
Systems biosynthesis of secondary metabolic pathways within the oral human microbiome member Streptococcus mutans†
Rostyslav
Zvanych
a,
Nikola
Lukenda
a,
Xiang
Li
b,
Janice J.
Kim
c,
Satheeisha
Tharmarajah
b and
Nathan A.
Magarvey
*ab
aDepartment Chemistry & Chemical Biology, McMaster University, M.G. DeGroote Institute for Infectious Disease Research, 1200 Main St. W, Hamilton, Ontario L8N 3Z5, Canada
bDepartment of Biochemistry & Biomedical Sciences, McMaster University, M.G. DeGroote Institute for Infectious Disease Research, 1200 Main St. W, Hamilton, Ontario L8N 3Z5, Canada. E-mail: magarv@mcmaster.ca; Tel: +1 905 5259140 ext 22244
cDepartment of Pathology and Molecular Medicine, Farncombe Family Digestive Health Research Institute, Health Sciences Centre, McMaster University, Hamilton, Ontario L8S 4K1, Canada
First published on 29th August 2014
Abstract
Streptococcus mutans, a Gram-positive human commensal and pathogen, is commonly recognized as a primary causative agent in dental caries. Metabolic activity of this strain results in the creation of acids and secreted products are recognized as pathogenic factors and agents that promote immunomodulation by stimulating the release of pro-inflammatory cytokines. Products of secondary metabolic pathways of microorganisms from the human microbiome are increasingly investigated for their immunomodulatory functions. In this study, we sought to explore the metabolomic output of nonribosomal peptide pathways within the model S. mutans strain, S. mutans UA159, using a systems metabolomic approach to gain in-depth analysis on products created by this organism and probe these molecules for their immunomodulatory function. Comparative metabolomics and biosynthetic studies using wild-type and nonribosomal peptide deletion strains (within the mutanobactin biosynthetic locus), precursor feedings (fatty acid derivatives) led to the identification of 58 metabolites, 13 of which were structurally elucidated. In addition to these, an assembly line derailment product, mutanamide, was also identified and used to assess immunomodulatory properties of mutanobactins and actions relating to their previously reported functions describing hyphal inhibitory profiles in Candida albicans. The results of this study demonstrate both the complexity and the divergent roles of products stemming from this unique biosynthetic assembly line.
Introduction
Bacteria often create bioactive products of intriguing chemical complexity and activity from biosynthetic assembly lines. Small molecule products from these often-multimodular assembly lines are frequently studied for their impacts on the producers and other microbes, as well as for their direct impacts on eukaryotic biology. Some examples of microbial polyketide (PK) and nonribosomal peptide (NRP) products from environmental bacteria with notable clinically relevant implications in eukaryotic biology are nystatin (PK), epothilone B (PK-NRP), cyclosporine (NRP) and rapamycin (PK-NRP).1–4 The latter two are noted immunomodulatory agents used to chemically control human immune pathways via altering cell communication and functions such as cytokine release (cyclosporine – IL-2, rapamycin – IL-2, IL-10).5–7 Interrogation of these bacterial assembly lines and their corresponding PK and NRP products is possible through metabolomic studies and facile biological testing methods such as multiplexed cytokine release assays to measure their cellular effects on eukaryotes.The human microbiome is a fascinating place to explore new biosynthetic products because of their possible evolved capacities towards human cellular targets and implications towards human disease. Streptococcus mutans UA159 bacterium is an interesting case, as it has a clear ability to function as a commensal, but also causes disease such as dental caries.8 Similarly to other Gram-positive bacteria, S. mutans is known to create complex peptidic products of nonribosomal origin, mutanobactin A and three of its analogs.9,10 Initial bioactivity assessment of mutanobactins demonstrated that they lack appreciable antibacterial activity, but do alter the physiology of another oral microbiome member, Candida albicans. More specifically, these lipopeptides were shown to blunt the hyphal formation in C. albicans, a critical step to its infectivity.11 However, the implications of these molecules in affecting eukaryotic cells, such as macrophages that are involved in the interaction between S. mutans and its host, have not been investigated. The biosynthetic machinery of mutanobactins features a hybrid nonribosomal peptide synthetase–polyketide synthase (NRPS–PKS) assembly line that is one of the few assembly lines that lead to the creation of specialized thioether ring architecture. Moreover, cross-comparative analysis indicates that these NRPS and PKS gene sets readily distinguish S. mutans from the other oral microbes.12 The presence of and unique arrangement and encoding properties of these gene sets make them of a particular interest. In this work we sought to develop a systems approach to biosynthetic assembly lines and used S. mutans as a case in point of how to explore biosynthesis in-depth with a platform we previously published called iSNAP.13 We make additions to this approach and demonstrate that the NRPS–PKS assembly from S. mutans is much more complicated by revealing the entirety of metabolites it can create and the immunomodulatory activity of mutanobactins.
Systematic evaluation of secondary metabolism on the whole is still in its infancy, as common reductionist approaches are aimed at investigating one or two agents at a time. It is widely known, however, that these assembly line systems have an imparted enzymatic promiscuity and naturally create a diverse suite of endproducts which may conceptually have alternate evolved functions for its producing organism.14 Few have defined the metabolomic approach to systematically interrogate metabolic outputs of biosynthetic clusters to define variability, infer structural changes, and capture small molecule diversity. Thus, we sought to define a systems approach that would capture the evolved flexibility, encompass a metabolomic framework to further query flexibility via precursor directed biosynthetic feedings, map the biosynthetic variation on metabolomic level, and facilitate structural assignment of new and minor constituents. Here, we present a systematic analysis of biosynthetic assembly line products within the human microbiome organism, S. mutans, from chemical and bioactivity perspectives to shed light on the diversity of products and activities arising from these microbiome chemistries.
