
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Micropatterning of TCR and LFA-1 ligands reveals complementary effects on cytoskeleton mechanics in T cells†
Erdem
Tabdanov
a,
Sasha
Gondarenko
b,
Sudha
Kumari
c,
Anastasia
Liapis
c,
Michael L.
Dustin
cd,
Michael P.
Sheetz
ef,
Lance C.
Kam
*a and
Thomas
Iskratsch‡
*e
aDepartment of Biomedical Engineering, Columbia University, New York, NY, USA. E-mail: lk2141@columbia.edu
bDepartment of Mechanical Engineering, Columbia University, New York, NY, USA
cDepartment of Pathology, New York University, New York, NY, USA
dKennedy Institute of Rheumatology, University of Oxford, Headington, Oxon, UK
eDepartment of Biological Sciences, Columbia University, New York, NY, USA. E-mail: t.iskratsch@kcl.ac.uk
fMechanobiology Institute, National University of Singapore, Singapore 117411, Singapore
First published on 3rd July 2015
Abstract
The formation of the immunological synapse between a T cell and the antigen-presenting cell (APC) is critically dependent on actin dynamics, downstream of T cell receptor (TCR) and integrin (LFA-1) signalling. There is also accumulating evidence that mechanical forces, generated by actin polymerization and/or myosin contractility regulate T cell signalling. Because both receptor pathways are intertwined, their contributions towards the cytoskeletal organization remain elusive. Here, we identify the specific roles of TCR and LFA-1 by using a combination of micropatterning to spatially separate signalling systems and nanopillar arrays for high-precision analysis of cellular forces. We identify that Arp2/3 acts downstream of TCRs to nucleate dense actin foci but propagation of the network requires LFA-1 and the formin FHOD1. LFA-1 adhesion enhances actomyosin forces, which in turn modulate actin assembly downstream of the TCR. Together our data shows a mechanically cooperative system through which ligands presented by an APC modulate T cell activation.
Insight, innovation, integrationDevelopment of tools to measure or apply forces resulted in our current knowledge of the mechano-regulation of cellular processes. It is increasingly important to understand how different receptor pathways interact with each other to influence the mechano-signalling. We focus here on the specific example of how TCR and LFA-1 interact in the context of the immunological synapse in T cells; augmentation of TCR signalling by LFA-1 provides efficient activation of these cells and the immune response. Microcontact printing of TCR and LFA-1 ligands revealed the specific contributions to the activation of actin assembly and generation of tensile forces. In conjunction with micropillar arrays used to quantify the cellular forces, we provide an integrative model of immune synapse formation. |
Introduction
T cell activation, a key regulatory point of the adaptive immune response, is initiated by engagement of an antigen presenting cell (APC) and formation of an immune synapse (IS) which focuses communication between these cells. Establishment of this specialized interface involves multiple membrane-associated signalling systems; binding of T cell receptor (TCR) to peptide-loaded major histocompatibility complex (pMHC) and Lymphocyte function-associated antigen 1 (LFA-1) to Intercellular cell adhesion molecule-1 (ICAM-1), which provide antigen-dependent activation and long-lasting cell–cell adhesion respectively, represents a minimalist set of signals that promote functional activation.1–5 In addition to molecular cues, it is increasingly clear that mechanical forces have important roles in IS signalling. Early studies demonstrated that the T cell cytoskeleton is important for formation of TCR- and LFA-1-containing microclusters along the distal edge of the expanding interface and their subsequent reorganization into the central- and peripheral-supramolecular activation clusters (cSMAC and pSMAC) that define the archetypal IS.6–9 Restricting centripetal motion of microclusters enhances early TCR signalling,10,11 potentially by delaying sorting of TCR into extracellular microvesicles at the cSMAC,12,13 suggesting that the response of the extracellular environment to cellular forces can modulate IS function. This concept is further supported by studies showing that cell activation is sensitive to the mechanical stiffness of a stimulatory substrate presenting ligands to CD3 and CD28,14,15 indicating a functional role of mechanosensing in T cell physiology.The cytoskeleton thus provides a mechanical platform for IS signalling, and understanding the factors that modulate this structure is critical for a complete model of T cell function. It is attractive to consider LFA-1 as the primary link between the cytoskeleton and extracellular environment, similar to the role that other integrins provide for adherent cells.16 However, LFA-1 signalling within the immune synapse is complex and intertwined with TCR signalling;17,18 full activation of LFA-1 is initiated by TCR function.17 Moreover, rigidity sensing is mediated by CD3 in the context of CD28 co-stimulation14 and TCR signalling leads to actin polymerization,5,19 suggesting direct, mechanical roles of this receptor. Clear delineation of how TCR and LFA-1 modulate cytoskeletal dynamics has been difficult since ligands to these receptors are normally presented to cells uniformly distributed across an activating surface. Micropatterning of surfaces with ligands to the TCR complex and LFA-1 has emerged as a powerful strategy for interrogating how IS structure influences intra-IS signalling.20–23 In this report, we use micropatterned surfaces and elastomer nanopillar arrays (in a traction force microscopy mode7,24) to delineate the specific roles of the TCR complex and LFA-1 in modulating the mechanics of the T cell cytoskeleton. Thereby, we find a separation of functions for both receptor systems that extend to a specification of actin assembly pathways. Actin nucleation downstream of TCR enables the actin network extension downstream of LFA-1, which provides the cytoskeletal tension to allow mechanical sensing, T cell spreading and to enhance TCR activation. Together our data provide new mechanistic insights into the processes that lead to the IS formation and T cell activation.
