
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selection of LNA-containing DNA aptamers against recombinant human CD73†
Ida C.
Elle
a,
Kasper K.
Karlsen
a,
Mikkel G.
Terp
b,
Niels
Larsen
c,
Ronni
Nielsen
d,
Nicola
Derbyshire
a,
Susanne
Mandrup
d,
Henrik J.
Ditzel
be and
Jesper
Wengel
*a
aNucleic Acid Center, Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense M, Denmark. E-mail: jwe@sdu.dk; Tel: +45-65502510
bInstitute of Molecular Medicine, University of Southern Denmark, J. B. Winsløwsvej 25, 5000 Odense C, Denmark
cDanish Genome Institute, Skt. Lucas Kirkeplads 8, 8000 Aarhus C, Denmark
dDepartment of Biochemistry and Molecular Biology, University of Southern Denmark, Campusvej 55, 5230 Odense M, Denmark
eDepartment of Oncology, Odense University Hospital, Sdr. Boulevard 29, 5000 Odense C, Denmark
First published on 17th February 2015
Abstract
LNA-containing DNA aptamers against CD73 (human ecto-5′-nucleotidase), a protein frequently overexpressed in solid tumours, were isolated by SELEX. A pre-defined stem-loop library, containing LNA in the forward primer region, was enriched with CD73 binding sequences through six rounds of SELEX with recombinant his-tagged CD73 immobilised on anti-his plates. Enriched pools isolated from rounds one, three and six were subjected to next-generation sequencing and analysed for enrichment using custom bioinformatics software. The software identified aptamer sequences via the primers and then performed several steps including sequence unification, clustering and alignment to identify enriched sequences. Three enriched sequences were synthesised for further analysis, two of which showed sequence similarities. These sequences exhibited binding to the recombinant CD73 with KD values of 10 nM and 3.5 nM when tested by surface plasmon resonance. Truncated variants of these aptamers and variants where the LNA nucleotides were substituted for the DNA equivalent also exhibited affinity for the recombinant CD73 in the low nanomolar range. In enzyme inhibition assays with recombinant CD73 the aptamer sequences were able to decrease the activity of the protein. However, the aptamers exhibited no binding to cellular CD73 by flow cytometry analysis likely since the epitope recognised by the aptamer was not available for binding on the cellular protein.
1. Introduction
Aptamers are short oligonucleotides, usually single-stranded and between 15 to 85 nucleotides in length. They form unique, sequence specific, three-dimensional shapes which enable them to bind to a wide range of targets with high specificity and affinity.1–3 Aptamers are generated through Systematic Evolution of Ligands by EXponential enrichment (SELEX), first demonstrated by the labs of Gold4 and Szostak5 in 1990. Several variations of the original selection strategies have been published and have expanded the possible range of aptamer targets and improved aptamer selectivity.6–8 Owing to their extraordinary affinity and specificity aptamers have been dubbed “chemical antibodies”. They possess several advantages over traditional antibodies such as smaller size, low immunogenicity, higher stability, potential of renaturing, as well as easier synthesis and lower batch-to-batch variability.9–11 However, aptamers often require chemical modification to ensure bioavailability and biostability.11,12LNA (Locked Nucleic Acid) is an RNA-mimic containing a 2′-O,4′-C-methylene bridge, which locks the furanose ring in a C3′-endo conformation.13–15 LNA nucleotides have been proven to improve thermal stability of nucleic acid duplexes and confer nuclease-resistance when incorporated into nucleic acid oligomers.16,17 Aptamers containing LNA nucleotides by post-SELEX modification have been reported (reviewed in16 and18), but the effects of these modifications on aptamer function can be difficult to predict. Therefore in investigating the properties of LNA nucleotides in aptamers, we and others, have focused on selecting aptamers using SELEX methods, front-loaded with LNA and other modified nucleotides.19–23
Commercially available polymerases are capable of reading and incorporating LNA nucleotides,22,24,25 but the efficacy of such reactions is often low. Despite this, Kasahara et al.19 recently reported the selection of aptamers containing LNA nucleotides in the primer and the random region. They used capillary electrophoresis and a modified SELEX protocol, which incorporated a primer extension step in each round. Following eleven rounds of selection, two groups of aptamer candidates were identified by cloning and sequencing of the enriched pool. The aptamer with the highest target affinity was investigated further for the effects of the LNA-nucleotides on its affinity and specificity. For this particular aptamer, the LNA-nucleotides were shown to be necessary for target binding,20 but previous work from the same group has demonstrated that this is not always the case.19
