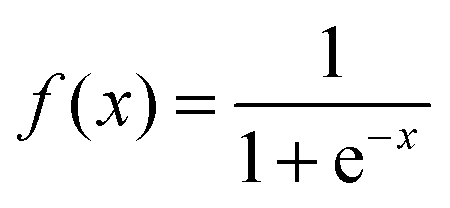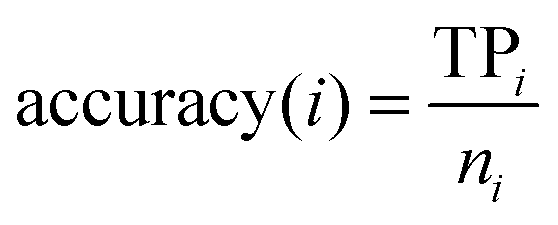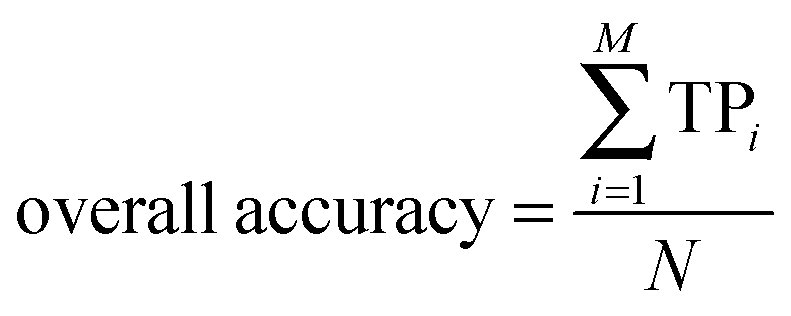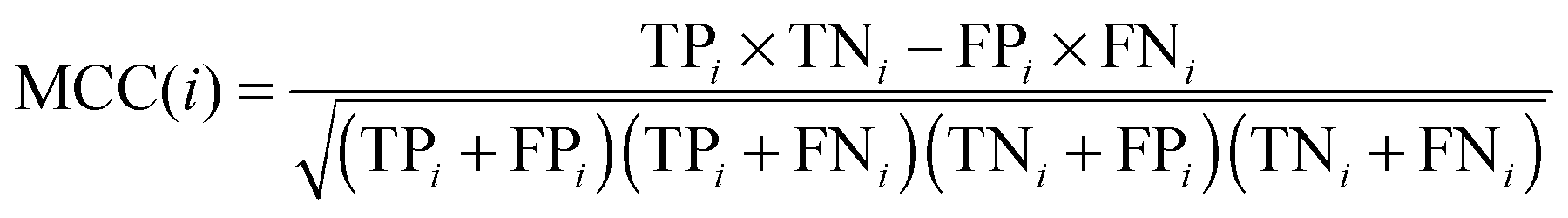DOI:
10.1039/C4MB00340C
(Paper)
Mol. BioSyst., 2015,
11, 170-177
Protein submitochondrial localization from integrated sequence representation and SVM-based backward feature extraction
Received
7th June 2014
, Accepted 16th October 2014
First published on 17th October 2014
Abstract
Mitochondrion, a tiny energy factory, plays an important role in various biological processes of most eukaryotic cells. Mitochondrial defection is associated with a series of human diseases. Knowledge of the submitochondrial locations of proteins can help to reveal the biological functions of novel proteins, and understand the mechanisms underlying various biological processes occurring in the mitochondrion. However, experimental methods to determine protein submitochondrial locations are costly and time consuming. Thus it is essential to develop a fast and reliable computational method to predict protein submitochondrial locations. Here, we proposed a support vector machine (SVM) based approach for predicting protein submitochondrial locations. Information from the position-specific score matrix (PSSM), gene ontology (GO) and the protein feature (PROFEAT) was integrated into the principal features of this model. Then a recursive feature selection scheme was employed to select the optimal features. Finally, an SVM module was used to predict protein submitochondrial locations based on the optimal features. Through the jackknife cross-validation test, our method achieved an accuracy of 99.37% on benchmark dataset M317, and 100% on the other two datasets, M1105 and T86. These results indicate that our method is economic and effective for accurate prediction of the protein submitochondrial location.
Introduction
Mitochondria plays an important role in various biological processes,1–3 including programmed cell death, oxidative phosphorylation, ion hemostasis and innate immune activation. A series of human diseases,4–6 such as Parkinson's disease, diabetes mellitus, epilepsy, cardiac ischemia/reperfusion injury, Alzheimer's disease and cancer, are associated with mitochondrial defects. Since the function of proteins is highly correlated with their locations, knowledge of the protein submitochondrial location can be very helpful for understanding mechanisms of mitochondrial defect related diseases and developing novel drugs. Biochemical experiments7 are time-consuming, tedious and costly. With a large number of protein sequences generated in the post-genomic age, it is highly desirable to develop effective computational systems to address this problem. To date, there have been only a few computational methods for identifying the protein submitochondrial location,7,8 and their efficiencies are still not satisfactory. Therefore, a novel method for accurate and reliable protein submitochondrial location prediction is essential.
As a typical classification task, a computational model for protein submitochondrial localization consists of the following three components: (i) protein feature representation; (ii) algorithm selection for classification; (iii) optimal feature selection. Formulating the protein sample by an effective mathematical expression is a critical factor to develop a powerful predictor for a protein system.9 Various methods have been proposed to extract features for protein localization prediction,10–13 which are commonly based on the protein sequence or sequence-related information, such as terminal signaling peptides, amino acid composition (AAC), pseudo amino acid composition (PseAAC), polypeptide composition, functional domain composition, Position-Specific Iterative Basic Local Alignment Search Tool (PSI-BLAST) profile, and amino acid sequence reverse encoding. Compared with traditional monolithic approaches based on a single feature, the methods based on fusing multiple features have been widely used to improve the prediction performance in the protein subcellular prediction. In this study, we attempted to represent the protein sample through the fusion of information obtained from PROFEAT, gene ontology (GO) and PSSM. PROFEAT is a web server for retrieving frequently used sequence-derived features of proteins, such as the amino acid composition, the Geary autocorrelation, or the sequence-order-coupling number.14 While the GO could provide a dynamic controlled vocabulary of terms for describing gene product characteristics and gene product annotation data from the GO consortium member,15 enhancing the success rate in prediction significantly.16 We previously applied GO annotation to improve the prediction of multi-location protein subcellular localization.17 Besides, the position-specific score matrix (PSSM),18 derived from the PSI-BLAST program, contains the evolutionary information as well as some essential signatures of the protein families. PSSM-based features were often used to detect distant homology, especially in low similarity datasets.