Results
TCR and LFA-1 have complementary roles in cytoskeleton anchorage and contractility
The dependence on both TCR and LFA-1 for promoting stable T cell interfaces is illustrated in Fig. 1. Primary human CD4+ T cells seeded on surfaces coated with a mix of OKT3 (an antibody that activates the TCR complex CD3ε chain) and ICAM-1 (hICAM-1-Fc, a fusion of the ICAM-1 extracellular domain with Fc, both present at the interface at comparable densities, Fig. S1A (ESI†)) exhibit extensive spreading with both actin and myosin IIa (basic molecular machinery for cytoskeletal contractility) present across the cell–substrate interface (Fig. 1A, left panel). Local enrichment of these proteins along the cell edge indicates that these primary cells develop lamella and lamellipodia structures (Fig. S2, ESI†), similar to T cell blasts and Jurkat cells.2,3,25 Cells on surfaces coated with OKT3 alone are more rounded with only thin, dendritic processes at the same 30 minute time point (Fig. 1A, right panel, Fig. S2, ESI†), while substrates presenting ICAM-1 alone do not support significant adhesion (data not shown). Tyrosine phosphorylation (as a measure for early T cell activation) on rigid surfaces (Sylgard 184, ∼2 MPa) coated with OKT3 and ICAM-1 was higher than on surfaces coated with OKT3 alone, indicating that engagement of LFA-1/ICAM-1 increases the level of TCR triggering (Fig. 1B) compared to the engagement of TCR/OKT3 alone. Moreover, similar to previous reports,14 a comparison between soft (Sylgard 527, ∼5 kPa)26 and rigid substrates (Sylgard 184), as well as blebbistatin treated cells indicated that TCR triggering depended on substrate compliance as well as the balancing cytoskeletal forces. While TCR and LFA-1 signalling cooperate to promote cell anchorage, these experiments illustrate the challenge in identifying the specific roles of each through conventional approaches. As such, we used micropatterning to spatially separate each receptor system. | ||
| Fig. 1 LFA-1 is necessary T cell spreading and enhances T cell activation. (A) Continuous presentation of TCR and LFA-1 ligands. 3D reconstruction of confocal micrographs of anti-myosin IIa and phalloidin stained human CD4+ cells on OKT3 + ICAM-1 (left panel) and OKT3 (right panel); note the dendritic vs. circular morphology of the T cells on the OKT3 vs. OKT3 + ICAM-1 substrates. (B) Phospho-tyrosine and phalloidin staining of human CD4+ cells on spin coated PDMS (50 μm thickness), coated with either OKT3 or OKT3 and ICAM-1. The mean phospho-tyrosine intensity is higher on rigid (Sylgard 184, 2 MPa) than on soft (Sylgard 527, 5 kPa) substrates. The presence of ICAM-1 further enhances phospho-tyrosine levels. p values from one-way ANOVA with Tukey's multiple comparison test: p < 0.005 (**); p < 0.0001 (***); not significant (ns). | ||
We first employed a grid pattern of OKT3 lines (1 μm in width, spaced 10 μm apart) overlaid with lines of ICAM-1 (OKT3/ICAM-1 grids), reorganizing the archetypal IS bulls eye pattern of TCR surrounded by LFA-1 along orthogonal, linear directions (Fig. 2A). Because ICAM-1 is sensitive to drying, we printed fluorescently labelled anti-human Fab fragments, blocked with bovine serum albumin and incubated with an ICAM-1 solution. Again we tested for equal ligand densities by using unlabelled Fab fragments and direct labelled ICAM-1 (Fig. S1B, ESI†).
 | ||
| Fig. 2 LFA-1 is necessary for the development of cell edge tension. (A) 3D reconstruction of phalloidin (blue) and myosin (green) in CD4+ T cells, spreading on OKT3 vs. ICAM-1 and OKT3 vs. OKT3 grids (1 μm wide lines, 10 μm pitch). (B) Population analysis of bi-directional spreading lengths along orthogonal OKT3 lines and along OKT3 vs. ICAM-1 grids in the control cells and after blebbistatin-treatment. Blebbistatin treatment decreases the spreading length along ICAM-1 lines and increases the spreading along OKT3 lines. (C) Free-cell edge curvature radii are significantly larger on OKT3 vs. ICAM-1 than on OKT3 vs. OKT3 grids. Blebbistatin treatment decreases the curvature radius on the OKT3 vs. ICAM-1 grids, indicating that LFA-1 regulates cytoskeletal tension via actomyosin contractility. Error bars indicate SD. n = 121 (OKT3 vs. ICAM-1 control), 92 (OKT3 vs. ICAM-1 blebbistatin), 130 (OKT3 vs. OKT3 control) and 152 (OKT3 vs. OKT3 blebbistatin); p values from student's t-test: p < 0.05 (*); p < 0.001 (**); p < 0.0001 (***). (D–F) PDMS pillar arrays. Jurkat cells were spread on PDMS pillar substrates coated with OKT3 + ICAM-1 or OKT3 and imaged with 1 frame per second from the initiation of spreading. Addition of ICAM-1 enables cell spreading (D) and increases (contractile) inwards-directed forces. (E) Quantification of displacement angles for all pillars over 60 s after first contact. (F) Contractile forces (vector component perpendicular to the cell edge of inwards displacements) are significantly higher in the presence of ICAM-1. n > 400 pillars for 5 cells per condition. Student's t test: p < 0.0001 (***). | ||
Primary CD4+ T cells extended preferentially along lines of OKT3 (Fig. 2A and B) indicating that the TCR complex promotes anchorage of the cytoskeleton to these surfaces. The role of ICAM-1 is seen in analysis of adhesion-free cell edges formed at the intersections between lines. The radius of curvature, R, is related to the extent of spreading along the grid lines as well as the cytoskeletal tension. Interestingly, while spreading along the OKT3 lines was unchanged and cells spread to a lesser extent along the perpendicular ICAM-1, the free-edge radii were significantly larger on the OKT3/ICAM-1 grids. This indicated that addition of LFA-1 signalling to the system was enhancing the cytoskeletal tension. This is in agreement with the idea of a contractile cytoskeleton across the IS (which is suggested by centripetal motion of microclusters for cells on lipid bilayers27,28) and was also confirmed by blebbistatin treatment, which reduced the free-edge radii on the OKT3/ICAM-1 but not the OKT3/OKT3 grids. Intriguingly, cell spreading along ICAM-1 decreased with blebbistatin treatment, suggesting that LFA-1 function, like that of other integrins, is reinforced by cell contractility. Notably, blebbistatin treatment increases spreading of cells on OKT3/ICAM-1 along OKT3 (Fig. 2B), further supporting the idea that OKT3 is associated with cell anchorage.
To directly test the effect of LFA-1 on cytoskeletal tension, we measured cell traction forces using sub-micrometre elastomer (polydimethylsiloxane, PDMS) pillar arrays.24,29,30 The pillars were modified with OKT3 and ICAM-1, and the arrays were seeded with Jurkat cells, which exhibit a similar pattern of spreading on surfaces as primary T cell blasts2 (see also Fig. 5). The larger, flatter lamellipodium of Jurkat cells allowed us to measure pillar deflection by bright-field microscopy,29 which offered better accuracy than the main alternative of fluorescence microscopy; the noise level through random fluctuations in position measured for fluorescent-labelled pillars not underlying any cells was three-fold higher than for bright-field (data not shown). Interaction of Jurkat cells with these arrays was similar to that with glass coverslips, consisting of an initial phase of rapid spreading and extension of flat protrusions, followed by stabilization of cell area over a period of several minutes (Movies 1 and 2). In both phases, pillars undergoing large deflections were localized to the cell edge (one to two rows of pillars spaced 1 μm centre-to-centre). Upon initial contact with a cell, individual pillars were initially deflected outward, but then contracted towards the cell centre as the membrane edge progressed across the surface (Fig. 2D and E). The distribution of contractile forces (calculated from only inward-deflected pillars) for cells on OKT3/ICAM-1 coated arrays is shown in Fig. 2F. Cells on arrays coated with OKT3 alone generated significantly smaller contractile forces. LFA-1 signalling thus augments cytoskeletal contractility, supporting the model developed using the micropatterned grids.