Materials and methods
Bacterial strains and reagents
Streptococcus mutans UA159 strain was used for all experiments in this study and Candida albicans (ATCC # 90028) was used for hyphal inhibition assays. Mutant strain of S. mutans UA159 with a complete deletion of biosynthetic genes (generated using in-frame allelic exchange, substituting the biosynthetic operon for an erythromycin resistance cassette) was constructed by Martha Cordova and generously provided by Dr Dennis G. Cvitkovitch (Department of Oral Microbiology, Faculty of Dentistry, University of Toronto, Toronto, ON, Canada). Unless otherwise stated, all reagents and chemicals were purchased from Sigma Aldrich.Fermentation, processing and analysis of S. mutans extracts
Streptococcus mutans strains were grown in Todd-Hewitt broth supplemented with 3 g L−1 yeast extract (THYE broth) at 37 °C with 5% CO2 without shaking. For the metabolomic comparison of the wild-type and the ΔNRPS mutant, the overnight cultures were inoculated (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100) into 50 mL of THYE medium (n = 8) in 250 mL flasks and grown for 12 hours. The supernatants were extracted using liquid–liquid partitioning with ethyl acetate (2:1 v/v, 3×) and the resulting organic phase was concentrated in vacuo, resuspended in methanol:water (1:1 v/v) and analyzed directly by Bruker micrOTOF II mass spectrometer (positive ESI, scanning m/z 100–1000) with an Agilent 1200 series HPLC with Ascentis Express C18 column (150 mm × 2.1 mm, 2.7 μm, Sigma) and A (acetonitrile with 0.1% formic acid) and B (water with 0.1% formic acid) as the mobile phases at 0.25 mL min−1. The solvent gradient was 0–5 min, 5% A; 5–45 min, linear to 100% A; 45–55 min, isocratic at 100% A, 55–60 min, linear to 5% A; 60–65 min, isocratic at 5% A. Cell extracts of S. mutans grown in a chemically defined medium (CDM) were used for the studies involving mutanolin and mutanobactin analog structure elucidations using MS/MS and isotopically labeled amino acids. The chemically defined medium was composed of five 0.22 μm filter-sterilized stock solutions that were added aseptically (per 1 L): 100 mL of 10× amino acid solution (per 2 L: L-glutamic acid, 50.8 g; L-cysteine, 4 g; L-leucine, 3 g; L-lysine, 3 g; L-arginine, 4.2 g; L-proline, 0.12 g; L-glycine, 0.08 g; adenine, 0.18 g; solubility was enhanced with HCl, then readjusted to pH = 7), 20 mL of 50× salt1 solution (per 2 L: K2HPO4, 207.25 g; KH2PO4, 292.6 g; NH4Cl, 53.6 g; urea, 30.0 g; sodium pyruvate, 60.5 g), 20 mL of 50× salt2 solution (per 2 L: MgCl2, 56.16 g; MnCl2 tetrahydrate, 1.98 g; FeSO4 heptahydrate, 2.78 g; CaCl2, 11.08 g), 10 mL of 100× vitamin solution (per 500 mL: riboflavin, 50 mg; thiamine hydrochloride, 25 mg; nicotinic acid, 50 mg, para-aminobenzoic acid, 5 mg; pyridoxine hydrochloride, 50 mg; calcium pantothenate, 5 mg; folic acid, 5 mg) and 10 mL of 100× glucose solution (per 1L: glucose or dextrose, 500 g).
:100) into 50 mL of THYE medium (n = 8) in 250 mL flasks and grown for 12 hours. The supernatants were extracted using liquid–liquid partitioning with ethyl acetate (2:1 v/v, 3×) and the resulting organic phase was concentrated in vacuo, resuspended in methanol:water (1:1 v/v) and analyzed directly by Bruker micrOTOF II mass spectrometer (positive ESI, scanning m/z 100–1000) with an Agilent 1200 series HPLC with Ascentis Express C18 column (150 mm × 2.1 mm, 2.7 μm, Sigma) and A (acetonitrile with 0.1% formic acid) and B (water with 0.1% formic acid) as the mobile phases at 0.25 mL min−1. The solvent gradient was 0–5 min, 5% A; 5–45 min, linear to 100% A; 45–55 min, isocratic at 100% A, 55–60 min, linear to 5% A; 60–65 min, isocratic at 5% A. Cell extracts of S. mutans grown in a chemically defined medium (CDM) were used for the studies involving mutanolin and mutanobactin analog structure elucidations using MS/MS and isotopically labeled amino acids. The chemically defined medium was composed of five 0.22 μm filter-sterilized stock solutions that were added aseptically (per 1 L): 100 mL of 10× amino acid solution (per 2 L: L-glutamic acid, 50.8 g; L-cysteine, 4 g; L-leucine, 3 g; L-lysine, 3 g; L-arginine, 4.2 g; L-proline, 0.12 g; L-glycine, 0.08 g; adenine, 0.18 g; solubility was enhanced with HCl, then readjusted to pH = 7), 20 mL of 50× salt1 solution (per 2 L: K2HPO4, 207.25 g; KH2PO4, 292.6 g; NH4Cl, 53.6 g; urea, 30.0 g; sodium pyruvate, 60.5 g), 20 mL of 50× salt2 solution (per 2 L: MgCl2, 56.16 g; MnCl2 tetrahydrate, 1.98 g; FeSO4 heptahydrate, 2.78 g; CaCl2, 11.08 g), 10 mL of 100× vitamin solution (per 500 mL: riboflavin, 50 mg; thiamine hydrochloride, 25 mg; nicotinic acid, 50 mg, para-aminobenzoic acid, 5 mg; pyridoxine hydrochloride, 50 mg; calcium pantothenate, 5 mg; folic acid, 5 mg) and 10 mL of 100× glucose solution (per 1L: glucose or dextrose, 500 g).
For the isotopically labeled amino acid experiments, the overnight THYE cultures of wild-type S. mutans were inoculated (1:100) into 50 mL of CDM (n = 2), directly supplemented (50 mg/50 mL) with one of glycine-2-13C, glycine-13C2, L-valine-1-13C, L-serine-3-13C, L-proline-1-13C, L-leucine-1-13C, L-cysteine-13C3, 15N, L-alanine-1-13C or sodium acetate-1-13C in 250 mL flasks and grown for 15 hours at 37 °C with 5% CO2 without shaking. In addition to isotopically labeled amino acids, 8-nonenoic and 7-octenoic fatty acids were also used directly as a separate supplementation (20 μL/100 mL of CDM). The cell pellets were extracted by vortexing for 5 min with 4 mL of solvent mixture containing methanol:ethanol:water (2:1:1 v/v) with 50 mg of 0.5 mm glass beads. Following centrifugation, samples were analyzed for incorporation using LC-MS as described before, but with mass spectrometer scanning m/z 100–2000 range.
The MS/MS experiments were carried out using Bruker amaZon X ion trap mass spectrometer (operating in auto-MS(2) positive ESI mode, scanning m/z 100–1800) coupled with a Dionex UltiMate 3000 HPLC, with an Ascentis Express C18 column (150 mm × 4.6 mm, 2.7 μ, Sigma Aldrich) and A (acetonitrile with 0.1% formic acid) and B (water with 0.1% formic acid) as the mobile phases at 1.2 mL min−1. The solvent gradient was 0–5 min, 5% A; 5–7 min, linear to 40% A; 7–30 min, linear to 65% A, 30–35 min, linear to 100% A, 35–38 min isocratic at 100% A, 38–40 min, linear to 5% A; 40–45 min, isocratic at 5% A.
