
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerilipin 5 mediated lipid droplet remodelling revealed by coherent Raman imaging†
Nils
Billecke
a,
Madeleen
Bosma‡
b,
William
Rock
a,
Frederik
Fleissner
a,
Gerrit
Best
a,
Patrick
Schrauwen
b,
Sander
Kersten
c,
Mischa
Bonn
a,
Matthijs K. C.
Hesselink
*d and
Sapun H.
Parekh
*a
aDepartment of Molecular Spectroscopy, Max Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: parekh@mpip-mainz.mpg.de
bDepartments of Human Biology, School for Nutrition, Toxicology and Metabolism, Maastricht University Medical Center, 6200 MD Maastricht, The Netherlands
cNutrition, Metabolism and Genomics Group, Division of Human Nutrition, Wageningen University, 6700 EV Wageningen, The Netherlands
dMovement Sciences, School for Nutrition, Toxicology and Metabolism, Maastricht University Medical Center, 6200 MD Maastricht, The Netherlands. E-mail: matthijs.hesselink@maastrichtuniversity.nl
First published on 17th March 2015
Abstract
Accumulation of fat in muscle tissue as intramyocellular lipids (IMCLs) is closely related to the development of insulin resistance and subsequent type 2 diabetes. Most IMCLs organize into lipid droplets (LDs), the fates of which are regulated by lipid droplet coat proteins. Perilipin 5 (PLIN5) is an LD coating protein, which is strongly linked to lipid storage in muscle tissue. Here we employ a tandem in vitro/ex vivo approach and use chemical imaging by label-free, hyperspectral coherent Raman microscopy to quantify compositional changes in individual LDs upon PLIN5 overexpression. Our results directly show that PLIN5 overexpression in muscle alters individual LD composition and physiology, resulting in larger LDs with higher esterified acyl chain concentration, increased methylene content, and more saturated lipid species. These results suggest that lipotoxic protection afforded by natural PLIN5 upregulation in muscle involves molecular changes in lipid composition within LDs.
Insight, innovation, integrationLipid droplets are crucial organelles in energy homeostasis for nearly all living organisms. LD coating proteins are strongly involved in lipid metabolism and metabolic disorders. The abundance of Perilipin 5 (PLIN5), a LD coating protein exclusively found in oxidative tissue, increases lipid content in skeletal muscle without negative metabolic effects. In this work, we use quantitative, label-free coherent anti-Stokes Raman scattering (CARS) microscopy to analyze lipid composition in LDs in muscle upon PLIN5 overexpression in vivo and in vitro. Hyperspectral imaging not only verified the previously observed size increase of LDs, but also revealed the fundamental impact PLIN5 has on the local lipid composition and density in droplets. Vibrational imaging is a powerful tool to directly reveal how LD coating proteins affect lipid composition in individual LDs. |
Introduction
Obesity, defined as excessive accumulation of body fat in the body, poses a major health threat in western countries and is strongly linked to pathologies such as hypertension, atherosclerosis and type 2 diabetes. While body fat is typically stored in white adipose tissue, in obesity fat deposition also occurs ectopically in non-adipose tissues. In these tissues, fat can be stored as neutral lipids in lipid droplets (LDs), which possess multiple functions.1 In skeletal muscle, the major organ for post-prandial glucose disposal, myocellular LDs are key organelles in lipid metabolism and energy homeostasis. Sequestering and release of fatty acids from LDs is a tightly regulated process involving lipases, and their (co)activators and inhibitors, as well as LD-specific coat proteins of the so-called perilipin family.2 Of these proteins, perilipin 5 (PLIN5) stands out, as PLIN5 is predominantly expressed in oxidative tissues like skeletal and cardiac muscle, liver and brown adipose tissue,3,4 all tissues with a pivotal role in the maintenance of glucose homeostasis. Excess accumulation of intramyocellular lipids (known as IMCLs) in LDs generally associates negatively with insulin-mediated glucose uptake except in endurance-trained athletes. IMCL content in these athletes is very high whilst paradoxically being extremely insulin sensitive.5 Interestingly, the LD coat protein PLIN5 is more abundant in muscles of trained athletes compared to BMI-matched normal and obese subjects.6 Moreover, we and others recently showed that the overexpression of PLIN5 strongly enhances fat storage in skeletal muscle,7 heart8,9 and CHO cells,10 while insulin sensitivity was maintained and oxidative gene expression was promoted. These observations indicate that of the LD specific coat proteins, PLIN5 in skeletal muscle may play an important role in modulating myocellular insulin sensitivity.Decreased insulin sensitivity as a result of elevated IMCLs is believed to arise from elevated specific lipid subtypes, like diacylglycerols (DAGs) and ceramides.11 An important question is therefore whether subjects with PLIN5 abundance – for whom lipotoxic insulin resistance is suppressed – exhibit modified IMCL composition within LDs. Indeed, fluorescence assays in cultured cells have indicated that even within a single cell, differential PLIN protein decoration on the LD surface affects the cholesterol or triacylglycerol species present in LDs.12 So far, nearly all studies examining the effect of PLIN protein levels on molecular lipid composition such as unsaturation or esterification have used cell or tissue lysates. From these lysates lipids are extracted and specific lipid species are typically examined using chromatographic techniques coupled to mass spectrometry or colorimetric digestion assays.13,14 While indisputably valuable, this approach does not readily permit examination of the lipid species composition in LDs with differential perilipin coating. To mechanistically understand if PLIN5 abundance in muscle affects insulin sensitivity via lipid storage, it is necessary to evaluate lipid composition at the individual LD level in muscle where it is possible to segregate LDs based on the PLIN5 protein content.
In situ label-free determination of LD composition is possible with chemically specific microscopy techniques such as hyperspectral Raman microscopy, which derives contrast based on the concentration chemical bonds in the sample, at diffraction-limited spatial resolution. More rapid and similarly quantitative chemical imaging can be achieved by hyperspectral coherent anti-Stokes Raman scattering (CARS) microscopy.15,16 Like Raman imaging, CARS microscopy exploits the intrinsic molecular vibrations of sample chemistry. However the nonlinear interaction of a pump, Stokes, and probe photon within the focal volume drastically reduces problems of autofluorescence, provides axial optical sectioning, and can boost the signal level substantially.17 Indeed, hyperspectral CARS (as well as other coherent Raman microscopies) has been successfully used to track LDs non-invasively in living cells18 and excised tissues19,20 as well as elucidate the local lipid chemical composition.21,22 CARS has recently been shown to quantitatively agree with GC-MS results for lipid profiling.22–24
Here we use hyperspectral CARS microscopy with in vitro myotubes and ex vivo myofibers expressing endogenous and elevated levels of PLIN5 to investigate whether PLIN5 overexpression modulates composition of individual LDs harboring IMCL in skeletal muscle. We directly measure changes in individual myocellular LD chemistry by quantification of different chemical species at sub-micron resolution. Our results from two independent and distinct experimental systems show that with PLIN5 overexpression, LDs exhibit more than 200% higher methylene concentration, contain at least 220% more esterified acyl chains or sterols, and preferentially store saturated moieties compared with LDs from muscle having endogenous PLIN5 levels. The consistency between our in vitro and in vivo approach demonstrates that modulating PLIN5, an LD coat protein, results in substantial remodeling of the constituents within myocellular LDs. This suggests that molecular changes in LD composition as a result of altered PLIN5 levels in muscle may alter physiological consequences, e.g. insulin signalling, associated with excessive lipid storage in myocellular LDs.
Materials and methods
PLIN5 overexpression in differentiated C2C12 myotubes
Passage 1 to 3 C2C12 cells were grown in low glucose DMEM supplemented with 10% FCS (both Gibco) to 80% confluence on cover slips coated with 1 μg mL−1 collagen (Roche) before differentiation. The formation of myotubes was achieved by exchanging FCS for 2% horse serum. After 5 days, C2C12 myotubes were transfected with a PLIN5-GFP expression vector (RG224783, OriGene) using Lipofectamine® LTX Plus™ (Life Technologies) according to manufacturer’s instructions. After 48 h, transfection agents were removed, and the differentiated myotubes were treated with a 20 μM fatty acid mixture (palmitic acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oleic acid, 1:3) complexed to 8 μM BSA (all from Sigma Aldrich) for 24 h. Lipofection and subsequent lipid loading led to transfection efficiencies of 10–30% as estimated by fluorescence microscopy. Results from three separate C2C12 myotube transfections were pooled for the data presented here.