We have attempted to exploit the potential benefits of LNA inclusion in aptamer sequences whilst maintaining a simple SELEX protocol with good pool regeneration at the end of each round. A library was designed containing six LNA nucleotides in the forward primer region (Fig. 1). The design requires the polymerase to read the modified nucleotides but not to incorporate them during library amplification. The primer regions were partially complementary forcing formation of a 7 bp stem containing two of the LNAs, while the remaining four LNAs were free to participate in formation of other secondary structures or in target binding (also Fig. 1). This type of library design promotes the involvement of the primer regions in the secondary structures and potentially in target binding. This means that post-selection removal of primer sequences for truncation would be expected to have detrimental consequences on the folding stability and target binding of the aptamer sequences.
 | ||
| Fig. 1 Library design used for the selection of CD73 aptamers. Primers were designed to form a pre-determined 7 bp stem containing two LNA-T nucleotides. Complementary regions forming this stem are underlined and LNA nucleotides are in red. The structure of the library was predicted by Mfold26 and is shown above. LNA nucleotides are marked by red circles. | ||
The specificity of aptamers makes them promising candidates for targeting sub-populations of cells displaying abnormal expression levels of certain proteins; e.g. cancer cells. The ecto-5′-nucleotidase, CD73, is a GPI-anchored, homodimeric, cell surface protein overexpressed in many solid tumours. CD73 hydrolyses extracellular AMP to adenosine, a potent immuno-suppressor. Therefore it has been suggested that tumour-derived CD73 is a mediator of tumour immune-escape.27 Antibody therapy against CD73 expressing cells inhibits breast tumour growth and metastasis,28,29 while RNAi-mediated suppression of CD73 expression induces cell-cycle arrest and apoptosis in the breast-cancer cell line MB-MDA-231.30 An aptamer binding to cellular CD73 could be useful as a direct inhibitor of enzymatic activity or as part of a delivery vehicle for cytotoxic drugs to cancer cells.
Here, LNA-containing aptamers recognising recombinant human CD73 were isolated via six rounds of SELEX coupled to screening for sequence enrichment with next-generation sequencing (NGS) and a customised bioinformatics platform. The isolated sequences exhibited low nanomolar affinity for the recombinant protein by surface plasmon resonance (SPR) analysis, and some also inhibited the recombinant protein's enzyme activity. The same was true of the truncated and modified sequences derived from the full length aptamers. However, none of the sequences displayed binding to cellular CD73 by flow cytometry analysis. SPRi was utilized to partially map the aptamer–protein interaction through a series of control and competition injections and suggests that the binding site of the aptamer sequences is sterically unavailable in the cellular protein.
2. Materials and methods
2.1. Oligonucleotides
The LNA-containing library, consisting of a 30 nt random region flanked by 20 nt primer regions, and all aptamer candidates were synthesised using an automated DNA synthesiser and standard phosphoramidite chemistry. Aptamer candidates were synthesised with 5′-FAM and 3′-biotin labels for analytical purposes. 3′-Biotin-TEG oligonucleotides were cleaved from the support matrix by treatment with saturated ammonium hydroxide at 55 °C for 12 h. Library purity and profile was evaluated by ion-exchange HPLC and MALDI-MS and broad peaks were identified reflecting the diverse library composition. LNA-containing forward primer (5′-FAM-C![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) CCAGACA
CCAGACA![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) AACTTC-3′ LNA bases underlined) was purchased from Exiqon A/S (Vedbæk, Denmark), and the biotin-tagged reverse primer (5′-biotin-CACAGTAGCGATACTCCATG-3′) was purchased from Sigma-Aldrich.
AACTTC-3′ LNA bases underlined) was purchased from Exiqon A/S (Vedbæk, Denmark), and the biotin-tagged reverse primer (5′-biotin-CACAGTAGCGATACTCCATG-3′) was purchased from Sigma-Aldrich.
2.2. Proteins and antibodies
Recombinant his-tagged human CD73 for selections and binding assays was purchased from NovoProtein (C446; New Jersey USA). His-tagged sortilin was used for negative selection steps and as a control in binding experiments. Human IgG and lysozyme used as controls for binding assays were purchased from Sigma-Aldrich. Recombinant N-terminal CD73 and anti-his tag antibody were purchased from Abcam. Other antibodies for binding assays were all commercially available, IE9 (Santa Cruz Biotechnologies), AD2 (BD Bioscience) and IgG1 (HYP 123-05; Bioporto).2.3. Selection
In selection round 1, 1000 pmol of library was dissolved in 100 μL selection buffer (1 × PBS + 0.005% Tween 20) with 4.5 mM MgCl2. The library was folded by incubation at 95 °C/10 min followed by 10 min on ice. Competitor RNA, Brewer's yeast tRNA (Roche Applied Science), was added at a 300 fold excess to reduce non-specific aptamer interactions. Recombinant his-tagged CD73 (5 pmol) was immobilised on anti-his-tag plate wells for positive selections. Negative selections were performed by pre-incubation of the library/pool with an empty anti-his-tag plate well for 1 h/RT/700 rpm. Positive selections were performed by incubation of supernatant, collected from negative selections, in CD73 coated anti his plate wells. Table 1 outlines the progression of stringency during the selections rounds which was achieved by adjusting the ratio of library to target, number of washes and additional negative selection steps against an unrelated his-tagged protein, sortilin. 200 pmol of the enriched library was used in rounds 2–6.| Round | Target![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :pool ratio :pool ratio |