TCR and LFA-1 recruit different actin assembly proteins
Seeking a molecular basis for the different roles of TCR and LFA-1 identified above, primary CD4+ T cell blasts on OKT3/ICAM-1 grids were stained for proteins involved in modulation of the actin cytoskeleton. Both Arp2/3 and HS1 (a stabilizer of Arp2/3 branching,28,31Fig. 3A) localized preferentially to the OKT3 lines, consistent with a role of TCR signalling in promoting cell anchorage and extension on these surfaces. Inhibition of Arp2/3 with CK-666 completely abrogated cell attachment (data not shown), illustrating the need of actin branching/polymerization in IS formation but limiting further exploration of the effect of TCR on IS cytoskeletal dynamics; a different approach to this issue is described in the following section. Complementarily to these proteins, FHOD1 (a formin needed for the organization of lamellar actin, integrin adhesion maturation and cell spreading32) accumulated around ICAM-1 adhesion lines (Fig. 3A and B). Other formin family proteins (mDia1, mDia2, FMNL1) lacked a clear localization to TCR and OKT3 lines (Fig. S3, ESI†), in agreement with previous studies suggesting these do not participate in assembly of IS actin.33 Furthermore, the pan-formin inhibitor smiFH2 selectively disabled spreading of T cell blasts along ICAM-1 lines, resulting in a concurrent T cell elongation along OKT3 lines (Fig. 3C and D). This effect was thus similar to that of blebbistatin, suggesting that formin-dependent elongation of actin fibres downstream of LFA-1 is required for cytoskeletal tension. | ||
| Fig. 3 Actin assembly at TCR and LFA-1 adhesions is regulated by ARP2/3 and FHOD1, respectively (A) CD4+ T cell blasts were stained for ARP2/3, HS1, or FHOD1. ARP2/3, as well as HS1 (quantification on the right) localize to OKT3 lines and FHOD1 is enriched at ICAM-1 lines. Quantification in (B). (C) CD4+ T cell blasts were pre-treated with 5 μM smiFH2 and spread on OKT3/ICAM-1 grids in the presence of the drug. (D) Measurement of the T cell bi-directional spreading lengths along orthogonal OKT3 and ICAM-1 lines. Formin inhibition disables T cell adhesion and spreading along ICAM-1 lines. n = 165 and 149 cells for control and smiFH2 treated cells respectively. (E–H) FHOD1 is needed for contractile forces. FHOD1 knockdown or treatment with smiFH2 decreases the forces to the level observed without ICAM-1. FHOD1-shRNA transfected cells were identified by the expression of GFP under control of a CMV promoter on the shRNA plasmid (E, inset), validation of knock down efficiency in (G). (F) Pillar displacement directions for all pillars over 60 s after first contact; (H) contractile forces (vector component perpendicular to the cell edge of inwards displacements); n > 400 pillars for 5 cells per condition. p values from student's t-test: p < 0.0001 (***). | ||
Finally, FHOD1 knockdown in Jurkat cells and formin inhibition with smiFH2 (Movies 3 and 4) reduced traction force profiles to levels comparable to cells on OKT3 alone, further supporting a role for FHOD1 in forming a contractile cytoskeleton (Fig. 3E–H) and coordinating intra-IS signalling of LFA-1 and the TCR complex.
The TCR complex provides a persistent source of actin nucleation
The separation of receptor systems provided by the grid patterns was effective in allowing study of their independent effects on the cytoskeleton as a mechanical platform, but does not capture the physiological configuration of the IS. As such, we examine in this section cells on arrays of 1 μm diameter dots containing OKT3 surrounded and separated by ICAM-1 (Fig. 4 and Fig. S1C (ESI†) for comparison of coating efficiencies). Within 30 min of attachment to these surfaces, primary CD4+ T cells developed dense clusters of actin over the OKT3 dots (Fig. 4, arrows), surrounded by continuous actin networks over the LFA-1 adhesion area. Omission of ICAM-1 or replacing it with poly-L-lysine reduced cell–substrate interaction to a series of small projections, reminiscent of protrusions reported to form at the interface between T cells and anti-CD3 coated beads19 and illustrating the need to study LFA-1 and TCR signalling together.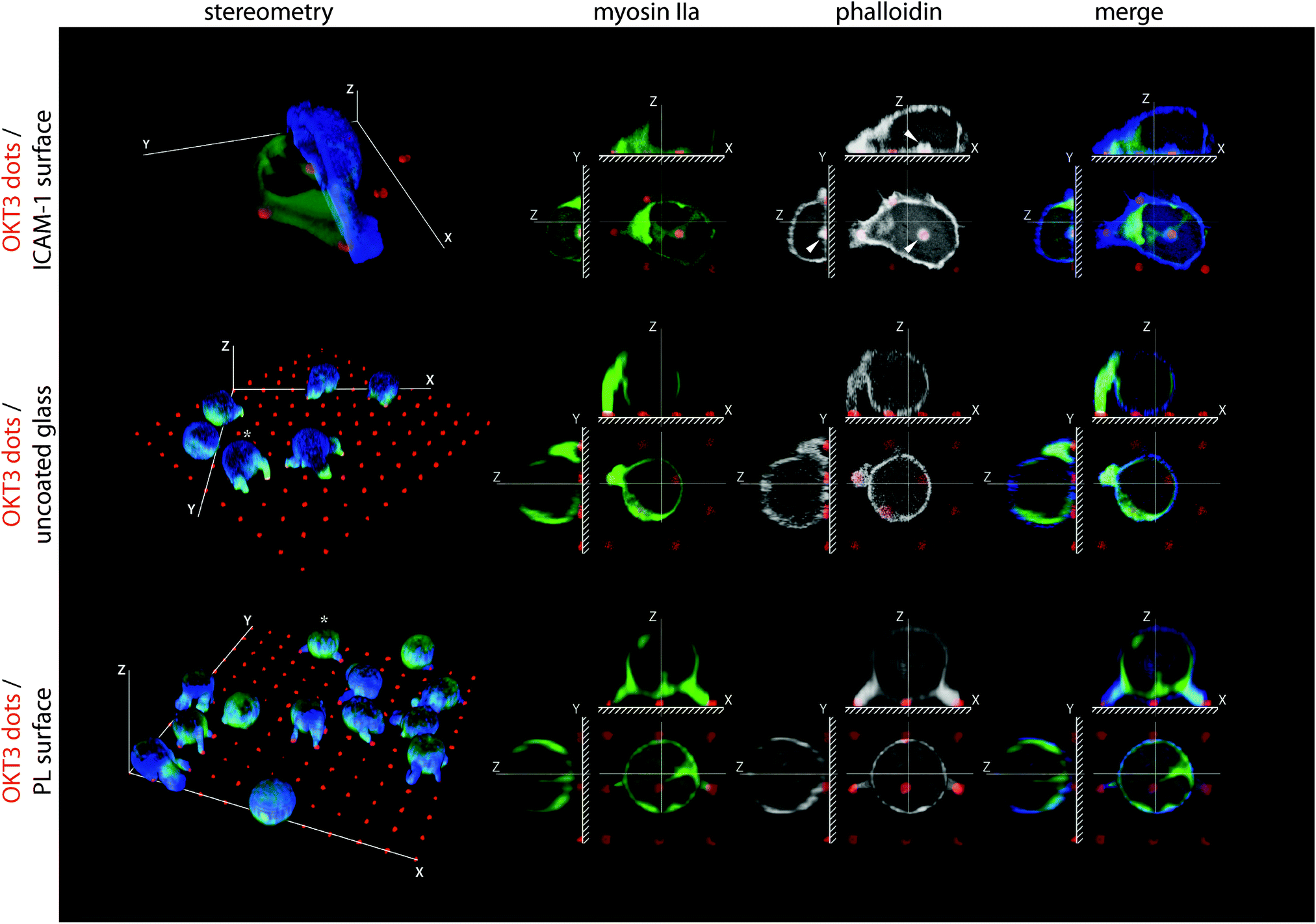 | ||
| Fig. 4 TCR-nucleated actin cluster are transformed into a continuous network by LFA-1. 3D reconstructions of the human CD4+ T cells on micropatterned surfaces of OKT3 dots on ICAM-1 background, BSA-blocked glass without ligands, or poly-L-lysine (PL) (1 μm dots, 5 μm pitch). CD4+ T cells spread out over multiple OKT3 dots on ICAM-1 background. Without ICAM-1 (uncoated glass) or when substituted with poly-L-lysine (PL) spreading was suppressed, and cells formed thin projections that connected to the OKT3 dots. | ||