:oleic acid, 1:3) complexed to 8 μM BSA (all from Sigma Aldrich) for 24 h. Lipofection and subsequent lipid loading led to transfection efficiencies of 10–30% as estimated by fluorescence microscopy. Results from three separate C2C12 myotube transfections were pooled for the data presented here.
Local PLIN5 overexpression in rodent tibialis muscle
Unilateral gene electroporation of PLIN5 has been described previously.25 Briefly, 8-week-old male Wistar rats were fed a high fat diet (45% energy from fat – soybean oil and lard, D01060502, Research Diets) for 2-weeks before overexpression of PLIN5 in either the right or left tibialis anterior (TA) muscle. The contralateral TA served as a sham-electroporated internal control. Rats were sacrificed 8 days post electroporation. TA muscles were excised and rapidly frozen in melting isopentane. The Animal Care and Use Committee of Maastricht University approved the experiments (approval number 2010-036) and the study complied with the principles of laboratory animal care. Results from four animals were pooled for the data presented here.Lipid staining and immunofluorescence
Following C2C12 differentiation, transfection, and FA incubation, myotubes were fixed in 4% paraformaldehyde (PFA) in PBS for 30 minutes and rinsed three times with PBS before mounting coverslips to standard glass slides using double sided tape, which created a thin channel. The channels were then filled with PBS and sealed with nail polish to avoid drying. Successfully transfected cells were identified by the green fluorescence of the PLIN5-GFP fusion construct and locations were physically marked for subsequent analysis by hyperspectral CARS microscopy.Frozen TA muscles were cut transversally on a cryostat at −20 °C into serial sections (5 μm), some of which were used for fluorescence imaging and others were used for CARS microscopy. After sectioning, the samples were mounted on uncoated glass slides and stored at −20 °C until imaging. The process used for locating PLIN5-overexpressing fibers and subsequent CARS imaging has been described previously.15 Briefly, “scout” sections were processed for immunofluorescence by fixing in 4% PFA prior to incubation for 1 hour with anti-PLIN5 (#GP31; Progen Biotechnik) diluted (1:40) in antibody dilution buffer (AbDil, 150 mM NaCl, 20 mM Tris, 0.1% NaN3, 2% BSA at pH 7.4). After three washing steps with PBS, sections were stained with 1 μg mL−1 Bodipy 493/503 (Life Technologies) and Alexa Fluor 594 goat (1:200) anti guinea pig IgG secondary antibody (Life Technologies) in AbDil for 1 hour. Following three additional washing steps with PBS, sections were mounted in fluorescence mounting medium (Dako, Glostrup). Fluorescence and phase contrast images were acquired on an IX81 inverted microscope (Olympus) using Cell F imaging software. Fluorescence images of the scout sections were scanned for regions containing muscle fibers with high PLIN5 levels. The subsequent section of the same muscle tissue was used for hyperspectral CARS microscopy.
Tissue sections for CARS were fixed in 4% PFA, washed with PBS, and covered with #1 coverslips. These sections were directly sealed with nail polish to avoid dehydration. Regions with high PLIN5 expression in the section for CARS microscopy were located by matching morphological features to the corresponding phase contrast images (with strong LD and PLIN5 fluorescence) from the scout.
CARS microspectroscopy
A dual-output laser source (Leukos-CARS, Leukos) provides the pump and Stokes beams. The source was a passively Q-switched 1064 nm microchip laser, delivering sub-nanosecond pulses at a 32 kHz repetition rate and ∼300 mW average power. The pump (and probe) was the fundamental beam at 1064 nm, and the Stokes was a fiber-generated super-continuum with a spectral density of more than 100 μW nm−1 from 1050 nm to 1600 nm. Both beams were provided from the Leukos-CARS source with the Stokes beam emerging from a fiber and the pump beam provided in free space. The Stokes and pump/probe beams were routed on the optical table and matched in time and space at the focus of our microscope as described previously.15Briefly, we used a reflective collimator (RC04APC-P01, Thorlabs) to collimate the Stokes beam and filter it through a longpass 700 nm filter (FEL0700, Thorlabs) and a longpass 830 nm filter (LP02-830RS-25, Semrock). The Stokes and pump beams were combined at a dichroic mirror (LP02-1064RU-25, Semrock) and introduced into a modified inverted microscope (Eclipse Ti, Nikon). The pump and Stokes pulses were tightly focused onto the sample with a near IR objective (PE IR Plan Apo 100X, NA 0.85, Olympus), resulting in ∼32.5 mW of total laser power at the sample. The sample was mounted on a stepper-motor stage for coarse positioning (Microstage, Mad City Labs) with a nested piezo stage (Nano-PDQ 375 HS, Mad City Labs) for sample scanning. Together, this provided 25 mm travel range with sub-nm resolution. The CARS signal generated by the sample was collected in the forward direction by another objective (M-10X, NA 0.25, Newport) and sent through notch (NF03-532/1064E-25, Semrock) and short-pass filters (FES1000, Thorlabs) to remove the pump and Stokes radiation. The filtered CARS signal was dispersed using a spectrometer (Shamrock 303i, 300 lines mm−1, 1000 nm blaze, Andor) and detected on a deep-depletion CCD (Newton DU920P-BR-DD, Andor). The sample was raster scanned across the focal volume with steps of 0.3 μm in plane. For each position in the sample, a CARS spectrum in the range between 600 and 3400 cm−1 was acquired (Fig. 1A and B). CARS data were acquired with pixel dwell times of 250–500 ms. The spatial resolution of the instrument was independently measured to be ∼0.5 × 0.5 × 3.5 μm3, and the spectral resolution was limited by the camera pixel pitch to ∼4 cm−1 per pixel. The entire CARS microscope is controlled with custom software written in Labview (National Instruments).
