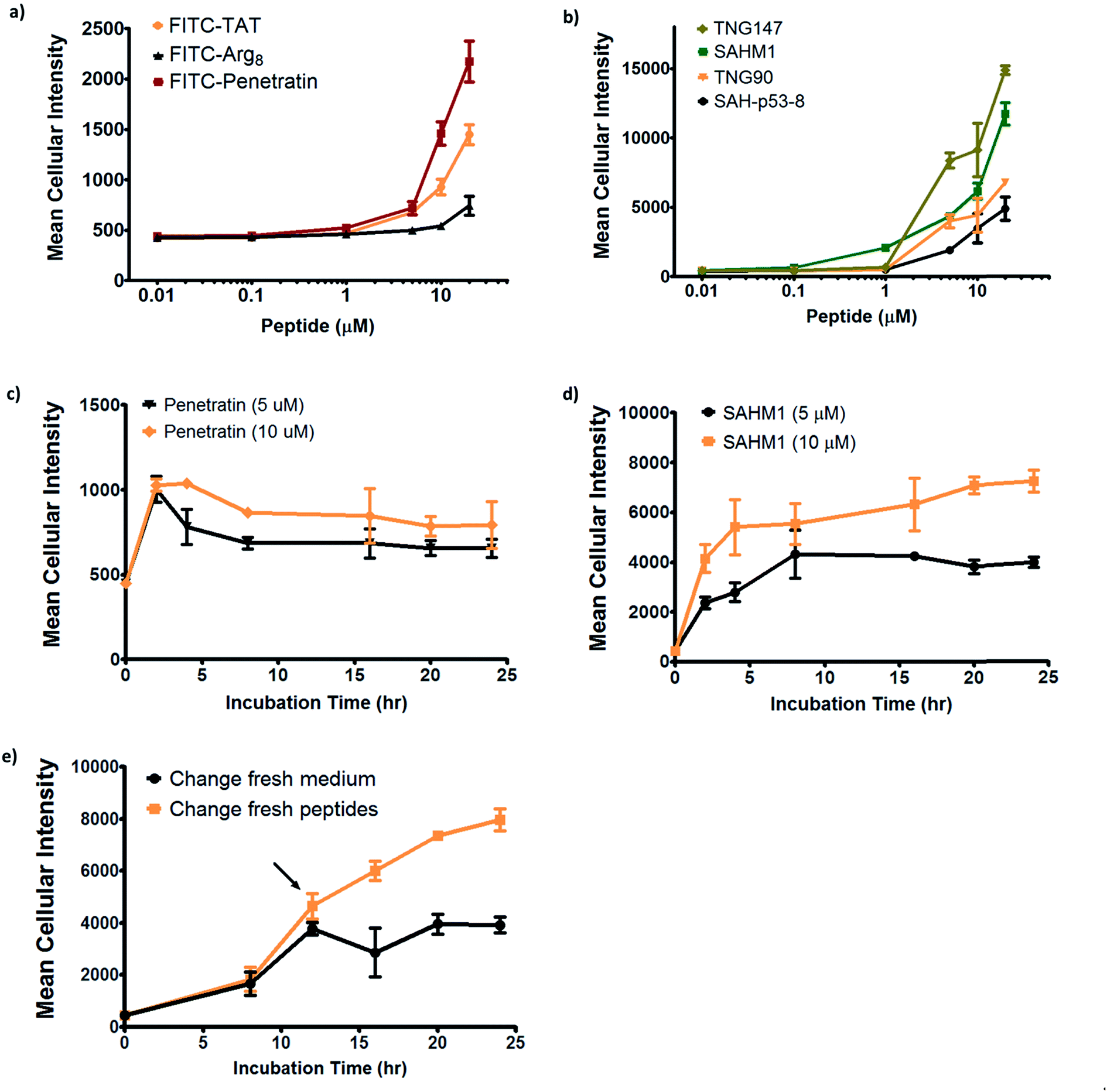 | ||
| Fig. 4 Effects of peptide concentration and incubation time on cellular uptake of stapled and wild-type peptides. (a) Wild-type and (b) stapled peptides showed a dose-dependent increase in cell internalization. Cellular uptake for (c) penetratin and (d) SAHM1 peptides over time at concentrations of 5 and 10 μM. (e) A pulse-chase penetration assay for SAHM1 peptide in which fresh medium containing either a new batch of peptide or DMSO vehicle were exchanged at 12 hours after initial treatment. Error bars represent the S.D. of triplicate samples. | ||
We next performed a time-course penetration assay to better understand the kinetics of peptide internalization using a representative CPP and stapled peptide. Penetratin and SAHM1 showed distinct kinetics of uptake and stabilization throughout a 24 hour time course. 5 μM and 10 μM penetratin peptide exhibited similar intracellular cellular fluorescence after 2 hours, which then decreased until approximately 8 hours and finally stabilized at different intracellular levels until 24 hours (Fig. 4c). On the other hand, the stapled peptide SAHM1 showed time- and dose-dependent cellular uptake, which stabilized after approximately 8 hours (Fig. 4d). Compared to the wild-type penetratin, the SAHM1 profile was unique in that dose-dependent accumulation was evident at all time points and no loss of signal was observed. One explanation for the loss of signal observed with penetratin could be attributed to an equilibrium between cell penetration and subsequent intracellular proteolysis followed by export of the fluorophore. The presence of the all-hydrocarbon crosslink in its peptide sequence and lower net charge of SAHM1 relative to penetratin, could contribute to enhanced cellular uptake and reduced intracellular proteolysis, leading to continuous accumulation in cells. To further explore the equilibrium observed for stapled peptides, we performed a pulse-chase experiment using SAHM1. After 12 hours of incubation with SAHM1, cell culture medium was aspirated and the cells were extensively washed with PBS to completely remove excess peptide. Then fresh medium containing either a new batch of 1 μM peptide or DMSO vehicle was added to cells and incubated for the indicated time points (Fig. 4e). As expected, the cellular uptake increased for the first 12 hours incubation. After