After representing protein sequence as a fixed-length numerical vector, a powerful classification algorithm should be used to operate the prediction. Many machine learning algorithms were developed for protein analysis in the last decade, such as the support vector machine (SVM), artificial neural network, fuzzy K-nearest neighbor (NN), optimized evidence-theoretic (OET)–KNN genetic algorithm and the Markov model. In this study, we used SVM to operate submitochondrial localization prediction for its flexibility, high computational efficiency and good generalization in high-dimensional input spaces in many classification tasks.19,20 However, the original SVM format lacks the ability to filter out irrelevant, redundant or noise features, which may affect the system performance, including classification accuracy and computational efficiency. Thus, selecting relevant features is an important task in protein submitochondrial localization prediction.
Commonly used feature selection techniques can be classified into three categories: filter, wrapper and embedded methods. Compared to filter and wrapper, embedded methods could avoid high risk of overfitting and ignorance of feature dependencies by taking feature correlations into account and discretely removing only one feature from the whole feature vectors. Thus it is much more robust to data overfitting than other feature selection approaches.21 Generally, with the ability to take feature dependencies into account, embedded methods can yield better performance than other methods. Recursive Feature Elimination (SVM–RFE)21 is one of the most popular embedded methods for SVMs. SVM–RFE conducts feature selection in a sequential backward elimination manner, which starts with the whole features and removes one feature each time. Some previous reports showed that features selected by SVM–RFE yield good classification performance in many applications, such as biomarker selection, gene selection, tissue detection22 and so on.
In this study, an SVM-based model was developed to improve the prediction of protein submitochondrial locations with recursively selecting features from the PSI-BLAST profile, physical–chemical properties and protein functional annotations. Before inputted to an SVM classifier to perform the prediction, critical features were selected by SVM–RFE and prediction quality was examined by jackknife tests on three datasets. The results of all prediction performances show that our proposed approach is superior to those methods8,23–27 ever reported.
Materials and methods
1. Datasets
In this study, three benchmark datasets26,27 were used to evaluate the performance of our method (Table 1): the M1105 dataset includes 1105 proteins distributed into 3 submitochondrial locations. The M317 dataset includes 317 proteins classified into 3 submitochondrial locations. The T86 dataset is an independent test dataset that includes 86 human mitochondrial proteins and also classified into 3 locations. None of the proteins in the three datasets has ≥40% sequence identity to any other in the same subset.
Table 1 Thedetailed information of three datasets in our predictor
| Submitochondrial location |
Number of proteins |
| M317 |
M1105 |
T86 |
| Inner membrane |
131 |
589 |
23 |
| Outer membrane |
41 |
236 |
15 |
| Matrix |
145 |
280 |
48 |
| Total |
317 |
1105 |
86 |
2. Feature preparation
To develop a powerful predictor for protein analysis, one of the most important problems is how to formulate a protein sample with an effective mathematical expression or a discrete model that could keep considerable sequence order information. To realize this, the concept of pseudo amino acid composition28 or Chou's PseAAC29 was proposed for representing the sample of a protein. Ever since the concept of PseAAC was introduced, it has been widely used in most of the areas of computational proteomics.30,31 After the web-server ‘PseAAC’32 was established, three effective open access software programs, i.e., ‘PseAAC-General’,33 ‘propy’,34 and ‘PseAAC-Builder’,35 were also built for the purpose. The first is for generating the general model of PseAAC, while the latter two for various modes of special PseAAC. In this work, we are to use a combination of evolutionary information, GO information and physicochemical/structural features to represent the protein samples via PseAAC.
2.1. Linear predictive coding of the PSI-BLAST profiles.
The evolutional information involved in PSSM is highly useful for evaluating relationships in database searches. In this study, PSSM extracted from sequence profiles generated by PSI-BLAST was selected as the feature descriptor. We used the PSI-BLAST tool and the NCBI non-redundant (NR) dataset on a local machine for creating PSSM for all proteins. The parameters j and h are set to 3 and 0.001, respectively. Every PSSM element was scaled to the range from 0 to 1 using the standard sigmoid function:| | 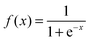 | (1) |
where x is the original PSSM value.
Then, the linear predictive coding (LPC) scheme,36 a tool used mostly in audio signal processing and speech processing, was employed to parameterize the optimal signal. LPC is one of the most powerful speech analysis techniques, and provides extremely accurate estimates of speech parameters. The derived coefficients were used as quantitative features replacing signal intensities. Here, we used the LPC analysis process to extract p features for each column of PSSM, and a 20 × p feature vector was transformed from the PSSM for each protein.
2.2. Gene function annotation features.
GO term data were obtained from http://ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/UNIPROT/ (released on May 7, 2014). All accession numbers in three datasets were searched against the GO database to find the corresponding GO numbers. As the current GO terms did not cover all proteins, BLAST was used to search homologous proteins of protein P without known GO terms under the expected parameter E ≤ 0.001, and collected proteins with ≥60% pairwise sequence similarity to P. Then, the geometrical center of these homologous GO features was used to represent protein P. Thus, we obtained 1569, 879 and 423 different GO terms for M1105, M317 and T86, respectively. Finally, a feature vector was created to represent the GO terms for each protein as described in ref. 22. Due to its low sequence similarity and large population size, M1105 was used to optimize the parameters in LIBSVM,37 and implemented to predict the submitochondrial location of a query protein.
2.3. Structural and physicochemical features computed by PROFEAT.
PROFEAT was designed for computing commonly-used structural and physicochemical features of proteins and peptides from their primary sequences.14,38 These features include the amino acid composition, dipeptide composition, Moran autocorrelation, sequence-order-coupling number, Geary autocorrelation, normalized Moreau–Broto autocorrelation and the composition, transition and distribution of various structural and physicochemical properties. Moreover, new feature groups such as pseudo-amino acid composition (PAAC), amphiphilic PAAC (APAAC), total amino acid properties (TAAPs), and atomic-level topological descriptors are added in the new version of PROFEAT. The enhancements facilitate prediction of proteins, peptides, small molecules of different properties and molecular interactions. In this study, for a query protein, the sequence was inputted and all the PROFEAT features were selected. As a result, we got a 1080-dimension vector of the PROFEAT feature.