 | ||
| Fig. 5 TCR and LFA-1 promote actin assembly by different mechanisms. (A) GFP-actin dynamics in primary human CD4+ (upper panels) and Jurkat T lymphocytes (lower panels) on OKT3-dotted ICAM-1 substrates (1 μm OKT3 dots, 5 μm pitch). From left to right – single capture of cell at the moment of spreading bifurcation (white arrow), time average density diagram, kymographs of the white line on the left panels showing OKT3 dot-induced bifurcation, T cell spreading area (black lines) and derivative plots (red). Sub-stable cell spreading areas – black asterisks, spreading bifurcations – black arrows, cell spreading acceleration peaks – red asterisks. (B) Actin-GFP density profile average (n = 17) across OKT3 dot and surrounding ICAM-1 area. (C) F-actin polymerization spiral unfolding from a nucleating TCR adhesion site in a Jurkat cell. Right panel: Kymograph and corresponding actin-GFP (black curve) and OKT3 dot (red curve) density profiles along the indicated line. (D–F) FRAP analysis of stable transfected GFP-actin Jurkat cells. (D) GFP-actin bleach areas and kymograph of recovery (right panels, asterisks mark the bleach event); (E) actin-GFP fluorescence recovery curves over OKT3 dot (top panel) and ICAM-1 adhesion areas (bottom panel) for control cells and CK-666 or smiFH2 treated cells. (F) Actin-GFP semi-recovery times (mean and standard deviation in red). Because of the lack of fluorescence recovery, curves could not be fitted for CK-666 treated cells. p values from student's t-test: p < 0.001 (**); p < 0.0001 (***); not significant (ns). | ||
To investigate the dynamics of this system, we followed spreading of primary CD4+ T cells and Jurkats expressing GFP-labelled actin on micropatterned surfaces. In both cell types, engagement of an OKT3 feature was immediately followed by F-actin polymerization (Fig. 5A, kymographs, white arrows; Movies 5 and 6) and acceleration of actin network extension, focused around these sites of TCR activation (note spreading acceleration on red dS/dt plots, asterisks). As a result, F-actin was highly concentrated at the OKT3 dots (Fig. 5B). This process repeated as the cell encountered new OKT3 features. Notably, actin polymerization occurred not only at OKT3 features along the cell edge, but also on those located within the central area of the cell–substrate interface. In addition, these internal features were frequently associated with rotating vortices of actin unfolding from the sites of TCR engagement (Fig. 5C, Movie 7), indicating that the polymerization was outpacing the transport of actin away from the nucleation site.
Since the spreading and bifurcation process was similar between both cell types we focused on Jurkat cells, which were stably transfected with GFP-actin, enabling a thorough analysis of actin dynamics by fluorescence recovery after photobleaching (FRAP) (Fig. 5D).
Thereby, we detected further differences in actin dynamics around TCR and LFA-1. Post-bleach recovery of GFP-actin fluorescence over OKT3 dots was faster than over ICAM-1 areas (half-time to full recovery, t1/2 = 7.4 s vs. 12.4 s, see Fig. 5E and F), suggesting that the site of TCR engagement promotes actin polymerization and turnover. Both recovery rates were slower than that predicted by free-diffusion (t1/2 of <0.074 s for reported G-actin diffusion rates of >3 μm2 s−1),34–37 indicating that actin assembly is regulated in both systems. The slower recovery over the ICAM-1 than over the OKT3 areas was consistent with the observations of higher actin polymerization over the OKT3 dots.
Since our analysis of the molecular components indicated that the actin nucleator Arp2/3 was mainly localized to the TCR, while the actin elongation and bundling protein FHOD1 was enriched at the LFA-1 lines, we hypothesized that Arp2/3 dependent actin nucleation at TCR sites drives the FHOD1 dependent network extension downstream of LFA-1. To investigate this, we turned again to FRAP experiments, using small molecule inhibitors of Arp2/3 and formins.
Indeed, inhibition of Arp2/3 actin nucleation with CK-666 blocked recovery over both OKT3 and ICAM-1-presenting areas (note: in this experimental setup the cells were already spread out at the timepoint of CK-666 treatment – unlike the pre-treatment with CK-666 above, which blocked cell attachment). In contrast, smiFH2 treatment mainly reduced the recovery over the LFA-1 area, while the effect on the TCR-based FRAP recovery curve was only small (Fig. 5E). Together, these results support the model that cytoskeletal polymerization at sites of TCR engagement acts as a structural primer feeding growth of lamellipodia structures, which requires LFA-1 signalling.