medium exchange, cells incubated with fresh medium containing DMSO vehicle retained the intracellular fluorescence intensity. Interestingly, the signal for cells treated with a new batch of staple peptide continued to increase up to 24 hours (Fig. 4e). This observation indicates that despite incubation over a time course previously shown to reach equilibrium, the mechanism(s) responsible for cellular uptake are not saturated, as evidenced by further uptake upon replacement with fresh stapled peptide. Taken together, these data indicate that the mechanism(s) underlying cellular uptake by both CPPs and stapled peptides exhibit time- and dose-dependency that is not saturable at early time points or low micromolar doses and, importantly, appears to be more robustly utilized by stapled peptides.15,31
Given that stapled peptides exhibit better cell penetration properties in general than parent unmodified peptides, we wondered whether the peptide stapling strategy could be applied generally to improve cellular uptake of parent unmodified peptides. To test this hypothesis, we designed a panel of stapled peptides based on Tat (48–60), penetratin and poly-Arg8 (Fig. 5a). These stapled peptides and their parent unmodified peptides were incubated in U2OS cells for 12 hours with a concentration range from 10 nM to 20 μM, mirroring the dose-dependent uptake studies shown in Fig. 4. As expected, all peptides showed dose-dependent cell penetration (Fig. 5b–d). Interestingly, stapled peptides derived from penetratin and poly-Arg8 showed improved cell permeability at concentrations starting from 1 μM for stapled penetratin and 5 μM for stapled poly-Arg8. It is noteworthy that the staple position also affected the cellular uptake as the two stapled penetratin peptides with different crosslink positions exhibited varied cell penetration, though both were superior to wild-type penetratin. In contrast, reduced cellular uptake was observed for both stapled peptide variants derived from the Tat sequence (Fig. 5b). This could result from several possible effects, including disruption of peptide secondary structure, masking of residues essential for surface recognition or altering peptide packing interactions involved in cell penetration. Further focused study of these variants is warranted to elucidate the source of altered cellular uptake, however these data clearly demonstrate