3. Feature extraction by SVM–RFE
Due to the limitation of training data, a small amount of features usually results in a better generalization of machine learning algorithms (Occam's razor).39 To select a set of key features for reliable prediction of protein submitochondrial locations, an SVM–RFE algorithm has been developed. Firstly, PSSM, PROFEAT and GO features of each protein were merged into a feature vector. All the feature vectors of proteins for each dataset were used to construct a feature matrix, where each row represents a sample and each column represented a feature. Then, training an SVM with a linear kernel, we ran the SVM–RFE algorithm to get a rank list of all features by removing only one feature with the smallest ranking criterion each time. The first item in the rank list was the most relevant to perform protein submitochondrial location prediction, and the last item had the least relevant feature. Finally, we were able to select different top K features according to the ranking list.
4. The SVM ensemble classifier
Due to excellent generalization capabilities to converge to a single globally optimal solution, SVM is widely used in the bioinformatics applications,40–42 including predictions of the protein subcellular location, membrane protein types, protein crystallization, zinc-binding sites and protein-binding RNA nucleotides. Compared to several other methods, SVM has some merits including the robustness against several types of model violations and outliers, the ability to learn well with only a few free parameters, and the computational efficiency.43 Due to the performance of SVM is decided by the type of kernel function, we used the most popular radial basis function (RBF) kernel44 for its good performance in different prediction tasks. When training an RBF kernel SVM, we considered the parameter γ and regularization parameter C, which could affect the performance of protein submitochondrial location prediction. In this study, the two parameters were also optimized based on the M1105 dataset by a grid search strategy.
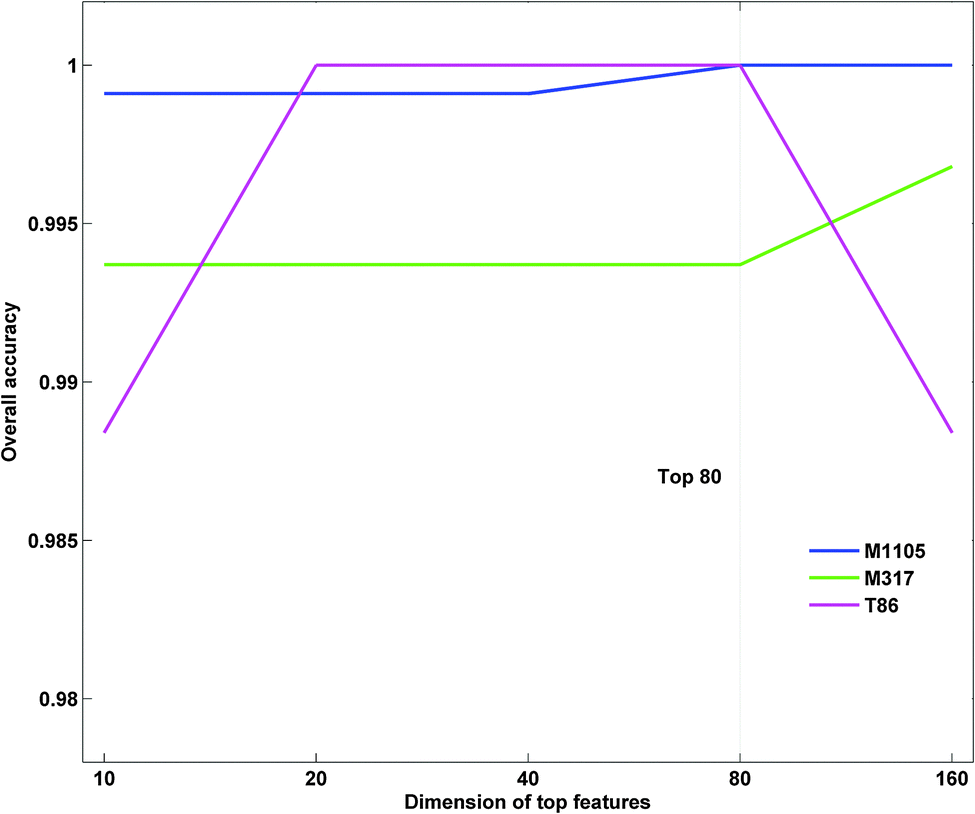
Prediction of protein submitochondrial locations is usually formulated as a multi-class classification problem. This requires a multi-class analysis be broken down into a series of binary classifications, following either the one-against-one or the one-against-rest approach.45 In this study, the one-against-one strategy was employed for its better symmetry than the one-against-rest strategy. Therefore, 3 × 2/2 = 3 binary classification tasks were constructed for each dataset. However, feature vectors optimized by different datasets showed a slight difference (Fig. 1). Finally, the SVM module predicted the submitochondrial location of a protein using the top features and the optimal combination of the two parameters.
