
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective modification of the β–β linkage in DDQ-treated Kraft lignin analysed by 2D NMR spectroscopy†
F.
Tran
,
C. S.
Lancefield
,
P. C. J.
Kamer
,
T.
Lebl
and
N. J.
Westwood
*
School of Chemistry and Biomedical Sciences Research Complex, University of St Andrews and EaStCHEM, North Haugh, St Andrews, Fife, Scotland KY16 9ST, UK. E-mail: njw3@st-andrews.ac.uk
First published on 11th August 2014
Abstract
The depolymerisation of the biopolymer lignin has the potential to provide access to a range of high value and commodity chemicals. However, research in this increasingly important area of green chemistry is hindered by the lack of analytical methods. The key challenge in using NMR is the throughput that can be achieved without the need for high field spectrometers fitted with cryoprobes. Here, we report the use of a relatively fast 2D HSQC NMR experiment performed on a 500 MHz spectrometer fitted with a BBFO+ probe to obtain high quality spectra. The use of the developed protocol to study the selective modification of the β–β linkage in Kraft lignin is also reported.
Introduction
The Kraft process, also known as Kraft pulping, is used in the pulp and paper industry to produce cellulose fibres from wood. Lignin is a by-product of this process. Global paper and paperboard production has increased by more than 50% since 1990,1 consequently providing a potentially large scale source of Kraft lignin worldwide. Although it is not typically being extracted from the black liquor,2 the recently developed LignoBoost technology can be used to recover high quality lignin from this waste product at an additional cost.3 According to the International Lignin Institute, approximately 50 million tons of lignin are produced every year worldwide. This lignin is mostly viewed, at present, as non-commercialisable waste.4Lignin is a complex cross-linked biopolymer derived from plants. Its structure consists of a series of aryl rings interconnected by various chemical linkages. Although the relative amount of each linkage is species-dependent, the key structures are common to all, with the main ones being the β-O-aryl ether unit (β-O-4), the phenylcoumaran unit (β-5) and the resinol unit (β–β, Fig. 1).
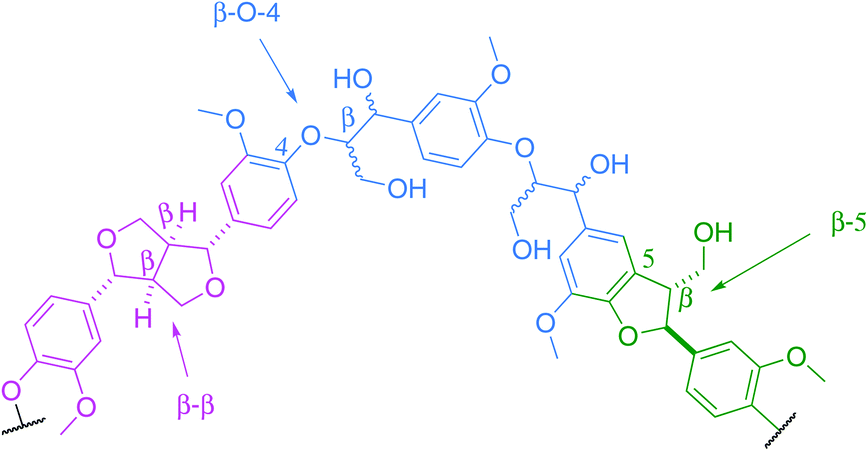 | ||
| Fig. 1 Structural representation of a fragment of lignin showing common linkages. | ||
With the depletion of fossil fuels as a source of chemicals and the widely accepted notion that biomass will play an important role in our Society, lignin is being regarded as a major future source of renewable aromatic chemicals, given its unique chemical structure.5,6 For example, lignin has been used in the production of vanillin7–12 and dyes13,14 and has shown potential as a renewable source of phenols in phenolic resins.15,16 However, at present lignin depolymerisation still constitutes a significant barrier to the exploitation of lignin as a green source of aromatic chemicals. This problem is a major obstacle for the economic viability of biorefineries, which need to valorize all components of lignocellulosic biomass including lignin.17
Nuclear magnetic resonance spectroscopy is widely used to study and understand the structure of lignin and lignin depolymerisation.18,19 Given its complex polymeric nature, the interpretation of traditional 1H and 13C spectra remains extremely challenging due to broad signals and extensive signal overlap. As a result, 2D HSQC experiments have become one of the most important analytical tools to study lignin, reflected in the large volume of papers published in this area in recent years. In the past decade, NMR analysis of lignin has been mainly conducted on relatively low field spectrometers,1,20–26 which suffer from long experiment times to produce good quality spectra. This has hampered the analysis of a large number of samples. With the advances in NMR spectroscopy in recent years, the state of the art in terms of lignin analysis using NMR relies heavily on the use of cryogenic probes27–34 or a 600–900 MHz spectrometer35–38 or a combination of both.6,39–50 This expensive technical requirement has limited the study of lignin degradation in many chemistry departments across the world.
Here we report the use of 2D HSQC experiments to characterise (30 min to 2 hours experiment time) and analyse Kraft lignin quickly using a 500 MHz spectrometer without the use of a cryoprobe. These studies enabled us to follow the selective modification of the β–β linkage in Kraft lignin upon treatment with the oxidant 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ).