that peptide stapling may be a general method to further improve the cell permeability of CPPs, which could serve as more efficient transduction domains for molecular cargoes. In addition, while increasing the helical content of stabilized peptides has been stated to be a guiding principle in the successful design of biologically active stapled peptides, it has not been shown to be generally correlated with cell penetration. To specifically address whether increasing the helical content of a peptide is correlated with augmented cell penetration, we have measured the relative helicity of hydrocarbon stapled variants of Tat, penetratin and poly-Arg8 (Fig. S5†). Notably, we did not observe a general correlation between increased helical character and cell penetration of these peptides. Peptide stapling increased the helical content of both Tat and poly-Arg8 peptide sequences, which were largely unstructured when unmodified. In contrast, the unmodified penetratin peptide had significant helical content (>50%), and the hydrocarbon stapled variants of this sequence largely retained their helicity, albeit lower overall helicity. Intriguingly, these species demonstrated the differing effect of hydrocarbon stapling and increased helical content on cell penetration since introduction of the hydrocarbon staple increased the cellular uptake of both penetratin and poly-Arg8 sequences, while it decreased uptake for Tat peptides. Therefore, we cannot conclusively state, a priori, that the incorporation of a hydrocarbon staple or increased α-helicity will lead to more productive cellular penetration, although in general stapling can increase the uptake of specific sequences (Fig. 5) and as a class stapled and stitched peptides are more cell penetrant (Fig. 3a). A more comprehensive follow-up study with CD analyses on a larger peptide library is needed to better address this question.
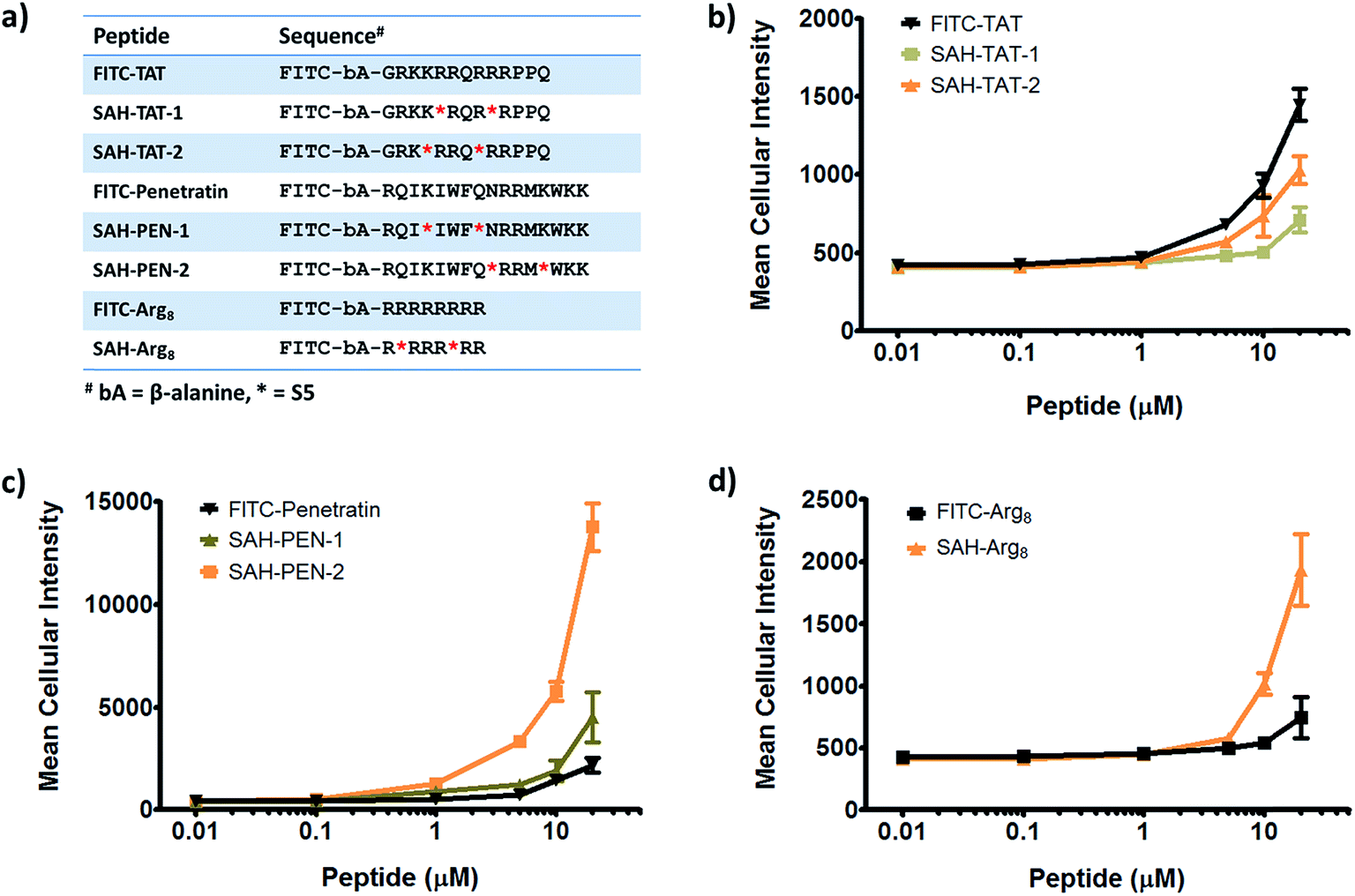 | ||
| Fig. 5 Effects of all-hydrocarbon staples on cell penetration by wild-type cell penetrating peptides. (a) List of wild-type cell penetrating peptides and their stapled derivatives investigated in this study. (b–d) Dose-dependent cell penetration assays showed that stapling strategy greatly improves the cellular uptake of penetratin and poly-Arg8 peptides. Experiments were performed in triplicate, and error bars represent S.D. of three measurements. | ||