Cytoskeletal contraction activated the TCR complex
As a counterpart to the effects of TCR and LFA-1 on cytoskeletal mechanics, this section focuses on modulation of cell signalling by actin contractility. Blebbistatin-based inhibition of myosin function suppressed actin turnover and polymerization in cells on patterns of OKT3 dots surrounded by ICAM-1 (Fig. 6A), indicating that these functions of the TCR complex can be initiated through mechanical forces generated by the cytoskeleton. | ||
| Fig. 6 Myosin contractility modulates TCR activity. (A) Examination of actin-GFP expressing Jurkat cells shows a reduction of actin polymerization after blebbistatin treatment. Top panel: Image taken immediately after bleaching; middle panel: actin density kymograph, starting after the bleach event (blebb: time point of blebbistatin addition); lower panel: actin density profiles at the time points indicated in the kymograph. (B) Calyculin A (ClcA) hyper-contractility-driven cell collapse is accompanied by formation of TCR-anchored retraction tethers. Note the continuous actin polymerization events (arrowheads) at TCR adhesions inside the retraction tether (white line). Addition of ClcA is marked on the kymograph. (C and D) Introduction of spatial anisotropy demonstrates the mechanosensitive nature of TCR. (C) Immunofluorescence analysis of CD4+ primary T cells: F-actin cluster formation at the apices of multi-apical OKT3 adhesions sites within ICAM-1 field. (D) Live cell imaging of CD4+: actin-GFP forms apical clusters with periodic density bursts (arrows) that consequently ignite F-actin polymerization waves (combs). Time average actin density map shows all three clusters at the tri-dent OKT3 adhesion patch. Note the apical actin periodic density bursts indicated in the kymograph. | ||
To test this hypothesis, we treated the cells with calyculin A to increase myosin contractility.38,39 The hypercontractility resulted in a rounding of the cells, which was previously reported in other cell types as well.38,40 In our system however, the collapse of the cell–substrate interface was accompanied by formation of retraction tethers between the cell body and the OKT3 dots (Fig. 6B). The change in cell morphology did not allow us to directly assess a potential influence of the contractility on the mechano-regulation of TCR actin assembly. However, the strong actin polymerization continued at the sites of OKT3 even after retraction of the cell body (Fig. 6B, kymographs: arrowheads; Movie 8), further supporting the dual roles of the TCR complex as both a point of cell anchorage and sites of actin polymerization.
As an alternative approach to test the effect of cytoskeletal contractility at the cellular level, we employed more extensive patterns of OKT3 consisting of stars, tridents, and spindles, surrounded by ICAM-1. Such adhesive features were previously demonstrated to concentrate mechanical tension at their apices in other cellular systems20,33
Indeed, CD4+ T cells formed dense F-actin clusters preferentially at the apices of these shapes (Fig. 6C), suggesting a tension sensitivity of TCR actin assembly. This was independent from the distance to the cell edge, excluding an enhancement of actin assembly due to the proximity to the cell periphery (Fig. 6C, star pattern). Live imaging showed repeated F-actin polymerization bursts at the apices and the concomitant propagation of the actin waves (Fig. 6D, Movie 9). Together, these results pose a provocative loop through which TCR signalling serves to sustain its own activity by promoting assembly and ultimately (together with LFA-1) contraction of the cytoskeleton.
Discussion
Mechanical forces are increasingly recognized as regulators of many cellular functions.41 In these processes, stimuli such as extracellular shear stress and/or intracellular actomyosin contractility are translated into downstream signals through mechanosensitive proteins.42 This has been studied predominantly in the context of interaction of adherent cells to an ECM matrix, in which integrins or linker proteins undergo force dependent conformational changes and display a catch bond behaviour, but more recently other cell surface receptors including N-cadherin, Netrin or the dystrophin–glycoprotein complex (DGC) have been suggested as mechanotransducers,43–46 expanding the range of physiological functions that are recognized to be influenced by mechanobiology. The identification of mechanosensing in T cells represents a new frontier that spans from fundamental principles of molecular biophysics to improving immunotherapy and treatment of disease.15 However, understanding these effects in the T cell system is challenging as signalling downstream of the TCR is tightly dependent on and interconnected with a wide array of additional cell surface receptors such as LFA-1 and CD28. Initial studies demonstrating an impact of mechanical forces on the TCR complex focused on this receptor alone,6,47,48 and were thus limited to comparatively early events. More recent studies have begun to integrate TCR and CD28 in the context of cellular mechanics. In particular, the ability of T cells to modulate activation in response to the rigidity of a substrate presenting antibodies to CD3 and CD28 is associated predominantly with the TCR complex.14 It was subsequently shown that CD28 co-stimulation increases cellular force generation on TCR adhesions.49 These results are consistent with the observation that soluble anti-CD28 is effective in stimulating T cells. Signalling by the integrin receptor LFA-1, in support of TCR, is anticipated to be more complicated, since both systems support cell spreading as illustrated throughout this report; previous studies using beads showed that LFA-1 signalling causes an early initiation of pulling forces following TCR activation.19 This transformation of polymerization-based forces into contractile forces agrees well with our current study. While, Husson et al. did not find a change in the magnitude of the pulling forces, compared to beads coated with OKT3 alone, this might have been masked by the higher probe stiffness (50 pN μm−1vs. 8.4 pN μm−1 for the pillars) and the spatial resolution of this assay. Similarly, LFA-1 alone led to a restricted pulling phase in the aspiration assays, but this data was limited by the co-presentation challenge discussed above.19In this study, we used surface micropatterning to dissect the effects of TCR and LFA-1, while maintaining stable attachment at the cellular level. We found that these systems serve different functions, namely that actin nucleation at sites of TCR activation lead to a sustained LFA-1 dependent actin network growth. The actin network, in response, enhances the cytoskeletal tension and the sensitivity to mechanical signals such as substrate rigidity. In line with previous studies2,50 our data suggests that on anti-CD3 coated substrates the contribution of myosin to the cellular forces and cell morphology is rather limited.
While the activation of early T cell signalling, measured here by total phospho-tyrosine levels, is still responding to substrate rigidity in the absence of LFA-1, this response is enhanced in its presence. Because the levels of actin assembly downstream of the TCR depend on myosin activity, we propose that it is the LFA-1 dependent increase in cytoskeletal forces that lead in turn to an enhanced TCR signalling on compliant substrates.
This separation of functions also extends to separation of actin assembly pathways, namely Arp2/3 dependent actin nucleation at the TCR and FHOD1 dependent network extension downstream of LFA-1. The different actin regulation by TCR and LFA-1 was clearly visible in dynamic spreading studies on the OKT3 dots, as well as the FRAP experiments. We attribute the particular actin-GFP recovery dynamics mainly to the different rates of nucleation or elongation of the respective assembly factors that associated with the receptors (i.e. Arp2/3 or FHOD1). In presence of a nucleation promotion factor (NPF), Arp2/3 strongly enhances actin nucleation,51 while the rate of actin incorporation into new or existing filaments is in fact lower in the presence of FHOD1 than in presence of actin alone.51,52 Additionally, Arp2/3 forms dendritic networks that are growing in three-dimensions (as can be seen with the actin clusters over the OKT3 dots in Fig. 4, and which has also been found in previous in vitro studies53), while in contrast FHOD1 has a strong actin bundling activity and forms linear actin structures that offer the bleached GFP-actin fewer degrees of freedom to exit the region of interest.32,52
A study that was published while this manuscript was under review found that the NPF WASp was responsible for the formation of dense actin foci at TCR microclusters on OKT3 and ICAM-1 functionalized coverslips and supported lipid bilayers that accounted for approx. 10% of the total actin.54 Silencing of the WASp pathway inhibited HS-1 recruitment and resulted in the loss of the actin foci. The presence of the same molecules (Arp2/3, HS-1) indicates that these foci correspond to the dense clusters that we find localized to the contact printed OKT3 areas. However, while we find that inhibition of Arp2/3 affects assembly of actin over of both OKT3 and ICAM-1 areas, the overall F-actin concentration is reportedly unchanged in absence of WASp activity.54,55 This suggests that different sources of actin assembly are localized to the periphery or the cortex of the cell, where we did not probe the actin dynamics. Indeed other NPFs, such as Scar/WAVE localize to the tip of lamellipodia in lymphocytes and other cells and could compensate for the loss of WASp.56,57 Interestingly, such compensation of actin nucleation – in combination with the mechanical anchoring function of the TCR, seen on our patterned surfaces may also explain the phenomenon of symmetry breaking, found in WASp deficient cells.56,57 WASp at the mechanically anchored TCR would allow to establish a symmetric immune synapse,58,59 which is lost after re-balancing towards other NPFs, such as Scar/WAVE at the cell periphery, – leading to symmetry breaking.