Metabolomic and chemoinformatic analyses
The volcano plot was generated using the Student's t-test model in the Bruker Daltonics ProfileAnalysis 2.1, by making pairwise comparisons between the wild-type and the mutant strains. More specifically, the bucket table for the t-test model was computed using rectangular bucketing parameters defined as ΔRt = 0.1 min, with a kernel size of 0.05 min, and Δm/z = 0.5 with a kernel size of m/z 0.15, and normalized by the sum of bucket values in the analysis. The fold change and p-value cut-offs used to construct the volcano plot were 5 and 0.05, respectively.To construct a 3D volcano plot, the data were imported from Bruker Daltonics ProfileAnalysis 2.1 into JMP 10.0.0 and the 1187 imported metabolite features were plotted using scatter plot 3D function, with nonpar density contour option selected. The data files obtained from the MS/MS experiments using unlabeled cell extract samples were converted to mzXML format and used for iSNAP analysis. For testing detection of mutanobactin A and B, the following parameters were utilized: precursor mass tolerance, ±1 Da; precursor charge, 1–2; search mode, precise search; P1 and P2 score threshold, 20; database, built-in NRP database and user-defined compounds (SMILES of mutanobactin A or B, accordingly), number of amide cleavages, 1–5; number of cyclized thioether cleavages, 0–1; theoretical fragment m/z tolerance, ±0.25 Da; theoretical fragment charge, 1–2. For the detection of mutanobactin-like compounds, the following parameters were utilized: precursor mass tolerance, ±400 Da; precursor charge, 1–2; search mode, precise search; P1 and P2 score threshold, 10; database, built-in NRP database and user-defined compounds (one of mutanobactin A, B and D SMILES), number of amide cleavages, 1–5; number of cyclized thioether cleavages, 0–1; theoretical fragment m/z tolerance, ±0.25 Da; theoretical fragment charge, 1–2. The hits obtained using all three seed structures were compiled and checked manually for the incorporation of isotopically labeled amino acids using data files from the feeding experiments. The accurate mass measurements of identified mutanobactin analogs and mutanolins were obtained using Bruker micrOTOF II mass spectrometer with internal calibration within Bruker Compass DataAnalysis 4.1 SR1 software, using mutanobactin A and spiked-in bradykinin and iturin A internal standards.
Isolation of biosynthetic products and absolute stereochemistry analysis of mutanamide
For the isolation of biosynthetic products, including mutanamide and mutanobactin A and B, ten 2.5 L THYE batches inoculated with 50 mL of wild-type S. mutans overnight seed culture were grown in 2.8 L Fernbach flasks at 37 °C with 5% CO2 without shaking for 12 hours. The supernatants were extracted using liquid–liquid partitioning with ethyl acetate (2:1 v/v, 3×) and the resulting organic phase concentrated in vacuo. The crude extract was resuspended in methanol and fractionated using a Combiflash® Rf reverse phase MPLC system (Teledyne Technologies Inc.) with a 50 g RediSep® Rf Gold HP C18 flash column and 40 mL min−1 flow rate. The following program of solvent A (acetonitrile) and B (water) was employed: 0–3 min, 5% A; 3–25 min, a linear gradient to 100% A; 26–30 min, isocratic 100% A; 30–35 min, linear gradient to 5% A. Resulting fractions were screened for the presence of compounds of interest by direct infusion using Bruker amaZon X ion trap mass spectrometer operating in positive ESI mode, scanning m/z 100–1000. Fractions that contained the compounds of interest were combined and subjected to preparatory HPLC-ESI-MS (Dionex UltiMate 3000 HPLC system with 160–600 nm variable UV detection connected in line with Bruker amaZon X ion trap mass spectrometer operating in positive ESI mode, and Phenomenex Luna 5μ C18(2) (250 mm × 15 mm, 4.6 μm) with 10 mL min−1 flow rate). Optimal separation was achieved using the following A (acetonitrile with 0.1% formic acid) and B (water with 0.1% formic acid) solvent gradient: 0–5 min, 5% A; 5–10 min, linear gradient to 55% A; 10–28 min, linear gradient to 60% A; 28–29 min, linear gradient to 100% A; 29–35 min, isocratic at 100% A, 35–39 min, linear to 5% A; 39–44 min, isocratic at 5% A. High-resolution mass spectral data for mutanamide were obtained using a Thermo Scientific LTQ Orbitrap XL. NMR experiments were performed using a Bruker Avance 700 MHz spectrometer with samples dissolved in 100% methanol-d4.
The absolute stereochemistry of mutanamide was determined using Marfey's reagent for chiral amino acid analysis.18 Briefly, approximately 0.5 mg of mutanamide were hydrolyzed with 1 mL of 6N HCl in an ACE high-pressure tube at 110 °C for 16 hours. The hydrolysate was evaporated to dryness and resuspended in 100 μL of water. To each 50 μL of above solution, 10 μL of 1M NaHCO3 and 100 μL of 1% L- or D-FDLA (1-fluoro-2,4-dinitrophenyl-5-leucinamide, solution in acetone) were added, respectively. The reaction vials were placed in a water bath at 40 °C for 1 hour and then were cooled to room temperature, quenched with 10 μL of 1N HCl, and evaporated to dryness. The residue was dissolved in 200 μL of methanol and analyzed on HPLC-UV system (Waters Alliance e2695 with Waters 2998 PAD set at 340 nm) together with the L-leucine, D-leucine, L-alanine and DL-alanine standards. The separation was achieved using with the following A (acetonitrile with 0.1% formic acid) and B (water with 0.1% formic acid) solvent gradient: 0–5 min, 20% A; 5–65 min, linear gradient to 60% A; 60–65 min, linear gradient to 100% A; 65–70 min, isocratic at 100% A, 70–75 min, linear to 20% A; 75–80 min, isocratic at 20% A.
Hyphal inhibition in C. albicans and HT-29 cell proliferation assay
Candida albicans pre-cultures were grown in YNB medium without amino acids and with ammonium sulfate (Bioshop® Canada Inc., YNB406.100), supplemented with 0.1% maltose and 0.2% glucose at 30 °C with agitation at 200 rpm for 16 hours. To assay hyphal formation, pre-cultures were used to inoculate hyphal promoting medium (1:100 dilution), consisting of YNB with 2.5 mM N-acetylglucosamine in a 96-well plate. Mutanobactin A, B and mutanamide were dissolved in DMSO and added at the noted concentrations. Importantly, no more than 1 μL of DMSO was added to the 100 μL cultures to avoid secondary effects. Cultures were grown under noted C. albicans conditions and were examined using light microscopy after 4 hours to assess cell morphology. More specifically, relative hyphal formation was calculated as the number of hyphal cells divided by the total number of counted cells. The examination of C. albicans cell morphology was performed using Nikon Eclipse TS100 microscope at a total magnification of 200× and iPhone 4S was used to capture the microscopy images. HT-29 cells (ATCC No. HTB-38TM) were routinely cultured in RPMI 1640 medium, supplemented with 10% (w/v) fetal bovine serum (FBS, PAA laboratories, A15-751), 2 mM L-glutamine (VWR, BDH), at 37 °C in a 5% CO2 incubator, keeping the passage below ten.