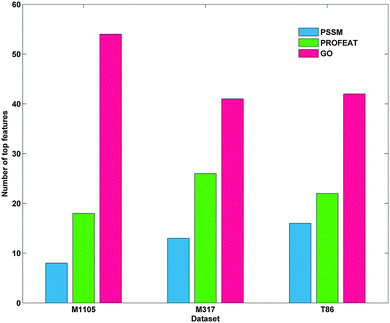 |
| | Fig. 1 Top80 features in the three datasets. | |
5. Assessment of prediction performance
In statistical prediction, the independent dataset test, the subsampling test and the jackknife test are three evaluation methods often used to examine a predictor for its prediction accuracy in practical applications.46 Among them, the jackknife test seems to be the most objective and rigid,47 and thus was adopted in this study. The accuracy, overall accuracy and Matthew's correlation coefficient (MCC)48 were defined by:| | 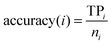 | (2) |
| |  | (3) |
| | 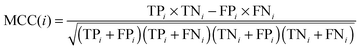 | (4) |
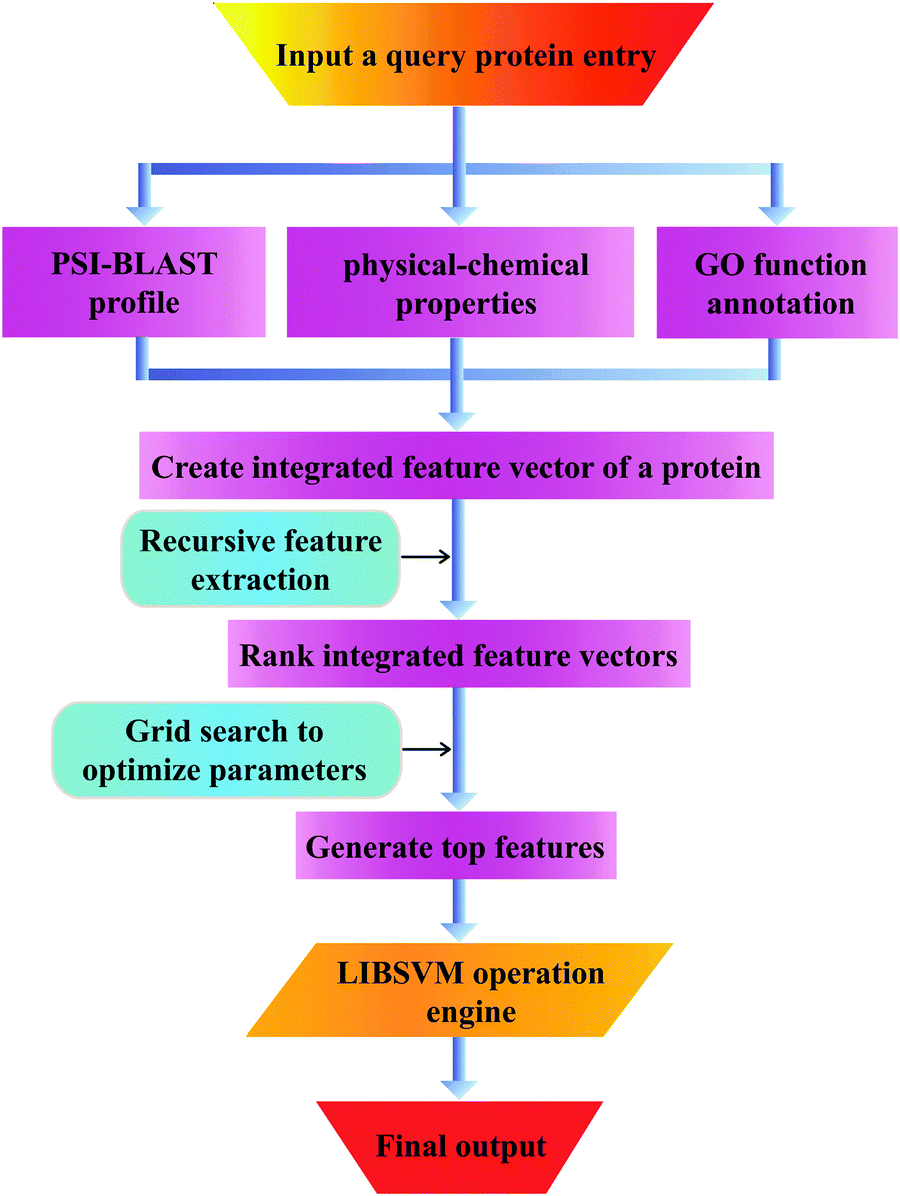
Here, N denotes the total number of proteins, M denotes the class number, ni is the number of proteins in class i. TPi, FPi, TNi and FNi denote true positives, false positives, true negatives, and false negatives in class i, respectively. It is instructive to point out that the above equation set is often used in the literature49–51 for examining the performance quality of a predictor. For an intuitive interpretation about these metrics, particularly for eqn (4), see the aforementioned papers. The set of metrics is efficacious only for the single-label systems. For the multi-label systems which were frequently existent in system biology,52 an absolutely different set of metrics was defined in ref. 53. A flowchart was provided in Fig. 2 to illustrate the prediction process of this method.
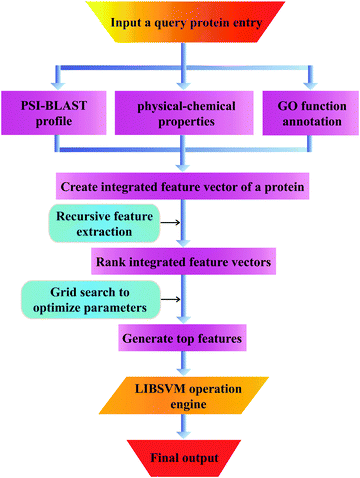 |
| | Fig. 2 The pipeline that goes from the query sequence to the final output and all intermediate steps. | |
Results and discussion
1. Parameter selection
The parameter γ of RBF kernel and regularization parameter C in LIBSVM should be determined to calculate the prediction accuracy. In this study, we utilized a grid search strategy in the M1105 dataset to select them via computing the best dimension Dim of the protein top feature vector. Firstly, we built up an initial feature vector, which was integrated by PSSM, PROFEAT and GO features. Secondly, according to their importance, a ranking list of all the features was returned based on SVM–RFE. According to the ranking list, we calculated the prediction accuracies for top N features, where N = 10 × 2n−1 (n = 1, 2, 3,…, 8). We found that the accuracy at top80 (n = 4) reached 100% for the M1105 dataset (Fig. 3). Finally, top80 features and the corresponding parameters (C = 512, γ = 1.221 × 10−4, and Dim = 80) were chosen as the optimal parameter group to calculate the accuracies for all three datasets.
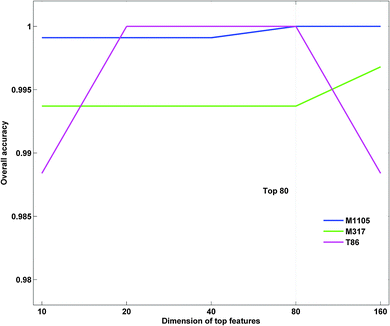 |
| | Fig. 3 Comparison of prediction accuracies of different top features. | |
As shown in Fig. 1, GO features consistently make up the majority of top80 features in each dataset, followed by PROFEAT and PSSM in turn. More than half of the top80 selected features were GO features for all three datasets. For instance, the number was up to 54 for M1105. These results indicated that the subcellular localization of a protein could be characterized by GO features.