Quick washes (μL) | 5 min washes (μL) | Negative selection |
|---|---|---|---|---|
| 1 | 1:200 |
5 × 200 | None | Empty plate |
| 2 | 1:40 |
8 × 200 | 2 × 200 | Empty plate |
| 3 | 1:200 |
4 × 200 | 1 × 200 | Empty plate and His-tagged sortilin |
| 4 | 1:500 |
8 × 200 | 2 × 200 | Empty plate |
| 5 | 1:1000 |
4 × 200 | 1 × 200 | Empty plate |
| 6 | 1:1000 |
8 × 200 | 2 × 200 | Empty plate and His-tagged sortilin |
Competition elutions were performed in selection buffer with a 10 fold excess of free CD73 compared to the immobilised protein and incubation at 37 °C/30 min/700 rpm. Eluted pools were amplified using KOD XL (Novagen, Merck Millipore, Darmstadt, Germany), FAM-labelled forward primer and biotin-labelled reverse primer in 25 cycles [98 °C/2 min followed by 25 cycles of 98 °C/15 s, 63 °C/15 s, 72 °C/1 min with a final extension step of 72 °C/2 min]. PCR products were verified on 10% denaturing acrylamide gels. Enriched pools were regenerated by strand separation using Dynabeads M-280 streptavidin (Invitrogen, Life Technologies) according to the manufacturer's protocol, and quantified by UV spectroscopy using a NanoDrop 2000 (Thermo Scientific).
2.4. Sequencing
Enriched pools from selection rounds one, three and six were amplified using unmodified all-DNA primers and the PCR-product purified from 4% agarose gels and prepared for Illumina HiSeq 1500 sequencing according to Nielsen and Mandrup.31 Data from individual samples were sorted based on Illumina index sequences prior to more complex bioinformatic analysis.2.5. Bioinformatics
Sequencing datasets were analysed using BION-apta package, custom software written by the Danish Genome Institute. During analysis sequential steps from a “pipeline” or “recipe” are performed drawing information from files containing raw sequences, a file defining the primers, library design, and the random region length and a file with output table parameters. The output from each step is input for the next and is available for inspection after analysis. Primer sequences are used to identify the sequences for analysis, then are cleaved resulting in a table of random regions only. Low quality sequences are filtered out and a maximum and minimum length filter is applied. Sequence unification collapses identical sequences into one and labels the sequence with a read count. Similarity-based clustering, alignment and consensus sequence extraction is then performed by the BION-apta software before sequence grouping and small conserved pattern discovery. This creates a reduced number of abundance tables displaying sequences sharing a common motif, making aptamer candidate and “family” discovery easier. Additional higher-level features can be added, such as covariance-discovery and graphics. BION-apta software also contains tools that can interactively search the output tables for user-specified primary and/or secondary structure motifs, and filter out unwanted sequences such as primer-repeats. The recipe and the outputs are available for download (see Availability below).2.6. Bio-layer interferometry
Aptamer candidates were qualitatively screened for target binding using a BLItz (FortéBio) and streptavidin-covered biosensors hydrated for 20 min in selection buffer before use. All binding assays were run in the “Advanced Kinetics” mode with an initial baseline of 120 s (tube), 120 s aptamer loading (drop), 120 s baseline (drop), 120 s association (drop), and 120 s dissociation (tube). The drop holder was cleaned three times in selection buffer between each step. 3′-Biotin-tagged aptamer candidates were diluted to 50 μM in selection buffer for immobilisation, and recombinant his-tagged CD73 was diluted to 1.7 μM in selection buffer prior to analysis.2.7. Surface plasmon resonance
Kinetic analyses of 3′-biotin-tagged aptamer candidates were carried out on a BIAcore X100 at room temperature with a CAP chip. Theoretical immobilisation levels yielding Rmax values of approximately 100 RU were calculated. The aptamer candidates were immobilised in selection buffer at the appropriate concentrations (0.025–0.1 μM) and a contact time of 60 s, except for the shorter candidates NAC6749, NAC6788, NAC6853 and NAC6970, which required a contact time of 180 s to yield reproducible KD values. Kinetic analyses using two-fold dilutions of analyte (recombinant his-tagged CD73; 0.0125–0.2 μM) were performed at least twice for each candidate. The analyte was injected with a flow-rate of 30 μL min−1 and a contact time of 180 s followed by 300 s dissociation. Regeneration of the CAP chip was achieved using the manufacturer supplied buffer and a 120 s injection. Sensorgrams were analysed and affinities calculated using BIAevalution software.2.8. Inhibition of CD73 enzymatic activity
Recombinant his-tagged CD73 was dissolved to 17 nM in 100 μL of glycine buffer (75 mM glycine, 5 mM MgSO4, pH 7.4) containing 1 μM aptamer candidate or α,β-methylene ADP (APCP) (Sigma Aldrich). After 1 h incubation with/without AMP at 37 °C, reactions were terminated by the addition of 50 μL of 0.5 M H2SO4. Conversion of AMP to adenosine was quantified by addition of 50 μL phosphate reaction solution (0.4% NH4-molybdate, 10% ascorbic acid) and incubation for 30 min at room temperature. Absorption measurements at 560 nm were acquired using a Victor3 Multilabel Plate Reader (PerkinElmer Life Sciences). Catalytic activity was determined by subtracting sample values in the absence of AMP from the corresponding values in the presence of AMP.2.9. Flow cytometry