To assay antiproliferative affects of mutanamide and mutanobactin A and B, cells were subcultured 1:8 and incubated in 96-well plate (BD Biosciences) with a total volume of 100 μL, treated with 1 μL of either each compound dissolved in DMSO (indicated 10 nM–100 μM range) and monitored using CellTiter 96® AQueous non-radioactive cell proliferation assay (Promega), following manufacturer's recommendations. DMSO was used as a negative control and the absorbance at 490 nm was measured after 1 hour and 15 hours of incubation at 37 °C in the 5% CO2 incubator.
Multiplex cytokine profiling
RAW 264.7 macrophage cell line (ATCC # TIB-71) was cultured in RPMI 1640 medium, supplemented with 10% (w/v) fetal bovine serum (FBS, PAA laboratories, A15-751), 2 mM L-glutamine (VWR, BDH), at 37 °C in a 5% CO2 incubator, keeping the passage below ten. Cells were subcultured 1:8 and incubated in 24-well plates (BD Biosciences) at a total volume of 2 mL. After reaching 50% confluence (∼48 hours), cells were stimulated with 100 ng mL−1 LPS (L3012-5mg, Sigma) and treated with mutanobactin A, B, and mutanamide dissolved in DMSO (20 μL, 100 μM final concentration). After 16 hours of incubation, supernatants were collected and stored at −20 °C. For analysis of the wide range of cytokines, samples were analyzed directly using the Bio-Plex Luminex system with a Bio-Plex Pro™ mouse cytokine 23-plex assay kit (Bio-Rad, M60-009RDPD). Fluorescence data were acquired and analyzed by Bio-Plex Manager software (version 5.0, Bio-Rad Laboratories), following the manufacturer's recommendations.
Statistical analysis
All data are presented as mean ± SEM. Unpaired t-test (for parametric data) was performed using GraphPad Prism version 6.0b for Mac OS X (GraphPad Software, La Jolla, CA). An associated p-value <0.05 was considered statistically significant.Results and discussion
Metabolomic and chemoinformatic analyses of S. mutans
Bacteria often create bioactive products of intriguing chemical complexity and activity from biosynthetic assembly lines. To define the chemical diversity arising from the NRPS–PKS assembly line of S. mutans, we employed a metabolomic analysis with a functional and a mutated NRPS cluster. A univariate analysis was performed on the LC-MS data acquired for the replicate samples from both sets of strains and plotted as a volcano plot to visualize the unique metabolite features (Fig. 1). From the volcano plot, the shear metabolomic complexity associated with the functional biosynthetic machinery illustrates why S. mutans is a great example to impart a systems biosynthetic metabolomic approach. Given the peptidic nature of four reported mutanobactins, we sought to make a connection between the metabolite features from the volcano plot and these compounds by analyzing the culture extracts of S. mutans using the iSNAP platform. To ensure accurate identification, we first defined that iSNAP was able to accurately identify mutanobactin A in the culture extracts of S. mutans using the mutanobactin A seed structure that was inputted into the iSNAP database. Using a tight precursor m/z mass tolerance of ±1.0 Da, both mutanobactin A and its isomer, mutanobactin C, were identified exclusively with retention times at 28.6 and 31.7 min, respectively (data not shown). Mutanobactin B was also accurately identified at its corresponding retention time at 35.6 min (data not shown). Due to its low abundance in the culture extract, mutanobactin D was not detected.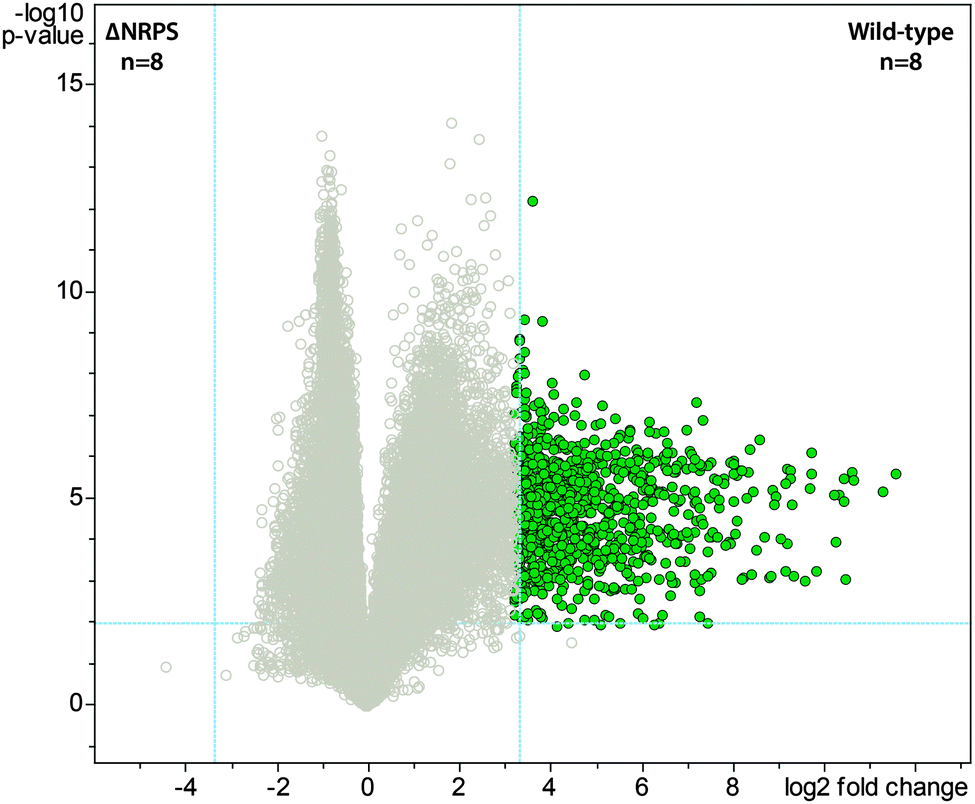 | ||
| Fig. 1 Volcano plot displaying metabolomic diversity arising from the NRPS–PKS biosynthetic machinery of Streptococcus mutans. Vertical and horizontal dashed blue lines indicate the p-value (p = 0.05) and fold-change (fold change = 5) cut-offs, respectively. Metabolite features (green circles) in the right top sector were considered statistically significant and pertain to wild-type, specifically due to the presence of the functional NRPS–PKS biosynthetic machinery. | ||
To explore the observed diversity of metabolite features observed from the volcano plot, the precursor m/z mass tolerance was expanded to ±400.0 Da. Using the chemical structures of mutanobactin A, B, and D as seed structures, a total of 69 hits were identified by iSNAP (Fig. 2). More specifically, the iSNAP analysis using mutanobactin A seed structure yielded 42 unique hits, whereas mutanobactin B and D seed structures yielded 17 and 10 unique hits, respectively. Although the total number of identified unique hits using three mutanobactin seed structures was 69, their structural similarity gave rise to some degree of overlap in the identified hits. Accounting for this overlap, a total of 58 unique hits identified by iSNAP were compiled. This provided an interesting exploration point to go forward and further probe the chemical identity of these molecules. To validate these 58 hits, we performed a feeding experiment using stable isotopically labeled amino acids, which were expected to be incorporated in these molecules at the conserved positions found in the mutanobactin A–D.10 These amino acids were proline, alanine and leucine. In three separate feeding experiments incorporation of isotopically labeled proline, alanine and leucine was observed in 44, 42 and 42 hits, respectively (Fig. 2). This conservation in labeling allowed us to define which hits were real and which ones were plausible false positives. Some of the hits including 12–14, 23–25, 41–43 and 47–48 were indeed false positives, as they lacked incorporation of all three labeled amino acids. In other instances, some hits were plausibly true, however, incorporation of some labeled amino acids was not definitive due to their intrinsically low abundance. Nonetheless, the ability of iSNAP to identify them as hits illustrates the applicability and overall fidelity of this platform to define a systems approach.