2. Comparison with other methods
To assess the performance of our predictor, we compared our method with several other previous methods on the three benchmark datasets with a jackknife test. Our method attained the best overall accuracy of 99.37%, which was higher than those achieved using methods8,23–26 listed in Table 2 (from 14.17% to 4.42%). Moreover, in terms of the accuracy and MCC of all the three sites, our method also ranked the first. We noticed that the second best method listed in Table 2 also used the combined features and the SVM algorithm, proving that the merged features and the SVM algorithm were powerful for inferring the submitochondrial location. We introduced dataset M1105 to further validate our predicting performance. We compared the results of our method and the method constructed by Fan.26 As can be seen from Table 3, our method achieved an overall accuracy of 100.00%, outperformed the latter in terms of the overall accuracy, as well as the accuracy and MCC of all the location sites. Of note, the accuracy of outer membrane in this method was 13.1% higher than that of the latter, suggesting that our method worked well in predicting the submitochondrial location. In fact, there are several GO terms describing submitochondrial locations. It could be a possible reason for our good performance. For example, top80 features in dataset M1105 consisted of 54 GO terms. There are only six cellular compartment GO terms, i.e., mitochondrial inner membrane GO:0005743, mitochondrial matrix GO:0005759, mitochondrial outer membrane GO:0005741, GO:0031307, GO:0045040, and GO:0005742. After removing the six GO terms from top80 features, we got an overall accuracy of 93.68%, which was still better than existing methods. Next, each individual feature type is removed from the integrated feature vector to test its prediction power. To facilitate the comparison, top80 selected features from any two groups of features based on SVM–RFE are input to the classifier for evaluating the contribution of the missing feature type. We found that the prediction accuracy based on PSSM + GO features was 74.73% for M1105, which was slightly lower than that by PSSM + PROFEAT and PROFEAT + GO features (75.55% and 75.82%). We also test the performance of the prediction based only on one group of features (also top80 features). The respective accuracies based on PSSM, PROFEAT and GO features were 72.99%, 75.09% and 76.37%, which are also significantly lower than the overall accuracy by integration of all three types of features. For a human mitochondrial protein dataset T86 with a small size, our method still achieved an overall accuracy of 100% (Table 4). The accuracies of the three subsets were 4.17–13.33% improvements over the method constructed by Shi et al.27 It is important to note that when the M1105 dataset was used to calibrate the parameters, the accuracy at top80 was the highest for M1105. While that was top40 and top20 for the two smaller datasets M317 and T86. It could explain why the prediction accuracies for the two small test datasets reached 100% based on top80 features.
Table 2 Prediction performance comparisons by the jackknife test for dataset M317
| Submitochondrial locations |
SUBMITO23 |
GP-LOC24 |
Predict_subMITO8 |
MitoLoc-LRSVM425 |
Method constructed by Fan and Li26 |
The proposed method |
| Accuracy (%) |
MCC |
Accuracy (%) |
MCC |
Accuracy (%) |
MCC |
Accuracy (%) |