Finally, our results show that the mechanical cooperative systems react to force in several ways: (1) LFA-1 adhesions are mechanosensitive and require force for stabilization. (2) Actin nucleation at the TCR is enhanced by cytoskeletal tension, which (3) is regulated downstream of LFA-1. Together, our data clarifies the distinct roles of TCR and LFA-1 signalling in the regulation of the T cell–APC interaction as well as in the force dependent T cell activation.
Materials and methods
Cell culture and transfections
Primary human CD4+ T cells were freshly isolated from the donor's blood samples using established techniques. Whole blood was diluted with cold RPMI in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 proportion and then sedimented through Ficoll layer (Ficoll-Paque PLUS, General Electric) at 800 g for 30 minutes. White blood cells were collected from the sediment atop of Ficoll layer and subjected to CD4+ T cells purification using Dynabeads Untouched Human CD4 T Cells kit (Invitrogen).
:1 proportion and then sedimented through Ficoll layer (Ficoll-Paque PLUS, General Electric) at 800 g for 30 minutes. White blood cells were collected from the sediment atop of Ficoll layer and subjected to CD4+ T cells purification using Dynabeads Untouched Human CD4 T Cells kit (Invitrogen).
For activation, RosetteSep-purified CD4+ human T cells were seeded in 24-well plates coated with anti-CD3 and anti-CD28 antibodies at a concentration of 1 × 106 cells per ml in complete RPMI medium and incubated at 37 degrees for 72 hours. At 72 hours blasted cells were diluted to 1 × 106 cells per ml, transferred to non-coated tissue culture flasks, and incubated for an additional 48 hours before freezing for long-term storage. Stably transfected actin-GFP Jurkat cells were a kind gift from Prof. Daniel Billadeau. Cells were transfected with the indicated plasmids using Amaxa nucleofection, according to the manufacturer's protocol.
Substrate preparation and micropatterning
Microcontact printing was performed as described elsewhere.22 Briefly, hPDMS stamps were cast on Ebeam-lithographed PMMA wafers. 20 μg ml−1 ligand proteins in PBS were deposited onto the stamps for 40 min. Micro-patterns were printed by stamping on plasma-treated glass for 1 min. OKT3/ICAM-1 micro-grids were printed in two steps: (1) fluorescent OKT3 lines deposition (BioLegend) followed by (2) printing of transverse fluorescent anti-human Fab lines (Jackson Inc. second lines set). After following 5% BSA (Sigma) blocking for 40 min the micro-patterns were incubated with 40 μg ml−1 solution of the hICAM-1/CD54 Fc chimera protein (R&D systems). OKT3/OKT3 grids were incubated and blocked in the same conditions to estimate the effect of non-specific ICAM-1 adsorption to the substrates. OKT3-dotted surfaces were prepared by fluorescent OKT3 deposition as described above for the lines, followed by incubation with ICAM-1 (10 μg ml−1) or 0.1 mg ml−1 poly-L-lysin (Advanced BioMatrix) for 1 hour.For direct labelling of antibodies to compare coating efficiencies (Fig. S1, ESI†), 10 μl of a 1 M sodium bicarbonate buffer at pH 8.3 were added to 100 μl of a 400 μg ml−1 solution of OKT3 or ICAM-1 in PBS and incubated with 10 μl of Atto 565 NHS ester (2 mg ml−1 in DMSO, Santa Cruz Biotechnology) for 1 hour at RT with continuous agitation before dialysis against PBS using Micro Float-A-Lyzer devices with a 100 kDa cut-off. The antibodies were used for coating and micropatterning at the concentrations specified above.
PDMS pillar (500 nm diameter, 1.3 μm height, 1 μm centre-to-centre) substrates were prepared and nanopillar assays were performed as described previously.32 Briefly, PDMS (Sylgard 184, Dow Corning) was mixed thoroughly with its curing agent (10:1), degassed, poured over the silicon mold, placed upside-down on a plasma-treated coverslip-dish (Mattek) and cured at 70 °C for 12 h. The mold was then peeled off and the pillars were incubated with a solution of the ligands (10 μg ml−1 OKT3 ±10 μg ml−1 ICAM-1) for 1 h in PBS.
Flat PDMS substrates were prepared by spin-coating Sylgard 184 or Sylgard 527 with a 150i spin processor (SPS), onto coverslips at 1000 rpm for 100 s and curing at 70 °C for 12 h. Before spin coating, Sylgard 527 was pre-cured at 70 °C for 30 minutes with intermitted mixing to achieve a comparable viscosity to the Sylgard 184 mixture. After curing a film thickness of 51.4 ± 2.2 μm (Mean ± SD) for Sylgard 184 and 52.6 ± 6.7 μm for Sylgard 527 was measured at a DektakXT Surface Profiler at the London Centre for Nanotechnology.