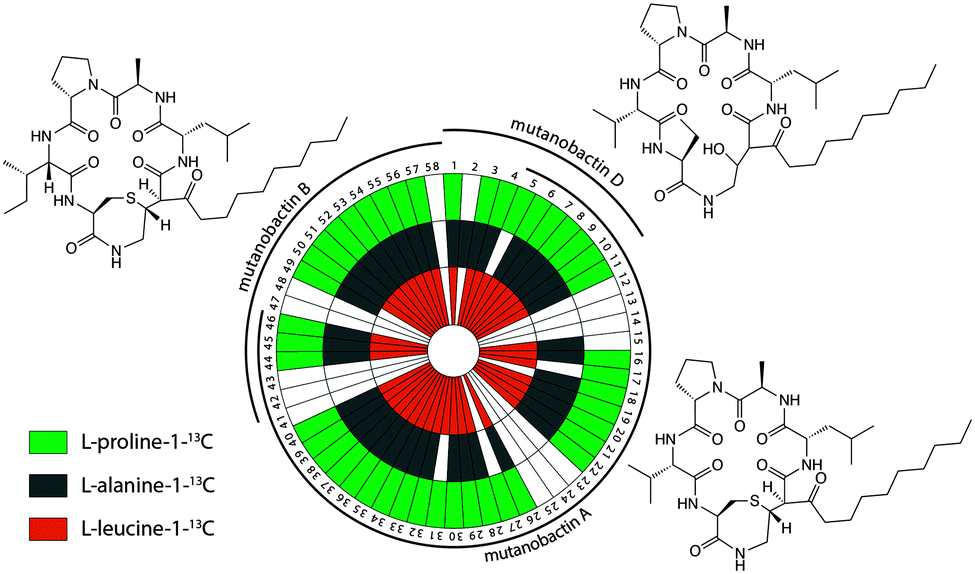 | ||
| Fig. 2 Targeted identification of biosynthetic products arising from the NRPS–PKS cluster of Streptococcus mutans using iSNAP. Metabolite features identified with iSNAP using mutanobactin A, B and D seed structures were validated with 13C-labeled amino acids. The white sectors indicate no observable incorporation. | ||
Structure elucidation of identified biosynthetic products
To explore the structural diversity of some of the hits that were identified by iSNAP a combination of detailed MS/MS and isotopically labeled precursor feeding experiments were performed. To better investigate the fragmentation pathway that mutanobactin A undergoes under collision-induced dissociation (CID) in positive ionization mode and obtain structural information of the fragments, seven separate feeding experiments were carried out using isotopically labeled leucine, alanine, proline, valine, cysteine and glycine and acetate. After assigning the dominant fragments of mutanobactin A (Fig. S1, ESI†), structures of the 13 most abundant compounds selected from the list of iSNAP hits were elucidated using further MS/MS and isotopically labeled precursor feeding experiments, and confirmed by high-resolution mass spectrometry (HRMS, Fig. S2–S14, ESI†). The existence of mutanobactin B2, an intramolecular cyclization isomer of mutanobactin B was expected (Fig. S2, ESI†), as such isomer of mutanobactin A, mutanobactin C, has been previously reported.10The biosynthetic patterns for known lipid-baring molecules suggest slight enzymatic promiscuity, reflected in the ability to incorporate acyl tails of varying length.15 The variants of mutanobactin A and B, mutanobactin E and F, are also produced and contain twelve-carbon long acyl tail (Fig. S3 and S4, ESI†). Interestingly, variants of mutanobactin A and B, mutanobactin G and H, have also been identified and contain alanine for serine substitution (Fig. S5 and S6, ESI†). In addition to HRMS experiments, the identities of mutanobactin G and H were validated using isotopically labeled serine. In both instances, only serine incorporation was observed and no alanine (data not shown). Semi-cyclized analogs of mutanobactin A and B also exist and were identified as mutanobactin I and J, respectively (Fig. S7 and S8, ESI†). Their existence was anticipated, given the stereochemical variability of cyclized biosynthetic products. In addition to the identified mutanobactin analogs, larger biosynthetic products were identified by iSNAP as mutanobactin-like compounds, named mutanolins. Further analysis revealed that mutanolins were indeed of mutanobactin ilk, and their increased molecular weight was due to the incorporation of additional amino acid monomers. For example, mutanolin A and B were found to have almost identical fragmentation profiles as mutanobactin A and B, and both incorporated an additional cysteine unit, which was also in agreement with the observable cysteine loss in their corresponding MS/MS spectra (Fig. S9 and S10, ESI†). This biosynthetic curiosity was unexpected, because there is no additional cysteine adenylation domain in the NRPS portion of this cluster. Perhaps, this is also related to malfunctioning of enzymes responsible for the intramolecular cyclization, which exposes the unfinished products to the reactive neighbouring cysteine, which then nucleophilically attacks the mutanobactin intermediate prior liberation from the assembly line. Interestingly, the existence of even larger mutanolins was observed. The complied analysis of their structures suggested that mutanolin C and D contain an additional glutamic acid that is seemingly attached via a peptide bond to the additional cysteine of mutanolin A and B, respectively (Fig. S11 and S12, ESI†). Together with cysteine, glutamic acid is the second most essential amino acid required for growth of S. mutans.16 Perhaps mutanolins act as a temporary storage of essential amino acids, as their relative abundance seems to decrease over time (data not shown). Lastly, the largest mutanolins, mutanolin E and F, were elucidated to contain a glycine monomer between cysteine and glutamic acid or mutanolin C and D, respectively (Fig. S13 and S14, ESI†). Although unexpected, the sequence of the connected monomers is in keeping with the original sequence observed in mutanobactins, where cysteine follows glycine. Having elucidated a number of mutanobactin analogs, it became evident which sites were prone to substitution. In addition, the identification of mutanolins was even more surprising and suggested that this hybrid NRPS–PKS assembly line does not only display wide substrate flexibility, but also has seemingly low fidelity, generating these two classes of biosynthetic products (Fig. S15, ESI†). Despite such vast and unexpected variation, guided by the iSNAP analysis these products were nonetheless identified, demystifying some of the vast diversity observed in the volcano plot.