MCC |
Accuracy (%) |
MCC |
Accuracy (%) |
MCC |
| Inner membrane |
85.50 |
0.79 |
83.21 |
0.80 |
91.8 |
0.79 |
89.31 |
0.84 |
94.70 |
0.91 |
100 |
0.99 |
| Outer membrane |
51.20 |
0.64 |
78.05 |
0.77 |
66.1 |
0.63 |
78.05 |
0.74 |
99.30 |
0.96 |
100 |
1.00 |
| Matrix |
94.50 |
0.78 |
97.24 |
0.85 |
96.4 |
0.79 |
93.79 |
0.87 |
80.50 |
0.84 |
98.61 |
0.99 |
| Total accuracy |
85.20 |
— |
89.00 |
— |
89.7 |
— |
89.90 |
— |
94.95 |
— |
99.37 |
— |
Table 3 Prediction performance comparisons by the jackknife test for dataset M1105
| Submitochondrial locations |
Method constructed by Fan and Li26 |
The proposed method |
| Accuracy (%) |
MCC |
Accuracy (%) |
MCC |
| Inner membrane |
96.1 |
0.891 |
100.00 |
1.0000 |
| Outer membrane |
86.9 |
0.890 |
100.00 |
1.0000 |
| Matrix |
93.9 |
0.901 |
100.00 |
1.0000 |
| Total accuracy |
93.57 |
— |
100.00 |
— |
Table 4 Prediction performance comparisons by the jackknife test for dataset T86
| Submitochondrial locations |
Method constructed by Shi and Qiu27 |
The proposed method |
| Accuracy (%) |
MCC |
Accuracy (%) |
MCC |
| Inner membrane |
86.96 |
0.7954 |
100.00 |
1.0000 |
| Outer membrane |
86.67 |
0.7427 |
100.00 |
1.0000 |
| Matrix |
95.83 |
0.8357 |
100.00 |
1.0000 |
| Total accuracy |
91.86 |
— |
100.00 |
— |
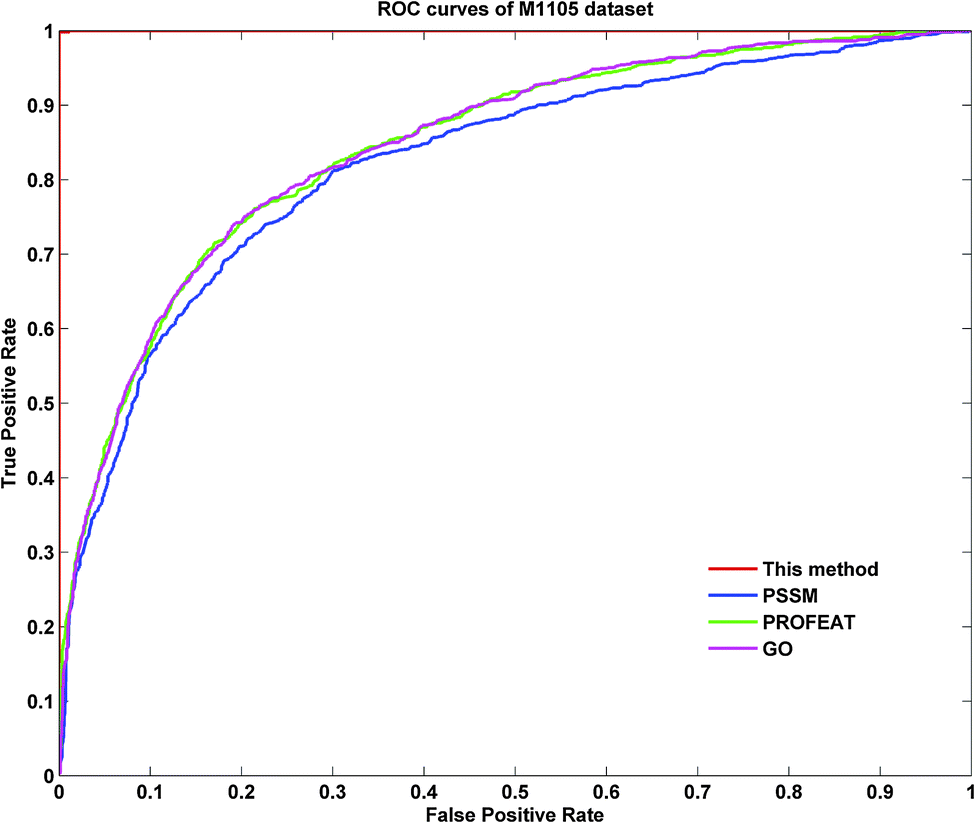
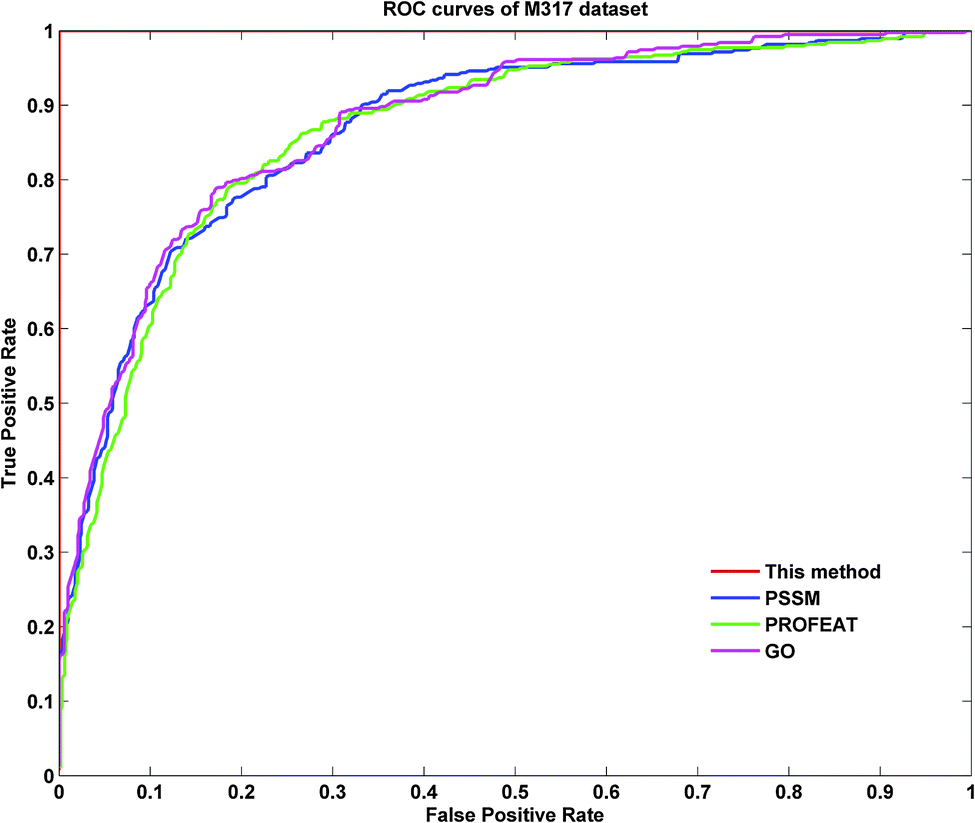
To further demonstrate the prediction power of our method, receiver operating characteristic (ROC) curves on three datasets were implemented here. However, protein submitochondrial location prediction was a multi-class prediction problem. To address this problem, we first transformed protein submitochondrial location prediction into multiple binary classifiers using the one-against-rest strategy, and then averaged all the binary ROC curves as the final output of a method. Fig. 4–6 showed the averaged ROC curves for three datasets by our method and the other three approaches. The area under curves (AUCs) of this method was 1 for all three datasets, which was significantly higher than those by PSSM, PROFEAT and GO features individually (e.g. AUCs were 0.8307, 0.8527 and 0.8547 for M1105, respectively).
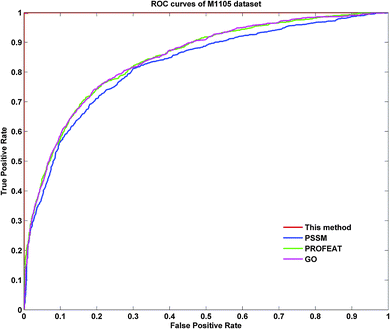 |
| | Fig. 4 The ROC curves for the M1105 dataset. | |
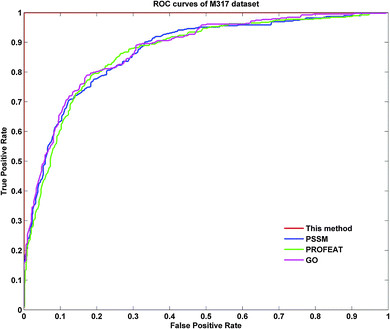 |
| | Fig. 5 The ROC curves for the M317 dataset. | |
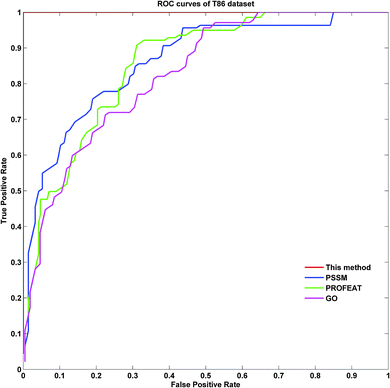 |
| | Fig. 6 The ROC curves for the T86 dataset. | |
3. Case study
To further illustrate our methods, we predicted the submitochondrial locations of 11 proteins, most of which were related to colorectal cancer. As shown in Table 5, 10 of 11 proteins were correctly predicted to the right submitochondrial locations by our predictor based on three datasets. For example, P00395 is a mitochondrion inner membrane protein, which is involved in colorectal cancer, a complex disease characterized by malignant lesions arising from the inner wall of the colon and the rectum.54 In this study, it was consistently predicted as a mitochondrion inner membrane protein by our predictor on three datasets. Another example was Q9BRQ8, a mitochondrion outer membrane protein, which played an important role in EB1 colon cancer cells. Our predictor trained by all three datasets also correctly predicted it as a mitochondrion outer membrane protein. These results imply that our method is suitable for protein submitochondrial location prediction.