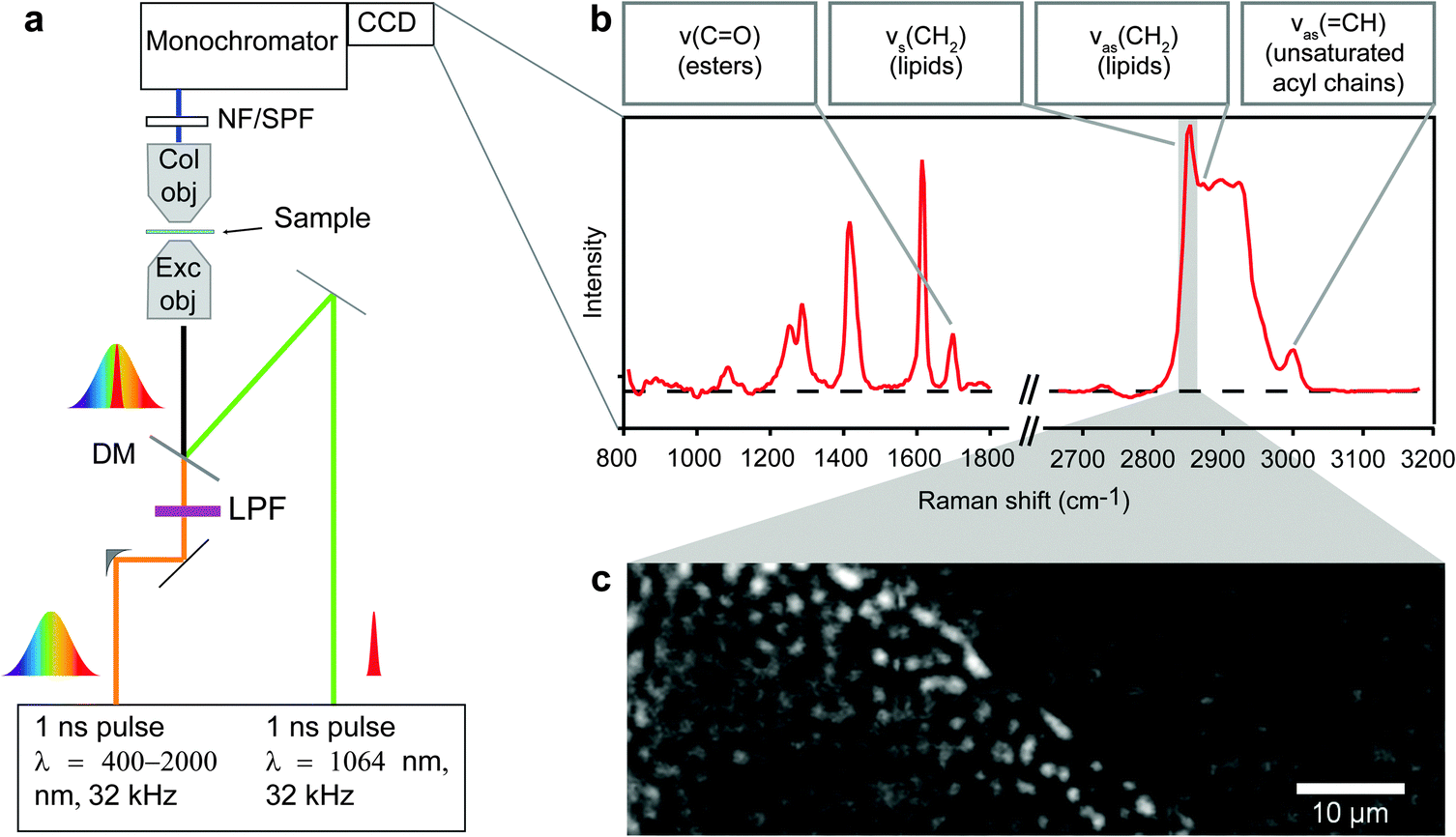 | ||
| Fig. 1 Hyperspectral CARS microscopy of myocellular LDs. (A) Schematic of the hyperspectral CARS microscope. The pump and probe photons are from the same laser, λ = 1064 nm, and the Stokes laser spans from λ = 1100–1600 nm after being spectrally filtered (LPF). These beams are combined by a dichroic mirror (DM) then focused by the excitation objective (Exc obj, 100X, 0.85 NA) into the sample. The CARS light is collected by the collection objective (Col obj, 10X, 0.25 NA), filtered (NF/SPF) from the pump and Stokes lasers, and detected by the monochromator and CCD. The sample is then moved in the X, Y, or Z direction to the next position and another spectrum is collected. (B) An RL CARS spectrum that shows a collection of peaks with selected vibrational modes relevant for this study (υs: symmetric vibration; υas: asymmetric vibration). (C) 2D concentration maps are created by plotting the intensity of a specific peak for each (X,Y) location. The image contrast is the value from integration over the CH2 vibration (marked by the gray shaded region in C) in a cross section of control rat tibialis muscle in which sub-micron diameter LDs are seen as bright regions. | ||
Hyperspectral data analysis
Raw CARS spectra were analyzed with custom routines in IgorPro 6.22A (Wavemetrics). Initially, data were normalized by exposure and signal counts from the non-resonant region of the spectra (2200–2300 cm−1) so datasets taken over multiple days and months could be compared. Treatment of the data with a modified Kramers–Kronig26 algorithm and background phase correction removes the non-resonant contribution to the signal and retrieves the imaginary component of the third order Raman susceptibility, here referred to as Raman-like CARS (RL CARS) spectra. These spectra are linear in concentration and can be quantitatively analysed similar to Raman spectra.26–28 Images of particular Raman frequencies were generated in IgorPro for subsequent analysis. Due to the abundance of acyl chains, and therefore CH2 moieties in lipids, Raman imaging of the CH2 symmetric stretching vibration has been often used for the identification of lipid-rich locations in biological samples. Concentration plots of the CH2 symmetric vibrations were constructed by integrating the intensity from RL CARS spectra from 2840–2856 cm−1, and these images (called CH2 images) were used for identifying LDs in all samples imaged with hyperspectral CARS microspectroscopy (Fig. 1C). For each sample, the concentration plots for the ester bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) and the asymmetric CH vibration were generated by integrating the intensity from RL CARS spectra from 1730–1750 cm−1 and 3001–3018 cm−1, respectively. All image processing and analysis was completed with ImageJ software as described below and in the Supplementary Information.
O stretching) and the asymmetric CH vibration were generated by integrating the intensity from RL CARS spectra from 1730–1750 cm−1 and 3001–3018 cm−1, respectively. All image processing and analysis was completed with ImageJ software as described below and in the Supplementary Information.
Image analysis
To demarcate LDs, CH2 images were used to highlight high methylene concentration regions using the following process: (1) the ImageJ unsharp mask tool (1–2 pixel radius 0.9 mask weight) was used to enhance edges, (2) empirical thresholding was used to separate high intensity image pixels, and (3) creation of regions of interest (ROIs) for all high intensity pixels. This process exclusively highlighted LDs in the hyperspectral images (see Fig. SI, ESI†). Note that any features smaller than two pixels were not counted in this analysis. The ROIs generated from LD identification from the CH2 images were superimposed on the concentration maps generated by plotting peak intensity of each specific wavenumber region mentioned in Hyperspectral Data Analysis section. The average CH2, CO, and CH intensities within- and sizes of the ROIs were extracted for every individual LD and quantitatively analyzed with OriginPro as described below. We verified the linear relationship of measured sizes to actual objects using monodisperse polystyrene microbeads with the same processing protocol (Fig. SII, ESI†).
Data analysis and statistics
All statistical analysis was performed with OriginPro 8.5.1 (OriginLab). All data were pooled by treatment (overexpressing PLIN5 or endogenous control) for cultured myotubes and similarly for in vivo tibialis tissue sections. Unpaired two sample t-tests were performed, and p ≤ 0.05 was considered significant.Results
We used two independent muscle systems: differentiated C2C12 myoblasts and muscle tissue from rat tibialis anterior. Myoblasts were differentiated into myotubes, transfected with PLIN5-GFP, and incubated with a fatty acid cocktail (3:1, oleic:palmitic acid) to induce LD formation (Fig. SIII, ESI†). High-fat fed rats were electroporated with PLIN5 and sacrificed 8 days later to obtain tibialis tissue sections. Fluorescence images of GFP from the GFP-PLIN5 fusion construct (Fig. 2A, red) and antibody-labelled PLIN5 (Fig. 2E, red) demarcated myotubes with PLIN5 overexpression in the in vitro and in vivo systems, respectively. Random myotubes not showing GFP-PLIN5 (for differentiated myotubes) and muscle fibers from the contralateral leg (for tibialis) were used as controls with endogenous PLIN5 expression. Hyperspectral CARS microscopy provided spatially resolved chemical information from these myocellular samples in situ.
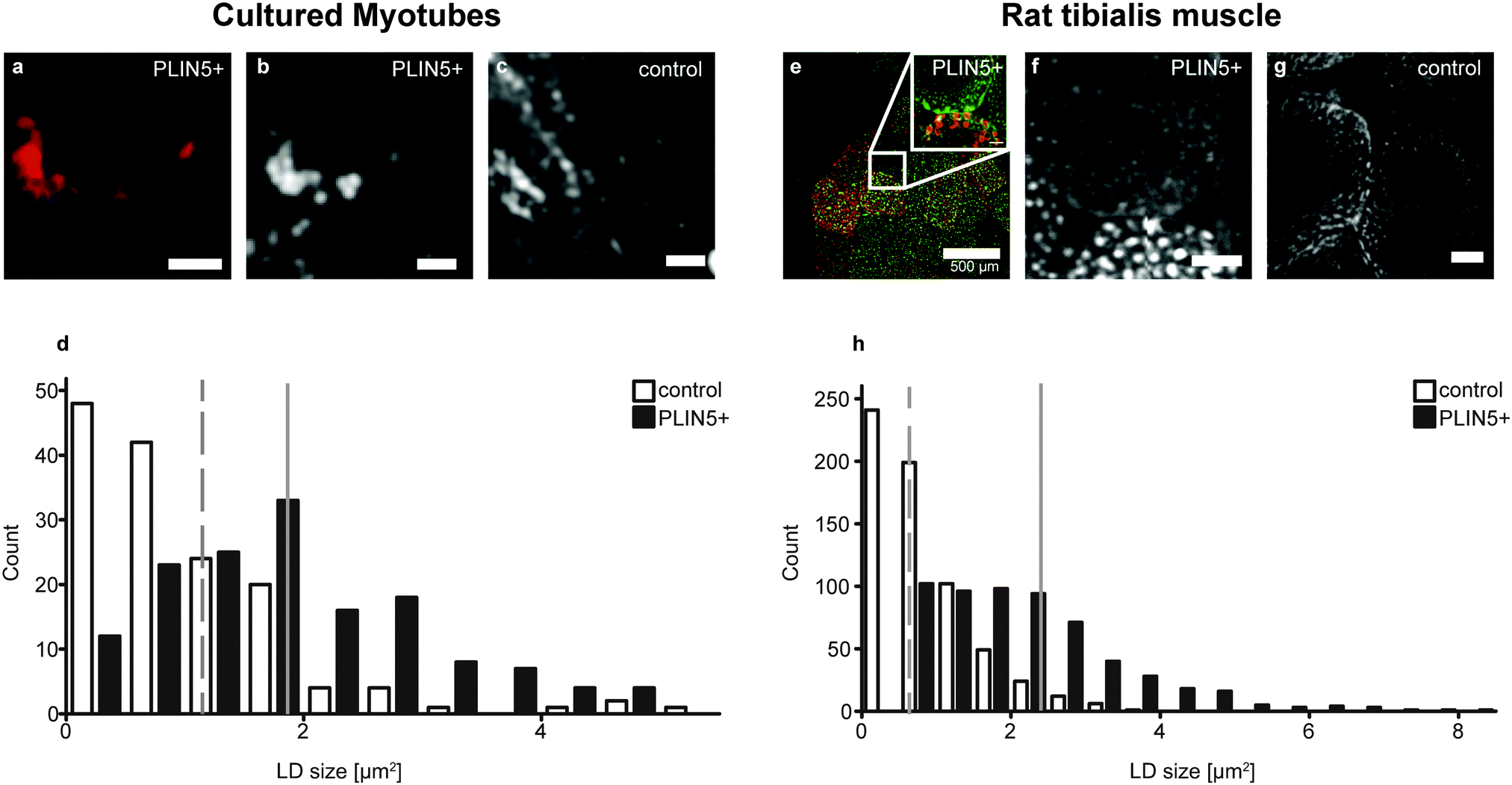 | ||
| Fig. 2 Ectopically induced PLIN5 locates to the LD shell and leads to an increase in LD size in myocellular samples after FA incubation/dietary intervention. Representative fluorescence images of (A) C2C12 myotubes and (E) rat tibialis muscle sections upon PLIN5 overexpression (PLIN5-red, lipids-green). E is a histological scout slice 5 μm removed from the actual slice that was used for hyperspectral CARS. Inset in E indicated by the white zoomed image was from the actual tissue section used for CARS imaging and corresponds to the image shown in F. (B), (F) Images of the CH2 symmetric intensity of the regions in A and E (inset), respectively. Smaller LDs are observed in (C) control C2C12 myotubes and (G) control rat tibialis muscle. (D) Histogram of LD area distribution in cultured myotubes (dashed line marks mean area for control LDs = 1.0 μm2, solid line marks mean area for PLIN5-overexpressing LDs = 1.9 μm2). (H) Histogram of LD area distribution in tibialis muscle (dashed line marks mean area for control LDs = 0.8 μm2, solid line marks mean area for PLIN5-overexpressing LDs = 2.2 μm2). Data are pooled from 150 LDs for PLIN5 overexpression and 147 LDs for endogenous controls in cultured myotubes and from 581 LDs for PLIN5 overexpression and 634 LDs for endogenous controls in tibialis muscle. Scale bars are 5 μm in cultured myotubes (A–C) and 20 μm in muscle sections (E–G) unless indicated otherwise. | ||
PLIN5 overexpression increases size and augments energy density in myocellular LDs
CH2 images of myotubes with elevated PLIN5 showed that the fluorescence signal was located around the rim of circular LDs having high CH2 content (Fig. 2A, B and E), which is consistent with PLIN5 localization to the LD border when overexpressed in other in vitro and in vivo systems.29 As expected in myotubes with endogenous PLIN5 expression, but fed the same diet (or fatty acid cocktail), LDs were observable, and CH2 images also allowed unambiguous identification of LDs (Fig. 2C and G). Because LDs were readily detectable from CH2 images in both control and PLIN5-overexpressing myotubes, a thresholding process was used to generate regions-of-interest (ROIs) to highlight LDs in all samples. The signals from different vibrational bands within individual LDs (above the diffraction limit) were then quantitatively compared within these ROIs (see ESI†).Size analysis of the CH2 images from the endogenous and PLIN5-overexpressing in vitro myotubes shows that LD area in PLIN5-overexpressing myotubes was 90% larger than in control myotubes (1.9 ± 1.1 μm2vs. 1.0 ± 0.9 μm2, mean ± S.D., p < 0.01) (Fig. 2D). Similar analysis from tibialis sections showed 180% larger area (2.2 ± 1.3 μm2vs. 0.8 ± 0.6 μm2, mean ± S.D., p < 0.01) upon PLIN5 overexpression. Histograms show a clear population shift to larger individual LDs upon PLIN5 overexpression (Fig. 2D and H). We note that the absolute sizes determined from our measurements are not directly comparable with those from electron microscopy (EM) due to diffraction and scattering. Nevertheless, the results show a similar LD size increase in tibialis muscle as was observed with EM.7
Still looking at the CH2 vibration, we plot the mean CH2 intensity within each LD and see that the LDs in PLIN5-overexpressing myotubes contained approximately 200–330% more CH2 groups per area within the droplets (Fig. 3A and C). Even with substantial biological variability and transfection efficiency, the increased CH2 content seen in PLIN5-overexpressing myotubes was statically significant (P < 0.001) compared to control myotubes in both systems. The robustness of these findings is further demonstrated in Fig. 3B and D, where total LD CH2 intensity is plotted versus LD area for the in vitro and in vivo system, respectively. Total CH2 content is strongly correlated with LD area in both PLIN5-overexpressing and control LDs in both systems, and the slope of the lines for LDs from PLIN5 – overexpressing myotubes is significantly larger than the slope for the control LDs. This independently confirms that PLIN5-overexpressing LDs contain more CH2 and have larger LD area, which ultimately means that PLIN5 overexpression results in bigger LDs with higher stored energy compared to LDs in muscle with endogenous PLIN5.
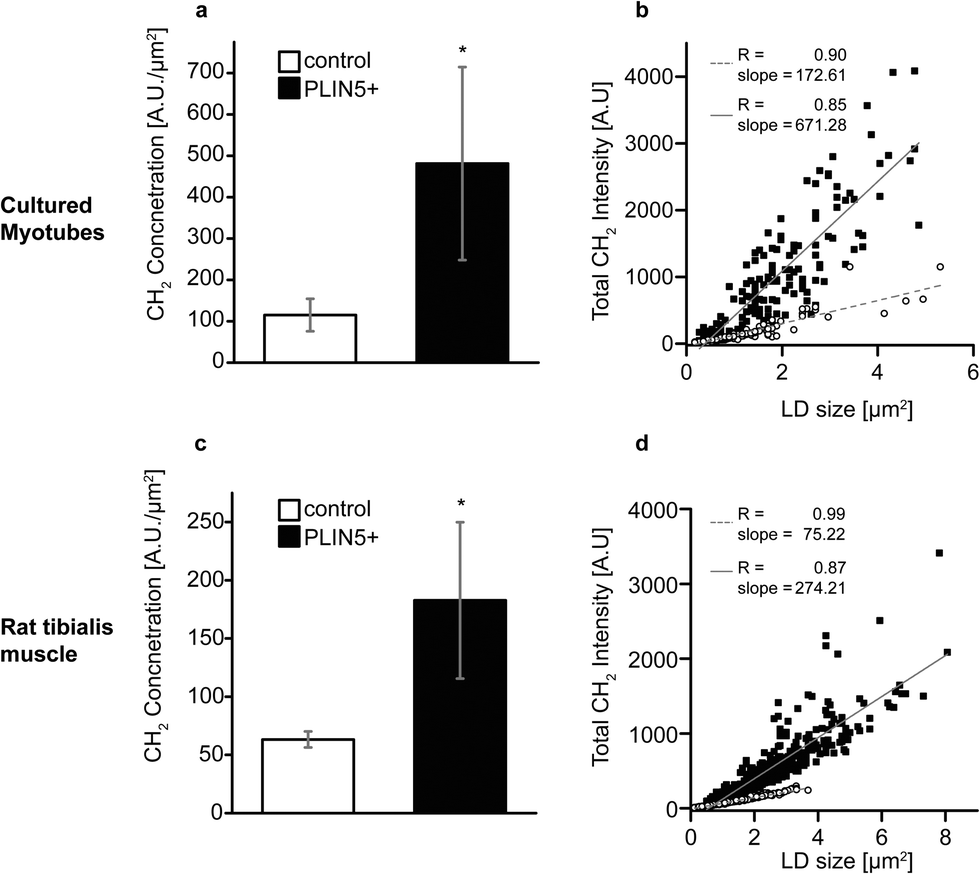 | ||
| Fig. 3 PLIN5 overexpression elevates methylene concentration in myocellular LDs. Average CH2 intensities per individual LD area were used to analyze methylene concentration. Methylene concentration was elevated in PLIN5-overexpressing (A) myotubes by 320% and (C) tibialis muscle by 200% when compared to the respective endogenous controls. Scatter plots of total CH2 intensity vs. LD area showed linear dependence for both (B) cultured myotubes and (D) tibialis muscle (open symbols – control LDs and closed symbols – PLIN5-overexpressing LDs). Data are pooled from 150 LDs for PLIN5 overexpression and 147 LDs for endogenous controls in cultured myotubes and from 581 LDs for PLIN5 overexpression and 634 LDs for endogenous controls in tibialis muscle. Error bars are SD, and * denotes p < 0.01. | ||
PLIN5 overexpression increases the amount of esterified lipid chains within myocellular LDs
In principle, the increased CH2 (methylene) concentration associated with PLIN5 overexpression could result from: (i) more dense acyl chain packing into LDs of PLIN5-overexpressing myotubes, (ii) increased carbon saturation of acyl chains within LDs of PLIN5-overexpressing myotubes, (iii) elongation of the acyl chains, or (iv) a combination of any of the above effects. With hyperspectral CARS microscopy, we obtain a full chemical fingerprint from each point in the sample, and we can directly quantify other chemical species within the LDs to explore the source of the increased CH2 concentration upon PLIN5 overexpression.Esterified lipids such as triacylglycerol (TAG) and cholesterol esters (CE), and to a lesser extent, DAG, constitute the majority of the lipid material within LDs. TAG, DAG, and CE species contain three, two, and one ester bond, respectively. Thus quantifying ester bonds in the LD should yield an indicative measure of the number of esterified acyl chains within the LD and allow us to determine if increased acyl chain packing contributes to the 200–330% increase in methylene groups in LDs of PLIN5-overexpressing muscle. The Raman mode at 1740 cm−1 is attributed to CO stretching vibration of ester bonds23,30 (Fig. 1B), so we created vibrational images of CO groups to investigate the ester content in LDs. Fig. 4B and G show ester images from PLIN5-overexpressing myotubes from cells and tibialis, respectively (corresponding methylene images are shown in Fig. 4D and I). Control myotubes show LDs (Fig. 4C and H) that contain substantially less ester content (Fig. 4A and F). From these ester images, it is evident that PLIN5 overexpression results in augmented ester content within LDs. These intense ester inclusions are not seen in the control myotubes. This is further highlighted by looking at representative vibrational spectra from locations within LDs in cultured and tibialis myotubes (Fig. 4, spectra and Fig. SV, ESI†). These spectra show that PLIN5-overexpressing LDs have an obvious peak at 1740 cm−1 whereas no peak is observable in the spectra of control LDs. Using the same ROIs generated for the size analysis of individual LDs from the CH2 images, we quantified the ester content in single LDs in both PLIN5-overexpressing and control myotubes. The local concentration of CO groups within LDs from PLIN5-overexpressing samples was more than 1000% larger than in LDs from control samples for cells (Fig. 4E) and 220% larger in PLIN5-overexpressing tibialis (Fig. 4J). This demonstrates that PLIN5 overexpression increases the amount of esterified species in a given area within LDs, consistent with higher CH2 concentration in LDs of PLIN5-overexpressing myotubes.
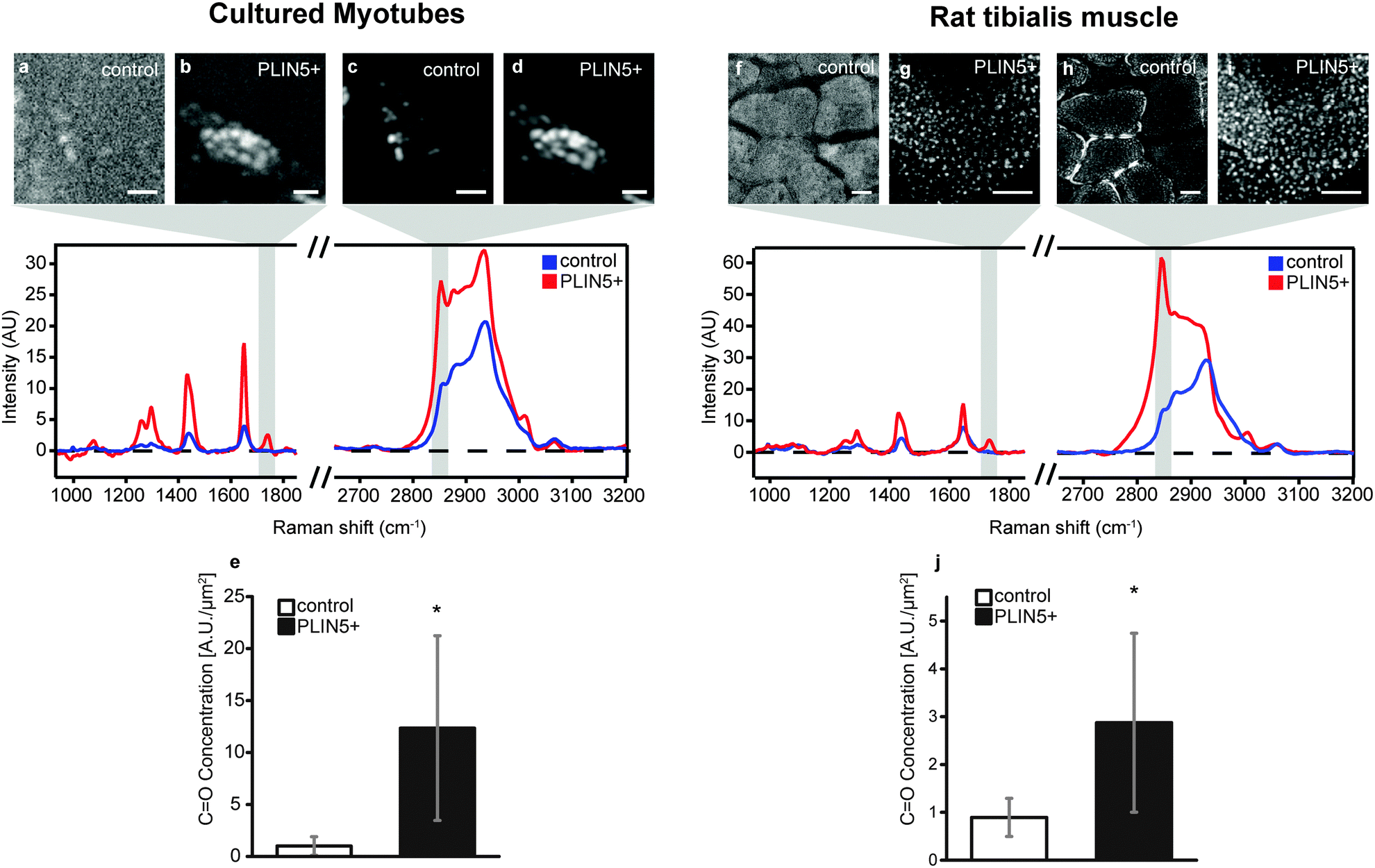 | ||
| Fig. 4 PLIN5 overexpression leads to increased ester bonds in myocellular LDs. Top panel shows representative paired images of ester (CO) and methylene (CH2) vibrational intensities. (A) CO intensity and (C) CH2 intensity of control myotubes. (B) CO intensity and (D) CH2 intensity of PLIN5-overexpressing myotubes. (F) CO intensity and (H) CH2 intensity of control tibialis muscle. (G) CO intensity and (I) CH2 intensity of PLIN5-overexpressing tibialis muscle. Center panel shows representative RL CARS spectra from within a single LD in the respective control and PLIN5 overexpression model. The gray shaded areas in the spectrum denote the respective spectral regions that were integrated to generate the intensity maps (A–D, F–I). Mean CO concentration was elevated in PLIN5-overexpressing (E) myotubes by 1140% and (J) tibialis muscle by 220%, respectively, when compared to the respective endogenous controls. Data are pooled from 150 LDs for PLIN5 overexpression and 147 LDs for endogenous controls in cultured myotubes and from 581 LDs for PLIN5 overexpression and 634 LDs for endogenous controls in tibialis muscle. Error bars are SD, and * denotes p < 0.01. Scale bars are 5 μm in cultured myotubes (A–D) and 20 μm in muscle sections (G–J). | ||
Saturated lipids are more abundant in LDs upon PLIN5 overexpression
In addition to esterified acyl chains, we explored if the lipid saturation in LDs was differentially modulated by PLIN5 overexpression. Principally we could do so by quantifying CC vibration at 1650 cm−1 or by using stretching vibrations from unsaturated carbon modes (CH) at 3005 cm−1 with intensity proportional to the number of carbon–carbon double bonds in the focal volume.23 We preferred using the latter (stretching vibrations of CH), as the CC vibration profoundly overlaps with protein contribution to the broad amide I band (1635–1665 cm−1) and hence is less specific for quantification of lipid unsaturation. Using neat fatty acid mixtures of palmitic and oleic acid, which have the same number of CH2 modes per chain, but with oleic acid having two additional CH modes, we tested the linearity of this metric. We find a linear dependence of ICH/ICH2 to the molar ratio of oleic acid in the mixture (Fig. SIV, ESI†). Using the established ROIs for the LDs, the