MDA-MB-231 cells (2 × 105) were harvested using scraping and incubated in 0.05 μM aptamer or 1 μg ml−1 Alexa Fluor-488 labelled anti-CD73 mAb (AD2 or 1E9) diluted in 1 × PBS 0.005% Tween 20 for 1 h at 4 °C. Following three washing steps in 1 × PBS 0.005% Tween 20 the cells were analysed by flow cytometry using a FACScalibur (BD Bioscience). The recorded data was analysed in Flowjo X software.2.10. Surface plasmon resonance imaging
Serial dilutions (62.5–2000 nM) of 3′-biotin-tagged aptamer candidates in 50 mM sodium acetate buffer pH 4.5 were immobilised on a G-Strep SensEye (Ssens, Enschede, The Netherlands) using 20 min contact time on a continuous flow microspotter (IBIS technologies, Enschede, The Netherlands). Aptamer analyte interactions spotted on SensEyes were monitored on a MX-96 SPR imager (IBIS technologies). All analytes were injected at 250 nM each. Selection buffer was used as the running and analyte dilution buffer, association was monitored for 20 min and dissociation for 30 min with a flow rate of 12 μL s−1. Regeneration was achieved using 5 M NaCl (1 min) followed by 0.5% SDS (1 min). Sensorgrams were analysed and kinetics determined using SPRint software.3. Results and discussion
3.1. Aptamer evolution and identification by NGS and bio-layer interferometry
Traditional SELEX consists of approximately ten rounds of selection followed by cloning and sequencing of the final pool. Typically, 50–200 bacterial clones would be randomly picked and sequenced. Over-represented sequences would then be synthesised and tested for binding. However, in pools that have not been highly enriched or where significant PCR bias exists, these numbers may prove insufficient to identify true binding sequences. The advent of NGS technologies make it feasible to have entire pools from multiple selection rounds analysed and to monitor enrichment of sequences or sequence families rather than copy number. This provides a much more complete picture of selection progress and, in principle, a greater success rate for identification of functional aptamer sequences.32 It has even been demonstrated that, with the right bioinformatic tools, it is possible to identify aptamers even after only one round of selection.33–35We developed custom software for the analysis of sequencing data from aptamer pools. This software cleaved off primer sequences, filtered out sequences of low quality and clustered unique sequences. Information from the resulting sequence tables for each pool analysed, such as sequences and read number were used to calculate enrichment. Using the number of reads for a unique sequence in each round and the total number of reads in that round it was possible to calculate the enrichment of a sequence using ordinary spreadsheet software. We analysed selection pools 1, 3 and 6 from SELEX of a library with a pre-defined stem-loop against the recombinant his-tagged protein CD73. Table 2 shows the ten, overall most abundant sequences and their enrichment factors over the course of selection. Since sequence read length was limited by reagent kits and fell short of a full sequence read, it was necessary to identify pairs of sequences (the complementary sequences to each other converted to the same orientation) in which the central 20 nucleotides were identical and the read counts comparable (Table 3). These pairs of sequences were merged into a single, full length sequence read (Table 4). Whilst this merging of shorter sequencing reads could have been avoided by use of an extra, costly reagent kit in a single sequencing run, the process also served as an additional quality control step.
| Read count | Enrichment factora | |||||||
|---|---|---|---|---|---|---|---|---|
| ID | Round 1 | Round 3 | Round 6 | Total | 1–3 | 3–6 | 1–6 | Sequence of random region |
| a Since it is not possible to calculate an enrichment factor from a zero read count, all zero values was substituted with one in the enrichment calculation. | ||||||||
| Total | 1084854 |
125333 |
53019 |
1263206 |
||||
| 1709 | 371 | 52 | 3 | 426 | 1 | 0 | 0 | CGTGGGTGGGGAGGGTGGCTAGTTA |
| 1157 | 222 | 21 | 0 | 243 | 1 | 0 | 0 | AGCGTCGTGGGTGGGGAGGGTGGCT |
| 1156 | 26 | 14 | 32 | 72 | 5 | 5 | 25 | GAGAGCGCATGGACCAACGCTAGTG |
| 1155 | 0 | 1 | 51 | 52 | 9 | 121 | 1044 | GGGTGGGTAGGGTGGGCAGGAACGT |
| 1154 | 14 | 17 | 16 | 47 | 11 | 2 | 23 | CGCATGGACCAACGCTAGTGCAAGA |
| 1153 | 0 | 1 | 44 | 45 | 9 | 104 | 900 | CGCACCGGCATTTGTTACAGGAAGC |
| 1152 | 0 | 0 | 39 | 39 | 9 | 92 | 798 | CGGCATTTGTTACAGGAAGCGCGGT |
| 1151 | 15 | 4 | 3 | 22 | 2 | 2 | 4 | ATGCAGGGAGAATGCAGCTACCCAG |
| 1150 | 0 | 0 | 18 | 18 | 9 | 43 | 368 | GGTGGGTGGGCGTCTCTGTCCGGGT |
| 1149 | 8 | 7 | 2 | 17 | 8 | 1 | 5 | AAGCCGACTGCAGTGTGGAATGCAG |
| Read count | Enrichment factor | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pair no. | ID | R 1 | R 3 | R 6 | Total | 1–3 | 3–6 | 1–6 | Sequence of random region (aligned) |
| Total | 1084854 |
125333 |
53019 |
1263206 |
|||||
| 1 | 1156 | 26 | 14 | 32 | 72 | 5 | 5 | 25 |