Table 5 Examples to show the predicted results by our predictor based on three datasets
| Accession number |
Entry name |
Submitochondrial location |
The proposed method |
| Trained by M1105 |
Trained by M317 |
Trained by T86 |
| P00395 |
COX1_HUMAN |
Inner membrane |
Inner membrane |
Inner membrane |
Inner membrane |
| Q9BRQ8 |
AIFM2_HUMAN |
Outer membrane |
Outer membrane |
Outer membrane |
Outer membrane |
| O14521 |
DHSD_HUMAN |
Inner membrane |
Inner membrane |
Inner membrane |
Inner membrane |
| P08074 |
CBR2_MOUSE |
Matrix |
Matrix |
Matrix |
Matrix |
| Q8IWA4 |
MFN1_HUMAN |
Outer membrane |
Outer membrane |
Outer membrane |
Outer membrane |
| O15239 |
NDUA1_HUMAN |
Inner membrane |
Inner membrane |
Inner membrane |
Inner membrane |
| P00156 |
CYB_HUMAN |
Inner membrane |
Inner membrane |
Inner membrane |
Inner membrane |
| P20000 |
ALDH2_BOVIN |
Matrix |
Matrix |
Matrix |
Matrix |
| Q96E52 |
OMA1_HUMAN |
Inner membrane |
Inner membrane |
Inner membrane |
Inner membrane |
| P22695 |
QCR2_HUMAN |
Inner membrane |
Outer membrane |
Outer membrane |
Matrix |
| P00403 |
COX2_HUMAN |
Inner membrane |
Inner membrane |
Inner membrane |
Inner membrane |
| Q969M1 |
TM40L_HUMAN |
Outer membrane |
Outer membrane |
Outer membrane |
Outer membrane |
Conclusions
In this work, an SVM-based model was constructed for the prediction of protein submitochondrial localizations by selecting the optimal features from three kinds of important features, i.e., protein GO function annotation, amino acid physical–chemical properties and the PSI-BLAST profile. The prediction performance of our method for three low similarity datasets was very promising (99.37% for M317 and 100% for M1105 and T86). It supported the assumption that an optimal combination of multi-features could improve the prediction accuracies for protein submitochondrial location prediction. Moreover, the recursive feature extraction strategy adopted here was highly powerful in getting the optimal features, thus it accelerated the computing procedure as well as improved the final prediction results. The good performances of our predictor for evaluating different datasets suggest that our method is adaptable to diverse datasets and can be applied as a useful tool in such predicting tasks.
Admittedly, there are still some challenges need to be addressed in submitochondrial localization prediction. Although our method suffered from a little high computational complexity for feature ranking and the inconsistent features chosen by different datasets, it could effectively catch the core features and improve the prediction of protein submitochondrial location. In addition, we mainly focused on the proteins with single location sites. Since proteins with multiple location sites might play a significant role in cellular metabolism, we will develop our method by addressing this problem.
Now that serviceable web-servers show the future direction for developing more useful methods, models and predictors,55,56 in our future work, we will attempt to provide a web-server for this method.
Competing interest
The authors declare that they have no competing interests.
Acknowledgements
This work was partially supported by grants from the National Natural Science Foundation of China (No. 81302134 and 31100953), and program for Changjiang scholars and innovative research team in University (IRT 13050 to HY), Shanghai Leading Academic Discipline Project (No. S30405), Innovation Program of Shanghai Municipal Education Commission (No. 12YZ088) and Supported by the Program of Shanghai Normal University (DZL121).
References
- R. Dhingra and L. A. Kirshenbaum, Circ. J., 2014, 78, 803–810 CrossRef CAS PubMed
 .
.
- M. J. Berardi, W. M. Shih, S. C. Harrison and J. J. Chou, Nature, 2011, 476, 109–113 CrossRef CAS PubMed .
- Q. Yang, S. Bruschweiler and J. J. Chou, Structure, 2014, 22, 209–217 CrossRef CAS PubMed .
- V. A. Morais, D. Haddad, K. Craessaerts, P. J. De Bock, J. Swerts, S. Vilain, L. Aerts, L. Overbergh, A. Grunewald, P. Seibler, C. Klein, K. Gevaert, P. Verstreken and B. De Strooper, Science, 2014, 344, 203–207 CrossRef CAS PubMed .
- A. Bilkei-Gorzo, Pharmacol. Ther., 2014, 142, 244–257 CrossRef CAS PubMed .
- E. Desideri, R. Vegliante and M. R. Ciriolo, Cancer Lett., 2014 DOI:10.1016/j.canlet.2014.02.023 .
- S. Mei, J. Theor. Biol., 2012, 293, 121–130 CrossRef CAS PubMed .
- Y. H. Zeng, Y. Z. Guo, R. Q. Xiao, L. Yang, L. Z. Yu and M. L. Li, J. Theor. Biol., 2009, 259, 366–372 CrossRef CAS PubMed .
- P. Du and L. Wang, PLoS One, 2014, 9, e86879 Search PubMed .
- S. W. Zhang, Y. F. Liu, Y. Yu, T. H. Zhang and X. N. Fan, Anal. Biochem., 2014, 449, 164–171 CrossRef CAS PubMed .
- A. S. Mer and M. A. Andrade-Navarro, BMC Bioinf., 2013, 14, 342 CrossRef PubMed .
- B. Liu, D. Zhang, R. Xu, J. Xu, X. Wang, Q. Chen, Q. Dong and K. C. Chou, Bioinformatics, 2014, 30, 472–479 CrossRef CAS PubMed .
- Y. L. Chen, Q. Z. Li and L. Q. Zhang, Amino Acids, 2012, 42, 1309–1316 CrossRef CAS PubMed .
- H. B. Rao, F. Zhu, G. B. Yang, Z. R. Li and Y. Z. Chen, Nucleic Acids Res., 2011, 39, W385–W390 CrossRef CAS PubMed .
- Z. Ramsak, S. Baebler, A. Rotter, M. Korbar, I. Mozetic, B. Usadel and K. Gruden, Nucleic Acids Res., 2014, 42, D1167–D1175 CrossRef CAS PubMed .
- G. Yachdav, E. Kloppmann, L. Kajan, M. Hecht, T. Goldberg, T. Hamp, P. Honigschmid, A. Schafferhans, M. Roos, M. Bernhofer, L. Richter, H. Ashkenazy, M. Punta, A. Schlessinger, Y. Bromberg, R. Schneider, G. Vriend, C. Sander, N. Ben-Tal and A. B. Rost, Nucleic Acids Res., 2014, 42, W337–W343 CrossRef CAS PubMed .