Identification of biosynthetic derailment product
It is noteworthy that some molecular features displayed in the volcano plot were much smaller than mutanobactins or mutanolins, which explains the likely reason why iSNAP was unable to detect them (Fig. S16, ESI†). Since these molecular features were exclusive to the wild-type strain, we reasoned that perhaps they were the spuriously generated derailment products from the biosynthetic assembly line. Detailed analysis revealed that the clustering observed in the 3D volcano plot around m/z 400.0 was due to one predominant ion – m/z 399.3. Driven by curiosity we set out to define this possibly truncated mutanobactin cluster-derived product. In doing so, we isolated mutanamide (Fig. S17A, ESI†). The chemical structure of mutanamide is interesting, as similarly to mutanobactins it contains the decanoic acid moiety attached to the first few amino acids – leucine and alanine (Fig. S21–S27, ESI†). To validate its biosynthetic origin, feeding experiments were performed using decanoic acid analogs to test for their incorporation in both mutanamide and mutanobactin A (Fig. S18, ESI†). As expected, both compounds incorporated 8-nonenoic and 7-octenoic fatty acids and their identity was confirmed with MS/MS analysis. It was curious what caused the actual derailment of mutanamide, so we decided to determine the absolute stereochemistry of its two amino acids (Fig. S17B and S19, ESI†). Based on the obtained results, the derailment seemingly arose from the incorporation of L-alanine and failure to epimerize to D-alanine, which is observed in all four previously reported mutanobactins.10 The opposite stereochemistry likely precluded the assembly line form going downstream. Therefore, the systems approach utilized to unravel the biosynthetic potential of NRPS–PKS cluster in S. mutans was not only successful at defining the sites of variable amino acid incorporation, but also led to the identification of the truncated intermediate with opposing stereochemistry (Fig. 3). The latter revealed the mechanism that must be in place in the assembly line to block the progression of the stereo-altered products, which are perhaps inactive with regards to their natural biological function.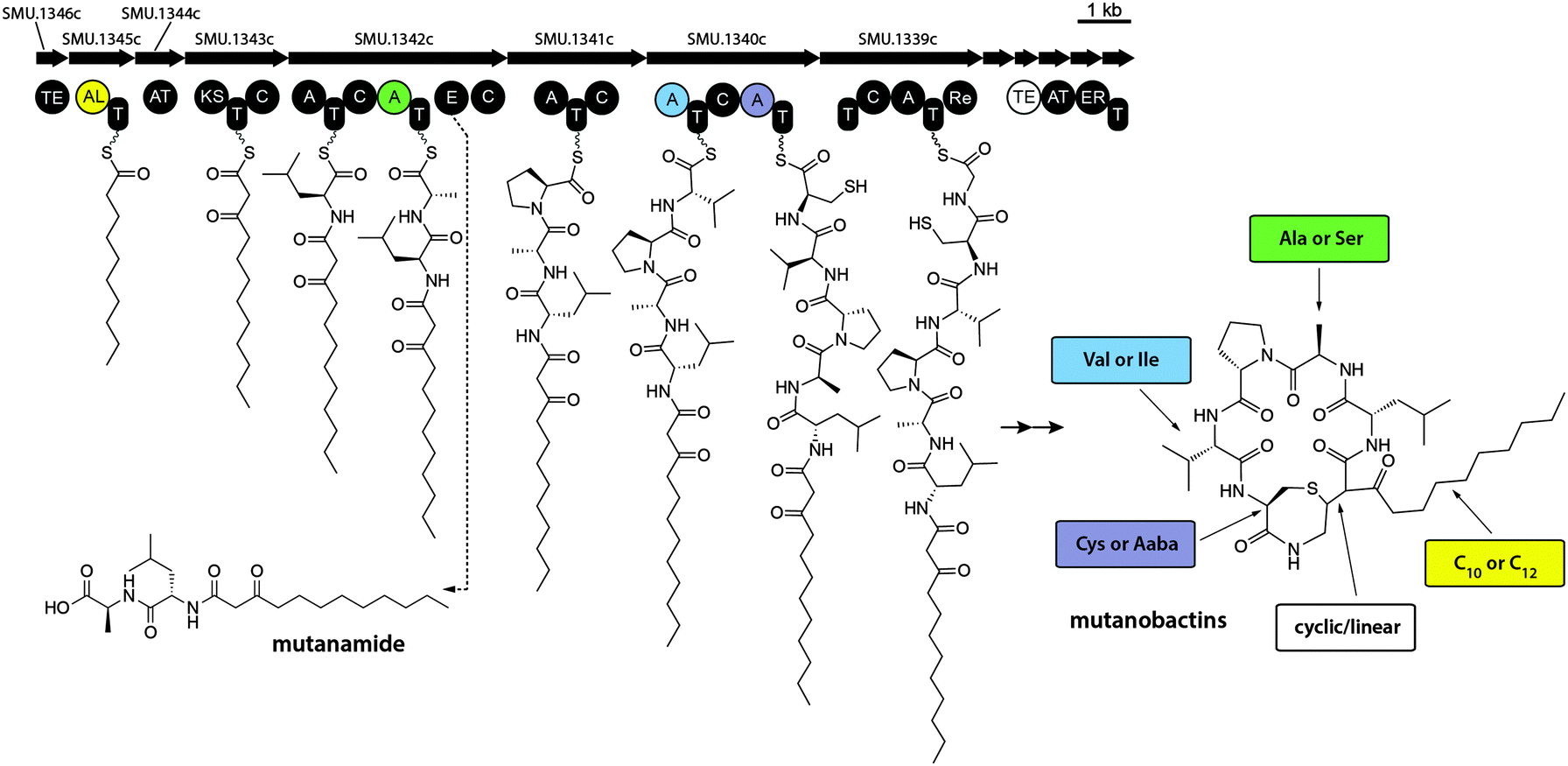 | ||
| Fig. 3 Biosynthetic assembly line that gives rise to mutanobactins. Mutanamide is a derailment product liberated from the assembly line due to the likely malfunctioning of epimerase. The diversity of mutanobactins is granted by the enzymatic flexibility in the colour-coded adenylation and thioesterase domains. | ||
Assessment of biological activity of against C. albicans
The desired stereochemistry is critical for natural products, especially in the complex structures like mutanobactins, and it is not surprising that the assembly lines would have some way to divert these unintended compounds so that they are not processed any further. However, this does not mean that these products are entirely biologically inactive. Due to the predominant acyl tail moiety in mutanamide, we noted that like the other fatty acids that have been previously tested to define activity against yeast signaling, mutanamide may also possess such activity.17 In addition, since this was the original interesting activity assigned to the