Results and discussion
2D HSQC NMR experiments
Given the literature precedent,20–50 we were pleasantly surprised at the quality of the 2D HSQC spectrum of Kraft lignin obtained in only 2 hours using a 500 MHz spectrometer equipped with a BBFO+ probe (Fig. 2A). A standard hsqcetgpsp.3 Bruker pulse sequence was used to obtain, reproducibly, spectra of sufficient quality to enable comparison of integral intensities in the aliphatic and aromatic regions. This subsequently allowed us to determine the relative ratios of the β-O-4 (1.94), β-5 (0.55) and β–β (0.89) linkages in Kraft lignin using the aromatic region (100) as an internal standard (Fig. 2D).26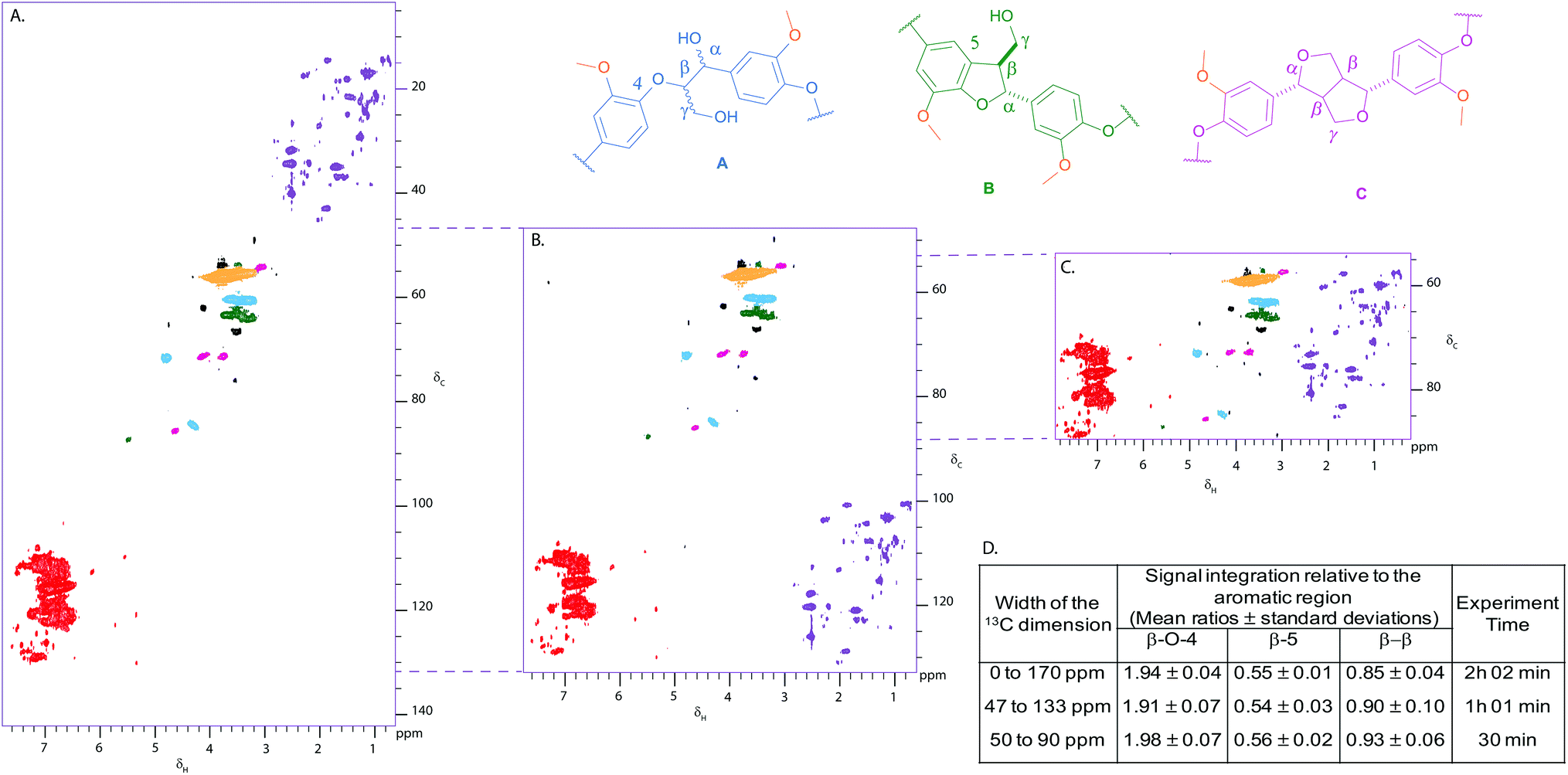 | ||
| Fig. 2 (A) Full width 2D HSQC NMR spectrum of Kraft lignin provided by MeadWestvaco Corporation; (B) 2D HSQC NMR spectrum obtained using a 86 ppm sweep width centred around 90 ppm (δc 47–133 ppm); (C) 2D HSQC NMR spectrum obtained using a 40 ppm sweep width centred around 70 ppm (δc 50–90 ppm); (D) Table comparing the ratios of integral intensities obtained for specific linkages relative to the aromatic region (coloured in red) calculated for each experiment. Each NMR analysis was run 3 times. Mean values ± SD are shown (n ≥ 3). Contours are colour coded according to the linkage they are assigned to. Black cross peaks currently correspond to unassigned signals. Folding in the indirect dimension of the non-oxygenated aliphatic and aromatic regions can be seen (purple and red regions respectively). NMR samples were run on a 500 MHz spectrometer at a concentration of 100 mg of substrate in 0.6 mL of DMSO-d6. | ||
With the cross-peaks characteristic of our linkages of interest i.e. the β-O-4, β-5 and β–β found between 50 and 90 ppm in the 13C dimension of the HSQC spectrum, we considered reducing the sweep width in that dimension in order to reduce experiment time and increase throughput. Gratifyingly, reducing the sweep width and the number of increments in the 13C dimension shortened the experiment time to 30 min, whilst maintaining the same resolution and sensitivity as the standard HSQC (Fig. 2C and 2D). Antiecho sampling mode was used, causing the signals below 50 ppm and above 90 ppm to be folded at the near end of the spectrum. It was shown that folding of cross peaks in the indirect dimension did not hamper using the aromatic region as an internal standard as the ratios of integral intensities obtained for each linkage remained practically unaffected compared to the standard HSQC (Fig. 2D). An experiment including the aromatic region, i.e. from 47 to 133 ppm, was also run (experimental time of one hour), which could be used to study interesting signals in the aromatic region (Fig. 2B and 2D). For comparison, 2D HSQC spectra were also obtained under more typical conditions (i.e. high field spectrometer fitted with a cryoprobe), which provided similar ratios to the ones obtained with our 500 MHz spectrometer equipped with a BBFO+ probe (see Fig. S1†).
Our modified NMR protocol was then used to analyse the outcome of the DDQ treatment of Kraft Lignin. Treatment of lignocelluloses with DDQ has previously been reported to characterise covalent bonds between lignin and carbohydrate in lignin–carbohydrate complexes.51–54 In addition, DDQ has been used to study the oxidation of pinoresinol moieties55 and the reactivity of furofuran56,57 but to the best of our knowledge, no work has been reported on the use of DDQ to modify lignin itself.
Analysis of the reaction of Kraft lignin with DDQ
Kraft lignin was treated with various weight equivalents of DDQ in DMF over 12 hours (see Fig. S2†). Following precipitation from Et2O and washing with H2O, the treated lignin samples were analysed using our 2D NMR methods. Fig. 3 shows the 2D HSQC analysis of a sample of Kraft lignin (typically soft wood lignin contains mainly guaiacyl units)58 before and after treatment with 0.5 and 0.75 weight equivalents of DDQ.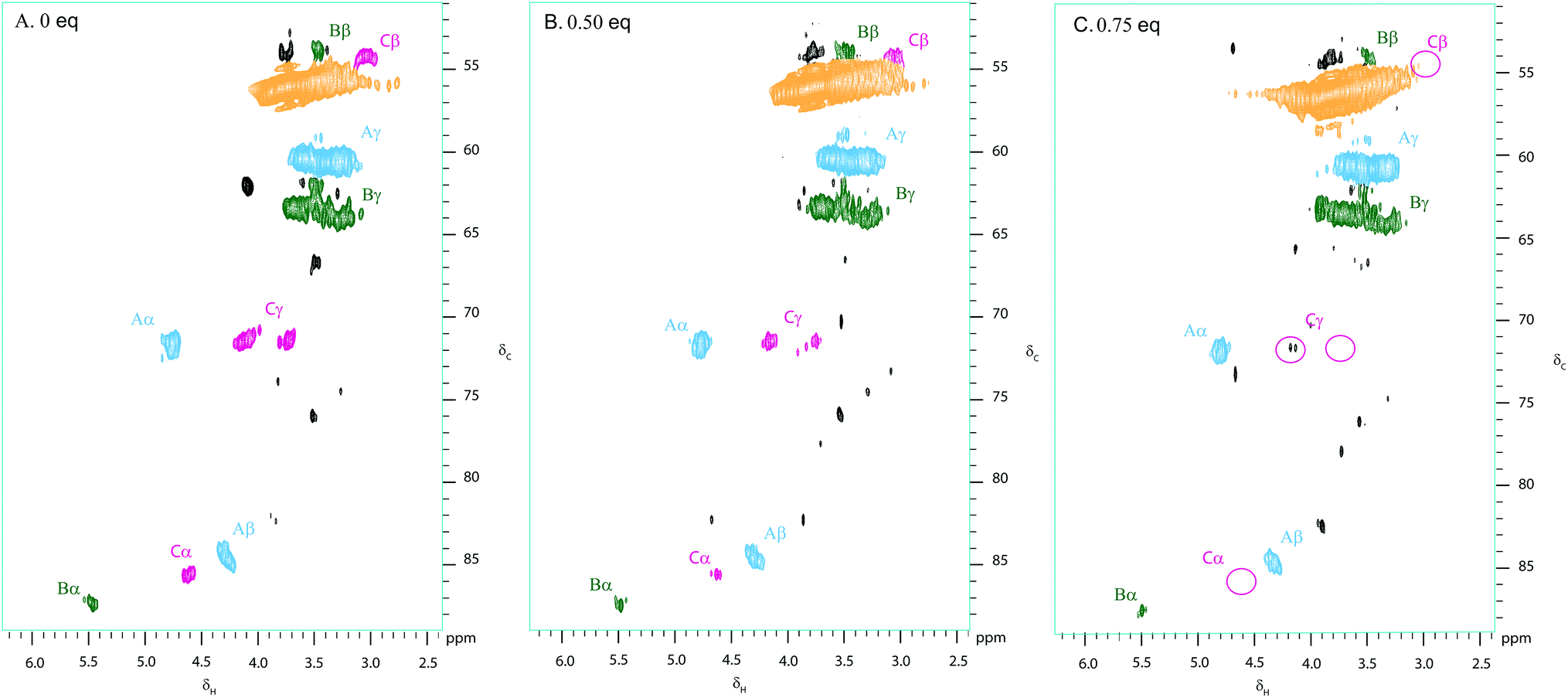 | ||
| Fig. 3 Partial 2D HSQC NMR spectra (δc/δH 50–95/2.5–6.0) of (A) commercially available Kraft lignin isolated after stirring in DMF overnight and precipitated from Et2O and Kraft lignin after treatment with (B) 0.5 and C. 0.75 weight equivalents of DDQ; Contours are colour coded according to the linkage they are assigned to (see Fig. 2 legend). The disappearance of the cross peaks corresponding to the β–β linkage is highlighted by pink circles. Black cross peaks currently correspond to unassigned signals. NMR samples were run on a 500 MHz spectrometer at a concentration of 100 mg of substrate in 0.6 mL of DMSO-d6. | ||
The treatment of Kraft lignin with DDQ appeared to modify selectively the β–β linkage at 0.5 weight equivalents of DDQ as judged by the reduction in volume of the cross-peaks assigned to this linkage (Fig. 3, 4 and S2 and Table S1†). At higher weight equivalents of DDQ, reaction of the β-O-4 units could also be detected (Fig. 4 and S2 and Table S1†). The data also suggested that the β-5 linkage remained essentially untouched under the reaction conditions investigated (Fig. 4 and S2 and Table S1†).
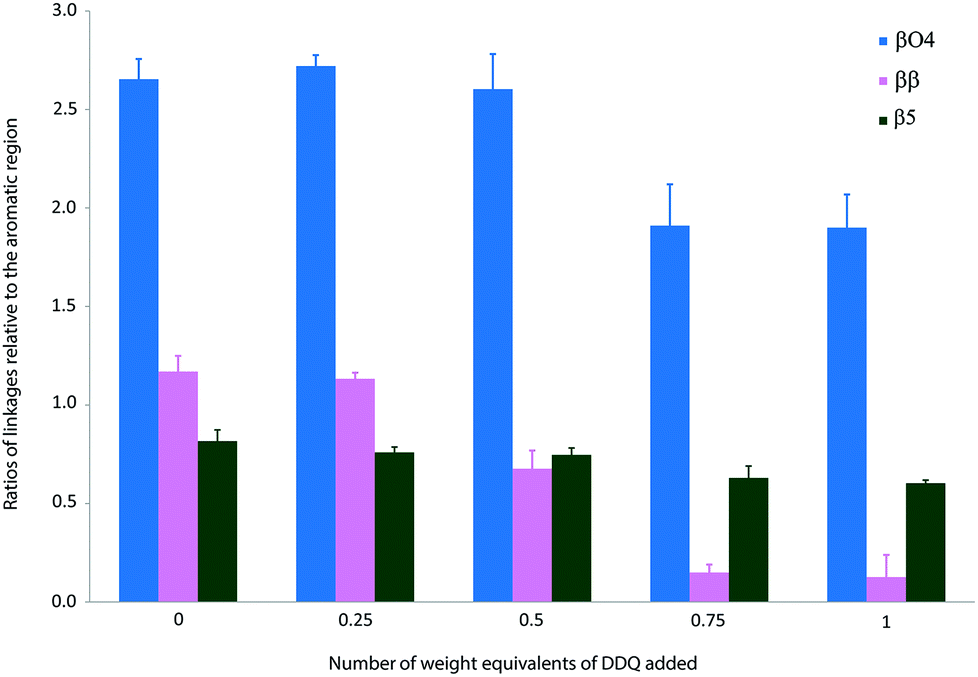 | ||
| Fig. 4 Ratios of linkages in precipitated Kraft lignin following treatment with different weight equivalents of DDQ. Each sample was stirred overnight in DMF in the absence or presence of DDQ and the lignin precipitated from Et2O. Each NMR analysis was run 3 times and quantification was performed by integration of the cross peaks and normalised relative to the aromatic region. Mean values ± SD are shown (n ≥ 3) (see Table S1†). | ||
The selective processing of the β–β linkage by DDQ prompted us to use a model compound to determine the chemical reaction taking place in this process.
Synthesis of β–β model compound, eudesmin (3)
Eudesmin (3) was selected as a β–β model compound. This was synthesised by dimerisation of coniferyl alcohol (1) in the presence of iron(III) chloride13 followed by methylation of the phenolic oxygens in 2 (Scheme 1). Epieudesmin (for structure, see S1 in ESI†) was also synthesised to validate the widely accepted relative configuration of the β–β linkage in lignin (Scheme S1 and Fig. S3† for more details).59–62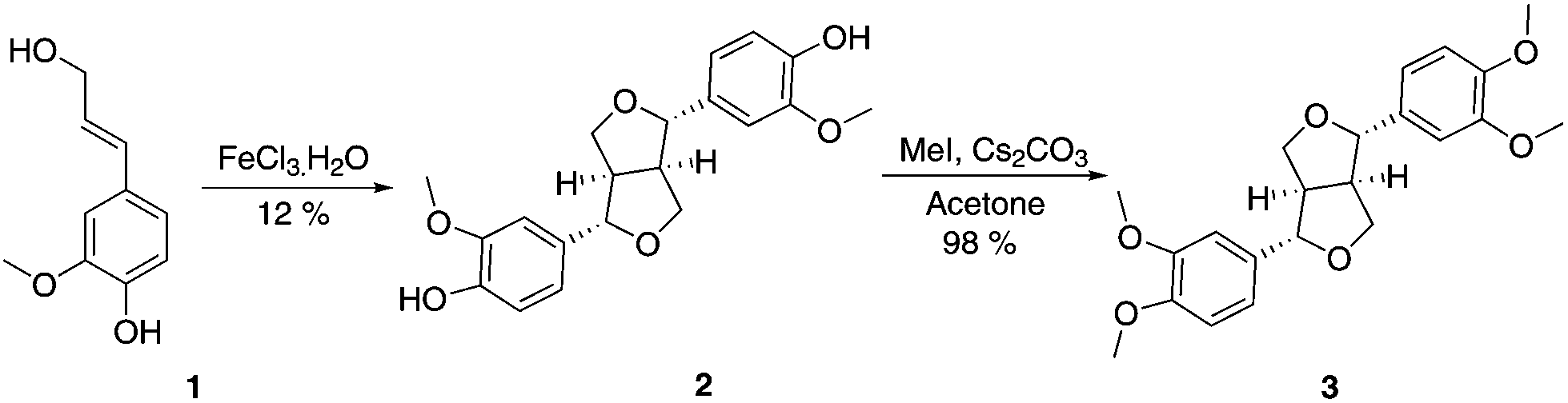 | ||
| Scheme 1 Synthesis of β–β model compound eudesmin (3).63–65 | ||
Treatment of 3 with DDQ
Treatment of eudesmin (3) with one mole equivalent of DDQ in DMF led to exclusive formation of alkene 4 (Scheme 2). However conversion was limited to 50% and 4 proved unstable to column chromatography. Interestingly, treatment of eudesmin (3) with 3 mole equivalents of DDQ led to pyran-4-one 5 in 45% isolated yield.56,57 The moderate yield of 5 obtained may be attributed