|
| 1 | 1154 | 14 | 17 | 16 | 47 | 11 | 2 | 23 |

|
| 2 | 1153 | 0 | 1 | 44 | 45 | 9 | 104 | 900 |

|
| 2 | 1152 | 0 | 0 | 39 | 39 | 9 | 92 | 798 |
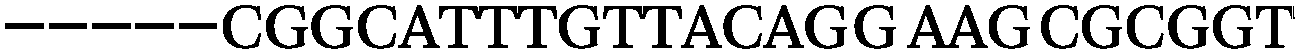
|
| 3 | 1155 | 0 | 1 | 51 | 52 | 9 | 121 | 1044 |

|
| 3 | 1183 | 0 | 0 | 8 | 8 | 9 | 19 | 164 |

|
| 4 | 1148 | 0 | 0 | 16 | 16 | 9 | 38 | 327 |

|
| 4 | 1145 | 0 | 0 | 8 | 8 | 9 | 19 | 164 |

|
| Name | Sequence pairs | Aptamer candidates |
|---|---|---|
| NAC6551 |

|

|

|
||
| NAC6552 |

|

|

|
||
| NAC6604 |
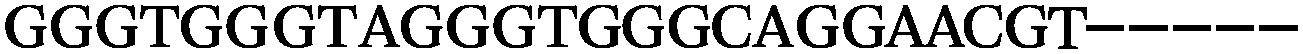
|

|

|
||
| NAC6772 |

|

|

|
||
This analysis yielded three potentially interesting aptamer candidates for qualitative analysis of target binding by BLItz bio-layer interferometry. Pre-folded aptamer candidates were immobilised on streptavidin-coated biosensors, while recombinant his-tagged CD73 and his-tagged sortilin were in solution. Both proteins exhibited no binding to empty streptavidin-coated biosensors. One of these candidates, NAC6552, bound specifically to the recombinant his-tagged CD73. NAC6551 exhibited no binding, and NAC6604 showed weak binding to both CD73 and his-tagged sortilin indicating that it might bind to the his-tag of both proteins (data not shown). Despite successful immobilisation of his-tagged CD73 on NTA biosensors, the reverse experiment, protein ligand and aptamer analyte, failed to identify binders. This is probably due to the relatively small size of the aptamer candidates vs. CD73, a phenomenon also seen in SPR experiments.36
The sequence of the CD73 binding aptamer, NAC6552, was used to further filter the sequencing data from these selection pools for sequences where the central 16 nucleotide segment was either identical or contained a maximum of three mismatches. This filtering process yielded an additional aptamer candidate (NAC6772) that resembled NAC6552 in the central region (also shown in Tables 3 and 4). Analysis by bio-layer interferometry showed that NAC6772 was also able to bind CD73 specifically.
The identification of candidate aptamer sequences after selection is clearly shifting from traditional cloning and Sanger sequencing to NGS-based approaches, and several groups have presented methods and software for analysis of NGS-data in the context of aptamer selection.11,32,35,37–40 Through the modification of the files defining the primers and library our software is applicable to all aptamer libraries. Several of the program steps are similar to steps in the program Aptaligner published by Lu et al.37 We believe that tandem use of NGS and well-designed analysis software allows a much more complete evaluation of the progress of a selection. It also enables the identification of genuinely enriched sequences from sequences resulting from PCR bias. The software described here is available free of charge at http://www2.sdu.dk/multi/bion-apta. Use of NGS and the associated bioinformatics to identify sequences of interest is limited only in its inability to calculate functionality of said sequences. As such it is necessary to have effective means of testing the sequences of interest for functionality. Bio-layer interferometry on the BLItz system appears to be a very effective means of screening the binding capabilities of individual aptamer candidates. The experimental demand on regents and time for a single aptamer is extremely low vs. methods such as SPR. With just a few μL of low μM aptamers and targets it is possible to discern the potential of an aptamer candidate in less than 15 minutes. However, with no automation, testing of batches of aptamer candidates can become tedious and time-consuming compared to more high throughput methods such as SPRi described below.
3.2. Folding and truncation
Following the successful identification of two binding sequences with high sequence homology, secondary structure predictions by Mfold26 were used to compile a selection of truncated sequences that maintained or partially maintained similar secondary structures in the random region (Fig. 2). | ||
| Fig. 2 Secondary structure predictions and KD values for CD73 binding aptamers and derived truncations. NAC6552 and NAC6772 were truncated step-by-step as indicated by the arrows and secondary structures predicted by Mfold.26 LNA nucleotides are marked by red circles. All-DNA versions of NAC6552, NAC6749, NAC6772, and NAC6853 are not shown but indicated by sequence number. “*” indicates that KD values could not be determined for this aptamer. | ||
Despite such sequence similarity the most favourable predicted fold for NAC6552, the originally identified sequence, was somewhat different to that of NAC6772 (Fig. 3). However, it was not surprising to see that it was possible for NAC6772 to also form a secondary structure that produced a similar stem-bulge-stem-loop, with only three bases in the loop section differing. Fig. 2 shows this more similar secondary structure in an attempt to illustrate the rationale for the specific truncations tested. NAC6746, NAC6747, NAC6749 and NAC6748 were composed of the random region of NAC6552 and gradually reduced primer regions. Likewise, NAC6853 was a truncation of the primers from NAC6772. The remaining candidates depicted were attempts to alter the loop of the secondary structures to partially define bases essential for target binding.
 | ||
| Fig. 3 Most favorable secondary structure predictions for NAC6772/NAC6750 (with and without LNA bases, respectively) and its truncated version NAC6853/NAC6970 suggested by Mfold.26 LNA nucleotides are marked by red circles. | ||
The conformational changes in an aptamer's secondary and tertiary structures that may be induced on target binding make it difficult to predict which bases and structures formed by a particular sequence are necessary for functionality. Despite this, secondary structure predictions often provide a useful starting point when considering truncation and mutation sites. Here, with the help of these predictions, we were able to make strategic modifications (truncations and LNA to DNA nucleotide substitutions) to the two CD73 binding sequences.
3.3. Surface plasmon resonance
Aptamer-target kinetics for all candidates were determined with SPR using a BIAcore X100. Aptamers were tested in the order that they were designed and the KD values are provided in Table 5. This permitted monitoring of the minimum length of a functional CD73 aptamer derived from the two original binding sequences (NAC6552 and NAC6772) and the necessity of the loop region in the secondary structure predictions. Substitution of LNA nucleotides with the DNA equivalents were analysed to determine if LNA was integral to CD73 binding.| Library | Sequence (5′–3′) | ||
|---|---|---|---|
| a Kinetic analyses were performed in at least duplicates and mean ± standard deviation is shown. Enzyme inhibition activity of recombinant CD73 was determined colorimetrically and normalized to background levels (no AMP present) for each candidate. A560 values of CD73 alone and with each aptamer were used to derive percentage inhibition values. | |||
| NAC5613 |