mutanobactins, we sought that mutanamide would be the ultimate product to test whether the true function of mutanobactins is actually the reported one. Thus, we tested mutanamide and mutanobactin A and B side by side in a yeast hyphal formation assay (Fig. 4). The ability of mutanamide to blunt hyphal formation was much the same as that of mutanobactin A and B, indicating that the functional moiety of these molecules is indeed the acyl tail. Despite their lipopeptide nature, both mutanamide and mutanobactin A had no affect on the proliferation of HT-29 cells (Fig. S20, ESI†). Therefore, we reasoned that the peptidic core that is exclusive to the human microbiome member may have another activity.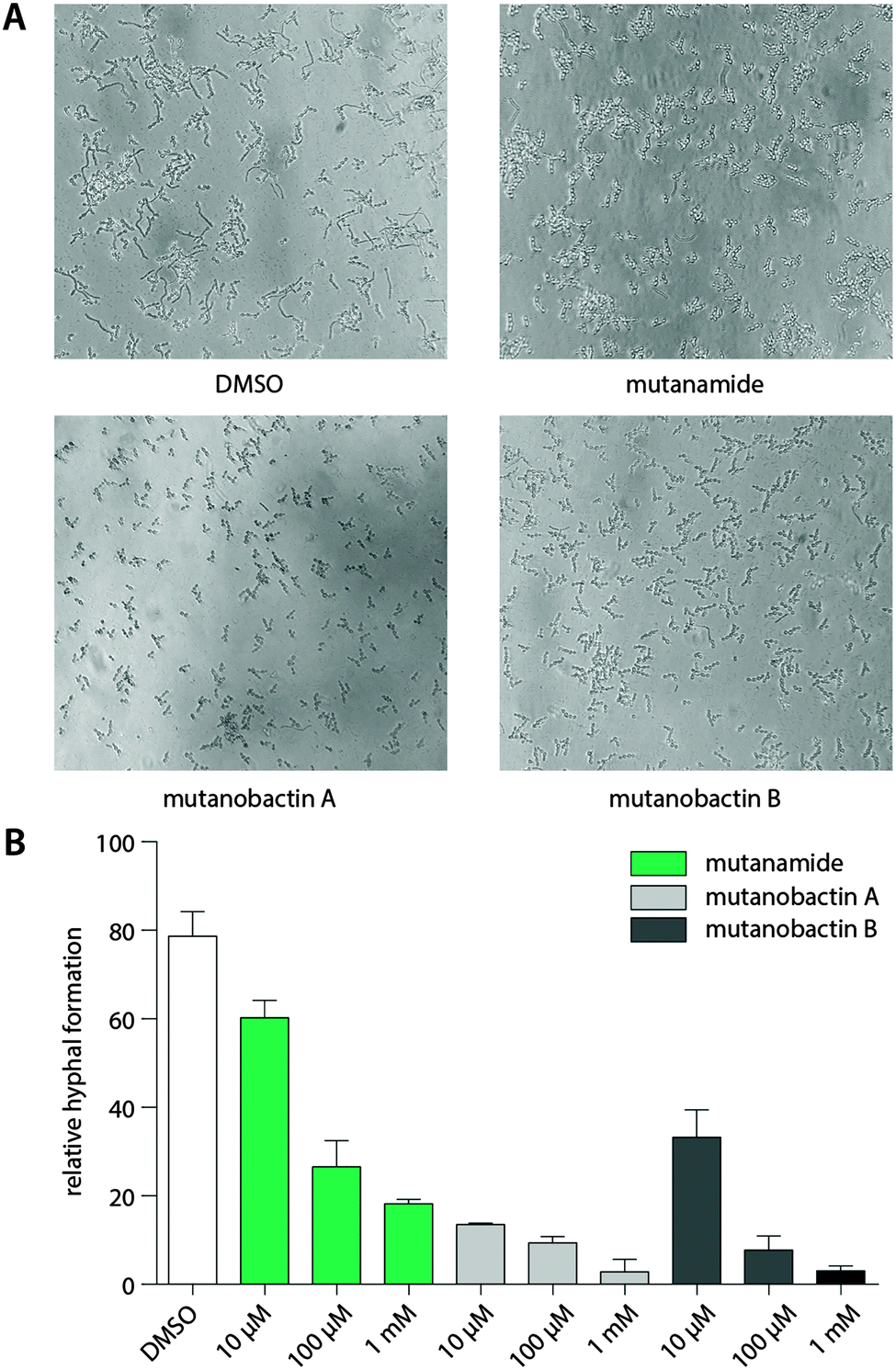 | ||
| Fig. 4 Bioactivity of mutanamide and mutanobactin A and B. (A) Inhibition of hyphal formation in C. albicans when treated with mutanamide and mutanobactins A and B (DMSO, 100 μM mutanamide, 100 μM mutanobactin A and 100 μM mutanobactin B). (B) Relative hyphal formation when treated with mutanamide, mutanobactins A and B. | ||
Immunomodulatory activity of biosynthetic products
Given the biological habitat of S. mutans and having identified mutanamide, we sought to test whether mutanobactins display any immunomodulatory activity and to determine whether the peptidic core is divergent from the acyl tail. To assess the immunomodulatory potential of mutanobactin A and B versus mutanamide, we chose RAW264.7 macrophage cell line and the assay that would read out multiple cytokines that would be produced by the immune cells upon activation. Compared to mutanamide, the affects of mutanobactins, especially mutanobactin B, were much more pronounced (Fig. 5).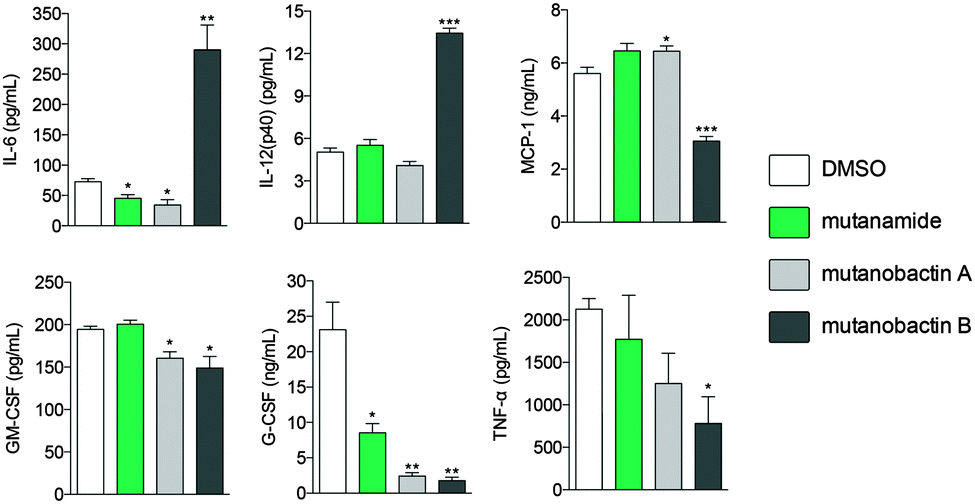 | ||
| Fig. 5 Distinct immunomodulatory activity of mutanobactins. The bar graphs represent the levels of secreted cytokines measured in the supernatant of LPS-stimulated RAW264.7 macrophages upon treatment with 100 μM mutanamide, mutanobactin A and B. Data are presented as mean ± SEM, asterisks (*) indicate significant differences between treatments (*p < 0.05, **p < 0.01, ***p < 0.001). | ||
Interestingly, in same instances, mutanobactin B caused a significant increase in pro-inflammatory cytokines, such as IL-6 and IL-12 and in others a decrease, as the case is for the other four measured cytokines. Although more research needs to be conducted to establish the most dominant immunomodulatory effect of mutanobactins, this preliminary data are significant. More specifically, these data suggest that mutanobactins may play an important role in modulating immune responses of human cells, rather than having a divergent activity against another organism like C. albicans.
Conclusions
From this complete systems approach to studying metabolic pathways it is evident that these types of biosynthetic assembly lines generate a multitude of products and we were able to create a platform to assign what those products are and to define what selective changes have gone on to impart their divergent chemistries. This platform also allows for a systems investigation of the assembly line and the ability to define what combinations and permutations are naturally possible. In addition, the importance of stereochemistry was highlighted by the discovery of