- L. Q. Li, Y. Zhang, L. Y. Zou, Y. Zhou and X. Q. Zheng, Protein Pept. Lett., 2012, 19, 375–387 CrossRef CAS .
- G. Prieto, A. Fullaondo and J. A. Rodriguez, Bioinformatics, 2014, 30, 1220–1227 CrossRef CAS PubMed .
- Z. Jagga and D. Gupta, PLoS One, 2014, 9, e97446 Search PubMed .
- B. Panwar, A. Arora and G. P. Raghava, BMC Genomics, 2014, 15, 127 CrossRef PubMed .
- C. Fernandez-Lozano, E. Fernandez-Blanco, K. Dave, N. Pedreira, M. Gestal, J. Dorado and C. R. Munteanu, Mol. BioSyst., 2014, 10, 1063–1071 RSC .
- L. Li, X. Cui, S. Yu, Y. Zhang, Z. Luo, H. Yang, Y. Zhou and X. Zheng, PLoS One, 2014, 9, e92863 Search PubMed .
- P. Du and Y. Li, BMC Bioinf., 2006, 7, 518 CrossRef PubMed .
- L. Nanni and A. Lumini, Amino Acids, 2008, 34, 653–660 CrossRef CAS PubMed .
- P. Zakeri, B. Moshiri and M. Sadeghi, J. Theor. Biol., 2011, 269, 208–216 CrossRef CAS PubMed .
- G. L. Fan and Q. Z. Li, Amino Acids, 2012, 43, 545–555 CrossRef CAS PubMed .
- S. P. Shi, J. D. Qiu, X. Y. Sun, J. H. Huang, S. Y. Huang, S. B. Suo, R. P. Liang and L. Zhang, Biochim. Biophys. Acta, 2011, 1813, 424–430 CrossRef CAS PubMed .
- K. C. Chou, Bioinformatics, 2005, 21, 10–19 CrossRef CAS PubMed .
- S. X. Lin and J. Lapointe, J. Biomed. Sci. Eng., 2013, 6, 435–442 CrossRef .
- S. Mondal and P. P. Pai, J. Theor. Biol., 2014, 356, 30–35 CrossRef CAS PubMed .
- M. Khosravian, F. K. Faramarzi, M. M. Beigi, M. Behbahani and H. Mohabatkar, Protein Pept. Lett., 2013, 20, 180–186 CrossRef CAS .
- H. B. Shen and K. C. Chou, Anal. Biochem., 2008, 373, 386–388 CrossRef CAS PubMed .
- P. Du, S. Gu and Y. Jiao, Int. J. Mol. Sci., 2014, 15, 3495–3506 CrossRef CAS PubMed .
- D. S. Cao, Q. S. Xu and Y. Z. Liang, Bioinformatics, 2013, 29, 960–962 CrossRef CAS PubMed .
- P. Du, X. Wang, C. Xu and Y. Gao, Anal. Biochem., 2012, 425, 117–119 CrossRef CAS PubMed .
- S. Agnihotri, P. V. Sundeep, C. S. Seelamantula and R. Balakrishnan, PLoS One, 2014, 9, e89540 Search PubMed .
- X. Wei, J. Ai, Y. Deng, X. Guan, D. R. Johnson, C. Y. Ang, C. Zhang and E. J. Perkins, BMC Genomics, 2014, 15, 248 CrossRef PubMed .
- A. N. Sarangi, M. Lohani and R. Aggarwal, Protein Pept. Lett., 2013, 20, 781–795 CrossRef CAS .
- L. Li, Y. Zhang, L. Zou, C. Li, B. Yu, X. Zheng and Y. Zhou, PLoS One, 2012, 7, e31057 CAS .
- X. Xu, A. Li, L. Zou, Y. Shen, W. Fan and M. Wang, Mol. BioSyst., 2014, 10, 694–702 RSC .
- Z. Chen, Y. Wang, Y. F. Zhai, J. Song and Z. Zhang, Mol. BioSyst., 2013, 9, 2213–2222 RSC .
- S. Choi and K. Han, Comput. Biol. Med., 2013, 43, 1687–1697 CrossRef CAS PubMed .
- D. E. Pires, D. B. Ascher and T. L. Blundell, Nucleic Acids Res., 2014, 42, W314–W319 CrossRef CAS PubMed .
- S. Yang, F. Zheng, X. Luo, S. Cai, Y. Wu, K. Liu, M. Wu, J. Chen and S. Krishnan, PLoS One, 2014, 9, e88825 Search PubMed .
- X. Li, L. Chen, F. Cheng, Z. Wu, H. Bian, C. Xu, W. Li, G. Liu, X. Shen and Y. Tang, J. Chem. Inf. Model., 2014, 54, 1061–1069 CrossRef CAS PubMed .
- W. Z. Lin, J. A. Fang, X. Xiao and K. C. Chou, Mol. BioSyst., 2013, 9, 634–644 RSC .
- L. Chen, J. Lu, N. Zhang, T. Huang and Y. D. Cai, Mol. BioSyst., 2014, 10, 868–877 RSC .
- C. Liu, Z. Wen, Y. Li and L. Peng, PLoS One, 2014, 9, e90163 Search PubMed .
- Y. Xu, J. Ding, L. Y. Wu and K. C. Chou, PLoS One, 2013, 8, e55844 CAS .
- W. Chen, P. M. Feng, H. Lin and K. C. Chou, Nucleic Acids Res., 2013, 41, e68 CrossRef CAS PubMed .
- S. H. Guo, E. Z. Deng, L. Q. Xu, H. Ding, H. Lin, W. Chen and K. C. Chou, Bioinformatics, 2014, 30, 1522–1529 CrossRef CAS PubMed .
- K. C. Chou, Z. C. Wu and X. Xiao, Mol. BioSyst., 2012, 8, 629–641 RSC .
- K. C. Chou, Mol. BioSyst., 2013, 9, 1092–1100 RSC .
- I. Namslauer and P. Brzezinski, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3402–3407 CrossRef CAS PubMed .
- H. Ding, E. Z. Deng, L. F. Yuan, L. Liu, H. Lin, W. Chen and K. C. Chou, BioMed Res. Int., 2014, 2014, 286419 Search PubMed .
- W. R. Qiu, X. Xiao and K. C. Chou, Int. J. Mol. Sci., 2014, 15, 1746–1766 CrossRef PubMed .
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.