Live imaging and FRAP
Briefly, the harvested cells were resuspended in the pre-warmed (37 °C) and CO2-preconditioned media (phenol red-free RPMI-1640 and 25 μM Hepes) and placed into the glass-bottomed micro-printed chambers (LabTek). Live cell imaging was performed on an Axiovert 200 (Zeiss) microscope, equipped with an UltraView ERS-6 spinning-disk confocal imaging system (PerkinElmer) and an environmental chamber (Solent Scientific), using a ORCA-ER camera (Hamamatsu Photonics) and Volocity software (v5.5; PerkinElmer) at the Children's Hospital of Philadelphia. FRAP was performed as described elsewhere.2Spreading assays, nanopillar assays and pharmacological treatments
T cell spreading assay was performed at 1 × 106 cells per ml concentration. Cells treatment with non-enantiomeric (−)-blebbistatin (Sigma) was performed in two different manners: for immunofluorescent analysis the treatment was continuous throughout pretreatment to the cell spreading stages: 15 minutes prior to the cells plating samples were resuspended in 100 μM blebbistatin solution in supplemented complete RPMI media and incubated on micro-chips. For live cell imaging of acto-myosin inhibition, blebbistatin was added at the moment of interest to its final concentration of 100 μM. Calyculin A (Cell Signaling Technology, Inc.), was added to the working concentration of 50 nM at the moment of interest. For formin and ARP2/3 inhibition experiments, cells were treated with 5 μM smiFH2 and 50 μM CK-666, respectively (both Sigma) for 15 minutes prior to cell plating and spread in presence of the drugs. For FRAP experiments, smiFH2 and ARP2/3 were added 30 minutes after plating and cells were imaged immediately after drug addition. Immunofluorescence staining was performed as described previously.60Antibodies and plasmids
The rabbit anti-non-muscle Myosin IIa, anti-mDia1 and anti-FMNL1 antibodies were obtained from Abcam, rabbit anti-FHOD1 and polyclonal goat anti ARP2/3 subunit 1b antibodies antibodies were from Sigma and rabbit anti pHS1 was from Cell Signaling Technology. Alexa Fluor 647 anti-Phosphotyrosine Antibody [Clone: PY20] was from Biolegend. Polyclonal rabbit anti-mDia2 was purchased from ECM Bioscience. Alexa-568 or 647-conjugated phalloidin was from Life technologies. HRP-conjugated anti mouse, anti-rabbit and anti-goat antibodies, DyLight 488, cy3, Alexa 647 or DyLight649 conjugated anti-rabbit, or anti-mouse antibodies were obtained from Jackson ImmunoResearch.Statistical testing
In the current study, two-tailed Student's t-test was used for comparison between two groups. Data sets were tested for normal distribution using the Shapiro–Wilk test. All statistical tests and curve fitting (FRAP) was performed with Graphpad Prism.Image processing
Original digital images obtained were assembled to the figures and labelled using Illustrator (Adobe). Only linear contrast adjustments were used and were always applied to the entire image.Competing interests statement
The authors declare no competing interests.Acknowledgements
We would like to acknowledge Prof. Daniel Billadeau for actin-GFP Jurkat cells, Prof. Roland Wedlich-Söldner for Lifeact plasmids, and Prof. Janis Burkhardt for assistance in imaging GFP-actin. We gratefully acknowledge the critical reading and helpful comments provided by Nicolas Biais, Luis Santos and Haguy Wolfenson. This work was supported by an American Heart Association fellowship and a British Heart Foundation Intermediate Basic Science Research Fellowship (to T.I.), a Cancer Research Institute fellowship (to S.K.) and NIH grants R01AI088377 (L.C.K.), PN2EY016586 (M.P.S., L.C.K. and M.L.D), AI043542 (M.L.D.). M.L.D. is a Principal Research Fellow of the Wellcome Trust and Senior Research Fellow of the Kennedy Trust.References
- A. Scholer, S. Hugues, A. Boissonnas, L. Fetler and S. Amigorena, Immunity, 2008, 28, 258–270 CrossRef CAS PubMed.
- A. Babich, S. Li, R. S. O'Connor, M. C. Milone, B. D. Freedman and J. K. Burkhardt, J. Cell Biol., 2012, 197, 775–787 CAS.
- J. Yi, X. S. Wu, T. Crites and J. A. Hammer, 3rd, Mol. Biol. Cell, 2012, 23, 834–852 CrossRef CAS PubMed.
- A. Grakoui, S. K. Bromley, C. Sumen, M. M. Davis, A. S. Shaw, P. M. Allen and M. L. Dustin, Science, 1999, 285, 221–227 CrossRef CAS.
- S. Kumari, S. Curado, V. Mayya and M. L. Dustin, Biochim. Biophys. Acta, 2014, 1838, 546–556 CrossRef CAS PubMed.
- S. T. Kim, K. Takeuchi, Z. Y. Sun, M. Touma, C. E. Castro, A. Fahmy, M. J. Lang, G. Wagner and E. L. Reinherz, J. Biol. Chem., 2009, 284, 31028–31037 CrossRef CAS PubMed.
- S. Ghassemi, N. Biais, K. Maniura, S. J. Wind, M. P. Sheetz and J. Hone, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.–Process., Meas., Phenom., 2008, 26, 2549–2553 CAS.
- Y. Klieger, O. Almogi-Hazan, E. Ish-Shalom, A. Pato, M. H. Pauker, M. Barda-Saad, L. Wang and M. Baniyash, Eur. J. Immunol., 2014, 44, 58–68 CrossRef CAS PubMed.
- C. R. Monks, B. A. Freiberg, H. Kupfer, N. Sciaky and A. Kupfer, Nature, 1998, 395, 82–86 CrossRef CAS PubMed.
- C. J. Hsu, W. T. Hsieh, A. Waldman, F. Clarke, E. S. Huseby, J. K. Burkhardt and T. Baumgart, PLoS One, 2012, 7, e32398 CAS.
- K. D. Mossman, G. Campi, J. T. Groves and M. L. Dustin, Science, 2005, 310, 1191–1193 CrossRef CAS PubMed.
- S. Y. Tseng, J. C. Waite, M. Liu, S. Vardhana and M. L. Dustin, J. Immunol., 2008, 181, 4852–4863 CrossRef CAS.
- K. Choudhuri, J. Llodra, E. W. Roth, J. Tsai, S. Gordo, K. W. Wucherpfennig, L. C. Kam, D. L. Stokes and M. L. Dustin, Nature, 2014, 507, 118–123 CrossRef CAS PubMed.
- E. Judokusumo, E. Tabdanov, S. Kumari, M. L. Dustin and L. C. Kam, Biophys. J., 2012, 102, L5–7 CrossRef CAS PubMed.
- R. S. O'Connor, X. Hao, K. Shen, K. Bashour, T. Akimova, W. W. Hancock, L. C. Kam and M. C. Milone, J. Immunol., 2012, 189, 1330–1339 CrossRef PubMed.
- R. O. Hynes, Cell, 2002, 110, 673–687 CrossRef CAS.
- M. L. Dustin and T. A. Springer, Nature, 1989, 341, 619–624 CrossRef CAS PubMed.
- B. Graf, T. Bushnell and J. Miller, J. Immunol., 2007, 179, 1616–1624 CrossRef CAS.
- J. Husson, K. Chemin, A. Bohineust, C. Hivroz and N. Henry, PLoS One, 2011, 6, e19680 CAS.
- J. Doh and D. J. Irvine, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5700–5705 CrossRef CAS PubMed.
- N. Martinez-Martin, E. Fernandez-Arenas, S. Cemerski, P. Delgado, M. Turner, J. Heuser, D. J. Irvine, B. Huang, X. R. Bustelo, A. Shaw and B. Alarcon, Immunity, 2011, 35, 208–222 CrossRef CAS PubMed.
- K. Shen, V. K. Thomas, M. L. Dustin and L. C. Kam, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7791–7796 CrossRef CAS PubMed.
- K. Shen M. C. Milone, M. L. Dustin and L. C. Kam, Materials Research Society Symposia Proceedings. Materials Research Society, 2009, 1209.
- J. L. Tan, J. Tien, D. M. Pirone, D. S. Gray, K. Bhadriraju and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1484–1489 CrossRef CAS PubMed.
- M. L. Dustin, Curr. Opin. Cell Biol., 2007, 19, 529–533 CrossRef CAS PubMed.
- R. N. Palchesko, L. Zhang, Y. Sun and A. W. Feinberg, PLoS One, 2012, 7, e51499 CAS.