|
— | — |

















The KD value of the parent aptamer candidate NAC6552 was 9.9 ± 0.04 nM. A minor truncation to NAC6552 of the 13 nucleotide 3′ “tail” of the reverse primer region (resulting sequence NAC6746) improved the KD slightly to 6.5 ± 0.79 nM. NAC6747, further reduced by five nucleotides in the 5′ and 3′ regions performed equally well to the parent aptamer with a KD value of 9.3 ± 0.04 nM. However, further truncation, removing all of the reverse primer and leaving only the last six nucleotides of the forward primer (sequence NAC6749) had a slightly detrimental effect of the KD 15.4 ± 0.35 nM. The final truncation, where NAC6552 was reduced to 26 nucleotides of the random region, lead to a molecule (NAC6748) which was unable to bind CD73 in SPR assays.
The KD value of parent sequence NAC6772 was 3.54 ± 0.59 nM. A truncated variant of this (NAC6853), similar in design to NAC6749 was determined to have a KD of 12.5 ± 1.14 nM suggesting that truncation of this particular sequence was tolerated to approximately the same extent as for NAC6552 and therefore further truncations were not tested.
Reduction of the loop region of the shortest functional variant of NAC6552, in sequences NAC6854 and NAC6789 eradicated CD73 binding. Whilst substitution of the LNA nucleotides with the DNA equivalent in both the parent strands (NAC6552 and NAC6772) and the shortest functional variants of these (NAC6749 and NAC6853, respectively) retained comparable CD73 binding. Data showed the KD of the all DNA version of NAC6552 to be 5.48 ± 0.04 nM vs. 9.94 ± 0.07 nM and the KD of the all DNA version of NAC6772 to be 4.04 ± 0.28 nM vs. 3.54 ± 0.59 nM. Data also showed the all DNA version of NAC6749 to have a KD of 13.20 ± 1.84 nM vs. 15.40 ± 0.34 nM and finally the all DNA version of NAC6853s KD to be 19.00 ± 0.94 nM vs. 12.50 ± 1.14 nM. This final variant did behave somewhat differently to the other sequences during SPR requiring higher aptamer concentrations (0.1 μM compared to 0.025 μM) to achieve the appropriate response levels. This indicated that the actual capture level may have deviated from the calculated one. It is possible that the secondary structure of this aptamer required for CD73 binding is less stable than for other candidates and therefore higher immobilisation concentrations are required to produce the sufficient response level.
Collectively, these affinity experiments suggest that only small parts of the primer regions adjacent to the random region and the loop seen in secondary structure predictions are requirements for CD73 binding. We hypothesise that whilst the loop forms the majority of aptamer–target interactions, the primer region stabilises the formed structure. The results also demonstrated that despite the design of the library to promote LNA involvement in the binding regions, LNA were not fundamental for target binding with very little difference in affinity seen when substituting LNA for DNA. They also show that only a minimal part of the 3′-end of the forward primer was required to keep the functionality intact. This finding is in line with the findings of Kasahara et al. who saw that target affinity was improved for certain structural motifs and reduced for others when a DNA-primer was replaced with an LNA-containing version for aptamer production.19,20 Similarly, we have found that single LNA nucleotide substitutions in an avidin-binding DNA aptamer can significantly impact aptamer binding.41
3.4. Inhibition of CD73 enzymatic activity
To elaborate on the binding results, the enzymatic inhibition of recombinant his-tagged CD73 by the aptamer candidates was measured. The AMP-mimic, APCP, was used as a positive control of inhibition and sample readings (CD73 plus aptamer and AMP) were normalized against background signals (CD73 plus aptamer). A560 readings of CD73 ± aptamers were used to calculate the percentage inhibition. Binding affinity does not always correlate with inhibitory effect, so it is not possible to accurately quantitate affinity based on inhibition. However, with all except one aptamer candidate (NAC6748) the enzymatic activity of the recombinant CD73 was reduced in the presence of aptamers proven to bind by bio-layer interferometry and SPR (Table 5).NAC6748 was an interesting case. This was the shortest aptamer candidate synthesised and CD73 binding was not detectable by bio-layer interferometry or SPR. The fact that it was able to inhibit the activity of CD73 to a similar level to sequences seen to bind by SPR suggests that it forms a similar interaction with CD73 to its longer counter parts. This postulates the question of why binding was not observed in earlier assays. It is possible that in ligand immobilised assays with such short aptamers the aptamer–target interaction could be sterically hindered by the proximity of the binding region to the capture surface. This could be confirmed by synthesis of the short sequence with a spacer prior to the biotin but this was not performed here.
3.5. Binding of cellular CD73 by aptamer candidate measured by flow cytometry
Flow cytometry was utilized to investigate whether the aptamer candidates were able to bind to CD73 expressed on live cells (MDA-MB-231 human breast cancer cells). None of the 17 aptamers showed any binding to CD73 on live breast cancer cells when compared to the negative control aptamer NAC6551 (Fig. 4A–C). Aptamer NAC6748, shown as non-binding in the affinity analysis although inhibiting the enzymatic activity of recombinant CD73, also showed no binding to CD73 on breast cancer cells (Fig. 4D). | ||
| Fig. 4 Flow cytometry analysis of aptamer binding to MDA-MB-231 breast cancer cells. (A) Each point represents the logarithm-transformed values of the geometric mean of the signal intensity from flow cytometry analysis of each of 17 different CD73 aptamers and two anti-CD73 mAb (AD2 and 1E9). Points representing unstained cells and cells stained with aptamer NAC6748, NAC6551, NAC6746 and anti-CD73 mAb AD2 and 1E9, respectively, are specified. (B–D). Staining profile of aptamers (NAC6746, NAC6551, NAC6748, respectively, gray tinted), unstained (black dashed), 1E9 (black solid line) and AD2 (black dotted line) on MDA-MB-231 cells. | ||
In light of this finding the enzymatic assay was repeated replacing the recombinant his-tagged CD73 with MDA-MB-231 cells. Unfortunately, no inhibition of the cellular protein's activity was seen with any of the aptamer candidates (data not shown), including NAC6748.
When trying to isolate aptamers against cellular proteins, compromises in the SELEX process are sometimes necessary. Direct selection against cellular proteins is possible42–45 but is challenging due to the complexity of cell surfaces and requires access to cell culture facilities. Using a purified recombinant protein as the target is a compromise. It simplifies the selection process but creates potential for evolution of aptamers that bind to parts of the protein that may be sterically unavailable in the cellular form. In this case, cellular CD73 is a GPI-anchored, cell-surface protein that forms a homodimer through non-covalent bonds,46 whilst the recombinant protein is a his-tagged protein expressed as a monomer without the GPI-anchor. This recombinant CD73 may or may not remain as a monomer after purification. Areas of CD73, such as the region around the GPI-anchor, near the cellular surface, may be inaccessible in the cellular vs. recombinant form. This may be the reason why we observed no binding of the aptamers to cellular CD73 in flow cytometry or inhibition of cellular CD73 activity. Though in this work a focus was on the technical aspects of the SELEX process and therefore only recombinant CD73 was used as the target for selection, we were interested to partially map the binding of the aptamer candidates to CD73 and utilised a series of analyte competition injections and SPRi to do so.
3.6 Aptamer–target interaction mapping and specificity by SPR imaging
SPRi was employed to verify the specificity of the aptamer candidates and attempt to narrow down the region of the protein the aptamers bind to. KD values for the aptamer–target interactions determined by SPRi were comparable to those determined by SPR (ESI,† Table S1). None of the aptamer candidates, with the exception of NAC6604 discussed previously, exhibited binding to the other proteins tested (lysozyme, thrombin, streptavidin and his-tagged sortilin) indicating that the aptamers were in fact specific for CD73 (Table S1, ESI†).No aptamer binding of AMP was observed and co-injection of CD73 with AMP showed no reduction in affinity of the aptamer candidates (example shown in Fig. 5). This would indicate that the aptamers do not bind directly to CD73′s active site, which is in the C-terminal portion of the protein, but instead inhibit the enzymatic activity by changing the conformation of the protein or limiting the conformational change required for activity.
 | ||
| Fig. 5 Partial NAC6552 aptamer-CD73 mapping by SPRi. (A) Sensorgrams of negative control injections. Buffer, AMP, lysozyme, his-tagged sortilin, CD73 binding antibodies (AD2 and IE9), anti-his tag antibody and non-CD73 binding antibodies (IgG1 and human IgG) showed no interaction with the immobilised aptamers. (B) Sensorgrams of competition injections. Aptamers showed specificity for CD73 but not the N-terminal portion of CD73 and did not bind competitively with any of the antibodies shown in (A) or AMP. (C) Summary table of SPRi injections, affinities and conclusions. Binding sensitivity changed only slightly between the co-injections of potentially competitive analytes (15.1–24.0 nM) and the CD73 alone (16.8 nM). | ||
It was expected that when performing co-injections of CD73 and anti-CD73 antibodies (AD2 and IE9) and negative control antibodies (human IgG, IgG1 and anti-his tag), that competitive binding would knock out binding. However, non-competitive binding would lead to an increased response owning to the larger analyte (protein plus antibody) but the KD would remain unchanged. None of the aptamers bound competitively to the AD2 binding site but it was not possible to comment on the interaction with IE9 owning to a buffer response obscuring the picture (Fig. 5). The negative controls also had no effect on binding of CD73. These data indicate that the aptamers bind recombinant his-tagged CD73 in regions distal to the antibodies which can bind cellular or recombinant CD73.
Finally, none of the aptamer candidates bound to a recombinant N-terminal CD73 fragment indicating that the aptamer candidates bound CD73 in the C-terminal domain of the protein. In cellular CD73, the GPI-anchor is at the C-terminal meaning that part of the C-terminal domain will be in contact with the cell surface and unavailable for binding. Further to this it is the C-terminal that is involved in formation of the cellular homodimer protein possibly restricting aptamer access.
Without complicated structural analysis of the recombinant his-tagged CD73 complexed with a binding aptamer candidate by crystallography or NMR it is impossible to discern the actual binding site. Doing so could determine the reason why the CD73 aptamer candidates were able to bind and inhibit the recombinant but not the cellular form of the protein. However, by using SPRi it has been determined that the aptamers bind to the C-terminal domain and within this domain we know that significant portions of the protein are occupied by interactions with the cell surface or protein dimerization which could hinder aptamer–target interactions.
4. Conclusions
Aptamers containing LNA nucleotides were selected against recombinant human CD73 using SELEX and NGS with custom NGS bioinformatics software. Two similar sequences where identified that bind CD73 with high affinity and good specificity. Several truncated and modified versions of these aptamers, the shortest of which was 34 nts, were shown to also bind to the target with low nanomolar affinities. It was shown that the LNA nucleotides had little impact on aptamer affinities but that reduction of the loop section predicted by Mfold was extremely detrimental to CD73 binding. These CD73 aptamers were also able to inhibit the enzyme activity of recombinant CD73, but were ineffective against cellular CD73 in the same assay and showed no binding during flow cytometry. These aptamers were shown to bind to the C-terminal domain of CD73 which may go some way to explain their lack of function with cellular CD73. Despite their lack of affinity for cellular CD73, these aptamers could find a use in research or as a capture agent in the purification of recombinant CD73. This work presents the first example of aptamers against CD73, the first evolution of LNA-containing aptamers selected using traditional SELEX, introduces the BION-apta software as a highly flexible tool for analyzing NGS data in the context of aptamer evolution, and introduces SPRi as a tool of partial mapping of aptamer binding sites.Abbreviations
| 3′-Biotin-TEG CPG | 1-Dimethoxytrityloxy-3-O-(N-biotinyl-3-aminopropyl)-triethyleneglycolglyceryl-2-O-succinyl long chain alkylamino-controlled pore glass |
| APCP | α,β-Methylene ADP |
| bp | Base pairs |
| CD73 | Ecto-5′-nucleotidase |
| GPI | Glycosylphosphatidylinositol |
| K D | Equilibrium dissociation constant |
| LNA | Locked nucleic acid |
| NGS | Next-generation sequencing |
| SELEX | Systematic evolution of ligands by exponential enrichment |
| SPR | Surface plasmon resonance |
Acknowledgements
We greatly appreciate support from the European Research Council under the European Union's Seventh Framework program (FP7/2007-2013)/ERC Grant Agreement No. 268776, Danish Cancer Society and Danish Research Council. The VILLUM Foundation is thanked for supporting the VILLUM Center for Bioanalytical Sciences at University of Southern Denmark. Joan Hansen, Tina Grubbe Hansen and Louise Johansson are acknowledged for oligonucleotide synthesis.Notes and references
- G. Mayer, Angew. Chem., Int. Ed., 2009, 48, 2672–2689 CrossRef CAS PubMed.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS PubMed.
- K. W. Thiel and P. H. Giangrande, Oligonucleotides, 2009, 19, 209–222 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Biomol. Eng., 2007, 24, 381–403 CrossRef CAS PubMed.
- G. Aquino-Jarquin and J. D. Toscano-Garibay, Int. J. Mol. Sci., 2011, 12, 9155–9171 CrossRef CAS PubMed.
- Z. Szeitner, J. András, R. E. Gyurcsányi and T. Mészáros, J. Pharm. Biomed. Anal., 2014, 101, 58–65 CrossRef CAS PubMed.
- B. Carlson, Biotechnology Healthcare, 2007, 4, 31–36 Search PubMed.
- K. M. Song, S. Lee and C. Ban, Sensors, 2012, 12, 612–631 CrossRef PubMed.
- R. Beier, E. Boschke and D. Labudde, BioMed Res. Int., 2014, 2014, 849743, DOI:10.1155/2014/849743.
- A. V. Lakhin, V. Z. Tarantul and L. V. Gening, Acta Naturae, 2013, 5, 34–43 CAS.
- S. K. Singh, A. A. Koshkin, J. Wengel and P. Nielsen, Chem. Commun., 1998, 455–456 RSC.
- S. Obika, D. Nanbu, Y. Hari, J.-I. Andoh, K.-I. Morio, T. Doi and T. Imanishi, Tetrahedron Lett., 1998, 39, 5401–5404 CrossRef CAS.
- H. Kaur, B. R. Babu and S. Maiti, Chem. Rev., 2007, 107, 4672–4697 CrossRef CAS PubMed.
- K. K. Karlsen and J. Wengel, Nucleic Acid Ther., 2012, 22, 366–370 CAS.
- R. Owczarzy, Y. You, C. L. Groth and A. V. Tataurov, Biochemistry, 2011, 50, 9352–9367 CrossRef CAS PubMed.
- J. Barciszewski, M. Medgaard, T. Koch, J. Kurreck and V. A. Erdmann, in Methods Mol. Biol., ed. G. Mayer, Humana Press, 2009, ch. 10, vol. 535, pp. 165–186 Search PubMed.
- Y. Kasahara, Y. Irisawa, H. Fujita, A. Yahara, H. Ozaki, S. Obika and M. Kuwahara, Anal. Chem., 2013, 85, 4961–4967 CrossRef CAS PubMed.
- Y. Kasahara, Y. Irisawa, H. Ozaki, S. Obika and M. Kuwahara, Bioorg. Med. Chem. Lett., 2013, 23, 1288–1292 CrossRef CAS PubMed.
- M. Kuwahara and S. Obika, Artif. DNA PNA XNA, 2013, 4, 39–48 CrossRef PubMed.
- R. N. Veedu, B. Vester and J. Wengel, Org. Biomol. Chem., 2009, 7, 1404–1409 CAS.
- N. Derbyshire, S. J. White, D. H. J. Bunka, L. Song, S. Stead, J. Tarbin, M. Sharman, D. Zhou and P. G. Stockley, Anal. Chem., 2012, 84, 6595–6602 CrossRef CAS PubMed.
- R. N. Veedu, B. Vester and J. Wengel, ChemBioChem, 2007, 8, 490–492 CrossRef CAS PubMed.
- M. Kuwahara, Y. Takano, Y. Kasahara, H. Nara, H. Ozaki, H. Sawai, A. Sugiyama and S. Obika, Molecules, 2010, 15, 8229–8240 CrossRef CAS PubMed.
- M. Zuker, Nucleic Acids Res., 2003, 31, 3406–3415 CrossRef CAS PubMed.
- L. Wang, X. Zhou, T. Zhou, D. Ma, S. Chen, X. Zhi, L. Yin, Z. Shao, Z. Ou and P. Zhou, J. Cancer Res. Clin. Oncol., 2008, 134, 365–372 CrossRef CAS PubMed.
- J. Stagg, U. Divisekera, N. McLaughlin, J. Sharkey, S. Pommey, D. Denoyer, K. M. Dwyer and M. J. Smyth, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1547–1552 CrossRef CAS PubMed.
- M. G. Terp, K. A. Olesen, E. C. Arnspang, R. R. Lund, B. C. Lagerholm, H. J. Ditzel and R. Leth-Larsen, J. Immunol., 2013, 191, 4165–4173 CrossRef CAS PubMed.
- X. Zhi, Y. Wang, X. Zhou, J. Yu, R. Jian, S. Tang, L. Yin and P. Zhou, Cancer Sci., 2010, 101, 2561–2569 CrossRef CAS PubMed.
- R. Nielsen and S. Mandrup, in Methods Enzymol., ed. O. A. MacDougald, Academic Press, 2014, ch. 14, vol. 537, pp. 261–279 Search PubMed.
- M. Cho, Y. Xiao, J. Nie, R. Stewart, A. T. Csordas, S. S. Oh, J. A. Thomson and H. Tom Soh, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15373–15378 CrossRef CAS PubMed.
- S. Hoon, B. Zhou, K. D. Janda, S. Brenner and J. Scolnick, Biotechniques, 2011, 51, 413–416 CrossRef CAS PubMed.
- R. Wilson, C. Bourne, R. R. Chaudhuri, R. Gregory, J. Kenny and A. Cossins, PLoS One, 2014, 9, e100572 Search PubMed.
- G. V. Kupakuwana, J. E. Crill 2nd, M. P. McPike and P. N. Borer, PLoS One, 2011, 6, e19395 CAS.
- E. González-Fernández, N. de-los-Santos-Álvarez, A. J. Miranda-Ordieres and M. J. Lobo-Castañón, Talanta, 2012, 99, 767–773 CrossRef PubMed.
- E. Lu, M.-A. Elizondo-Riojas, J. T. Chang and D. E. Volk, Biochemistry, 2014, 53, 3523–3525 CrossRef CAS PubMed.
- P. Jiang, S. Meyer, Z. Hou, N. E. Propson, H. Tom Soh, J. A. Thomson and R. Stewart, Bioinformatics, 2014, 30, 2665–2667 CrossRef CAS PubMed.
- T. Schutze, B. Wilhelm, N. Greiner, H. Braun, F. Peter, M. Morl, V. A. Erdmann, H. Lehrach, Z. Konthur, M. Menger, P. F. Arndt and J. Glökler, PLoS One, 2011, 6, e29604 Search PubMed.
- B. Zimmermann, T. Gesell, D. Chen, C. Lorenz and R. Schroeder, PLoS One, 2010, 5, e9169 Search PubMed.
- F. J. Hernandez, N. Kalra, J. Wengel and B. Vester, Bioorg. Med. Chem. Lett., 2009, 19, 6585–6587 CrossRef CAS PubMed.
- W. H. Thiel, T. Bair, A. S. Peek, X. Liu, J. Dassie, K. R. Stockdale, M. A. Behlke, F. J. Miller, Jr. and P. H. Giangrande, PLoS One, 2012, 7, e43836 CAS.
- K. Sefah, D. Shangguan, X. O. Xiong, M. B. Donoghue and W. Tan, Nat. Protoc., 2010, 5, 1169–1185 CrossRef CAS PubMed.
- K. T. Guo, G. Ziemer, A. Paul and H. P. Wendel, Int. J. Mol. Sci., 2008, 9, 668–678 CrossRef CAS PubMed.
- T. Janas and T. Janas, Cell. Mol. Biol. Lett., 2011, 16, 25–39 CAS.
- K. Knapp, M. Zebisch, J. Pippel, A. El-Tayeb, C. E. Müller and N. Sträter, Structure, 2012, 20, 2161–2173 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mb00045a |
| This journal is © The Royal Society of Chemistry 2015 |