stereochemistry-altered derailment product, mutanamide. More importantly, having identified and isolated this truncated product and use it as a comparative control, we were able to define the roles of mutanobactins in greater depth. We present this entirety and highlight the importance of integrating both metabolomic and chemoinformatic platforms such as iSNAP to gain a systems view of the products that would arise from the assembly line. We are curious to see what this platform and systems metabolomics biosynthetic analysis can offer for other assembly lines to define the roles of those chemicals and the diversity of structures that occur naturally and define them. In future work, we hope that this is a mechanism that can be deployed in the system metabolomics of other natural biosynthetic clusters and specifically those from the human microbiome that are of strategic importance with regards to defining the roles of their bioactive compounds in human biology.Acknowledgements
This work was supported by grant 10832 from Canadian Institutes of Health Research to NAM, Canada Research Chair.Notes and references
- H. P. R. Martins, M. C. da Silva, L. C. F. Paiva, T. I. E. Svidzinski and M. E. L. Consolaro, Acta Derm.-Venereol., 2012, 92, 78–82 CrossRef CAS PubMed.
- C. M. Starks, Y. Zhou, F. Liu and P. J. Licari, J. Nat. Prod., 2003, 66, 1313–1317 CrossRef CAS PubMed.
- L. M. Shaw, Clin. Chem., 1989, 35, 1299–1308 CAS.
- C. Vézina, A. Kudelski and S. N. Sehgal, J. Antibiot., 1975, 28, 721–726 CrossRef.
- S. Ferrini, A. Moretta, R. Biassoni, A. Nicolin and L. Moretta, Clin. Immunol. Immunopathol., 1986, 38, 79–84 CrossRef CAS.
- N. Feuerstein, D. Huang and M. B. Prystowsky, J. Biol. Chem., 1995, 270, 9454–9458 CrossRef CAS PubMed.
- M. E. B. Foldenauer, S. A. McClellan, E. A. Berger and L. D. Hazlett, J. Immunol., 2013, 190, 5649–5658 CrossRef CAS PubMed.
- D. Ajdić, W. M. McShan, R. E. McLaughlin, G. Savić, J. Chang, M. B. Carson, C. Primeaux, R. Tian, S. Kenton, H. Jia, S. Lin, Y. Qian, S. Li, H. Zhu, F. Najar, H. Lai, J. White, B. A. Roe and J. J. Ferretti, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14434–14439 CrossRef PubMed.
- P. M. Joyner, J. Liu, Z. Zhang, J. Merritt, F. Qi and R. H. Cichewicz, Org. Biomol. Chem., 2010, 8, 5486 CAS.
- X. Wang, L. Du, J. You, J. B. King and R. H. Cichewicz, Org. Biomol. Chem., 2012, 10, 2044 CAS.
- P. E. Sudbery, Nat. Rev. Microbiol., 2011, 9, 737–748 CrossRef CAS PubMed.
- C. Wu, R. Cichewicz, Y. Li, J. Liu, B. Roe, J. Ferretti, J. Merritt and F. Qi, Appl. Environ. Microbiol., 2010, 76, 5815–5826 CrossRef CAS PubMed.
- A. Ibrahim, L. Yang, C. Johnston, X. Liu, B. Ma and N. A. Magarvey, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19196–19201 CrossRef CAS PubMed.
- S. A. Vanner, X. Li, R. Zvanych, J. Torchia, J. Sang, D. W. Andrews and N. A. Magarvey, Mol. BioSyst., 2013, 9, 2712–2719 RSC.
- L. Robbel and M. A. Marahiel, J. Biol. Chem., 2010, 285, 27501–27508 CrossRef CAS PubMed.
- B. Terleckyj and G. D. Shockman, Infect. Immun., 1975, 11, 656–664 CAS.
- J. Shareck and P. Belhumeur, Eukaryotic Cell, 2011, 10, 1004–1012 CrossRef CAS PubMed.
- R. Bhushan and H. Brückner, Amino Acids, 2004, 27, 231–247 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mb00406j |
| This journal is © The Royal Society of Chemistry 2015 |