- T. Ilani, G. Vasiliver-Shamis, S. Vardhana, A. Bretscher and M. L. Dustin, Nat. Immunol., 2009, 10, 531–539 CrossRef CAS PubMed.
- D. Kumari, P. Bandyopadhyay and N. Suryaprakash, J. Org. Chem., 2013, 78, 2373–2378 CrossRef CAS PubMed.
- S. Ghassemi, G. Meacci, S. Liu, A. A. Gondarenko, A. Mathur, P. Roca-Cusachs, M. P. Sheetz and J. Hone, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5328–5333 CrossRef CAS PubMed.
- T. Iskratsch, H. Wolfenson and M. P. Sheetz, Nat. Rev. Mol. Cell Biol., 2014, 15, 825–833 CrossRef CAS PubMed.
- T. Uruno, P. Zhang, J. Liu, J. J. Hao and X. Zhan, Biochem. J., 2003, 371, 485–493 CrossRef CAS PubMed.
- T. Iskratsch, C. H. Yu, A. Mathur, S. Liu, V. Stevenin, J. Dwyer, J. Hone, E. Ehler and M. Sheetz, Dev. Cell, 2013, 27, 545–559 CrossRef CAS PubMed.
- T. S. Gomez, K. Kumar, R. B. Medeiros, Y. Shimizu, P. J. Leibson and D. D. Billadeau, Immunity, 2007, 26, 177–190 CrossRef CAS PubMed.
- M. Kang, C. A. Day, A. K. Kenworthy and E. DiBenedetto, Traffic, 2012, 13, 1589–1600 CrossRef CAS PubMed.
- T. Kiuchi, T. Nagai, K. Ohashi and K. Mizuno, J. Cell Biol., 2011, 193, 365–380 CAS.
- J. L. McGrath, Y. Tardy, C. F. Dewey, Jr., J. J. Meister and J. H. Hartwig, Biophys. J., 1998, 75, 2070–2078 CrossRef CAS.
- D. Zicha, I. M. Dobbie, M. R. Holt, J. Monypenny, D. Y. Soong, C. Gray and G. A. Dunn, Science, 2003, 300, 142–145 CrossRef CAS PubMed.
- L. Chartier, L. L. Rankin, R. E. Allen, Y. Kato, N. Fusetani, H. Karaki, S. Watabe and D. J. Hartshorne, Cell Motil. Cytoskeleton, 1991, 18, 26–40 CrossRef CAS PubMed.
- H. Ishihara, H. Ozaki, K. Sato, M. Hori, H. Karaki, S. Watabe, Y. Kato, N. Fusetani, K. Hashimoto and D. Uemura, et al., J. Pharmacol. Exp. Ther., 1989, 250, 388–396 CAS.
- A. Inutsuka, M. Goda and Y. Fujiyoshi, Biochem. Biophys. Res. Commun., 2009, 390, 1160–1166 CrossRef CAS PubMed.
- V. Vogel and M. Sheetz, Nat. Rev. Mol. Cell Biol., 2006, 7, 265–275 CrossRef CAS PubMed.
- P. Roca-Cusachs, T. Iskratsch and M. P. Sheetz, J. Cell Sci., 2012, 125, 3025–3038 CrossRef CAS PubMed.
- S. W. Moore, N. Biais and M. P. Sheetz, Science, 2009, 325, 166 CrossRef CAS PubMed.
- S. W. Moore, X. Zhang, C. D. Lynch and M. P. Sheetz, J. Neurosci., 2012, 32, 11574–11585 CrossRef CAS PubMed.
- A. Chopra, E. Tabdanov, H. Patel, P. A. Janmey and J. Y. Kresh, Am. J. Physiol.: Heart Circ. Physiol., 2011, 300, H1252–1266 CrossRef CAS PubMed.
- A. Kumar, N. Khandelwal, R. Malya, M. B. Reid and A. M. Boriek, FASEB J., 2004, 18, 102–113 CrossRef CAS PubMed.
- A. Lanzavecchia, G. Lezzi and A. Viola, Cell, 1999, 96, 1–4 CrossRef CAS.
- Y. C. Li, B. M. Chen, P. C. Wu, T. L. Cheng, L. S. Kao, M. H. Tao, A. Lieber and S. R. Roffler, J. Immunol., 2010, 184, 5959–5963 CrossRef CAS PubMed.
- K. T. Bashour, A. Gondarenko, H. Chen, K. Shen, X. Liu, M. Huse, J. C. Hone and L. C. Kam, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2241–2246 CrossRef CAS PubMed.
- K. L. Hui, L. Balagopalan, L. E. Samelson and A. Upadhyaya, Mol. Biol. Cell, 2015, 26, 685–695 CrossRef CAS PubMed.
- J. Zalevsky, L. Lempert, H. Kranitz and R. D. Mullins, Curr. Biol., 2001, 11, 1903–1913 CrossRef CAS.
- A. Schonichen, H. G. Mannherz, E. Behrmann, A. J. Mazur, S. Kuhn, U. Silvan, C. A. Schoenenberger, O. T. Fackler, S. Raunser, L. Dehmelt and M. Geyer, J. Cell Sci., 2013, 126, 1891–1901 CrossRef PubMed.
- R. Galland, P. Leduc, C. Guerin, D. Peyrade, L. Blanchoin and M. Thery, Nat. Mater., 2013, 12, 416–421 CrossRef CAS PubMed.
- S. Kumari, D. Depoil, R. Martinelli, E. Judokusumo, G. Carmona, F. B. Gertler, L. C. Kam, C. V. Carman, J. K. Burkhardt, D. J. Irvine and M. L. Dustin, eLife, 2015, 4, e04953 Search PubMed.
- J. L. Cannon and J. K. Burkhardt, J. Immunol., 2004, 173, 1658–1662 CrossRef CAS.
- P. A. Zipfel, S. C. Bunnell, D. S. Witherow, J. J. Gu, E. M. Chislock, C. Ring and A. M. Pendergast, Curr. Biol., 2006, 16, 35–46 CrossRef CAS PubMed.
- P. Hahne, A. Sechi, S. Benesch and J. V. Small, FEBS Lett., 2001, 492, 215–220 CrossRef CAS.
- M. L. Dustin, Annu. Rev. Cell Dev. Biol., 2008, 24, 577–596 CrossRef CAS PubMed.
- T. N. Sims, T. J. Soos, H. S. Xenias, B. Dubin-Thaler, J. M. Hofman, J. C. Waite, T. O. Cameron, V. K. Thomas, R. Varma, C. H. Wiggins, M. P. Sheetz, D. R. Littman and M. L. Dustin, Cell, 2007, 129, 773–785 CrossRef CAS PubMed.
- T. Iskratsch, S. Lange, J. Dwyer, A. L. Kho, C. dos Remedios and E. Ehler, J. Cell Biol., 2010, 191, 1159–1172 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ib00032g |
| ‡ Present address: Randall Division of Cell and Molecular Biophysics, King's College London, London, UK |
| This journal is © The Royal Society of Chemistry 2015 |