Mechanistic studies of cell penetration by stapled peptides
The aforementioned studies indicate that stapled peptides exhibit better cellular uptake properties than wild-type peptides in general, and that internalization correlated primarily with hydrocarbon staple type and formal peptide charge. However, the mechanism(s) utilized by peptides to translocate across the cell membrane are still unclear. Therefore, we sought to investigate the uptake mechanism(s) for stapled peptides. The uptake mechanism(s) of wild-type CPPs have been extensively studied. Some evidence indicates that they enter cells via energy-dependent endocytosis, which is an active transport process, however data suggesting passive diffusion for CPPs have also been reported; hence, the mechanism(s) of cell uptake by CPPs remains ambiguous.32–34 We first sought to determine whether cell penetration by stapled peptides and wild-type CPPs occurs via ATP-dependent endocytosis.2 Cells were pre-treated with NaN3 and 2-deoxyglucose (2-DG) to reduce cellular ATP levels, and then incubated with FITC-labeled peptides (wild-type and stapled) for 4 hours and compared to normal cells for intracellular fluorescence. Cellular ATP levels were confirmed to be decreased by approximately 90% after NaN3 and 2-DG treatment (Fig. S6†), but Tat and poly-Arg8 exhibited almost identical cellular uptake in ATP-depleted and normal cells, supporting the model that they utilize passive diffusion to translocate across the cell membrane. However, penetratin and all stapled peptides showed 20–50% lower accumulation in ATP-depleted cells, indicating an active trans-membrane process requiring cellular ATP (Fig. 6a). These data indicate that there may be more than one uptake mechanism for CPPs and stapled peptides, but that for the most robust cell penetrating peptides (penetratin and stapled peptides studied here), the internalization mechanism(s) involves ATP-dependent endocytosis.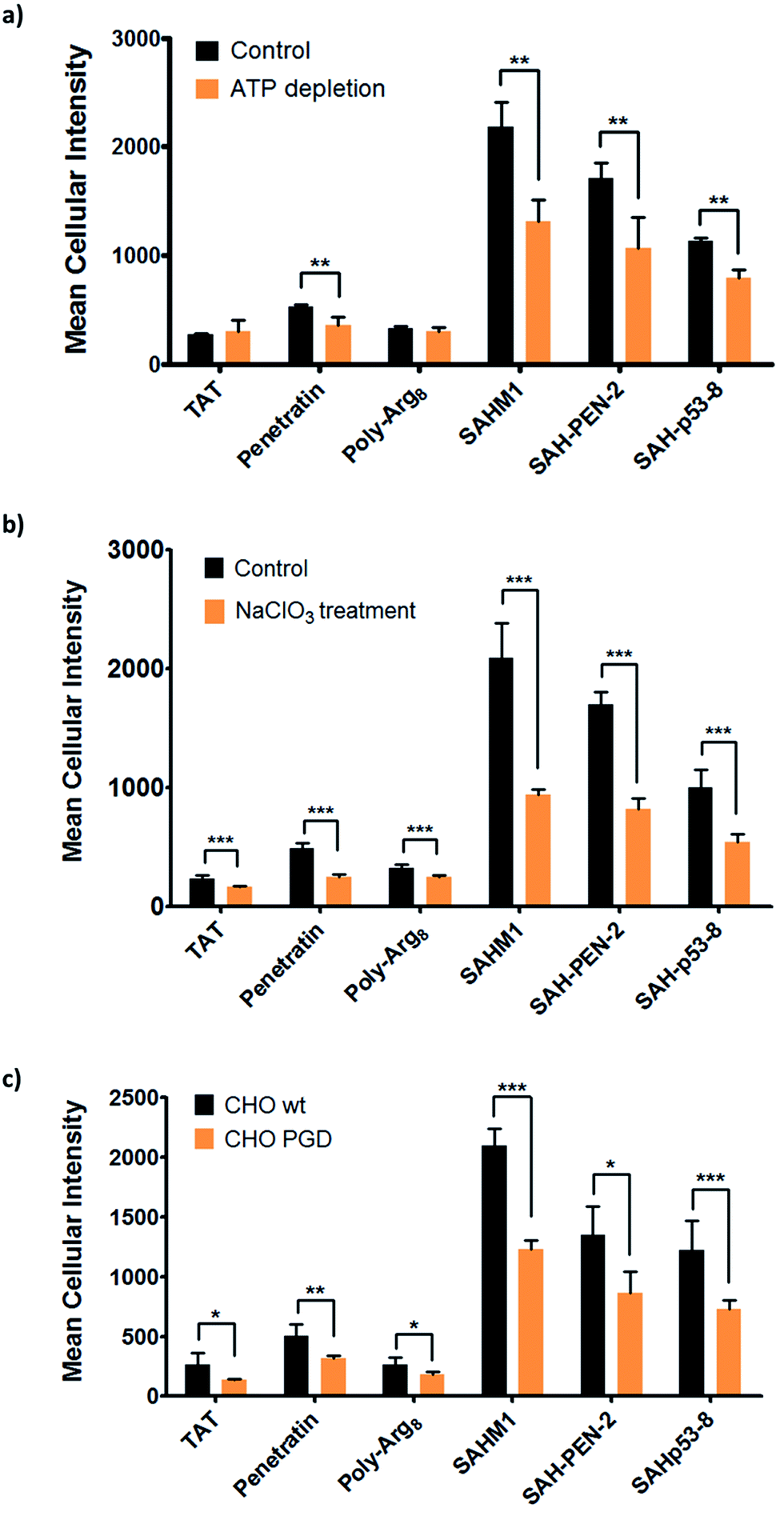 | ||
| Fig. 6 Mechanistic study of cell penetration by stapled peptides and wild-type cell penetrating peptides. (a) Cellular uptake in normal and ATP-depleted cells indicated that stapled peptides penetrate cells via an ATP-dependent endocytosis. (b) Impaired uptake was observed in NaClO3 treated cells, which inhibit proteoglycan biosynthesis. (c) Cell penetration of wild-type and stapled peptides in wild-type CHO and proteoglycan-deficient CHO cells. Experiments were performed in triplicate, and error bars represent S.D. of three measurements.*P < 0.05, **P < 0.01, ***P < 0.001. | ||