ICH/ICH2 ratio within individual LDs was calculated. This denotes the relative unsaturation per methylene group. In LDs from PLIN5-overexpressing myotubes, unsaturation showed a 31% decrease compared to LDs in control myotubes (0.18 ± 0.07 vs. 0.26 ± 0.12, mean ± S.D., p < 0.01) (Fig. 5A). In PLIN5-overexpressing tibialis muscle, the ratio CH/CH2 was more prominently reduced by 83% compared endogenous controls (0.05 ± 0.02 vs. 0.29 ± 0.07, mean ± S.D., p < 0.01) when mean values were compared. Scatter plots of total CH vs. total CH2 values for each individual LD show a correlation between these variables for both experimental systems under endogenous and PLIN5-overexpressing conditions (Fig. 5B and D). PLIN5 overexpression in vitro did not lead to a substantial (although statistically significant) difference in the unsaturation level (31% decrease in PLIN5 compared to control), and the slopes of the CH vs. CH2 lines for the control and PLIN5-overexpressing cases were almost identical (Fig. 5B). For tibialis LDs, the unsaturation difference was much larger (83% decrease in PLIN5 compared to control), and the slope of the control LDs was correspondingly much steeper than for PLIN5-overexpressing LDs (Fig. 5D). The decreased unsaturation per methylene group in both muscle model systems demonstrates that LDs contain a larger fraction of saturated carbons per methylene when PLIN5 is overexpressed.
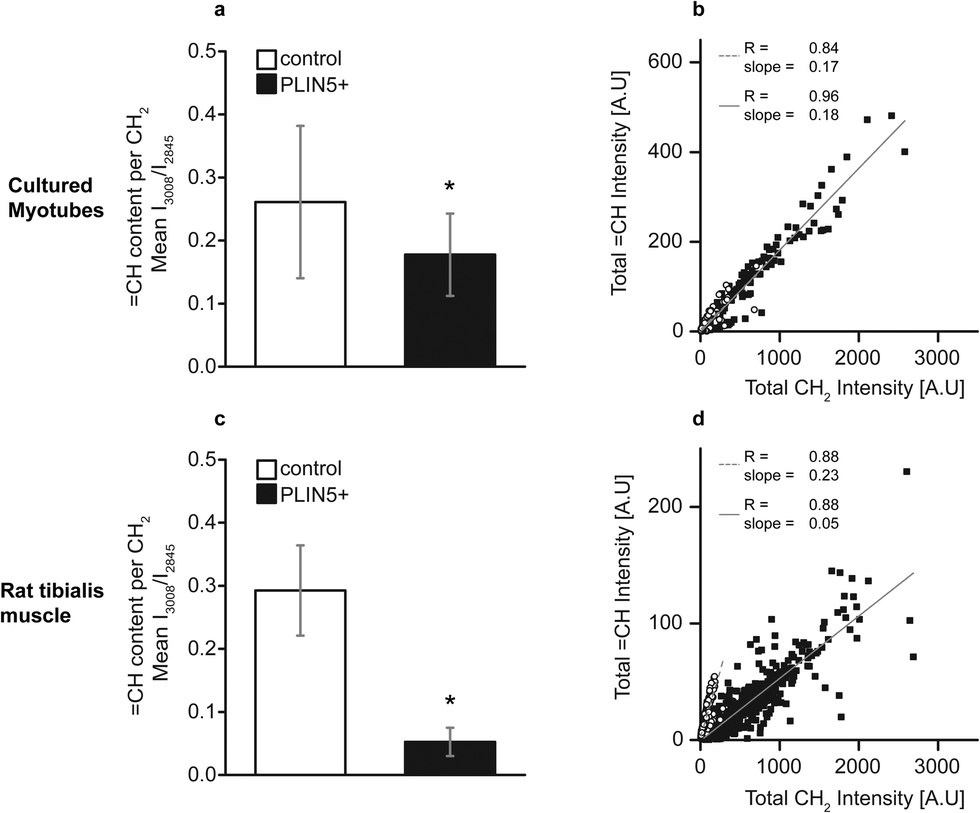 | ||
| Fig. 5 Lipid unsaturation is decreased upon PLIN5 overexpression in myocellular LDs. Unsaturated carbon (CH) content per CH2 was lowered in LDs in PLIN5-overexpressing (A) myotubes by 31% and (C) tibialis muscle by 83%, respectively, when compared to the respective endogenous controls. (B), (D) Scatter plots of CH vs. CH2 content in PLIN5-overexpressing LDs show linear dependence of the two variables (open symbols – control LDs and closed symbols – PLIN5-overexpressing LDs). Data are pooled from 150 LDs for PLIN5 overexpression and 147 LDs for endogenous controls in cultured myotubes and from 581 LDs for PLIN5 overexpression and 634 LDs for endogenous controls in tibialis muscle. Error bars are SD, and * denotes p < 0.01. | ||
Discussion
Ectopic neutral lipids are stored as LDs, which are coated by numerous proteins that modulate the LD phenotype by regulating lipolysis and subsequent fatty acid oxidation and signalling as well as overall cellular lipid distribution. Of these proteins, PLIN proteins have been shown to critically affect lipid metabolism in a wide range of species and tissue. PLIN5 is predominantly expressed in oxidative tissue and plays a critical role in LD dynamics7,29,31,32 that help maintain glucose homeostasis in e.g. muscle. By using in vitro and in vivo independent muscle model systems, we show that PLIN5 abundance is capable of altering the internal composition of intramyocellular LDs. Information on the local chemistry within LDs was obtained using hyperspectral CARS microscopy and data processing to obtain Raman-like spectra from in situ samples with sub-micron spatial resolution that could be quantitatively analysed. LDs were easily identified by hyperspectral CARS through the abundance of methylene groups in their core,15i.e. by integrating the signal intensity at 2840–2856 cm−1, corresponding to the molecular vibration of CH2. With CARS microscopy, we confirmed elevated IMCL levels in muscle upon feeding with a high-fat cocktail (in vitro) or high-fat diet (in vivo) as expected33 and increased LD size when PLIN5 was overexpressed.9,15,34 In addition to increased size, LDs in PLIN5-overexpressing samples showed a 200–330% increase in CH2 content per LD area, more esterified lipids and more saturated lipid species when compared to the endogenous control muscle.Taking into account that the LD core is primarily composed of TAGs and to a lesser extent sterol esters, increased CH2 concentration can be attributed to an augmented number of acyl chains, increased carbon saturation, increased acyl chain length, or combinations of these modifications. Most acyl chains in LDs are ester-bound to a glycerol backbone (non-esterified acyl chains typically partition to the LD–sarcoplasm interface), so the ester content of individual LDs from the 1740 cm−1 vibration is roughly proportional to the number of acyl chains. The ester concentration in LDs was 1140% and 220% higher in cultured myotubes and rat tibialis muscle, respectively, in the PLIN5-overexpressing cases when compared to endogenous controls. The increase in esterification upon PLIN5 overexpression we observe agrees well with previous studies reporting an accumulation of TAG in whole skeletal muscle7 and heart tissue8,35 upon PLIN5 overexpression using mass spectrometry. Surprisingly, spectra from LDs in control in vitro myotubes as well as control tibialis muscle showed a virtually undetectable signal at 1740 cm−1 and corresponding image plots of the ester bond intensity did not overlap with LD morphology (Fig. 4 and Fig. SV, ESI†). Whether this is truly due to the absence of ester bonds (and therefore esterified acyl chains) is difficult to judge based on the current dataset. Our results additionally show that the marked increase in esterification is located within LDs (Fig. SVI, ESI†). Previous mass spectrometry approaches could not make this observation as this requires an LD-specific approach. A potential mechanism for the increase in esterified species in myocellular LDs upon PLIN5 overexpression is increased activity of diacylglycerol-O-acyltransferase 1 (DGAT1), the rate limiting enzyme in synthesis of TAG from DAG and acyl-CoA. We explored this possibility by examining DGAT1 gene and protein expression in cells and tissues (Fig. SVII, ESI†). While still imperfect, these data seem to agree with the LD esterification changes from CARS. Further studies are certainly required to clarify the relationship between increased lipid esterification under high PLIN5 conditions and DGAT1.
Saturated fats have repeatedly been reported to impede insulin sensitivity,36,37 whereas unsaturated fats can improve insulin sensitivity in diabetic patients.38 Saturated NEFAs have been shown to induce oxidative stress and mitochondrial dysfunction39 and trigger a pro-inflammatory phenotype40 associated with myocellular insulin resistance. Thus, sequestering saturated fats into LDs is beneficial by segregating them from participating in detrimental signaling. This observation is further substantiated by in vitro studies, where fully saturated palmitic acid impaired insulin mediated glucose uptake, while monounsaturated oleic acid did not.41 The ratio of CH/CH2 (quantifying lipid unsaturation) was decreased in LDs in PLIN5-overexpressing cultured myotubes (31% compared to endogenous controls) and tibialis muscle (83% compared to endogenous controls) (Fig. 5). This indicates that the abundance of PLIN5 promotes an LD phenotype in which more saturated moieties per methylene are sequestered into LDs. Interestingly, increased sequestering of saturated lipids in LDs upon PLIN5 overexpression links to our previous observation that the overexpression of PLIN5 in muscle resulted in the down regulation of inflammatory genes in muscle.7
An interesting deduction from our data relates to the large increase in methylene and ester concentration in PLIN5-overexpressing LDs. In both models (in vitro and in vivo), we observed that the acyl chain concentration (approximately equal to ester bonds per μm2) is at least 220% higher and that methylene concentration is more than 200% higher upon PLIN5 overexpression. This implies that under conditions of PLIN5 abundance, more than three times as many ester bonds and CH2 groups reside within an LD. Given the comparatively small variability in the density of oil (800–1100 mg mL−1), the only way to explain this observation is that under endogenous PLIN5 conditions, LDs contain fewer esterified TAGs and CE (Fig. SV, ESI†). Therefore, our data suggest that LDs in muscle with endogenous PLIN5 expression potentially contain additional lipophilic material that is not esterified and has relatively low CH2-content, which presumably maintains the physical chemical properties of the LD (oil-like) core. Based upon freeze fracture electron microscopy, it has been suggested that PLIN family proteins themselves (PLIN2 or ADRP) can be found within the lipid core of LDs.42 While augmented CH2 intensity is clearly observed in LDs in the control muscle when compared to the surrounding sarcoplasm, the overall spectral shapes from these LDs do exhibit protein-like features with strong contributions from CH modes of higher energy around 2920 cm−1 (Fig. 4 and Fig. SV, ESI†). From our results, the most we can say is that in muscle with endogenous PLIN5 expression additional species other than TAG or CE appear to exist in the LD, which are less present when PLIN5 levels are elevated.
Conclusion
Our study shows that remodelling of LD composition and content in skeletal muscle (the major organ for post-prandial glucose disposal) can occur as a result of the overexpression of PLIN5, a LD coat protein predominantly present in oxidative tissues like cardiac and skeletal muscle and brown adipose tissue. We employed two separate muscle model systems: cultured myotubes and in vivo tibialis anterior muscle tissue, and the notable agreement between these two systems for the observed LD compositional phenotypes underscores the robustness of our observations. Apart from morphology, hyperspectral CARS datasets provide localized quantitative information on lipid species within LDs. Our data confirmed that myocellular PLIN5 overexpression leads to an increase in LD size in both experimental systems and additionally showed that LDs in PLIN5-overexpressing myotubes exhibited elevated methylene concentration, ester concentration, and lipid saturation compared to endogenous controls. These findings indicate that LDs present in muscle with abundant PLIN5 are biochemically distinct and perhaps correspond to a benign phenotype wherein augmented myocellular lipid content is observed while insulin-mediated glucose uptake is still maintained.7 This is in contrast to obese and insulin resistant subjects with endogenous PLIN5 where augmented myocellular lipid content is known to impede insulin-mediated glucose uptake, thereby connecting the PLIN5 level with LD composition and insulin sensitivity.Acknowledgements
NB, MB, and MKCL acknowledge financial support from the NanoNextNL, a micro and nanotechnology consortium of the Government of the Netherlands and 130 partners. MaB was financially supported by NUTRIM and the Graduate School VLAG. A Vici (Grant 918.96.618) grant for innovative research from the Netherlands Organization for Scientific Research supports the work of PS. SHP acknowledges financial support from the Marie Curie Foundation #CIG322284. The authors wish to thank J. Hunger and G. Waschatko for stimulating discussions and S. Pütz for technical support with cell handling and transfection.References
- A. R. Thiam, R. V. Farese and T. C. Walther, Nat. Rev. Mol. Cell Biol., 2013, 14, 775–786 CrossRef CAS PubMed.
- N. A. Ducharme and P. E. Bickel, Endocrinology, 2008, 149, 942–949 CrossRef CAS PubMed.
- R. Minnaard, P. Schrauwen, G. Schaart, J. A. Jorgensen, E. Lenaers, M. Mensink and M. K. C. Hesselink, J. Clin. Endocrinol. Metab., 2009, 94, 4077–4085 CrossRef CAS PubMed.
- N. E. Wolins, B. K. Quaynor, J. R. Skinner, A. Tzekov, M. A. Croce, M. C. Gropler, V. Varma, A. Yao-Borengasser, N. Rasouli, P. A. Kern, B. N. Finck and P. E. Bickel, Diabetes, 2006, 55, 3418–3428 CrossRef CAS PubMed.
- B. H. Goodpaster, J. He, S. Watkins and D. E. Kelley, J. Clin. Endocrinol. Metab., 2001, 86, 5755–5761 CrossRef CAS PubMed.
- F. Amati, J. J. Dube, E. Alvarez-Carnero, M. M. Edreira, P. Chomentowski, P. M. Coen, G. E. Switzer, P. E. Bickel, M. Stefanovic-Racic, F. G. Toledo and B. H. Goodpaster, Diabetes, 2011, 60, 2588–2597 CrossRef CAS PubMed.
- M. Bosma, L. M. Sparks, G. J. Hooiveld, J. A. Jorgensen, S. M. Houten, P. Schrauwen, S. Kersten and M. K. Hesselink, Biochim. Biophys. Acta, 2013, 1831, 844–852 CrossRef CAS PubMed.
- N. M. Pollak, M. Schweiger, D. Jaeger, D. Kolb, M. Kumari, R. Schreiber, S. Kolleritsch, P. Markolin, G. F. Grabner, C. Heier, K. A. Zierler, T. Rulicke, R. Zimmermann, A. Lass, R. Zechner and G. Haemmerle, J. Lipid Res., 2013, 54, 1092–1102 CrossRef CAS PubMed.
- H. Wang, U. Sreenivasan, D. W. Gong, K. A. O'Connell, E. R. Dabkowski, P. A. Hecker, N. Ionica, M. Konig, A. Mahurkar, Y. Sun, W. C. Stanley and C. Sztalryd, J. Lipid Res., 2013, 54, 953–965 CrossRef CAS PubMed.
- S. R. Bartholomew, E. H. Bell, T. Summerfield, L. C. Newman, E. L. Miller, B. Patterson, Z. P. Niday, W. E. T. Ackerman and J. T. Tansey, Biochim. Biophys. Acta, 2012, 1821, 268–278 CrossRef CAS PubMed.
- M. J. Watt and A. J. Hoy, American journal of physiology. Endocrinology and metabolism, 2012, 302, E1315–E1328 CrossRef CAS PubMed.
- K. Hsieh, Y. K. Lee, C. Londos, B. M. Raaka, K. T. Dalen and A. R. Kimmel, J. Cell Sci., 2012, 125, 4067–4076 CrossRef CAS PubMed.
- Y.-S. Cheng, Y. Zheng and J. VanderGheynst, Lipids, 2011, 46, 95–103 CrossRef CAS PubMed.
-
H. Nygren, T. Seppänen-Laakso, S. Castillo, T. Hyötyläinen and M. Ore
![[s with combining breve]](https://www.rsc.org/images/entities/char_0073_0306.gif) ič, in Metabolic Profiling, ed. T. O. Metz, Humana Press, 2011, vol. 708, ch. 15, pp. 247–257 Search PubMed.
ič, in Metabolic Profiling, ed. T. O. Metz, Humana Press, 2011, vol. 708, ch. 15, pp. 247–257 Search PubMed. - N. Billecke, G. Rago, M. Bosma, G. Eijkel, A. Gemmink, P. Leproux, G. Huss, P. Schrauwen, M. K. Hesselink, M. Bonn and S. H. Parekh, Histochem. Cell Biol., 2014, 141, 263–273 CrossRef CAS PubMed.
- T. Hashimoto, H. Segawa, M. Okuno, H. Kano, H. O. Hamaguchi, T. Haraguchi, Y. Hiraoka, S. Hasui, T. Yamaguchi, F. Hirose and T. Osumi, J. Cell Sci., 2012, 125, 6127–6136 CrossRef CAS PubMed.
- W. M. Tolles and R. D. Turner, Appl. Spectrosc., 1977, 31, 96–103 CrossRef CAS.
- C. Jungst, M. Klein and A. Zumbusch, J. Lipid Res., 2013, 54, 3419–3429 CrossRef PubMed.
- T. T. Le, A. Ziemba, Y. Urasaki, S. Brotman and G. Pizzorno, PLoS One, 2012, 7, e51092 CAS.
- M. C. Wang, W. Min, C. W. Freudiger, G. Ruvkun and X. S. Xie, Nat. Methods, 2011, 8, U135–U152 CrossRef PubMed.
- M. N. Slipchenko, T. T. Le, H. T. Chen and J. X. Cheng, J. Phys. Chem. B, 2009, 113, 7681–7686 CrossRef CAS PubMed.
- H. A. Rinia, K. N. Burger, M. Bonn and M. Muller, Biophys. J., 2008, 95, 4908–4914 CrossRef CAS PubMed.
- C. Di Napoli, F. Masia, I. Pope, C. Otto, W. Langbein and P. Borri, J. Biophotonics, 2014, 7, 68–76 CrossRef CAS PubMed.
- T. T. Le, H. M. Duren, M. N. Slipchenko, C. D. Hu and J. X. Cheng, J. Lipid Res., 2010, 51, 875 CrossRef CAS.
- M. Bosma, R. Minnaard, L. M. Sparks, G. Schaart, M. Losen, M. H. de Baets, H. Duimel, S. Kersten, P. E. Bickel, P. Schrauwen and M. K. Hesselink, Histochem. Cell Biol., 2012, 137, 205–216 CrossRef CAS PubMed.
- Y. Liu, Y. J. Lee and M. T. Cicerone, Opt. Lett., 2009, 34, 1363–1365 CrossRef.
- J. P. R. Day, K. F. Domke, G. Rago, H. Kano, H. O. Hamaguchi, E. M. Vartiainen and M. Bonn, J. Phys. Chem. B, 2011, 115, 7713–7725 CrossRef CAS PubMed.
- S. H. Parekh, Y. J. Lee, K. A. Aamer and M. T. Cicerone, Biophys. J., 2010, 99, 2695–2704 CrossRef CAS PubMed.
- J. G. Granneman, H. P. Moore, E. P. Mottillo and Z. Zhu, J. Biol. Chem., 2009, 284, 3049–3057 CrossRef CAS PubMed.
- C. Krafft, L. Neudert, T. Simat and R. Salzer, Spectrochim. Acta, Part A, 2005, 61, 1529–1535 CrossRef PubMed.
- K. T. Dalen, T. Dahl, E. Holter, B. Arntsen, C. Londos, C. Sztalryd and H. I. Nebb, Biochim. Biophys. Acta, 2007, 1771, 210–227 CrossRef CAS PubMed.
- J. G. Granneman, H. P. Moore, E. P. Mottillo, Z. Zhu and L. Zhou, J. Biol. Chem., 2011, 286, 5126–5135 CrossRef CAS PubMed.
- V. B. Schrauwen-Hinderling, M. K. C. Hesselhik, P. Schrauwen and M. E. Kooi, Obesity, 2006, 14, 357–367 CrossRef CAS PubMed.
- H. Li, Y. Song, L. J. Zhang, Y. Gu, F. F. Li, S. Y. Pan, L. N. Jiang, F. Liu, J. Ye and Q. Li, PLoS One, 2012, 7, e36712 CAS.
- K. Kuramoto, T. Okamura, T. Yamaguchi, T. Y. Nakamura, S. Wakabayashi, H. Morinaga, M. Nomura, T. Yanase, K. Otsu, N. Usuda, S. Matsumura, K. Inoue, T. Fushiki, Y. Kojima, T. Hashimoto, F. Sakai, F. Hirose and T. Osumi, J. Biol. Chem., 2012, 287, 23852–23863 CrossRef CAS PubMed.
- J. W. Hunnicutt, R. W. Hardy, J. Williford and J. M. Mcdonald, Diabetes, 1994, 43, 540–545 CrossRef CAS.
- L. H. Storlien, A. B. Jenkins, D. J. Chisholm, W. S. Pascoe, S. Khouri and E. W. Kraegen, Diabetes, 1991, 40, 280–289 CrossRef CAS.
- P. Schrauwen and M. K. C. Hesselink, Diabetes, 2004, 53, 1412–1417 CrossRef CAS.
- L. Yuzefovych, G. Wilson and L. Rachek, Am. J. Physiol., 2010, 299, E1096–E1105 CAS.
- N. J. Pillon, K. Arane, P. J. Bilan, T. T. Chiu and A. Klip, Cell Commun. Signaling, 2012, 10 Search PubMed.
- J. A. Chavez and S. A. Summers, Arch. Biochem. Biophys., 2003, 419, 101–109 CrossRef CAS PubMed.
- H. Robenek, I. Buers, O. Hofnagel, M. J. Robenek, D. Troyer and N. J. Severs, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2009, 1791, 408–418 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ib00271g |
| ‡ Current address: Department of Cell and Molecular Biology, Karolinska Institutet, P.O. Box 285 SE-171 77 Stockholm, Sweden. |
| This journal is © The Royal Society of Chemistry 2015 |