to the instability of 5 on the silica gel column since the crude reaction mixture only appeared to contain predominantly 5 with only traces of 4 (see Fig. S4† for 1H NMR analysis). Further treatment of the mixture of 3 and 4 with DDQ also resulted in the formation of 5 (Scheme 2), suggesting 4 is formed en route from 3 to 5.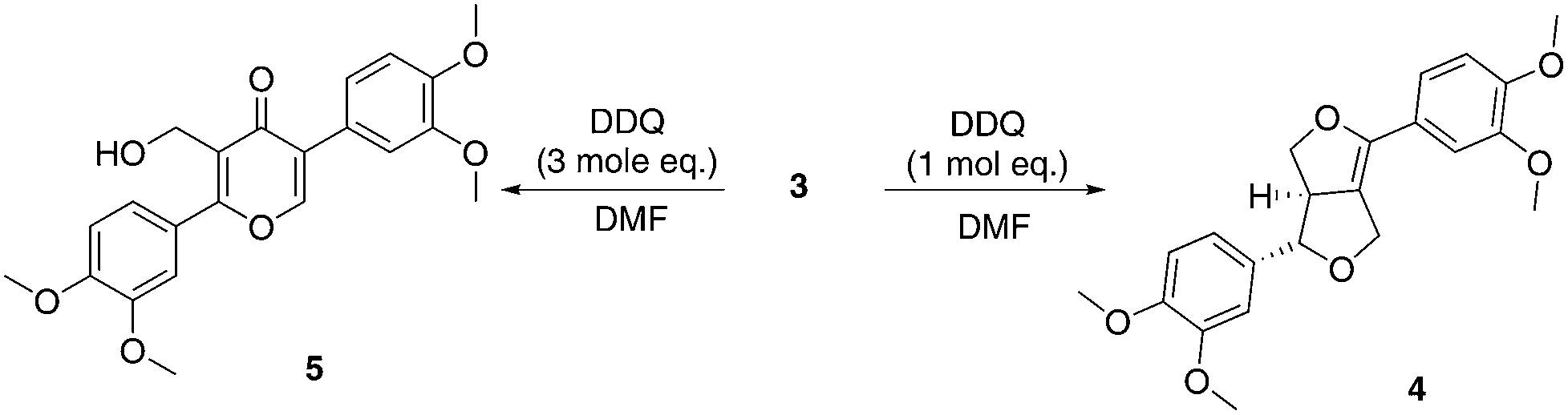 | ||
| Scheme 2 Synthesis of pyran-4-one 5 from eudesmin (3). | ||
Confident that eudesmin (3) was converted to pyran-4-one 5 under our reaction conditions, we were interested in determining whether the same process was also taking place in Kraft lignin. Gratifyingly, comparison of the 2D HSQC spectrum of an authentic sample of 5 with the 2D HSQC spectrum of Kraft lignin treated with 0.75 weight equivalents of DDQ revealed a close proximity between the cross peak corresponding to the methylene group in 5 at δc/δH 52.9/4.65 ppm (shown in red, Fig. 5B) and a new cross peak in the DDQ-treated lignin spectrum (shown in black, Fig. 5B). An additional close proximity of a cross peak at δc/δH 147.3/8.21 ppm corresponding to the pyran-4-one CH proton in 5 and a new cross peak was observed in the Kraft lignin spectrum after treatment with DDQ (Fig. 5A). These observations were confirmed by a doping experiment, in which an authentic sample of 5 was added to the sample of Kraft lignin treated with 0.75 weight equivalents of DDQ. This showed a clear overlap between the 2 sets of cross peaks (Fig. S5†). Taken together, these data strongly suggest that the reactivity of the monomeric β–β linkage translates to polymeric Kraft lignin.
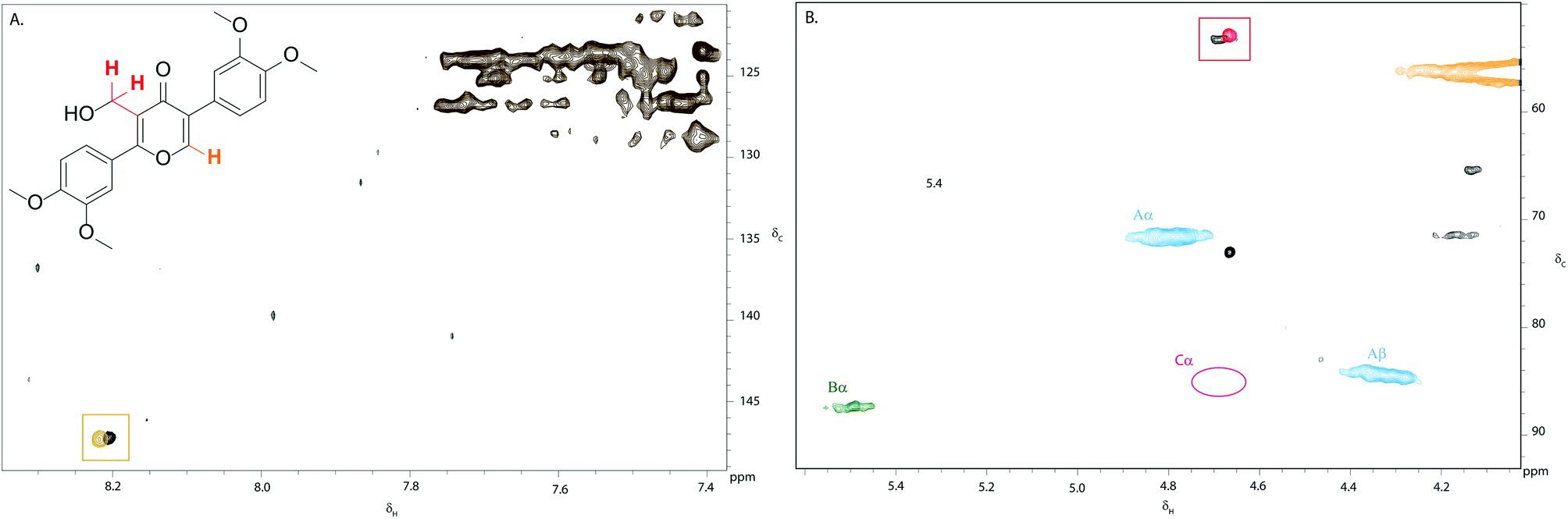 | ||
| Fig. 5 Overlay of partial 2D HSQC NMR spectra of (i) Kraft lignin after treatment with 0.75 weight equivalents of DDQ and (ii) of an authentic sample of pyran-4-one 5: (A) aromatic region (δc/δH 120–150/7.4–8.4) and (B) aliphatic region (δc/δH 50–90/4.0–5.6). The close proximity of the aromatic and methylene cross peaks in 5 with new cross peaks present in the DDQ-treated lignin spectrum are shown in orange and red boxes respectively. Contours are colour coded according to the linkage they are assigned to (see Fig. 2 legend). | ||
Conclusions
This work has demonstrated that high quality 2D HSQC spectra of Kraft lignin can be acquired using a 500 MHz spectrometer in the absence of a cryoprobe. The relatively short experimental time (between 30 and 120 minutes depending on the selected width of the 13C dimension) enabled the analysis of a large number of samples and allowed us to follow the selective modification of the β–β linkage in Kraft lignin upon treatment with DDQ. To the best of our knowledge, this is the first time the selective modification of this linkage has been reported in the literature. This may prove important in ultimately depolymerising Kraft lignin since the Kraft pulping process is known to both cleave and modify the β-O-4 linkage20,37 resulting in other linkages being relatively more abundant in this type of lignin compared to, for example, organosolv lignin. We are currently working on the selective modification of the pyran-4-one moiety that results from DDQ treatment of Kraft lignin.Experimental
NMR methods
NMR spectra were acquired on a Bruker Avance III 500 MHz spectrometer equipped with a BBFO+ probe. The central DMSO solvent peak was used as internal reference (δC 39.5, δH 2.49 ppm). The 1H, 13C-HSQC experiment was acquired using standard Bruker pulse sequence ‘hsqcetgpsp.3’ (phase-sensitive gradient-edited-2D HSQC using adiabatic pulses for inversion and refocusing). Composite pulse sequence ‘garp4’ was used for broadband decoupling during acquisition. 2048 data points was acquired over 12 ppm spectral width (acquisition time 170 ms) in F2 dimension using 24 scans with 1 s interscan delay and the d4 delay was set to 1.8 ms (1/4J, J = 140 Hz). The spectrum was processed using squared cosinebell in both dimensions and LPfc linear prediction (32 coefficients) in F1. Volume integration of cross peaks in the HSQC spectra was carried out using MestReNova software.General procedure for the treatment of Kraft lignin with DDQ
DDQ (X weight equivalent) was added to a solution of a Kraft lignin (200 mg) in anhydrous DMF (20 mL). The mixture was then stirred at room temperature overnight before being concentrated in vacuo to a 2 mL solution. This was then added to Et2O (300 mL) to induce the precipitation of lignin. The suspension was then left to stir at room temperature for 3 hours before lignin was collected. The lignin obtained was subsequently stirred in water for 1 hour to remove any residual DMF and DDQ-H. The solid collected was finally dried in a vacuum oven for 3 hours at 80 °C to yield 150 to 175 mg of treated lignin. This was then analysed by 2D NMR spectroscopy.Acknowledgements
This work was funded by the EPSRC grants EP/J018139/1 and EP/K00445X/1 and through the International Training Network Subicat. We are also grateful to MeadWestvaco Corporation, Richmond, VA for kindly providing the Kraft lignin used and the University of Edinburgh and especially Mr Juraj Bella for allowing us to use their high field spectrometer fitted with a cryoprobe. Finally we would like to thank Professors Bob Tooze and Derek Stewart for extensive discussions on lignin processing.Notes and references
- The Industrial Efficiency Technology Database, http://ietd.iipnetwork.org/content/pulp-and-paper#key-data (accessed May 2014).
- P. Stenius, in Forest Products Chemistry, Papermaking Science and Technology 3, Fapet OY, Helsinki, Finland, 2000 Search PubMed.
- F. Ohman, H. Theliander, P. Tomani and P. Axegard, Methods for separating lignin from black liquor, a lignin product for the production of fuels or materials, US20100325947A1, 2010 Search PubMed.
- The International Lignin Institute, http://www.ili-lignin.com/aboutlignin.php (accessed May 2014).
- F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227 CrossRef CAS PubMed.
- J. Zakzeski, A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602 CrossRef CAS PubMed.
- J. D. P. Araújo, C. A. Grande and A. E. Rodrigues, Chem. Eng. Res. Des., 2010, 88, 1024 CrossRef PubMed.
- C. Eckert, C. Liotta, A. Ragauskas, J. Hallett, C. Kitchens, E. Hill and L. Draucker, Green Chem., 2007, 9, 545 RSC.
- W. A. Herrmann, T. Weskamp, J. P. Zoller and R. W. Fischer, J. Mol. Catal. A: Chem., 2000, 153, 49 CrossRef CAS.
- P. Parpot, A. P. Bettencourt, A. M. Carvalho and E. M. Belgsir, J. Appl. Electrochem., 2000, 30, 727 CrossRef CAS.
- P. C. Rodrigues Pinto, E. A. Borges da Silva and A. E. Rodrigues, Ind. Eng. Chem. Res., 2010, 50, 741 CrossRef.
- F. G. Sales, L. C. A. Maranhão, N. M. L. Filho and C. A. M. Abreu, Chem. Eng. Sci., 2007, 62, 5386 CrossRef CAS PubMed.
- R. Saad, Z. Radovic-Hrapovic, B. Ahvazi, S. Thiboutot, G. Ampleman and J. Hawari, J. Environ. Sci., 2012, 24, 808 CrossRef CAS.
- D. Suteu, C. Zaharia, A. Muresan, R. Muresan and A. Popescu, Environ. Eng. Manage. J., 2009, 8, 1097 CAS.
- A. Tejado, G. Kortaberria, C. Peña, M. Blanco, J. Labidi, J. M. Echeverría and I. Mondragon, J. Appl. Polym. Sci., 2008, 107, 159 CrossRef CAS PubMed.
- A. Tejado, G. Kortaberria, C. Peña, J. Labidi, J. M. Echeverría and I. Mondragon, J. Appl. Polym. Sci., 2007, 106, 2313 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS PubMed.
- R. John and L. L. Larry, in Lignin and Lignans, CRC Press, 2010, p. 137 Search PubMed.
- J.-L. Wen, S.-L. Sun, B.-L. Xue and R.-C. Sun, Materials, 2013, 6, 359 CrossRef CAS PubMed.
- C. Fernández-Costas, S. Gouveia, M. A. Sanromán and D. Moldes, Biomass Bioenergy, 2014, 63, 156 CrossRef PubMed.
- D. D. Laskar, J. Zeng, L. Yan, S. Chen and B. Yang, Ind. Crops Prod., 2013, 50, 391 CrossRef CAS PubMed.
- A. J. Simpson, B. Lefebvre, A. Moser, A. Williams, N. Larin, M. Kvasha, W. L. Kingery and B. Kelleher, Magn. Reson. Chem., 2004, 42, 14 CrossRef CAS PubMed.
- J.-L. Wen, S.-L. Sun, T.-Q. Yuan, F. Xu and R.-C. Sun, J. Agric. Food Chem., 2013, 61, 11067 CrossRef CAS PubMed.
- M. Wu, J. Pang, F. Lu, X. Zhang, L. Che, F. Xu and R. Sun, Ind. Crops Prod., 2013, 50, 887 CrossRef CAS PubMed.
- T.-T. You, J.-Z. Mao, T.-Q. Yuan, J.-L. Wen and F. Xu, J. Agric. Food Chem., 2013, 61, 5361 CrossRef CAS PubMed.
- L. Zhang and G. Gellerstedt, Magn. Reson. Chem., 2007, 45, 37 CrossRef CAS PubMed.
- J. C. del Río, J. Rencoret, P. Prinsen, Á. T. Martínez, J. Ralph and A. Gutiérrez, J. Agric. Food Chem., 2012, 60, 5922 CrossRef PubMed.
- A. Eudes, A. George, P. Mukerjee, J. S. Kim, B. Pollet, P. I. Benke, F. Yang, P. Mitra, L. Sun, Ö. P. Çetinkol, S. Chabout, G. Mouille, L. Soubigou-Taconnat, S. Balzergue, S. Singh, B. M. Holmes, A. Mukhopadhyay, J. D. Keasling, B. A. Simmons, C. Lapierre, J. Ralph and D. Loqué, Plant Biotechnol. J., 2012, 10, 609 CrossRef CAS PubMed.
- H. Kim and J. Ralph, Org. Biomol. Chem., 2010, 8, 576 CAS.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415 CrossRef CAS PubMed.
- J. Rencoret, G. Marques, A. Gutiérrez, J. Jiménez-Barbero, Á. T. Martínez and J. C. del Río, Ind. Eng. Chem. Res., 2013, 52, 4695 CrossRef CAS.
- J. Talbot, D. Yelle, J. Nowick and K. Treseder, Biogeochemistry, 2012, 108, 279 CrossRef CAS.
- A. Wagner, Y. Tobimatsu, G. Goeminne, L. Phillips, H. Flint, D. Steward, K. Torr, L. Donaldson, W. Boerjan and J. Ralph, Plant Mol. Biol., 2013, 81, 105 CrossRef CAS PubMed.
- F. Yue, F. Lu, R. Sun and J. Ralph, Chem. – Eur. J., 2012, 18, 16402 CrossRef CAS PubMed.
- S. Bauer, H. Sorek, V. D. Mitchell, A. B. Ibáñez and D. E. Wemmer, J. Agric. Food Chem., 2012, 60, 8203 CrossRef CAS PubMed.
- S. Heikkinen, M. M. Toikka, P. T. Karhunen and I. A. Kilpeläinen, J. Am. Chem. Soc., 2003, 125, 4362 CrossRef CAS PubMed.
- T. M. Liitiä, S. L. Maunu, B. Hortling, M. Toikka and I. Kilpeläinen, J. Agric. Food Chem., 2003, 51, 2136 CrossRef PubMed.
- J.-L. Wen, T.-Q. Yuan, S.-L. Sun, F. Xu and R.-C. Sun, Green Chem., 2014, 16, 181 RSC.
- M. Balakshin, E. Capanema, H. Gracz, H.-m. Chang and H. Jameel, Planta, 2011, 233, 1097 CrossRef CAS PubMed.
- M. Brennan, J. P. McLean, C. Altaner, J. Ralph and P. Harris, Cellulose, 2012, 19, 1385 CrossRef CAS.
- J. M. W. Chan, S. Bauer, H. Sorek, S. Sreekumar, K. Wang and F. D. Toste, ACS Catal., 2013, 3, 1369 CrossRef CAS.
- K. Cheng, H. Sorek, H. Zimmermann, D. E. Wemmer and M. Pauly, Anal. Chem., 2013, 85, 3213 CrossRef CAS PubMed.
- J. C. del Río, P. Prinsen, J. Rencoret, L. Nieto, J. Jiménez-Barbero, J. Ralph, Á. T. Martínez and A. Gutiérrez, J. Agric. Food Chem., 2012, 60, 3619 CrossRef PubMed.
- X. Du, M. Pérez-Boada, C. Fernández, J. Rencoret, J. Río, J. Jiménez-Barbero, J. Li, A. Gutiérrez and A. Martínez, Planta, 2014, 1 Search PubMed.
- P. Prinsen, A. Gutiérrez, J. Rencoret, L. Nieto, J. Jiménez-Barbero, A. Burnet, M. Petit-Conil, J. L. Colodette, Á. T. Martínez and J. C. del Río, Ind. Crops Prod., 2012, 36, 572 CrossRef CAS PubMed.
- P. Prinsen, J. Rencoret, A. Gutiérrez, T. Liitiä, T. Tamminen, J. L. Colodette, M. Á. Berbis, J. Jiménez-Barbero, Á. T. Martínez and J. C. del Río, Ind. Eng. Chem. Res., 2013, 52, 15702 CrossRef CAS.
- J. Ralph, T. Akiyama, H. Coleman and S. Mansfield, BioEnergy Res., 2012, 5, 1009 CrossRef CAS.
- N. Sathitsuksanoh, K. M. Holtman, D. J. Yelle, T. Morgan, V. Stavila, J. Pelton, H. Blanch, B. A. Simmons and A. George, Green Chem., 2014, 16, 1236 RSC.
- M. Sette, R. Wechselberger and C. Crestini, Chem. – Eur. J., 2011, 17, 9529 CrossRef CAS PubMed.
- A. Socha, S. Plummer, V. Stavila, B. Simmons and S. Singh, Biotechnol. Biofuels, 2013, 6, 61 CrossRef CAS PubMed.
- T. B. T. Lam, K. Kadoya and K. Iiyama, Phytochemistry, 2001, 57, 987 CrossRef CAS.
- T. Watanabe, Wood Res., 1989, 76, 59 CAS.
- T. Watanabe and T. Koshijima, Agric. Biol. Chem., 1988, 52, 2953 CrossRef CAS.
- T. Watanabe, J. Ohnishi, Y. Yamasaki, S. Kaizu and T. Koshijima, Agric. Biol. Chem., 1989, 53, 2233 CrossRef CAS.
- I. R. Jack, R. S. Ward and A. Pelter, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1989, 28, 184 Search PubMed.
- P. Satyanarayana, P. K. Rao, K. Sethuramu, K. N. Viswanatham and S. Venkateswarlu, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1991, 30, 825 Search PubMed.
- R. S. Ward, A. Pelter, I. R. Jack, P. Satyanarayana, B. V. G. Rao and P. Subrahmanyam, Tetrahedron Lett., 1981, 22, 4111 CrossRef CAS.
- D. N.-S. Hon and N. Shiraishi, in Wood and Cellulosic Chemistry, Revised, and Expanded, CRC Press, 2nd edn, 2000 Search PubMed.
- K. Lundquist and R. Stomberg, Holzforschung, 1988, 42, 375 CrossRef CAS.
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. Schatz, J. Marita, R. Hatfield, S. Ralph, J. Christensen and W. Boerjan, Phytochem. Rev., 2004, 3, 29 CrossRef CAS.
- M. Bardet, D. R. Robert and K. Lundquist, Sven. Papperstidn., 1985, 88, R61 CAS.
- Y. Z. Lai and K. V. Sarkanen, in Isolation and structural studies in Lignins, ed. K. V. Sarkanen and C. H. Ludwig, Wiley-Interscience, New York, 1971 Search PubMed.
- R. Brežný and J. Alföldi, Chem. Zvesti, 1982, 36, 267 Search PubMed.
- M. Tanahashi, H. Takeuchi and T. Higuchi, Wood Res., 1976, 61, 44 CAS.
- X. Wang, X. Li, J. Xue, Y. Zhao and Y. Zhang, Tetrahedron Lett., 2009, 50, 413 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc01012d |
| This journal is © The Royal Society of Chemistry 2015 |