Next, we sought to identify the specific pathway(s) utilized for cellular uptake, since energy-dependent endocytosis can be accomplished by several different pathways including caveolin- and clathrin-mediated endocytosis. We repeated the cell penetration experiments under a variety of conditions that each blocked a different endocytosis pathway (Table S3†).35–37 We found that uptake was partially blocked in cells treated with sodium chlorate (Fig. 6b), which aborts the decoration of cells with sulfated proteoglycans, but was unaffected by inhibitors of other endocytic pathways (Fig. S7†). It thus appears that interaction with sulfated proteoglycans is responsible for some, but not all, endocytic uptake of stapled peptides and wild-type CPPs. It is reasonable to connect this result with the previous discovery that peptide charge is a key factor determining cell penetration. Proteoglycans are negatively charged under physiologic conditions due to the occurrence of sulfate groups, and these might form electrostatic pairs with positively charged peptides to facilitate anchoring on the cell membrane.38–40 To further confirm that sulfated proteoglycans are important to mediate cellular uptake for peptides, we performed a secondary assay using wild-type CHO cells (CHO-K1) and proteoglycan-deficient CHO cells (pgsA-745) which harbor a defect in xylosyltransferase, thereby preventing glycosaminoglycan biosynthesis. All peptides showed similar penetration properties in wild-type CHO cells, but uptake was decreased by approximately 50% in proteoglycan-deficient CHO cells, consistent with the experiment using a small molecule inhibitor (Fig. 6c). Taken together, our data suggest that CPPs and stapled peptides penetrate cells through a clathrin- and caveolin-independent endocytosis pathway that is in part mediated by interaction with anionic cell surface proteoglycans. This result is very similar to the previous reports on the mechanism of cellular uptake for supercharged GFP (scGFP), which likewise does not utilize clathrin- or caveolin-mediated endocytosis.41 Notably, scGFP internalization requires actin polymerization, which may not be required for peptide penetration (Fig. S7c†).
In conclusion, we sought to investigate the cell penetration properties of stapled peptides, which is one of the most significant yet poorly understood aspects of peptide stapling technology and cellular transduction technologies in general. In order to address this problem, we developed a high-throughput assay to quantitatively measure stapled peptide intracellular accumulation. Using this assay, we analyzed more than 200 discrete peptides with various sequences, staple positions and types, and distinct physicochemical properties. As a result, we found that stapled peptides penetrate cells more efficiently than unmodified peptides, including well-characterized cell penetrating peptides. For the panel of peptides used in this study, only staple type and formal charge were significantly correlated with cell penetration potential, whereas the other physical parameters did not appear to have a significant effect. We further studied the relationships between cellular uptake and peptide concentration or incubation time, revealing that stapled peptides accumulate in cells in a dose-dependent fashion and reach steady intracellular levels over a course of a few hours. These studies revealed similar time- and dose-dependent behavior for CPPs and stapled peptides, but stapled peptides, including stapled versions of CPPs, were shown to be 10- to 20-fold more penetrant, measured by intracellular fluorescence level at a given dose, than the most potent CPP. We also propose that the specific intracellular accumulation and stabilization kinetics of stapled peptides or unmodified CPPs may be a consequence of equilibria between peptide penetration, cellular proteolysis and/or retrograde transport of the species. Finally, we investigated the mechanism(s) involved in the internalization of stapled peptide and unmodified CPPs and demonstrated that cell penetration occurs through a clathrin- and caveolin-independent, energy-dependent endocytosis pathway that utilizes, in part, sulfated cell surface proteoglycans. This dataset provides significant insight into the physicochemical properties correlated with productive cellular penetration as well as a more detailed understanding of the mechanism(s) utilized by stapled peptides to access intracellular compartments, which together should aid in the design of and characterization of novel stapled peptides in the future.
Acknowledgements
This research was supported by GlaxoSmithKline. We thank J. McGee, R.-J. Sung and S. Berkovitch for useful discussions and comments on the manuscript. We also thank N. Tolliday and T. Hasaka at the Broad Institute for the access to instrumentation. We are grateful for Professor David R. Liu at Harvard University for kindly providing the wild-type CHO cells (CHO-K1) and proteoglycan-deficient CHO cells (pgsA-745). T.N.G. is grateful for a fellowship from Deutsche Akademie der Naturforscher Leopoldina (LPDS 2009-2). J.T.-H.Y. is the recipient of a Susan G. Komen for the Cure fellowship. R.E.M was supported by an AACR Centennial Pre-doctoral Research Fellowship in Cancer Research and an HHMI Fellowship from the Damon Runyon Cancer Research Foundation.References
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed.
- G. L. Verdine and G. J. Hilinski, Methods Enzymol., 2012, 503, 3–33 CAS.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- L. K. Henchey, A. L. Jochim and P. S. Arora, Curr. Opin. Chem. Biol., 2008, 12, 692–697 CrossRef CAS PubMed.
- M. J. P. de Vega, M. I. Garcia-Aranda and R. Gonzalez-Muniz, Med. Res. Rev., 2011, 31, 677–715 Search PubMed.
- P. S. Kutchukian, J. S. Yang, G. L. Verdine and E. I. Shakhnovich, J. Am. Chem. Soc., 2009, 131, 4622–4627 CrossRef CAS PubMed.
- G. H. Bird, N. Madani, A. F. Perry, A. M. Princiotto, J. G. Supko, X. Y. He, E. Gavathiotis, J. G. Sodroski and L. D. Walensky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14093–14098 CrossRef CAS PubMed.
- Y. Q. Long, S. X. Huang, Z. Zawahir, Z. L. Xu, H. Y. Li, T. W. Sanchez, Y. Zhi, S. De Houwer, F. Christ, Z. Debyser and N. Neamati, J. Med. Chem., 2013, 56, 5601–5612 CrossRef CAS PubMed.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K. H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3445–E3454 CrossRef CAS PubMed.
- G. H. Bird, W. C. Crannell and L. D. Walensky, Curr. Protoc. Chem. Biol., 2011, 3, 99–117 Search PubMed.
- Y. W. Kim, T. N. Grossmann and G. L. Verdine, Nat. Protoc., 2011, 6, 761–771 CrossRef CAS PubMed.
- R. E. Moellering, M. Cornejo, T. N. Davis, C. Del Bianco, J. C. Aster, S. C. Blacklow, A. L. Kung, D. G. Gilliland, G. L. Verdine and J. E. Bradner, Nature, 2009, 462, 182-U157 CrossRef PubMed.
- T. N. Grossmann, J. T. H. Yeh, B. R. Bowman, Q. Chu, R. E. Moellering and G. L. Verdine, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17942–17947 CrossRef CAS PubMed.
- W. Kim, G. H. Bird, T. Neff, G. J. Guo, M. A. Kerenyi, L. D. Walensky and S. H. Orkin, Nat. Chem. Biol., 2013, 9, 643–650 CrossRef CAS PubMed.
- Y. W. Kim and G. L. Verdine, Bioorg. Med. Chem. Lett., 2009, 19, 2533–2536 CrossRef CAS PubMed.
- F. Bernal, A. F. Tyler, S. J. Korsmeyer, L. D. Walensky and G. L. Verdine, J. Am. Chem. Soc., 2007, 129, 2456–2457 CrossRef CAS PubMed.
- D. Derossi, A. H. Joliot, G. Chassaing and A. Prochiantz, J. Biol. Chem., 1994, 269, 10444–10450 CAS.
- J. M. Gump and S. F. Dowdy, Trends Mol. Med., 2007, 13, 443–448 CrossRef CAS PubMed.
- S. Futaki, Adv. Drug Delivery Rev., 2005, 57, 547–558 CrossRef CAS PubMed.
- F. Heitz, M. C. Morris and G. Divita, Br. J. Pharmacol., 2009, 157, 195–206 CrossRef CAS PubMed.
- M. Zorko and U. Langel, Adv. Drug Delivery Rev., 2005, 57, 529–545 CrossRef CAS PubMed.
- J. S. Appelbaum, J. R. LaRochelle, B. A. Smith, D. M. Balkin, J. M. Holub and A. Schepartz, Chem. Biol., 2012, 19, 819–830 CrossRef CAS PubMed.
- R. Fischer, K. Kohler, M. Fotin-Mleczek and R. Brock, J. Biol. Chem., 2004, 279, 12625–12635 CrossRef CAS PubMed.
- M. Rodrigues, B. G. de la Torre, D. Andreu and N. C. Santos, Biochim. Biophys. Acta, 2013, 1830, 4554–4563 CrossRef CAS PubMed.
- L. Zhang, J. Yu, H. Pan, P. Hu, Y. Hao, W. Cai, H. Zhu, A. D. Yu, X. Xie, D. Ma and J. Yuan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19023–19028 CrossRef CAS PubMed.
- A. E. Carpenter, Nat. Chem. Biol., 2007, 3, 461–465 CrossRef CAS PubMed.
- S. Stalmans, E. Wynendaele, N. Bracke, B. Gevaert, M. D'Hondt, K. Peremans, C. Burvenich and B. De Spiegeleer, PLoS One, 2013, 8, e71752 CAS.
- D. B. Thompson, R. Villasenor, B. M. Dorr, M. Zerial and D. R. Liu, Chem. Biol., 2012, 19, 831–843 CrossRef CAS PubMed.
- I. Nakase, T. Takeuchi, G. Tanaka and S. Futaki, Adv. Drug Delivery Rev., 2008, 60, 598–607 CrossRef CAS PubMed.
- K. Takada, D. Zhu, G. H. Bird, K. Sukhdeo, J. J. Zhao, M. Mani, M. Lemieux, D. E. Carrasco, J. Ryan, D. Horst, M. Fulciniti, N. C. Munshi, W. Xu, A. L. Kung, R. A. Shivdasani, L. D. Walensky and D. R. Carrasco, Sci. Transl. Med., 2012, 4, 148ra117 Search PubMed.
- S. Deshayes, M. C. Morris, G. Divita and F. Heitz, Cell. Mol. Life Sci., 2005, 62, 1839–1849 CrossRef CAS PubMed.
- I. M. Kaplan, J. S. Wadia and S. F. Dowdy, J. Controlled Release, 2005, 102, 247–253 CrossRef CAS PubMed.
- M. Silhol, M. Tyagi, M. Giacca, B. Lebleu and E. Vives, Eur. J. Biochem., 2002, 269, 494–501 CrossRef CAS.
- J. E. Schnitzer, P. Oh, E. Pinney and J. Allard, J. Cell Biol., 1994, 127, 1217–1232 CrossRef CAS.
- D. Vercauteren, R. E. Vandenbroucke, A. T. Jones, J. Rejman, J. Demeester, S. C. De Smedt, N. N. Sanders and K. Braeckmans, Mol. Ther., 2010, 18, 561–569 CrossRef CAS PubMed.
- C. Lamaze, L. M. Fujimoto, H. L. Yin and S. L. Schmid, J. Biol. Chem., 1997, 272, 20332–20335 CrossRef CAS PubMed.
- N. A. Alhakamy, A. Kaviratna, C. J. Berkland and P. Dhar, Langmuir, 2013, 29, 15336–15349 CrossRef CAS PubMed.
- P. A. Gurnev, S. T. Yang, K. C. Melikov, L. V. Chernomordik and S. M. Bezrukov, Biophys. J., 2013, 104, 1933–1939 CrossRef CAS PubMed.
- N. Schmidt, A. Mishra, G. H. Lai and G. C. Wong, FEBS Lett., 2010, 584, 1806–1813 CrossRef CAS PubMed.
- B. R. McNaughton, J. J. Cronican, D. B. Thompson and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6111–6116 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4md00131a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |