
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inhibition of low-grade inflammation by anthocyanins from grape extract in an in vitro epithelial-endothelial co-culture model
Sabine
Kuntz
*ab,
Heike
Asseburg
b,
Sebastian
Dold
c,
Andreas
Römpp
c,
Bettina
Fröhling
d,
Clemens
Kunz
b and
Silvia
Rudloff
ab
aDepartment of Pediatrics, Feulgenstrasse 12, University of Giessen, Germany. E-mail: Sabine.Kuntz@ernaehrung.uni-giessen.de; Fax: +49-641-9939049; Tel: +49-641-9939044
bInstitute of Nutritional Science, Wilhelmstrasse 20, University of Giessen, Germany
cInstitute of Inorganic and Analytical Chemistry, Schubertstrasse 60, University of Giessen, Germany
dInstitute of Enology and Beverage Research, Section of Wine Analysis and Beverage Technology, Von-Lade-Strasse 1, Geisenheim Research Center, USA
First published on 9th February 2015
Abstract
Background: Anthocyanins (ACNs) are the most prevalent flavonoids in berries and their health promoting effects on vascular functions are still discussed. The aim of the present study was to identify the anti-inflammatory effect of ACNs on activated human umbilical vein endothelial cells (HUVECs) after their transport across an epithelial monolayer. Study design: We established a transwell epithelial-endothelial co-culture system with Caco-2/HT29-B6 cells mimicking the intestinal layer and HUVECs as endothelial cells mimicking the vascular layer. Caco-2 were seeded alone (100%) or together with HT29-B6 cells (10 and 20%) on transwell inserts in order to simulate different metabolization sides of the gut. ACNs as well as malvidin-3-glucoside (M3G) were applied to the luminal compartment of the transwell-system. Transport and degradation rates were determined by high performance liquid chromatography with ultraviolet detection (HPLC-UV) or by ultra-PLC coupled to mass spectrometry (UPLC-MS). After 4 hours incubation time, co-cultured HUVECs were used immediately (short-term incubation) or after 20 hours (long-term incubation). Thereafter, HUVECs were stimulated for 3 hours with 1 ng mL−1 TNF-α to mimic a low-grade or 10 ng mL−1 to mimic a high-grade inflammation. Afterwards, (1.) leukocyte adhesion, (2.) expression of cell adhesion molecules (ICAM-1, VCAM-1 and E-selectin) and (3.) cytokine expression and secretion (IL-6 and IL-8) were determined using flow cytometry and real-time PCR. Results: Degradation and incubation studies revealed that ACNs were differently degraded depending on the ACN structure and the seeding densities. Incubation of ACNs and M3G to Caco-2 cells (100%) led to a fast decrease, which was not observed when HT29-B6 cells were co-cultured (10 and 20%). Concomitantly, anti-inflammatory effects were only observed using 100% Caco-2 cells, whereas mixtures of Caco-2 and HT29-B6 cells failed to induce an effect. ACN extract and M3G significantly attenuated TNF-α-stimulated low-grade leukocyte adhesion, expression of adhesion molecules E-selectin, VCAM-1 and ICAM-1 and cytokine expression and secretion (IL-8 and IL-6) as well as NF-κB mRNA expression. No effects were observed with high TNF-α (10 ng mL−1) or after short-term incubation (4 hours). Conclusions: ACNs in physiological concentrations reached the serosal compartment and reduced inflammation-related parameters, which were related to the initial steps during the pathogenesis of atherosclerosis.
Introduction
Cardiovascular diseases (CVD) are the leading cause of morbidity and mortality in the Western population and it was postulated that the consumption of fruit and vegetables has been postulated to be associated with reduced risk for CVD.1,2 Despite the controversial results from epidemiological observations or intervention studies, current research has been focusing on the so-called low-grade inflammation (LGI), which is considered to be the first step in the pathogenesis of several inflammation associated diseases such as atherosclerosis, obesity, diabetes and cancer.3,4 There are many causes for a LGI with lifestyle factors playing a major role.5–7 At first, dietary factors such as a high fat diet, low dietary fiber or polyunsaturated fat are associated with LGI.8 LGI is characterized by enhanced circulating cytokines (TNF-α, IL-6), chemokines (IL-8) or acute phase proteins such as CRP (C-reactive peptide) leading to an activation of vascular endothelium and impairment. The activation of endothelial cells during inflammatory processes is a well-known multistep process in which multiple cell adhesion molecules are expressed in a time- and concentration-dependent manner.9 Thus, after endothelial activation, cell adhesion molecules such as E-selectin, mediating the first cell–cell interactions between leukocytes and the vascular endothelium and molecules of the immunoglobulin superfamily (intercellular cell adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1)), responsible for the firm adhesion and transmigration of the cells to the extravasal space, have been reported to play a major role in the initial steps of atherosclerotic pathogenesis.10–12 As a consequence, prolonged and chronic activation of the vascular epithelium may lead to thrombotic vessel closure and plaque rupture.13,14 Recently, it was shown that consumption of blueberries improved endothelial dysfunction and reduced inflammatory markers in subjects with increased cardiovascular risk.15 Furthermore, ingestion of bilberries by individuals with features of metabolic syndrome was associated with a reduction of LGI.16 Such effects of fruits, especially berries have been mainly attributed to the presence of polyphenols and their anti-oxidative, anti-inflammatory and anti-adhesive effects, and may exert positive effects on vascular function.3,17 Flavonoids are the most prevalent group of polyphenols in fruits, with flavonols, flavanones and ACNs as the most important subgroups.18 For example, the flavanones naringenin and hesperitin as well as their chemically synthesized glucuronides and sulfates were able to reduce monocyte adhesion to HUVEC monolayers under low-grade inflammation.19 This could also be observed using flavonol glucuronides like quercetin-3′-sulfate, quercetin-3-glucuronide or 3′-methylquercetin-3-glucuronide.20 In contrast to flavonoids, effects of ACNs were only measured using single compounds and/or used in supraphysiological concentrations. However, ACNs can be directly absorbed in the intestine without prior chemical modification or after deglycosylation and could be further metabolized by phase II enzyme into methyl, glucuronide, or sulfate conjugates in the small intestine and in the liver.21 Keeping in mind that the concentration of glycosides of ACNs was extremely low or absent in plasma and that intense metabolization in the intestinal epithelial cells generated phase II metabolites (glucuronidation, sulphation and methylation), it seems conceivable that metabolites are the relevant bioactive components. Furthermore, there is increasing evidence that degradation products rather than the parental compounds were responsible for the health effects. This involves the formation of phenolic and aromatic acids such as hydroxybenzoic or hydroxycinnamic acids, which could also have an endothelium protecting effect.22,23 The aim of the present study was to investigate degradation, transport and metabolization of ACNs from an ACN-rich grape extract as well as malvidin-3-glucoside (M3G) and protocatechuic acid (PCA) using Caco-2 cells alone as an intestinal monolayer or in combination with HT29-B6 cells to mimic different absorption sites of the gut for ACNs. The establishment of an in vitro co-culture model (epithelial and endothelial cells) allowed to investigate effects of ACNs on low- and high-grade inflammation events such as changes in adhesion, or the expression of adhesion molecules (E-selectin, VCAM-1 and ICAM-1), cytokines (IL-6 and IL-8) and regulator proteins (NF-κB) after crossing the intestinal layer.Methods and materials
Materials
M3G was purchased from Carl Roth GmbH (Karlsruhe, Germany), PCA from Sigma-Aldrich (Taufkirchen, Germany), TNF-α from R&D (Heidelberg, Germany), Calcein-AM and human plasma fibronectin from Invitrogen (Heidelberg, Germany). Cell culture media and supplements for human umbilical vein endothelial cells (HUVECs) were obtained from Promocell (Heidelberg, Germany), and those for Caco-2 cells and HT29-B6 cells from Invitrogen (Heidelberg, Germany). All chemicals were with analytical grade and cell culture tested.Methods
| Extract [mg L−1] | Extract [μmoL L−1] | |
|---|---|---|
| Malvidin-3-glucoside | 767.6 | 1555.6 |
| Malvidin-3-(6′′-O-acetyl)-glucoside | 13.0 | 24.3 |
| ∑Malvidin | 780.6 | 1579.9 |
| Peonidin-3-glucoside | 345.2 | 744.9 |
| Peonidin-3-(6′′-O-acetyl)-glucoside | 137.9 | 272.8 |
| ∑Peonidin | 483.1 | 1017.7 |
| Petunidin-3-glucoside | 164.5 | 343.1 |
| Petunidin-3-(6′′-O-acetyl)-glucoside | 22.6 | 43.3 |
| Petunidin-3-rutinoside | 74.3 | 118.8 |
| ∑Petunidin | 261.4 | 505.2 |
| Delphinidin-3-glucoside | 173.2 | 372.2 |
| Delphinidin-3-(6′′-O-acetyl)-glucoside | 20.6 | 40.6 |
| ∑Delphinidin | 193.8 | 412.8 |
| Cyanidin-3-glucoside | 47.2 | 105.0 |
| ∑Cyanidin | 47.2 | 105.0 |
| Sum ACNs | 1766.1 | 3620.6 |
UPLC separation was performed using an UltiMate 3000 RSLC (Rapid Separation LC) (Dionex, Idstein, Germany) system equipped with a pump (HPG 3200 RS), an autosampler (WPS-3000 TRS) and a 100 mm × 2.1 mm ID, 2.6 μm particle size, Kinetex reversed phase (C18) column (Phenomenex, Aschaffenburg, Germany). Mobile phases consisted of 0.7% v:v formic acid in water (solvent A) and methanol (solvent B). The solvent gradient was 2% B at 0 to 0.5 min, 7% B at 1 min, 15% B at 5 min, 40% B at 10 min, 70% B at 13 to 16 min at flow rate of 400 μL min−1 (p(max) = 550 bar). Injection volume was 40 μL. The UPLC system was coupled to an FT-ICR MS (Fourier Transform Ion Cyclotron Resonance Mass Spectrometer, Thermo Fisher Scientific, Bremen, Germany) by an electrospray ion source. Measurements were performed in positive ion mode. Capillary temperature was set to 350 °C spray voltage in positive ion mode was 4.4 kV. Mass spectra were acquired in the range of m/z = 200–850 for positive ion mode.
Epithelial and endothelial cells and culture conditions
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 and 80:20%. Cells were allowed to grow to confluence (2 days) and thereafter to differentiate to absorptive enterocytes within 21 days (n = 3). The culture medium was changed every 2 or 3 days at the apical (0.5 mL) and basolateral side (1.5 mL).
:3 split ratio with trypsin/EDTA (0.05%/0.01%) solution and were used at passage number 2–4. Trypsinized cells (1 × 105) were seeded on fibronectin coated 24-well plates (Biochrom KG, Berlin, Germany) and allowed to growth until they reached 100% confluence (2 days). Confluent HUVECs on fibronectin-coated 24-well plates were used for co-culture experiments.
:10 and 80:20%) on semipermeable filters over 21 days to differentiate and develop an enterocyte-like phenotype. After differentiation, transwell filters were inserted onto a 24-well plate where endothelial cells were cultivated on the bottom of the 24-well cavity. The upper compartment (insert) with epithelial cells was exposed to target substrates for 4 hours. In order to ensure an intact intestinal monolayer during the time of the experiment, the transepithelial electrical resistance (TEER) was monitored before and at the end of the incubation experiments. Only epithelial monolayers exerting TEER values above 550 Ωm cm−2 were used for incubation experiments. Thereafter, inserts were removed and HUVECs were stimulated immediately (short-term incubation) or after 20 h of cultivation (long-term incubation) with TNF-α. To mimic low-grade and high-grade inflammation, HUVECs were incubated for 3 hours with 1 ng mL−1 and 10 ng mL−1 TNF-α, respectively. After TNF-α stimulation, we determined (a) leukocyte adhesion to HUVECs, (b) expression of cell adhesion molecules, (c) expression of NF-κB and (d) expression and secretion of the cytokines IL-6 and IL-8 in the supernatants or in HUVECs.
:10 and 80:20%. Cells were allowed to grow to confluence (2 days) and thereafter to differentiate to absorptive enterocytes within 21 days (n = 3). The culture medium was changed every 2 or 3 days at the apical (0.5 mL) and basolateral side (1.5 mL).
:3 split ratio with trypsin/EDTA (0.05%/0.01%) solution and were used at passage number 2–4. Trypsinized cells (1 × 105) were seeded on fibronectin coated 24-well plates (Biochrom KG, Berlin, Germany) and allowed to growth until they reached 100% confluence (2 days). Confluent HUVECs on fibronectin-coated 24-well plates were used for co-culture experiments.
:10 and 80:20%) on semipermeable filters over 21 days to differentiate and develop an enterocyte-like phenotype. After differentiation, transwell filters were inserted onto a 24-well plate where endothelial cells were cultivated on the bottom of the 24-well cavity. The upper compartment (insert) with epithelial cells was exposed to target substrates for 4 hours. In order to ensure an intact intestinal monolayer during the time of the experiment, the transepithelial electrical resistance (TEER) was monitored before and at the end of the incubation experiments. Only epithelial monolayers exerting TEER values above 550 Ωm cm−2 were used for incubation experiments. Thereafter, inserts were removed and HUVECs were stimulated immediately (short-term incubation) or after 20 h of cultivation (long-term incubation) with TNF-α. To mimic low-grade and high-grade inflammation, HUVECs were incubated for 3 hours with 1 ng mL−1 and 10 ng mL−1 TNF-α, respectively. After TNF-α stimulation, we determined (a) leukocyte adhesion to HUVECs, (b) expression of cell adhesion molecules, (c) expression of NF-κB and (d) expression and secretion of the cytokines IL-6 and IL-8 in the supernatants or in HUVECs.
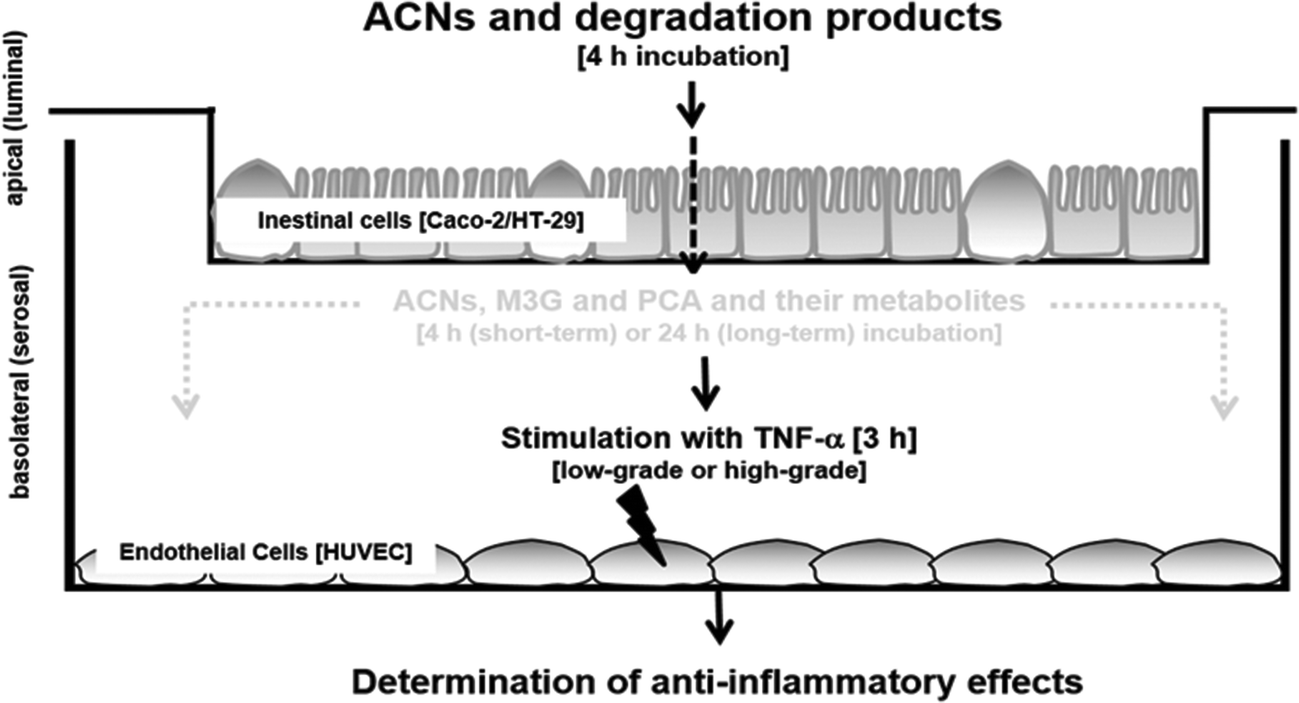 | ||
| Fig. 1 Determination of the anti-inflammatory effects of ACNs and metabolites in an in vitro epithelial-endothelial co-culture cell system. | ||
Cell viability and cytotoxicity
:1) or 12.43 ± 0.67 h−1 per 106 cells (ratio 8:1). In contrast, confluent HT29-B6 cells on filters for 21 days revealed AP activity levels of 1.07 ± 0.24 h−1 per 106 cells, reflecting the lower differentiation grade in comparison to Caco-2 cells; however, they were not able to generate an intact monolayer [data not shown], (2) nearly identical population doubling times of HT29-B6 and Caco-2 cells (21.3 ± 1.3 h and 24.5 ± 2.1 h) assured that after 24 hours a confluent monolayer and stable cell ratios could be attained, (3) cytotoxicity was not induced in HT29-B6 cells since cell viability measurements using the Guava® ViaCount™ assay showed that there was no significant difference in the number of non-viable cells (apoptotic and necrotic) between Caco-2 and HT29-B6 cells cultured alone (21 days) or in a ratio of 80:20 [data not shown].
In a second set of experiments, intestinal cell inserts were removed after 240 min of incubation and basolateral compartments containing HUVECs grown on fibronectin were immediately stimulated for 3 hours using 1 ng mL−1 or 10 ng mL−1 TNF-α to mimic low-grade or high grade inflammation (short-term incubation). To determine long-term effects of ACNs, HUVECs were cultured for further 20 hours and thereafter stimulated with both TNF-α concentrations (see Fig. 1).
| Gene | GenBank accession no | Primer and TaqMan probe sequence |
|---|---|---|
| E-selectin | M30640 | Forward primer: 5′-CCCGTGTTTGGCACTGTGT-3′ |
| Reverse primer: 5′-GCCATTGAGCGTCCATCCT | ||
| TaqMan probe: 5′-CAAGTTCGCCTGTCCTG-Tamra-3′ | ||
| VCAM-1 | M60335 | Forward primer: 5′-GGGAAGCCGATCACAGTCAA-3′ |
| Reverse primer: 5′-ATGAGATGATCTCCTTTCAGTAAGTCTATC-3′ | ||
| TaqMan probe: 5′-TCAGTTGCTGATGTATACCCATTTGACAGGC-Tamra-3′ | ||
| ICAM-1 | NM_000201 | Forward primer:5′-GCAGACAGTGACCATCTACAGCTT-3′ |
| Reverse primer: 5′-CTTCTGAGACCTCTGGCTTCGT-3′ | ||
| TaqMan probe: 5′-CCGGCGCCCAACGTGATTCT-Tamra-3′ | ||
| IL-6 | M54894 | Forward primer:5′-GGTACATCCTCGACGGCATCT-3′ |
| Reverse primer: 5′-GTGCCTCTTTGCTGCTTTCAC-3′ | ||
| TaqMan probe: 5′-Fam-TGTTACTCTTGTTACATGTCTCCTTTCTCAGGGCT-Tamra-3′ | ||
| IL-8 | M28130 | Forward primer: 5′-AGCTGGCCGTGGCTCTCT-3′ |
| Reverse primer: 5′-TTTAGCACTCCTTGGCAAAACTG-3′ | ||
| TaqMan probe: 5′-Fam-CAGCCTTCCTGATTTCTGCAGCTCTGTG-Tamra-3′ | ||
| NF-κB | NM_62399 | Forward primer: 5′-AGCACAGATACCACCAAGACCC-3′ |
| Reverse primer: 5′-CCAGGGAGATGCGCACTG-3′ | ||
| TaqMan probe: 5′-Fam-CATCAAGATCAATGGCTACACGGACCAGG-Tamra-3 | ||
| β-actin | NM_002046 | Forward primer: 5′-CCACATCGCTCAGACACCAT-3′ |
| Reverse primer: 5′-GTGACCAGGCGCCCAATA-3′ | ||
| TaqMan probe: 5′-Fam-AGGTCGGAGTCAACGGATTTGG-Tamra-3 |
Results
ACNs degradation and incubation studies with culturing Caco-2 cells (100%) on transwell inserts
The ACN-rich extract as well as M3G were tested for degradation without (at pH 2.0 and pH 7.4) and with cells (pH 7.4) in order to distinguish between the cell-mediated degradation rates, i.e. cellular uptake and the chemically induced degradation rates. As shown in Fig. 2, all ACNs from the extract were stable at pH 2.0, whereas at pH 7.4 (without cells) ACNs from the extract were differently degraded (Fig. 2B–F). After 240 min incubation, the loss was 5.4 ± 0.0% for C3G, 11.8 ± 0.1% for Peo3G, 22.9 ± 3% for M3G (p < 0.05), 40.8 ± 3.5% for D3G (p < 0.05) and 46.2 ± 5.5% for Pet3G (p < 0.05) compared to their initial concentrations. The loss of M3G, as a single compound (Fig. 2A) was 37.4 ± 6.1% (p < 0.05) after incubation for 240 min. In all cases, degradation rates in the presence of cells were significantly higher than the degradation rates without cells at pH 7.4. After 240 min incubation with cells, the loss was 96.5 ± 5.8% for M3G (*#p < 0.05), 85.8 ± 7.1% for Peo3G (*#p < 0.05), 84.3 ± 6.2% for Pet3G (*#p < 0.01), 78.5 ± 11.1% for D3G (*#p < 0.05) and 77.4 ± 7.4% for C3G (*#p < 0.01) compared to their initial concentrations. The loss of M3G as a single compound (Fig. 2A) was 59.1 ± 3.8% (*#p < 0.05). | ||
| Fig. 2 Degradation rates of ACNs from grape (Dakapo) extract and M3G on as a function of incubation time and pH in the presence of Caco-2 cells. 50 μmoL L−1 M3G (A) and 50 μmoL L−1 M3G equivalents of ACN extract (B–F) were given to the apical compartment of transwell inserts (with or without cells). After 30, 60, 90, 120 and 240 min incubation time at pH 2.0 or 7.4, ACNs were quantified by HPLC-UV. Differences were significant at *p < 0.05 compared to the initial concentration or #*p < 0.05 compared to the degradation rates without cells. Values are given as mean ± SEM (n = 3). | ||
ACN incubation studies with cultured Caco-2 cells (100%) and Caco-2 cells mixed with HT29-B6 cells (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :10 and 80:20) on transwell inserts
:10 and 80:20) on transwell inserts
In order to investigate the cell-mediated uptake efficiencies of ACNs from Dakapo extract and of M3G, both were given to the apical compartment of the transwell system, and basolateral enrichment was quantified with UPLC-MS after SPE. Furthermore, ACNs (extract and single compounds) were given to different intestinal cell mixtures in order to imitate different metabolization sites of the intestine, which could be responsible for the uptake of ACNs and degradation products such as phenolic acids. As shown in Fig. 3, the apical loss of ACNs was associated with their enrichment in the basolateral compartment. The most occurring ACN M3G in the ACN-rich extract, which rapidly decreased in the apical compartment, was also the most prevalent compound in the basolateral solution indicating that M3G was transported across the intestinal epithelium (100% Caco-2) (Fig. 3A and B). After 240 min incubation time, M3G concentration was 157 ± 41 nmoL L−1 in the basolateral compartment representing 0.35% of the initial apical concentration. In addition, Peo3G (89 ± 22 nmoL L−1), Pet3G (0.6 ± 0.2 nmoL L−1) and C3G (5.3 ± 1.2 nmoL L−1) but no D3G of the extract were accumulated. Using transwell inserts with mixed intestinal cells (Caco-2:HT29-B6 90:10 and 80:20) the degradation rates of ACNs were lower than those in the Caco-2 (100%) model (Fig. 3C–F). However, at the basolateral side M3G, Peo3G, Pet3G and C3G could be determined using the Caco-2:HT29-B6 (90:10) as well in the Caco-2:HT29-B6 (80:20) cultured cells with concentrations of 36 ± 15, 25 ± 12, 15 ± 3.5 and 0.3 ± 0.2 nmol L−1 (90:10), and 33 ± 5, 26 ± 14, and 1 ± 0.3 nmol L−1 for M3G, Peo3G, and C3G (80:20), respectively. Similar to the results with the extract, M3G as a single compound was accumulated at the basolateral side in the mixed cell models, albeit in lower concentrations than in the Caco-2 model.
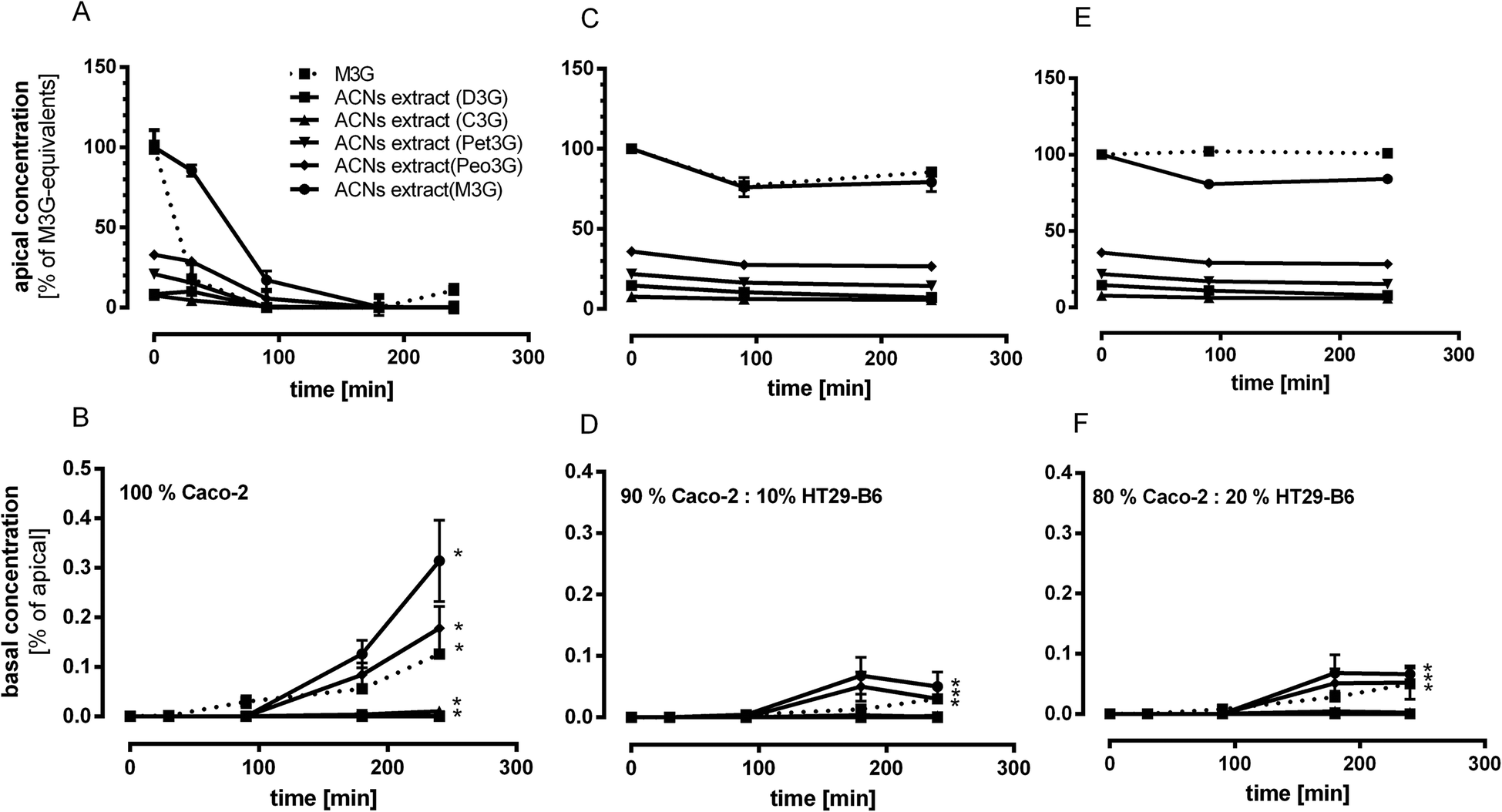 | ||
| Fig. 3 Concentrations of ACNs from Dakapo extract and M3G in apical and basolateral solutions using various cell mixtures and incubation times. Cell mixtures were incubated with 50 μmoL L−1 M3G equivalents. Concentrations were determined in the apical and basolateral compartment of the transwell system at various time points. Upper panel: Apical concentrations (% of M3G equivalents) of ACNs using Caco-2 cells alone (A) and with 10% (C) or 20% HT29-B6 cells (E). Lower panel: basolateral concentrations (% of apical) of ACNs using Caco-2 cells alone (B) and with 10% (D) or 20% HT29-B6 cells (F). Values are given as mean ± SEM (n = 3). Differences were significant at *p < 0.05 compared to the initial concentrations. | ||
Establishment of low-grade and high-grade inflammatory conditions in HUVECs stimulated with TNF-α
In order to distinguish between low-grade inflammation to mimic an in vivo LGI and a chronic (high-grade) inflammation, HUVECs were stimulated with different concentrations of TNF-α for 3 hours. Thereafter, inflammation-related parameters were measured as described in “Materials and methods”. The determination of leukocyte adhesion to HUVECs with the adhesion assay showed that adhesion was significantly increased by low-grade (1 ng mL−1, p < 0.001) and high-grade (10 ng mL−1, p < 0.001) inflammatory conditions in comparison to control cells without TNF-α stimulation (Table 3). Similar to leukocyte adhesion, low-grade inflammation significantly increased the expression of E-Selectin (p < 0.01), VCAM-1 (p < 0.001) and ICAM-1 (p < 0.001) in endothelial cells concomitant with enhanced protein levels measured by FACS analysis (E-selectin and ICAM-1 (p < 0.001) and VCAM-1 (p < 0.01)). This was also observed at high-grade inflammatory conditions. Leukocyte adhesion, mRNA- and protein expression of the adhesion molecules were significantly higher in stimulated cells using 10 ng mL−1-TNF-α compared to non-stimulated cells (p < 0.001). Gene expression of IL-6 and IL-8 in HUVECs was determined by quantitative RT-PCR using β-actin as internal standard and was expressed as 2−ddCT values after normalization of the target gene with the housekeeper gene β-actin. TNF-α stimulation of HUVECs induced a significant up-regulation of IL-6 (p < 0.01) and IL-8 mRNA (p < 0.01) as well as IL-6 (p < 0.001) and IL-8 protein (p < 0.001) levels compared to non-stimulated cells (NC). The inflammation induced effects of TNF-α correlated with the mRNA expression of NF-κB. The amount of NF-κB was significantly induced after exposure to 1 ng mL−1 TNF-α (p < 0.01) or 10 ng mL−1 TNF-α (p < 0.01).a
| Negative control | Low-grade [1 ng mL−1 TNF-a] | High-grade [10 ng mL−1 TNF-a] | |
|---|---|---|---|
| a *,**,*** indicate p < 0.01, p < 0.01 and p < 0.001, respectively, compared to non-stimulated cells (values are given as mean ± SD (n = 3). | |||
| Leukocyte adhesion | 5802 ± 256 | 49796 ± 647*** |
106854 ± 9491*** |
| Adhesion molecules | |||
| E-selectin mRNA | 0.190 ± 0.035 | 0.527 ± 0.023** | 4.610 ± 0.572*** |
| E-selectin protein | 17.33 ± 2.50 | 105.67 ± 10.17*** | 236.66 ± 20.13*** |
| VCAM-1 mRNA | 0.273 ± 0.140 | 1.313 ± 0.183*** | 3.603 ± 0.476*** |
| VCAM-1 protein | 9.33 ± 1.52 | 25.33 ± 2.08** | 87.33 ± 8.08*** |
| ICAM-1 mRNA | 0.163 ± 0.015 | 1.037 ± 0.074*** | 5.837 ± 0.492*** |
| ICAM-1 protein | 10.66 ± 5.13 | 121.66 ± 19.55*** | 348.00 ± 38.98*** |
| Cytokines | |||
| IL-8 mRNA | 0.553 ± 0.059 | 1.673 ± 0.322*** | 6.623 ± 0.366*** |
| IL-8 protein | 3.51 ± 0.51 | 6.61 ± 1.261*** | 15.88 ± 2.01*** |
| IL-6 mRNA | 0.34 ± 0.06 | 0.91 ± 0.11** | 1.7 ± 0.26*** |
| IL-6 protein | 0.64 ± 0.18 | 1.27 ± 0.23*** | 2.66 ± 0.45*** |
| Regulator protein | |||
| NF-kB mRNA | 0.823 ± 0.065 | 2.17 ± 0.246** | 2.19 ± 0.28** |
To compare the anti-inflammatory effects of ACNs with controls, values of the positive controls (TNF-α stimulated cells, low-grade and high-grade) were set to 100% and inhibition by ACNs, M3G or PCA was compared to the positive control (n = 3).
Inhibition of leukocyte adhesion to HUVECs by ACNs and possible degradation products under different inflammatory conditions
To determine whether ACNs exert anti-inflammatory effects on human vascular cells, we first examined the effects of leukocyte adhesion on TNF-α-activated HUVECs. After incubation of ACNs from extract, M3G as a single compound and PCA as a possible degradation product of C3G with intestinal cells cultured on transwell inserts (Caco-2 (100%) or Caco-2/HT29-B6 cell mixtures (90:10 and 80:20) for 240 min, basolateral solutions were used immediately or remained on the co-cultured HUVECs for further 20 hours. Thereafter, HUVECs were stimulated with 1 ng mL−1 or 10 ng mL−1 TNF-α (3 h). Maximal leukocyte adhesion to HUVECs with different TNF-α concentrations was set to 100% (PC, positive control) and possible ACN effects were related to the positive control. Fig. 4 shows that leukocyte adhesion to HUVECs was significantly reduced by ACNs from the extract (56%, p < 0.01) and M3G (70%, p < 0.05) when Caco-2 (100%) cells were pre-incubated for 24 hours prior to low-grade TNF-α stimulation of HUVECs. No difference in leukocyte adhesion was observed after short-term pre-incubation (4 hours) or under high-grade inflammatory conditions (Fig. 4A).
 | ||
| Fig. 4 Effect of ACNs, M3G and PCA on leukocyte adhesion to HUVECs under different inflammatory conditions in various cell mixtures. After incubation of different cell mixtures Caco-2 without (A) or with co-cultured HT29-B6 cells (10% or 20%, (B and C)) with ACNs, M3G and PCA for 4 hours, transwell inserts were removed and the co-cultured HUVECs were used immediately (4 h) or were further incubated in the basolateral compartment (20 h). Thereafter, cells were stimulated for 3 hours using 1 ng mL−1 TNF-α (low-grade inflammation) or 10 ng mL−1 TNF-α (high-grade inflammation). After TNF-α stimulation, leukocyte adhesion was measured as described in the Material and methods section. Corresponding controls (maximal adhesion to HUVECs after low-grade or high-grade TNF-α stimulation) were set to 100%. Values are given as mean ± SD (n = 3). Differences were significant at *p < 0.05 and **p < 0.01. | ||
Using the basolateral media from the intestinal cell mixtures (Caco-2/HT29-B6 mixed cells (90:10 and 80:20)), neither an effect on inflammation (low-grade and high-grade) nor an effect of pre-incubation (4 h and 24 h) was observed (Fig. 4B and C).
Inhibition of adhesion molecules expression on HUVECs under low-grade inflammatory conditions
Since our results from the adhesion assays were significant for inhibiting low-grade inflammation and were only observed using 100% Caco-2 cells, these conditions were chosen for subsequent experiments. For the quantification of cell adhesion molecules on mRNA and protein levels, we either used the media from the basolateral compartments from the co-culture system or directly incubated HUVECs with ACNs from extract, M3G and PCA. Direct incubation was performed with ACNs (0.1 or 1 μmoL L−1 M3G equivalents) or M3G and PCA (0.1 or 1 μmoL L−1). Fig. 5 shows that 24-hour pre-incubation of HUVECs with the basolateral solutions from the transport studies with ACNs, M3G and PCA resulted in a decrease of all three adhesion molecules in comparison to TNF-α stimulated cells (Fig. 5A and B). ACNs significantly inhibited the TNF-α induced adhesion molecule mRNA and protein expression with a greater effect on ICAM-1 and VCAM-1 than on E-selectin. ACNs of basolateral concentrations could significantly inhibit protein levels of adhesion molecules to 66 ± 5, 52 ± 7 and 59 ± 9% of TNF-α stimulated controls and 76 ± 29, 40 ± 10 56 ± 14% of the TNF-α stimulated mRNA levels of E-selectin, ICAM-1 and VCAM-1, respectively. Similar to ACNs, M3G significantly reduced the expression of the three adhesion molecules to 86 ± 6, 72 ± 6 and 71 ± 12% of the protein level for E-selectin, ICAM-1 and VCAM-1 as well as to 50 ± 10, 66 ± 12 and 60 ± 10% of the TNF-α-induced mRNA levels for E-selectin, ICAM-1 and VCAM-1. PCA could also significantly alter TNF-α-induced protein and mRNA expression levels, with 83 ± 9, 61 ± 7 and 61 ± 4% of the protein levels and 79 ± 10, 80 ± 10 and 64 ± 13% of the mRNA levels for E-selectin, ICAM-1 and VCAM-1. | ||
| Fig. 5 Effect of ACNs, M3G and PCA on the expression of cell adhesion molecules in HUVECs under long-term and low-grade inflammatory conditions. After incubation of Caco-2 with ACNs, M3G and PCA for 4 hours, transwell inserts were removed and the co-cultured HUVECs were further incubated with the basolateral compartment (20 h) or were incubated directly with 0.1 or 1 μmoL L−1 M3G equivalents, respectively PCA. Thereafter, cells were stimulated for 3 hours using 1 ng mL−1 TNF-α (low-grade inflammation). After TNF-α stimulation, expression of adhesion molecules (E-selectin, ICAM-1 and VCAM-1) were measured on protein (upper level) and mRNA levels (lower level) as described in material and methods. Inserts (C and D): leukocyte adhesion to HUVECs after direct incubation with ACNs, M3G and PCA under long-term (20 h) and low-grade TNF-α stimulation. Corresponding controls were set to 100%. Values are given as mean ± SD (n = 3). Differences were significant at *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
Using ACNs, M3G and PCA in approximately equimolar concentrations to incubate the HUVECs directly, no effect on leukocyte adhesion (inserts) and expression of adhesion molecules was observed (Fig. 5C and D). Only concentrations of 1 μmoL L−1 ACNs, M3G and PCA were able to induce a significant inhibition of protein and mRNA expression of cell adhesion molecules (Fig. 5E and F).
Inhibition of cytokine expression in HUVECs under low-grade inflammatory conditions by ACNs/ACN metabolites
We used ELISAs and real-time PCR to measure the concentrations of selected chemokines (IL-6 and IL-8) and NF-κB as regulator protein of inflammatory-related proteins to further validate some of the anti-inflammatory effects. Fig. 6 shows that 24-hour pre-incubation of HUVECs with the basolateral solutions from the transport studies using ACNs, M3G and PCA resulted in a decrease of IL-8 secretion and mRNA expression in TNF-α-stimulated cells (Fig. 6A and B). ACNs significantly inhibited TNF-α-induced IL-8 mRNA and protein levels. ACNs of basolateral concentrations significantly reduced protein levels of IL-8 to 65 ± 12% and mRNA levels of IL-8 to 23 ± 11% in TNF-α-stimulated HUVECs. Similar to ACNs, M3G and PCA significantly decreased the expression of IL-8 to 75 ± 10 and 55 ± 14% of TNF-α-induced IL-8 protein levels as well as to 41 ± 16 and 56 ± 21% of the TNF-α-induced IL-8 mRNA levels. Using ACNs, M3G and PCA in approximately equimolar concentration to incubate the HUVECs directly, no effect on IL-8 protein and mRNA expression was observed (Fig. 6C and D). Again, only concentrations of 1 μmoL L−1 ACNs, M3G and PCA were able to induce a significant inhibition of IL-8 protein and mRNA expression [ACNs (51 ± 13 and 25 ± 17% TNF-α-induced IL-8 expression), M3G (69 ± 15 and 52 ± 11% TNF-α-induced IL-8 expression) and PCA (40 ± 9 and 63 ± 18% TNF-α-induced IL-8 expression)] (Fig. 6E and F). No effect was observed on IL-6 mRNA and protein expression.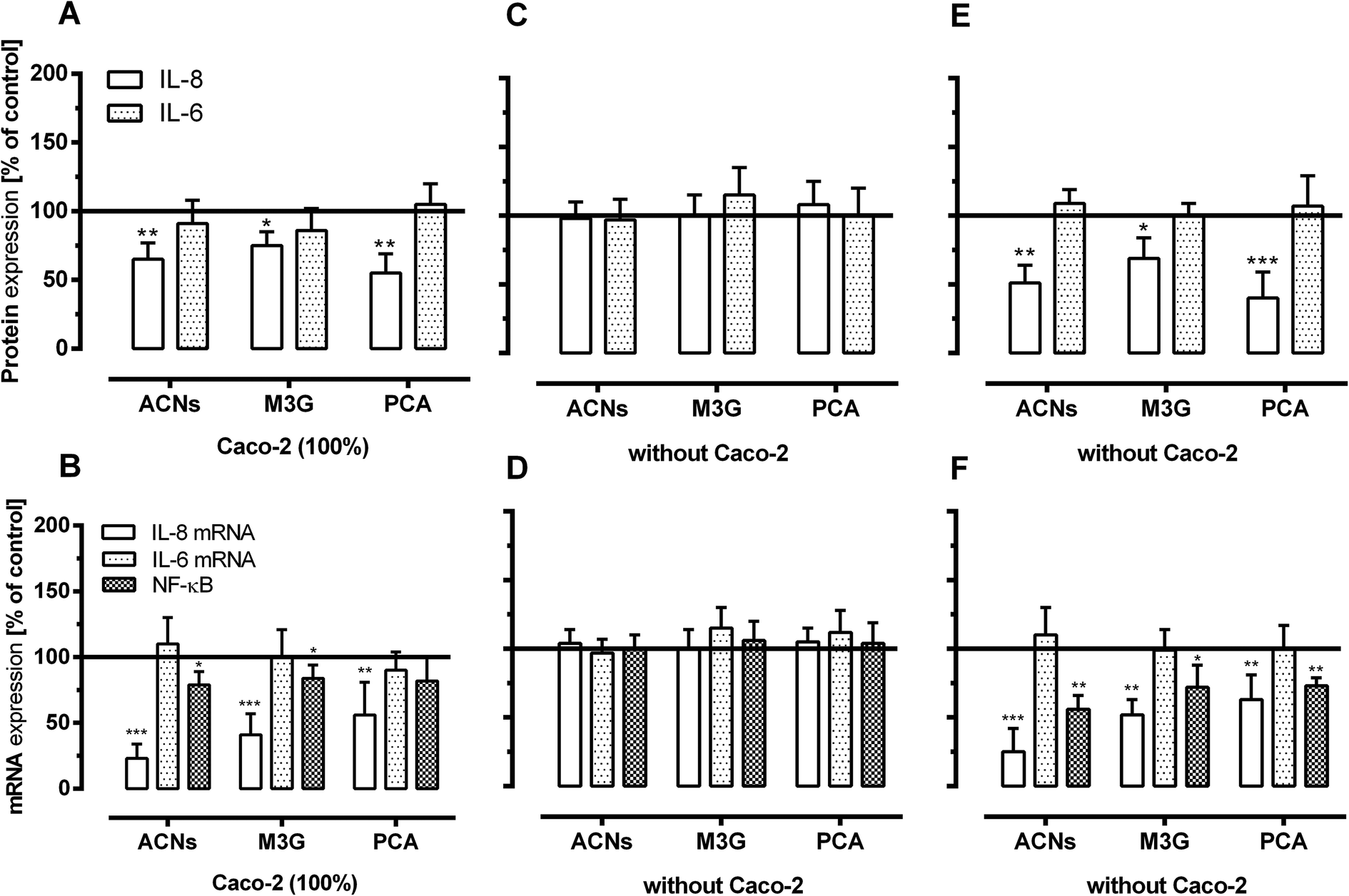 | ||
| Fig. 6 Effect of ACNs, M3G and PCA on expression of inflammation-related molecules in HUVECs under long-term and low-grade inflammatory conditions. After incubation of ACNs, M3G and PCA for 4 hours with Caco-2, transwell inserts were removed and the co-cultured HUVECs were further incubated with the basolateral compartment (20 h) or were incubated directly with 0.1 or 1 μmoL L−1 M3G or PCA equivalents. Thereafter, cells were stimulated for 3 hours using 1 ng mL−1 TNF-α (low-grade inflammation). After TNF-α stimulation, cytokine (IL-8 and IL-6) protein (upper panel) and mRNA levels (lower panel) as well as NF-kB mRNA gene expression was measured as described in the Material and methods section. Corresponding controls were set to 100%. Values are given as mean ± SD (n = 3). Differences were significant at *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
The adhesion and anti-inflammatory effects of ACNs, M3G and PCA correlated with the effects on NF-κB mRNA levels. The amount of NF-κB mRNA was significantly reduced by ACNs from the basolateral compartment of the transwell systems. They reduced TNF-α-stimulated mRNA levels to 69 ± 10%. Similar to ACNs, M3G and PCA significantly decreased the expression of NF-kB mRNA levels to 84 ± 10% and 81 ± 9% of TNF-α-stimulated mRNA levels. This effect was greater, when HUVECs were directly incubated. ACNs, M3G and PCA reduced the mRNA levels to 56 ± 15% and 72 ± 16% and 73 ± 6% of the TNF-α-induced expression when using 1 μmoL L−1 equivalents, but no effect was observed when 0.1 μmoL L−1 was used for pre-incubation.
Discussion
The link between atherogenesis and health promoting effects of ACNs has not yet been completely clarified; open questions concern (a) the identification and quantification of metabolites found in plasma in vivo after an ACN-rich diet, (b) the role of intestinal metabolization and microbial fermentation in the gut and (c) the molecular mechanisms in target cells. Therefore, our aim was to establish an in vitro co-culturing system with epithelial endothelial cells representing the intestinal epithelial layer and the adjacent endothelium to investigate effects of dietary ACNs. We have shown for the first time that ACNs from a grape extract as well as M3G as single ACN and PCA as a phenolic acid (degradation product of C3G) significantly inhibited leukocyte adhesion when being transported across the intestinal layer. This effect was only observed when (a) Caco-2 cells as single cell type served as an absorptive cell model, (b) ACNs, M3G and PCA had been pre-incubated for 24 hours and (c) low-grade inflammatory conditions were simulated. In addition, inhibition of leukocyte adhesion was associated with significant down-regulation of adhesion molecules as well as inhibition of chemokine secretion and expression in endothelial cells.It has been shown recently that the bioavailability of ACNs was extremely low compared to other flavonoids. Available data from bioavailability studies suggested plasma concentrations to range between 1 and 550 nmoL L−1 for parenteral ACNs and their conjugates, and phenolic metabolites concentration between 10–2000 nmol L−1 post-consumption.34–36 These physiological concentrations were able to induce systemic effects.37,38 However, it is well-known, that ACNs are relatively instable and undergo chemical degradations at physiological pH (pH 7.4), a pH value existing in the intestine and colon.39,40 Furthermore, ACNs which reach the intestine, could be absorbed without modification or after enzymatic hydrolyzation of the attached sugar.41 Once absorbed, they could be further metabolized by phase-II-enzymes.42 This metabolization step was suggested to be dependent on the distribution pattern with enzyme-dependent differences along the intestine.43 Thus, we investigated ACN transport using Caco-2 cells alone (100%) or Caco-2 cells mixed with HT29-B6 (10 and 20%). Our results showed that the decrease in apical ACN concentrations was not a result of chemically induced degradation because of the greater reduction in ACN concentrations in the presence of cells (100% Caco-2) in transport studies compared to experiments without cells. This implicated that ACNs were taken up, an observation which was confirmed by the basal occurrence of major ACNs present in the extract, i.e., M3G, Peo3G, C3G and Pet3G. Using mixtures of intestinal cells (Caco-2/HT29-B6 cells: ratio 90/10 or 80/20) in the transport model we also observed a basal occurrence of M3G and Peo3G, albeit to a lesser extent, but no minor ACNs like Pet3G and D3G were detected after 240 min of incubation. Despite the occurrence of ACNs in the basolateral compartment, the rates do not correlate with their apical decline in concentration. For example, less than 0.3% of the initial M3G concentration could be found in the basolateral solution. These differences in the apical disappearance respectively basolateral occurrence of intact glucosides could be a consequence of β-glucosidase activities or lower transport capacities. Enzymes such as the brush border associated LPH (lactase phlorizin hydrolase) act on β-glucosidic cleavage to release the aglycone as shown for flavonols like quercetin.44 Both, the aglycone of quercetin as well as quercetin-3-glucoside were able to inhibit GLUT2 mediated glucose transport.45 Similar results were obtained with the ACNs pelargonidin-3-glucoside or M3G, which inhibit glucose transport primarily by inhibition of GLUT2 with less inhibition of SGLT1.41,46 This, however, must remain speculative, since we have not measured enzyme activities or inhibition of glucose transport. Nevertheless, in all three intestinal models we did not only measure the occurrence of ACNs from grape extract in basal compartments, but also that of M3G when applied as a single compound. In both cases, the transport efficiencies were low and recovery rates were less than 1%; this was similar to other recently published results. Toydemir et al. observed cyanidin-3-(2G-glucosylrutinoside) transport from morello cherries across Caco-2 cells with transport efficiencies of 3–4%.46 Similar transport efficiencies (1%) were reported by Faria et al. who tested the uptake of ACNs from grape skin extracts as well as of M3G as single compound.24 Notably, Steinert et al. could not observe any transport of ACNs when supplied to Caco-2 cells.47 Apart from intact glycosides in the basolateral compartment, we did not find any further metabolites, neither degradation nor phase-II products. This was similar to data from Zhou et al. who demonstrated that conjugated metabolites were predominant in rat in situ intestinal perfusion and rat in vivo pharmacokinetic studies but not when using Caco-2 monolayers.48 Such differences could be explained by the different expression patterns of e.g. UDP-glucuronosyltransferase (UGT) in the intestine and intestinal cell lines as well as by differences between species.49,50
However, although we detected ACN glycosides in the basolateral compartment in all three intestinal models, anti-inflammatory effects could only be observed using the basolateral compartments from the studies with 100% Caco-2 cells on the transwell filters under LGI conditions and long-term pre-incubation (24 hours). The inhibition of pro-inflammatory cytokines such as TNF-α and the reduction of leukocyte adhesion to endothelial cells are key mechanisms in the control of atherogenesis, especially in situations of LGI. Our results indicate that ACNs in an intestinal-endothelial cell co-culture model lead to a significant reduction of the TNF-α-induced leukocyte adhesion under LGI conditions. This could also be observed for M3G or PCA, albeit to a lesser extent. Interestingly, direct incubation of endothelial cells with ACNs from extract or M3G and PCA in equivalent concentrations found in the basal compartment failed to induce this anti-adhesive effect; only higher concentrations were able to do so. Thus, we speculated that unknown intermediate metabolites were generated when ACNs, M3G and PCA cross the intestinal cell layer. Furthermore, these unknown metabolites were only effective in LGI and long-term incubation (24 hours pre-incubation).
The inhibitory effects on cell adhesion induced by ACNs from extract or as single compound were found to be associated with changes in the expression of adhesion molecules such as E-selectin, ICAM-1 and VCAM-1. In previous studies others have also shown that ACNs and their degradation products were able to influence ICAM-1 and VCAM-1 expression in vitro and in vivo.51–54 However, in vitro experiments were usually performed (a) with direct incubation of endothelial cells with ACNs and (b) by stimulation with high TNF-α concentrations to induce a pro-inflammatory situation. For example, Huang et al. have demonstrated that pre-incubation of HUVECs with 1–100 μM M3G attenuated TNF-α-induced ICAM-1 and VCAM-1 expression and secretion.55 Although they used a high concentration of TNF-α of 10 μg mL−1, they observed effects of M3G already at low concentrations (1 μmoL L−1). The reason might have been that they reduced serum concentrations in HUVEC culture media prior to their pre-incubation experiments with M3G. Chao et al. on the other hand showed that 100 μg mL−1 purple sweet potato leaf extract (PSPLE) and its major ACN cyanidin attenuated TNF-α- induced (2 ng mL−1) VCAM-1 expression in human aortic endothelial cells (HAECs).56 Hidalgo et al. reported that the TNF-α-induced ICAM-1 and VCAM-1 expression levels were dose-dependently reduced when using high concentrations of possible breakdown products of ACNs such as gallic acid (10–100 μM).57 This also applies to PCA for which Zhou et al. have shown that high concentrations (150 μM) inhibited TNF-α-induced (10 ng mL−1) up-regulation of lymphoma cell (U937) adhesion to HUVECs.58 Furthermore, PCA inhibited TNF-α-stimulated VCAM-1 and ICAM-1 expression through mechanisms involving the transcription factors NF-κB and AP1. Keeping in mind that PCA or gallic acid concentrations in plasma were found to be low under physiological conditions, concentrations used in vitro were much higher.59 In our study, the expression of NF-κB mRNA in TNF-α-stimulated HUVECs was decreased with ACNs as well as with M3G and PCA, but only under LGI conditions and long-term incubation. However, inhibition of the NF-κB signaling pathway to reduce the expression of pro-inflammatory mediators is a well-known process and ACNs or their phenolic acids may influence these signaling pathways in multiple ways.60 The reduced NF-κB mRNA expression due to ACN, M3G and PCA exposure correlated with the reduced expression of cell adhesion molecules and cytokines. Under our conditions, leukocyte adhesion to HUVECs was concomitant with the up-regulation of IL-6 and IL-8 expression, chemoattractant cytokines acting as potent regulators of inflammation-induced atherogenesis. This was also observed in previous studies showing that ACNs and phenolic acid exerted anti-atherogenic effects by modulation of cytokine secretion.57
In conclusion, we have shown for the first time that physiological concentrations of ACNs from an extract or applied as a single compound crossed the intestinal cell layer and ameliorated the inflammatory response in endothelial cells by down-regulating the expression of pro-inflammatory cytokines, particularly IL-8, reducing the levels of adhesion molecules and subsequently leukocyte adhesion. Under the conditions tested (LGI and long-term incubation) these changes were significant, whereas no effect was observed with short-term exposure and high-grade inflammation. A limitation of the study was that the observed effects may not only be a result of parenteral anthocyanins, but rather an effect of unidentified metabolites. In further studies, a focus should be put on the identification and characterization of anthocyanins and their metabolites which would give information on which ACN metabolites were responsible for the effects. Although it is difficult to directly transfer these data to the in vivo situation, our results support the hypothesis that ACNs or their degradation products protect against inflammation-induced atherogenesis.
Acknowledgements
This research was funded by the German Ministry of Education and Research (BMBF) as part of the research consortium ANTHONIA (grant # 0315379A); the funders had no role in the study design, the data collection and analysis, the decision to publish, or the preparation of the manuscript. SK, CK and SR were the principal investigators of the study responsible for the study design and the writing of the manuscript. HA performed the intestinal Caco-2/HT29-B6 study, BF prepared the ACN-rich extract and determined its ACN content, SD and AR were responsible for measuring ACNs and their metabolites (Financial support by the Deutsche Forschungsgemeinschaft (DFG Sp314/12-1) is gratefully acknowledged). The authors would like to thank Bianca Walber and Nicole Tscherney for their excellent technical assistance. All authors approved the final version of the manuscript before submission and declared no conflict of interest.Notes and references
- O. Oyebode, V. Gordon-Dseagu, A. Walker and J. S. Mindell, J. Epidemiol Commun. Health, 2014, 68, 856–862 CrossRef PubMed.
- K. T. Teng, C. Y. Chang, L. F. Chang and K. Nesaretnam, Nutr. J., 2014, 13, 1–34 CrossRef PubMed.
- C. A. Cunha, F. S. Lira, J. C. Rosa Neto, G. D. Pimentel, G. I. Souza, C. M. da Silva, C. T. de Souza, E. B. Ribeiro, A. C. Sawaya, C. M. Oller do Nascimento, B. Rodrigues, P. de Oliveira Carvalho and L. M. Oyama, Mediators Inflammation, 2013, 635470, 1–24 CrossRef PubMed.
- O. R. Wright, G. A. Netzel and A. R. Sakzewski, Can. J. Physiol. Pharmacol., 2013, 91, 480–488 CrossRef CAS PubMed.
- E. Dean and R. G. Hansen, Arthritis., 2012, 560634, 1–29 CrossRef PubMed.
- D. Calçada, D. Vianello, E. Giampieri, C. Sala, G. Castellani, A. de Graaf, B. Kremer, B. van Ommen, E. Feskens, A. Santoro, C. Franceschi and J. Bouwman, Mech. Ageing Dev., 2014, 136–137, 138–147 CrossRef PubMed.
- S. M. Heringa, E. van den Berg, Y. D. Reijmer, G. Nijpels, C. D. Stehouwer, C. G. Schalkwijk, T. Teerlink, P. G. Scheffer, K. van den Hurk, L. J. Kappelle, J. M. Dekker and G. J. Biessels, Psychoneuroendocrinology, 2014, 40, 108–118 CrossRef CAS PubMed.
- B. C. van Bussel, S. S. Soedamah-Muthu, R. M. Henry, C. G. Schalkwijk, I. Ferreira, N. Chaturvedi, M. Toeller, J. H. Fuller and C. D. Stehouwer, Nutr. Metab. Cardiovasc. Dis., 2013, 23, 758–764 CrossRef CAS PubMed.
- B. Petri, M. Phillipson and P. Kubes, J. Immunol., 2008, 180, 6439–6446 CrossRef CAS.
- E. Jubeli, L. Moine, J. Vergnaud-Gauduchon and G. Barratt, J. Controlled Release, 2012, 158, 194–206 CrossRef CAS PubMed.
- M. B. Lawrence and T. A. Springer, Cell, 1991, 65, 859–873 CrossRef CAS.
- M. I. Cybulsky, K. Iiyama, H. Li, S. Zhu, M. Chen, M. Iiyama, V. Davis, J. Gutierrez-Ramos, P. W. Connelly and D. S. Milstone, J. Clin. Invest., 2001, 107, 1255–1262 CrossRef CAS PubMed.
- D. H. Kusters, J. Tegtmeier, L. J. Schurgers and C. P. Reutelingsperger, Mol. Imaging Biol., 2012, 14, 523–533 CrossRef PubMed.
- S. Kaptoge, S. R. Seshasai, P. Gao, D. F. Freitag, A. S. Butterworth, A. Borglykke, E. Di Angelantonio, V. Gudnason, A. Rumley, G. D. Lowe, T. Jørgensen and J. Danesh, Eur. Heart J., 2014, 35, 578–589 CrossRef CAS PubMed.
- A. Rodriguez-Mateos, C. Rendeiro, T. Bergillos-Meca, S. Tabatabaee, T. W. George, C. Heiss and J. P. Spencer, Am. J. Clin. Nutr., 2013, 98, 1179–1191 CrossRef CAS PubMed.
- M. Kolehmainen, O. Mykkänen, P. V. Kirjavainen, T. Leppänen, E. Moilanen, M. Adriaens, D. E. Laaksonen, M. Hallikainen, R. Puupponen-Pimiä, L. Pulkkinen, H. Mykkänen, H. Gylling, K. Poutanen and R. Törrönen, Mol. Nutr. Food Res., 2012, 56, 1501–1510 CAS.
- M. Monagas, N. Khan, C. Andres-Lacueva, R. Casas, M. Urpí-Sardà, R. Llorach, R. M. Lamuela-Raventós and R. Estruch, Am. J. Clin. Nutr., 2009, 90, 1144–1150 CrossRef CAS PubMed.
- S. Kumar and A. K. Pandey, Sci. World J., 2013, 162750, 1–52 Search PubMed.
- A. Chanet, D. Milenkovic, S. Claude, J. A. Maier, M. Kamran Khan, N. Rakotomanomana, S. Shinkaruk, A. M. Bérard, C. Bennetau-Pelissero, A. Mazur and C. Morand, Br. J. Nutr., 2013, 110, 587–598 CrossRef CAS PubMed.
- S. Tribolo, F. Lodi, C. Connor, S. Suri, V. G. Wilson, M. A. Taylor, P. W. Needs, P. A. Kroon and D. A. Hughes, Atherosclerosis, 2008, 197, 50–56 CrossRef CAS PubMed.
- C. Felgines, S. Talavera, O. Texier, A. Gil-Izquierdo, J. L. Lamaison and C. Remesy, J. Agric. Food Chem., 2005, 53, 7721–7727 CrossRef CAS PubMed.
- G. Gross, D. M. Jacobs, S. Peters, S. Possemiers, J. van Duynhoven, E. E. Vaughan and T. van de Wiele, J. Agric. Food Chem., 2010, 58, 10236–10246 CrossRef CAS PubMed.
- A. M. Aura, P. Martin-Lopez, K. A. O'Leary, G. Williamson, K. M. Oksman-Caldentey, K. Poutanen and C. Santos-Buelga, Eur. J. Nutr., 2005, 44, 133–142 CrossRef CAS PubMed.
- A. Faria, D. Pestana, J. Azevedo, F. Martel, V. de Freitas, I. Azevedo, N. Mateus and C. Calhau, Mol. Nutr. Food Res., 2009, 53, 1430–1437 CAS.
- M. Amasheh, J. Luettig, S. Amasheh, M. Zeitz, M. Fromm and J. D. Schulzke, Ann. N. Y. Acad. Sci., 2012, 1258, 100–107 CrossRef CAS PubMed.
- M. Pinto, S. Robine-Leon and M. D. Appay, Biol. Cell, 1983, 47, 323–330 Search PubMed.
- A. Blais, P. Aymard and B. Lacour, Pflugers Arch., 1997, 434, 300–305 CrossRef CAS.
- A. Le Bivic, A. Quaroni, B. Nichols and E. Rodriguez-Boulan, J. Cell Biol., 1990, 111, 1351–1361 CrossRef CAS.
- M. Yoshioka, R. H. Erickson, H. Matsumoto and E. Gum, Int. J. Cancer, 1991, 47, 916–921 CrossRef CAS.
- K. M. Kreusel, M. Fromm, J. D. Schulzke and U. Hegel, Am. J. Physiol., 1991, 261, 574–582 Search PubMed.
- C. Augeron and C. L. Laboisse, Cancer Res., 1984, 44, 3961–3969 CAS.
- G. Chiva-Blanch, M. Urpi-Sarda, M. Rotchés-Ribalta, R. Zamora-Ros, R. Llorach, R. M. Lamuela-Raventós, R. Estruch and C. Andrés-Lacueva, J. Chromatogr., A, 2011, 1218, 698–705 CrossRef CAS PubMed.
- I. Reiss, S. Kuntz, R. Schmidt, C. Kunz, L. Gortner and S. Rudloff, Immunobiology, 2004, 209, 235–244 CrossRef CAS PubMed.
- M. Garcia-Alonso, A. M. Minihane, G. Rimbach, J. C. Rivas-Gonzalo and S. Pascual-Teresa, J. Nutr. Biochem., 2009, 20, 521–529 CrossRef CAS PubMed.
- C. D. Kay, G. Mazza and B. J. Holub, J. Nutr., 2005, 135, 2582–2588 CAS.
- T. Miyazawa, K. Nakagawa, M. Kudo, K. Muraishi and K. Someya, J. Agric. Food Chem., 1999, 47, 1083–1091 CrossRef CAS PubMed.
- R. M. de Ferrars, C. Czank, Q. Zhang, N. P. Botting, P. A. Kroon, A. Cassidy and C. D. Kay, Br. J. Pharmacol., 2014, 171, 3268–3282 CrossRef CAS PubMed.
- A. Jennings, A. A. Welch, S. J. Fairweather-Tait, C. Kay, A. M. Minihane, P. Chowienczyk, B. Jiang, M. Cecelja, T. Spector, A. Macgregor and A. Cassidy, Am. J. Clin. Nutr., 2012, 96, 781–788 CrossRef CAS PubMed.
- Y. Cahyana and M. H. Gordon, Food Chem., 2013, 141, 2278–2285 CrossRef CAS PubMed.
- Y. Liu, D. Zhang, Y. Wu, D. Wang, Y. Wei, J. Wu and B. Ji, Int. J. Food Sci. Nutr., 2014, 65, 440–448 CrossRef CAS PubMed.
- S. Manzano and G. Williamson, Mol. Nutr. Food Res., 2010, 54, 1773–1780 CAS.
- U. Hribar and N. P. Ulrih, Curr. Drug Metab., 2014, 15, 3–13 CrossRef CAS.
- G. M. Pacifici, M. Franchi, P. G. Gervasi, V. Longo, P. di Simplicio, A. Temellini and L. Giuliani, Pharmacology., 1989, 38, 137–145 CrossRef CAS PubMed.
- A. J. Day, F. J. Canada, J. C. Diaz, P. A. Kroon, R. Mclauchlan, C. B. Faulds, G. W. Plumb, M. R. Morgan and G. Williamson, FEBS Lett., 2000, 468, 166–170 CrossRef CAS.
- J. Song, O. Kwon, S. Chen, R. Daruwala, P. Eck, J. B. Park and M. Levine, J. Biol. Chem., 2002, 277, 15252–15260 CrossRef CAS PubMed.
- G. Toydemir, D. Boyacioglu, E. Capanoglu, I. M. van der Meer, M. M. Tomassen, R. D. Hall, J. J. Mes and J. Beekwilder, J. Agric. Food Chem., 2013, 61, 11434–11441 CrossRef CAS PubMed.
- R. E. Steinert, B. Ditscheid, M. Netzel and G. Jahreis, J. Agric. Food Chem., 2008, 56, 4995–5001 CrossRef CAS PubMed.
- Z. Zuo, L. Zhang, L. Zhou, Q. Chang and M. Chow, Life Sci., 2006, 79, 2455–2462 CrossRef CAS PubMed.
- B. Xu, S. Gao, B. Wu, T. Yin and M. Hu, J. Pharm. Biomed. Anal., 2014, 88, 180–190 CrossRef CAS PubMed.
- T. Furukawa, Y. Naritomi, K. Tetsuka, F. Nakamori, H. Moriguchi, K. Yamano, S. Terashita, K. Tabata and T. Teramura, Xenobiotica, 2014, 44, 205–216 CrossRef CAS PubMed.
- Y. Zhu, W. Ling, H. Guo, F. Song, Q. Ye, T. Zou, D. Li, Y. Zhang, G. Li, Y. Xiao, F. Liu, Z. Li, Z. Shi and Y. Yang, Nutr. Metab. Cardiovasc. Dis., 2013, 23, 843–849 CrossRef CAS PubMed.
- R. Kalin, A. Righi, A. Del Rosso, D. Bagchi, S. Generini, M. M. Cerinic and D. K. Das, Free Radical Res., 2002, 36, 819–825 CrossRef.
- K. Miyazaki, K. Makino, E. Iwadate, Y. Deguchi and F. Ishikawa, J. Agric. Food Chem., 2008, 56, 11485–11492 CrossRef CAS PubMed.
- C. K. Sen and D. Bagchi, Mol. Cell. Biochem., 2001, 216, 1–7 CrossRef CAS.
- W. Y. Huang, J. Wang, Y. M. Liu, Q. S. Zheng and C. Y. Li, Eur. J. Pharmacol., 2014, 723, 67–72 CrossRef CAS PubMed.
- P. Y. Chao, Y. P. Huang and W. B. Hsieh, Cell Adhes. Migr., 2013, 7, 237–245 CrossRef PubMed.
- M. Hidalgo, S. Martin-Santamaria, I. Recio, C. Sanchez-Moreno, B. de Pascual-Teresa, G. Rimbach and S. de Pascual-Teresa, Genes Nutr., 2012, 7, 295–306 CrossRef CAS PubMed.
- Z. Zhou, Y. Liu, A. D. Miao and S. Q. Wang, Eur. J. Pharmacol., 2005, 513, 1–8 CrossRef CAS PubMed.
- S. Shahrzad, K. Aoyagi, A. Winter, A. Koyama and I. Bitsch, J. Nutr., 2001, 131, 1207–1210 CAS.
- L. Yi, C. Y. Chen, X. Jin, T. Zhang, Y. Zhou, Q. Y. Zhang, J. D. Zhu and M. T. Mi, Biochimie, 2012, 94, 2035–2044 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |