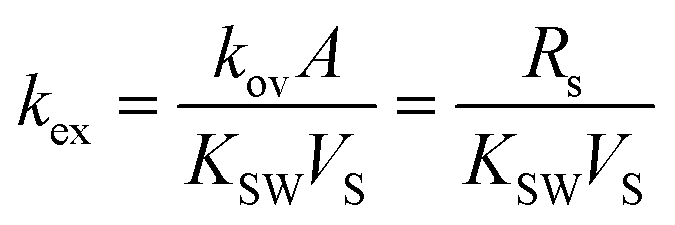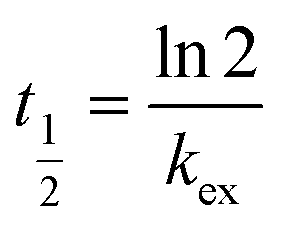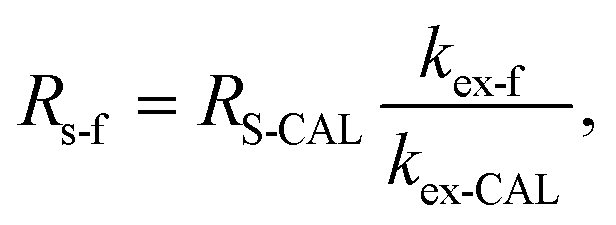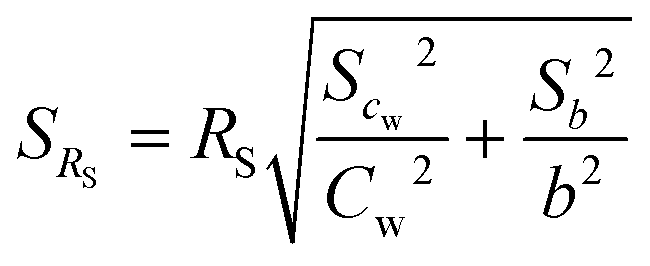Calibration of Chemcatcher® passive sampler for selected highly hydrophobic organic substances under fresh and sea water conditions†
Received
18th August 2014
, Accepted 9th January 2015
First published on 16th January 2015
Abstract
This study presents sampling rates (Rs) of the nonpolar Chemcatcher® for selected polybrominated diphenylethers (PBDEs), polychlorinated biphenyls (PCBs) and organochlorine pesticides measured in two calibration experiments with fresh water and salt water (2.5% NaCl), respectively. The uptake experiments were performed with samplers on a carousel in a flow-through tank system at constant concentration of test substances and a temperature of 13 °C. It was found that the linear flow velocity at the sampler surface calculated based on the rotation speed of the carousel (0.4 m s−1) disagreed with the actual measured velocity using a vane anemometer (0.11 m s−1). The sampling rates Rs of the test compounds were in the range of 0.15 to 0.89 L d−1 and were slightly lower in salt water than in fresh water, showing the minor effect of the salt matrix on the uptake of highly hydrophobic organic compounds. Five performance reference compounds (PRC) were also tested during the tank experiments. A higher offload of PRCs was found in salt water (with elimination rates between 0.034 d−1 and 0.051 d−1) compared to fresh water (elimination rates between 0.017 d−1 and 0.028 d−1). Our study demonstrates the suitability of the Chemcatcher® device for time-integrated monitoring of highly hydrophobic micropollutants dissolved in fresh and salt water down to the lower ng-per-litre range.
Water impact
Water monitoring of priority organic pollutants as required e.g. under the European Water Framework Directive include polybrominated diphenylethers (PBDEs) and DDT and its metabolites. This is a challenging task because of their low concentration in the water phase. Passive samplers accumulate such aqueous micro-pollutants and can, after calibration, be used to determine time-weighted average water concentrations for periods of weeks to months. The paper describes calibration experiments with the non-polar Chemcatcher as passive sampler for selected PBDEs, DDT and its metabolites, and some polychlorinated biphenyls in order to determine the sampler uptake rates in fresh and salt water. The results are useful for monitoring tasks in different aqueous environments.
|
1. Introduction
1.1 General facts
Contaminants show great distribution variability in natural aqueous environments due to their diverse chemical characteristics, different input pathways or their continuous remobilization from sediments caused by anthropogenic activities. To ensure compliance with the European Water Framework Directive (WFD),1 knowledge of contaminant concentration is mandatory. This requires accurate monitoring efforts with sufficient coverage regarding time scale and a close-meshed monitoring network. The current monitoring approaches are, however, often limited to low frequency spot sampling, which provides only snapshot information about the current contamination status, in particular, when working in dynamic environments. One solution could be the use of time-integrated sampling approaches, namely passive sampling. In comparison to conventional spot sampling the application of passive samplers allows for the detection of very low analyte concentrations.2 Moreover, the determination of time-weighted average concentration of pollutants makes passive sampling more representative than spot sampling strategies. In addition, passive sampling provides information about the bioavailability of target pollutants.3 For these reasons, passive samplers are becoming increasingly important. In 2011 the European Commission approved a guidance document of the task group on chemical monitoring where passive sampling techniques were named as alternative monitoring tools under the WFD regulations.4 In June 2011 an ISO standard5 was published to give guidance for passive sampling in surface waters which can help to broaden the acceptance of passive sampling devices among water management authorities.
Many passive sampling systems have been developed for sampling organic and inorganic substances and some of them are commercially available. The most widely used and investigated passive sampling system is the semi-permeable membrane devices (SPMD)6,7 for lipophilic substances, the diffusive gradients in thin films (DGT) for metal and metal species8 and the polar organic chemical integrative sampler (POCIS) for polar organic substances.9 Other passive sampling systems for collecting nonpolar contaminants were developed, such as the membrane-enclosed sorptive coating (MESCO),10,11 low-density polyethylene membrane12 or the Chemcatcher®.13
The aim of this study was to test experimentally the influence of dissolved salts on the uptake of nonpolar substances using the Chemcatcher.
The present paper shows the results of two different calibration experiments for the Chemcatcher® with a C18 Empore™ disk as receiving phase and a low-density polyethylene membrane as diffusion-limiting barrier for nonpolar substances, including polybrominated diphenylethers (PBDEs), polychlorinated biphenyls (PCBs) and organochlorine pesticides. The calibration experiments were conducted in a laboratory flow-through system with a carousel made from polytetrafluoroethylene (PTFE), described previously by Vrana, et al.14 The samplers were exposed for 14 days, in spiked fresh water and in 2.5% salt water simulating Northern Sea conditions. Moreover, the applicability of various performance reference compounds was tested.
2. Theory
The mass transfer of an analyte from water to the sampler includes diffusion and interfacial transport steps across several barriers (compartments), including the stagnant aqueous boundary layer, possible biofilm layer, the diffusion-limiting membrane, and finally the receiving phase, which is in this case a n-octanol-saturated C18Empore™ disk.15 The accumulation of pollutants in the receiving phase of a passive sampler follows 1st-order kinetics:16| | 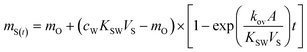 | (1) |
where ms(t) [kg] is the mass of substance in the receiving phase, mo [kg] the substance mass in the receiving phase at the start of exposure, cw [kg m−3] the water concentration during the exposure time, Ksw [L kg−1] the receiving phase-water distribution coefficient, Vs [m3] the volume of the receiving phase, kov [m s−1] the overall mass transfer coefficient, A [m2] the membrane surface area, and t [s] the exposure time.
The coefficient of the exponential function can be combined to the constant kex [s−1], which is defined as the overall exchange rate. Its relation to the so-called sampling rate Rs [m3 s−1], which represents formally an equivalent volume of water extracted by the sampler per unit of time is given in eqn (2).
| | 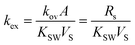 | (2) |
The accumulation of substances can be approximately regarded as linear until the amount of analyte reaches half of its equilibrium level:17
| | 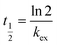 | (3) |
With substances similar to the target analytes such as deuterated or isotope-labelled substances, the overall exchange rate kex can be estimated. By using such performance reference compounds (PRCs), the influence of biofouling, temperature, water flow and other environmental parameters can be approximately taken into account and a field sampling rate Rs-f can be calculated.
The experimental PRC exchange rate can be determined by eqn (4):
| | | mPRC(t) = m0PRC × exp(−kext) | (4) |
where
mPRC is the mass of PRC in the receiving phase [ng] and
m0PRC the amount of PRC added to the receiving phase before exposure (this amount is known).
With the help of the experimental exchange rate, the field sampling rate Rs-f can be adjusted, when the PRC has similar physicochemical properties as the monitored compound:16
| | 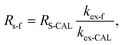 | (5) |
where
kex-CAL is the experimental exchange rate of a PRC, which is determined in the laboratory,
kex-f is the exchange rate of a PRC observed under field conditions and
RS-CAL is the sampling rate determined in the laboratory using
eqn (4).
3. Material and methods
3.1. Physicochemical properties of substances
The WFD include PBDE 47, 100 and total DDT (sum of p,p′-DDT; o,p′-DDT; p,p′-DDD and p,p′-DDE) as priority substances. The European Union defines environmental quality standards in water for these compounds in a recent directive.18 Some important physiochemical properties of all test substances are summarized in Table 1.
Table 1 Physiochemical properties of test substances
3.2 Material and chemicals
The Chemcatcher® body parts were provided by the University of Portsmouth, UK. C18 Empore® disks (47 mm diameter) were purchased from Varian Inc., Walton-on-Thames, UK. LDPE membrane material (40 μm thick) was obtained from Fisher Scientific, Loughborough, UK. Polydimethylsiloxane (PDMS)-Twister™ (10 mm long, coated with PDMS, 0.5 mm film thickness) were provided by Gerstel, Mülheim, Germany. The solvents (HPLC grade quality or higher) dichloromethane, ethylacetate, isopropyl alcohol, trimethylpentane, methanol and n-octanol were purchased from Merck, Darmstadt, Germany. All PCB standards were obtained from Dr. Ehrenstorfer, Augsburg, Germany. PBDE standards were obtained from Chiron AS, Trondheim, Norway. DDT and its metabolites were purchased from Riedel-de Haën®, Seelze, Germany. All labeled standards were obtained from Cambridge Isotope Laboratories (Andover, MA, USA). Salt for the calibration experiment was purchased in the form of table salt (99.8% NaCl, no additives) from the European Salt Company (Hannover, Germany).
3.3 Evaluation of the analyte recovery
To determine the loss of substances during the extraction method, described in 3.4, a recovery experiment was conducted. 400 μL of a standard solution (1 ng μL−1) including each test compound was added to each of six Empore disks. After air drying, 1 mL methanol in acetone (45% v/v) was carefully dropped on each disk, following the extraction method described in section 3.4. Finally the extracts were measured together with external calibration standards using GC-MS.
3.4 Preparation and processing of the sampler
The Chemcatcher® device was used with the reusable body made from polytetrafluoroethylene (PTFE) as described by Kingston et al.13 The non-polar Chemcatcher type imbeds a C18-Empore disk as the receiving phase which is covered by low-density polyethylene (LDPE) as the diffusion-limiting membrane. Before exposure, the receiving phases were conditioned by soaking overnight in methanol and after passing 50 mL of methanol and 150 mL tap-water through the disk. PRCs were loaded on the Empore disk by filtration of an aqueous solution of 250 mL containing 2 μg of PCB 202, PCB 54, D10-Benz(a)antracene, 13C6-HCB and D10-Fluoranthene.28 The diffusion membrane soaked overnight in n-hexane. Before assembly the Empore Disk was applied on the lower part of the Chemcatcher housing and 1 mL of n-octanol in methanol (45% v/v) was added between the receiving phase and the diffusion-limiting membrane, to minimize the initial resistance between this layers for ensuring higher sampling rates.15 After exposure the Chemcatcher® body was rinsed with tap-water and the body carefully disassembled. The C18 disk was taken out of the PTFE-body, dried under vacuum and extracted with 5 mL of acetone in an ultrasonic bath followed by 5 mL of mixture of ethylacetate and isooctane (1/1). The combined extract was passed over a NaSO4 cartridge to remove water traces. Finally the extract was evaporated under nitrogen. Approximately 450 μL of the extract remained as a result of the low volatility of n-octanol. Afterwards the extract was transferred to a 2 mL vial and 50 μl of 10 ng μL−1 internal standard D10-anthracene was added to the sample.
3.5 Extraction of test-substances from water
As described by De la Cal et al.29 there is a problem associated with the extraction of PBDEs and DDX from water using solid phase extraction (SPE) with C18 cartridges. For this reason it was decided to use the Gerstel-PDMS-Twister™ for extracting the analytes from water samples.
The Twister™ provides evidence of accumulation of trace organic substances in aqueous media by employing the concept of stir bar sorptive extraction.30 The Twister absorbs the analytes according to their partition coefficient between aqueous sample and Twister receiving phase. The water concentration during the flow-through exposure experiments was set to 25 ng L−1 by continuous injection of appropriate volume of spiking solution. During the experiments samples of 200 mL of the effluent from the tank were collected daily and were stirred after adding 40 mL methanol at 1200 U min−1 for 16 h with a PDMS-Twister™. Addition of methanol increases the extraction recovery, as a result of the decreasing absorption of the analytes on the glass ware.31 It is known, that the presence of methanol influences the distribution coefficient between the aqueous and extraction phase,32 demanding an external calibration using the same extractions method to graduate the effect of methanol. After extraction the Twister™ was measured with thermodesorption-GC-MS together with an external calibration. The external calibration was accomplished first with Twisters in spiked tap water (+20% methanol) and second in spiked tap water with 2.5% salt content (+20% methanol) over a concentration range of 1.6 ng L−1 to 35 ng L−1. Each water sample was taken and extracted in duplicate and every calibration level in triplicate. The analytical performances of the Twister were evaluated for both matrices (tap water and 25 PSU NaCl). The LOD and LOQ are shown in Table S1.†
3.6 Experimental setup of Chemcatcher calibration
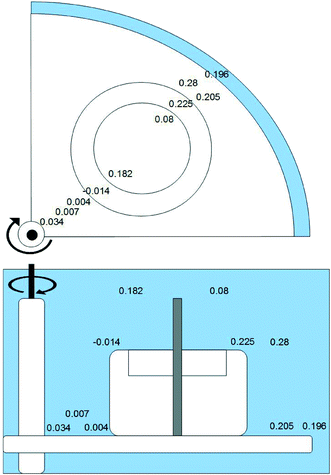
Two 14-day calibration experiments were included in the study. Each experiment employed a constant concentration flow-through exposure system. The experimental setup is shown in Fig. 1. Using a flow-through exposure system the passive samplers could be calibrated under controlled condition of temperature, water flow, salinity and analyte concentration. The experimental setup consisted of a 20 L glass tank with an overflow to waste. The tank contained a Teflon carousel, described by Vrana et al.14 with places for 14 samplers and twist controlled by a RZA 2051 electronic stirrer (Heidolph Instruments GmbH & Co.KG, Schwabach, Germany). The water was stirred at 40 rpm. The water turbulences were measured by a MiniAir20 (Schiltknecht Messtechnik AG, Gossau, Schweiz). To obtain a constant analyte concentration in the exposure system test analytes, dissolved in methanol, were pumped by a micro pump (ProMinent®, Heidelberg, Germany). The fresh water was pumped by ProMinert Gamma 5 with 5 L h−1 in the tank. For generating salt water (2.5%/(25 PSU) NaCl content) two peristaltic pumps were used, one for pumping tap water with 4 L h−1 and the second peristaltic pump for adding with 1 L h−1 high concentrated salt solution (12.5% NaCl). The experiments were done in a chromatographic refrigerator TC 105-1 from Tritec (Hannover/Germany) at a constant temperature of 13 °C (ESI† Fig. S1). The content of the salinity was measured with a density meter (Anton Paar DMA48, Graz, Austria). Before the start of the calibration, the experimental setup was running 48 h without sampler to stabilize the concentration of the test substances and to equilibrate the system. At the beginning of each experiment, 12 Chemcatcher®, prepared and spiked with PRC, were placed in the flow-through system on the carousel. After defined time periods two samplers were removed. Additionally three Chemcatcher controls were analyzed to determine initial contamination from assembly processes and background contamination during sampler handling as well as the initial PRC concentration before exposure.
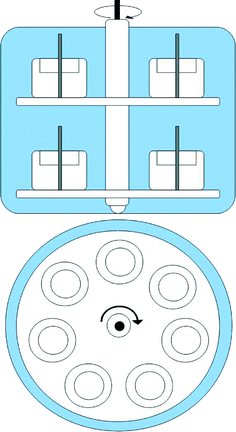 |
| | Fig. 1 Exposure tank and carousel device used in calibration experiments. | |
3.7 Instrumental analysis
The Chemcatcher® samples were measured by using an Agilent 6890N gas chromatograph, Agilent Technologies, Waldbronn, Germany, with an HP-5MS-ultraInert column (15 m × 0, 25 mm × 0, 25 mm) associated with an Agilent 5975C mass spectrometer. 1 μL of sample was injected with pulsed splitless mode. The oven program started at 140 °C for 2 min, heating 2 °C per minute to 170 °C, hold 2 minute, then heating 15 °C per minute to end at 320 °C.
The Twister™ were measured directly after thermodesorption by GC-MS using a Gerstel Twister™ Desorption Unit linked to an Agilent 6890N gas chromatograph with a HP-5MS-ultraInert column (15 m × 0, 25 mm × 0, 25 mm) with a oven program, beginning at 60 °C for 2 minute, heating 10 °C to 310 °C, held for 5 min. The gas chromatograph was coupled with an Agilent 5973 mass spectrometer.
3.8 Data processing
The accurate n-octanol extract volume was calculated from the external calibration with the standard component D10-anthracene and its known internal standard concentration (added to the extract before volume reduction). The mass of the individual test compounds extracted from the Empore disk were calculated using eqn (6) , where csample is the analyzed specific analyte sample concentration, Vsample is the accurate sample volume and fe specific analyte recovery (determined in 4.1). The accumulated masses of test substances on the receiving phases were plotted by linear regression model using eqn (7):33| |  | (6) |
with cw as average of the water concentration in the exposure system determined by Twister™ measurements. The standard derivation of the sampling rate was calculated by the law of error propagation (eqn (8)), where b is the value of the slope of the linear regression curve, sb is the standard derivation of the slope and scw the standard derivation of the tank water concentration of the analytes.| | 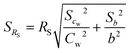 | (8) |
4. Results and discussion
4.1 Analyte recovery
A recovery experiment was conducted to determine the loss of selected compounds during the extraction method. For this propose the Empore disk was spiked with 400 μL of a standard solution (1 ng μL−1 of each test substance). Afterwards it was handled as a sample described above. The analyte recovery of spiked C18-Empore disk (see 3.5) was between 34.55% for p,p′-DDT to 137% for PBDE 100. As a result of the low recovery of p,p′-DDT, this analyte was removed from all following considerations. The summary of all measured analyte recovery is shown in Table 2. The value of extraction recovery was used later to calculate the correct mass of the test substances on the Empore Disk in the calibration experiments using eqn (6).
Table 2 Analyte recovery of spiked C18Empore disk with standard deviation (n = 6)
| Substance |
Recovery in % |
| PBDE 47 |
121.64 ± 17.76 |
| PBDE 100 |
136.59 ± 18.79 |
| PCB 28 |
60.76 ± 9.90 |
| PCB 52 |
66.03 ± 10.44 |
| PCB 101 |
72.76 ± 10.62 |
| PCB 153 |
67.18 ± 10.62 |
|
p,p′-DDT |
34.55 ± 4.34 |
|
o,p′-DDE |
52.21 ± 9.63 |
|
p,p′-DDE |
50.99 ± 8.66 |
|
o,p′-DDD |
69.12 ± 11.62 |
4.2 Concentration of test substances in water during the experiments
The analyte concentration resided in the range from 9.73 ng L−1 to 18.38 ng L−1 during the calibration experiment with tap water and between 13.14 ng L−1 and 18.38 ng L−1 for the salt water experiment. The respective coefficient of variation was between 1.90 ng L−1 and 5.00 ng L−1 and between 1.13 and 5.65, respectively. The highest variation was reported for PBDE 47 and PBDE 100 in the tap water calibration experiment and for PBDE 100 and PCB 153 in the 2.5% NaCl content calibration experiment. Both experiments indicated less analyte water concentration than expected. The lower analyte water concentration is likely the result of reduced pumped volume of the chemical pump or increased pumped volume of the water pump. Overall the analyte water concentration was less for all test substances than expected, but quite stable on this level during the individual calibration experiments.
4.3 Measurement of flow velocity
In previous papers,14,33 the linear velocity in the calibration tank system was calculated with eqn (9), where r is the radius of the circular path of the passive sampler on the carousel and f the rotation speed of the carousel
By using eqn (9) a linear flow velocity of 0.4 m s−1 for the passive samplers inside the flow-through system was estimated. But it could be seen that the water moved partly with the carousel in a round exposure tank system and as a consequence the velocity was expected to differ from the calculated value. Hence the flow velocity was measured with a MiniAir20 system directly. The MiniAir20 is a vane anemometer and can measure flow velocity of gaseous and liquid media. For a precise determination of the flow velocity, the sensor was held diagonally to the direction of rotation. This indicated that the velocity of the Chemcatcher was lower than the linear velocity measured with the sensor, as a result of the round tank design. The effective velocity was calculated by subtracting the calculated value (eqn (9)) from the measured velocity. The effective velocity results are shown in Fig. 2. The average of the effective velocity was 0.11 m s−1 and thus much lower than the velocity estimated (0.4 m s−1).
 |
| | Fig. 2 Effective velocity [m s−1] measured with MiniAir20 in flow-through exposure tank system including 14 Chemcatcher. | |
4.4 Uptake kinetics
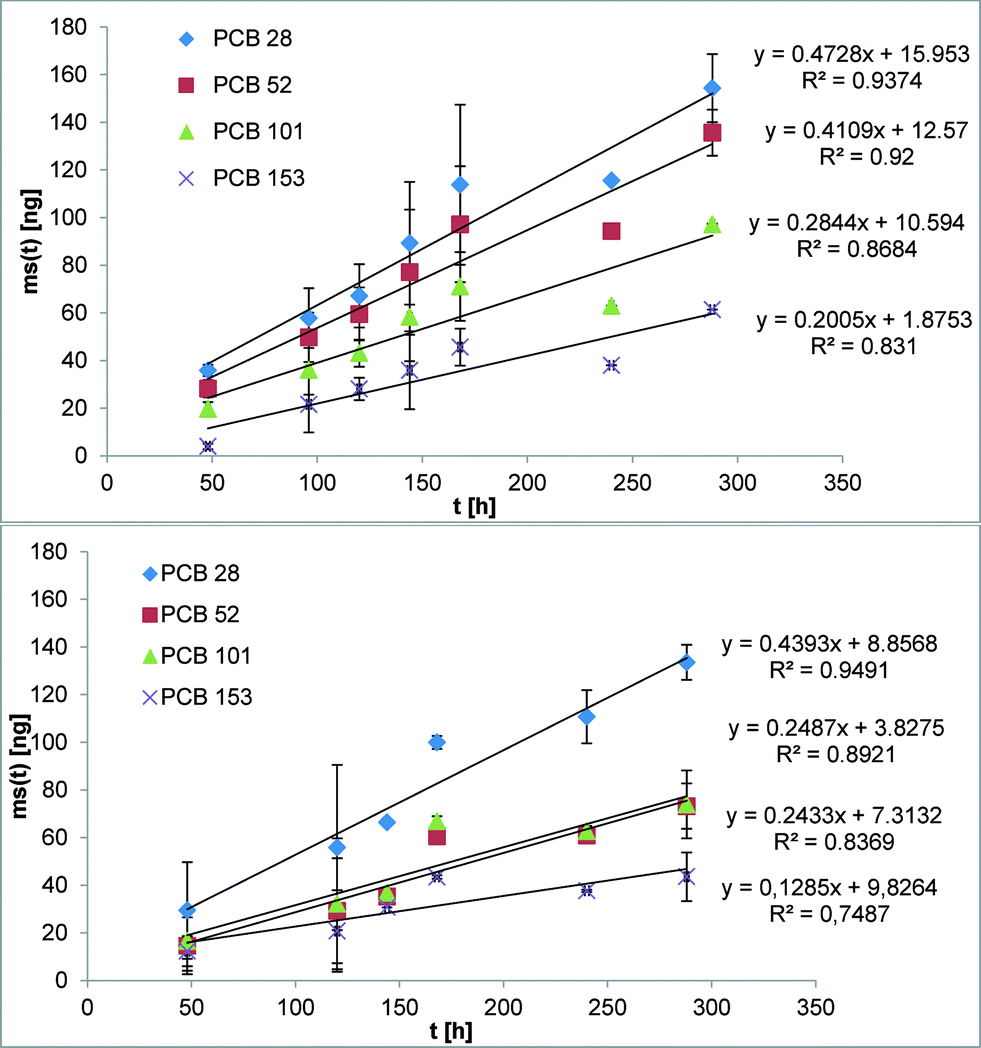
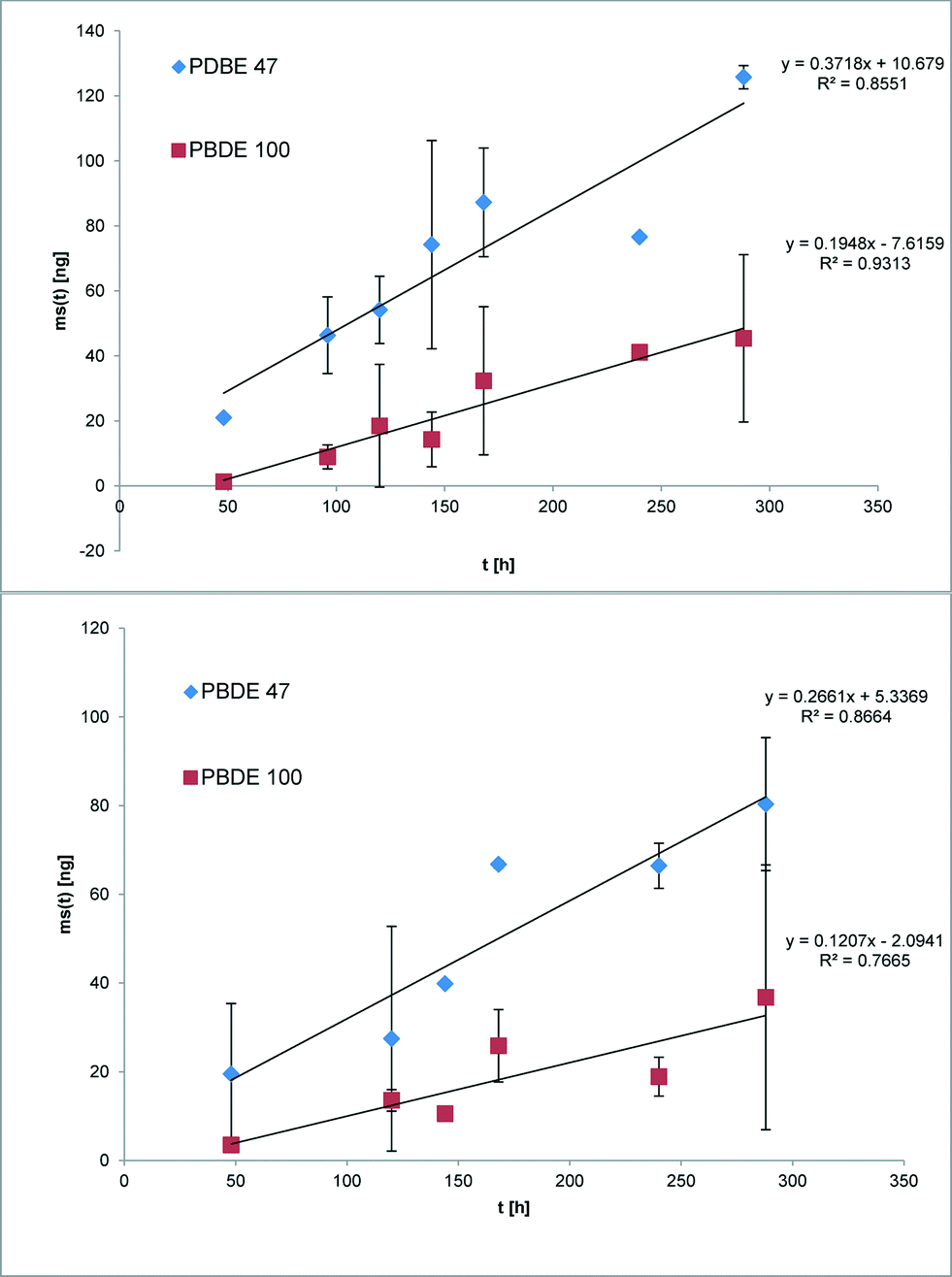
The suitability of the nonpolar version of the Chemcatcher® was tested in calibration experiments in fresh- and salt water. Both experiments were carried out at constant temperature at 13 °C and a mean linear flow velocity of 0.1 m s−1 (as described above). The salinity of the water in the second exposure experiment ranged from 21.29 to 28.43 PSU NaCl. The initial start concentration of the test substances in the three Chemcatcher control samples were below limit of quantification. The uptake curves of test substances are presented in Fig. 3 (PCBs), Fig. 4 (PBDEs) and Fig. 5 (DDX).
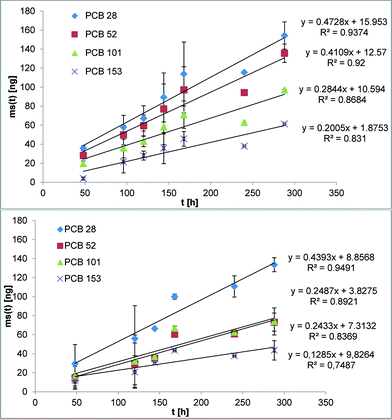 |
| | Fig. 3 Uptake curve of selected polychlorinated biphenyls in fresh (up) and seawater (down) by effective velocity of 0.1 m s−1 and 13 °C. | |
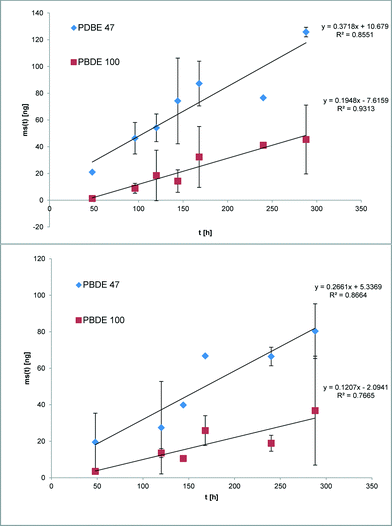 |
| | Fig. 4 Uptake curve of selected polybrominated diphenylether in fresh (up) and seawater (down) by effective velocity of 0.1 m s−1 and 13 °C. | |
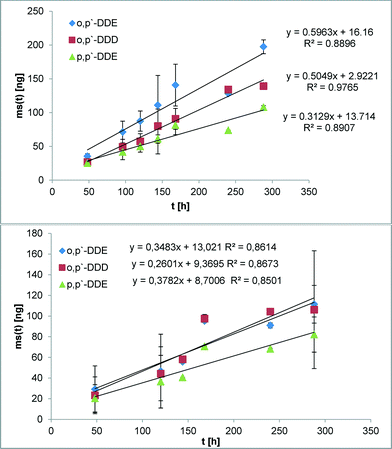 |
| | Fig. 5 Uptake curve of organochlorine pesticides in fresh (up) and seawater (down) by effective velocity of 0.1 m s−1 and 13 °C. | |
The sampling rate Rs was calculated with eqn (7) and the standard derivation of the sampling rate with eqn (8), summarized in Table 3:
Table 3 Uptake rates Rs [L d−1] estimated in this study, cw measured during the experiments and value of slopes b and p-values of the linear regression curves of the test substances (the standard deviations are given in the respective ± columns)
| Substance |
Molecular weight g mol−1 |
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kow Kow |
R
s [L d−1] |
c
w [ng L−1] |
b [ng h−1] |
p-value |
| Tap water 14 days calibration experiment |
| PCB 28 |
257.54 |
5.87 |
0.89 ± 0.18 |
12.94 ± 2.09 |
0.48 ± 0.06 |
3.40 × 10−04 |
| PCB 52 |
291.99 |
5.97 |
0.76 ± 0.14 |
13.32 ± 1.90 |
0.42 ± 0.05 |
6.34 × 10−04 |
|
o,p′-DDE |
318.03 |
5.43 |
0.81 ± 0.18 |
18.38 ± 3.40 |
0.62 ± 0.08 |
1.43 × 10−03 |
|
p,p′-DDE |
318.03 |
5.69 |
0.57 ± 0.15 |
12.70 ± 2.59 |
0.30 ± 0.05 |
1.40 × 10−03 |
|
o,p′-DDD |
320.04 |
6.42 |
0.65 ± 0.15 |
18.38 ± 3.40 |
0.50 ± 0.07 |
2.88 × 10−05 |
| PCB 101 |
326.43 |
6.38 |
0.89 ± 0.21 |
13.21 ± 2.62 |
0.29 ± 0.04 |
2.24 × 10−03 |
| PCB 153 |
360.88 |
6.58 |
0.76 ± 0.41 |
9.84 ± 5.00 |
0.20 ± 0.04 |
4.25 × 10−03 |
| PDBE 47 |
485.79 |
6.81 |
0.58 ± 0.15 |
15.90 ± 3.25 |
0.39 ± 0.06 |
2.87 × 10−03 |
| PBDE 100 |
564.68 |
7.24 |
0.29 ± 0.10 |
15.58 ± 3.97 |
0.19 ± 0.04 |
4.31 × 10−04 |
| 25 PSU water 14 days calibration experiment |
| PCB 28 |
257.54 |
5.87 |
0.78 ± 0.17 |
13.14 ± 1.70 |
0.43 ± 0.07 |
9.88 × 10−04 |
| PCB 52 |
291.99 |
5.97 |
0.41 ± 0.12 |
13.50 ± 1.75 |
0.23 ± 0.06 |
4.53 × 10−03 |
|
o,p′-DDE |
318.03 |
5.43 |
0.57 ± 0.17 |
13.59 ± 1.13 |
0.32 ± 0.09 |
7.57 × 10−03 |
|
p,p′-DDE |
318.03 |
5.69 |
0.40 ± 0.13 |
14.48 ± 2.61 |
0.24 ± 0.07 |
6.92 × 10−03 |
|
o,p′-DDD |
320.04 |
6.42 |
0.44 ± 0.14 |
19.49 ± 2.24 |
0.35 ± 0.11 |
8.89 × 10−03 |
| PCB 101 |
326.43 |
6.38 |
0.39 ± 0.13 |
13.74 ± 2.04 |
0.23 ± 0.07 |
1.06 × 10−02 |
| PCB 153 |
360.88 |
6.58 |
0.19 ± 0.08 |
14.93 ± 3.33 |
0.12 ± 0.04 |
2.60 × 10−02 |
| PDBE 47 |
485.79 |
6.81 |
0.35 ± 0.10 |
17.02 ± 1.58 |
0.25 ± 0.07 |
7.02 × 10−03 |
| PBDE 100 |
564.68 |
7.24 |
0.15 ± 0.06 |
17.39 ± 4.14 |
0.11 ± 0.04 |
2.23 × 10−02 |
The sampling rate Rs varies between 0.29 L d−1 for PBDE 100 and 0.89 L d−1 for PCB 28 and PCB 101 in fresh water. The Rs value in salt water varies between 0.15 L d−1 for PBDE 100 and 0.78 L d−1 for PCB 28.
The comparison of both experiments shows only a small influence of salinity on the sampling rates for the highly hydrophobic organic test substances. Recently, Lohmann et al.34 have estimated a similar trend with LDPE passive samplers by using the Setschenow equation. Booij et al.35 predicted that the sampling rate is expected to decrease with increasing molecular mass. The experimental data in this work (highest sampling rate for the lowest molecular mass and vice versa) are in close agreement with this theoretical model.
The obtained uptake rates for several test substances can be compared with results reported by De la Cal et al.29 In the present work much higher sampling rates for these substances (Table 4) were found. This may result from a different experimental setup used by De la Cal et al.29 where no carousel device was used to ensure similar hydrodynamic conditions for all Chemcatchers. They placed the Chemcatchers at the bottom of the exposure tank and used a stirrer to mix the solution above. Furthermore, problems arose as a result of the extraction of the water from the tank because of the high uncertainty and the low recovery of the test substances, which influence the value of the sampling rate.
Table 4 Comparison of experimental and literature Rs for nonpolar Chemcatcher® for PBDEs and DDT and its metabolites
| Substance |
R
s experimentala [L d−1] |
R
s literatureb [L d−1] |
|
Determined by 13 °C. 0.1 m s−1.
Determined by 11 °C. 20 rpm (no carousel device).29
|
|
o,p′-DDE |
0.81 ± 0.18 |
0.002 |
|
p,p′-DDE |
0.57 ± 0.15 |
0.001 |
|
o,p′-DDD |
0.65 ± 0.15 |
0.003 |
| PBDE 47 |
0.58 ± 0.15 |
0.32 |
Some Chemcatcher sampling rates from our fresh water experiment can be compared with rates reported for the semipermeable membrane device (SPMD) Table 5 shows these data, for comparison normalized to 1 cm2 for both sampler types. This was necessary due to the very different surface areas of SPMD (460 cm2) and Chemcatcher (17 cm2).35 The uptake of highly hydrophobic substances is flow-dependent, i.e. increases with water turbulences. As expected, the uptake rates in this study are higher than Rs values for the SPMD at 0.004 cm s−1 but lower than Rs for SPMD at 90 cm s−1.
Table 5 Surface area normalized sampling rates (Rs [L d−1] per 1 cm2) of test substances determined with the Chemcatcher (present study) and the SPMD (literature data)
| Compound |
R
s [L d−1] per 1 cm2 |
| Chemcatcher (present study) |
SPMD35 |
SPMD36 |
SPMD37 |
|
|
13 °C. 10 cm s−1 |
13 °C. 90 cm s−1 |
12 °C. 0.004 cm s−1 |
10 °C. 0.004 cm s−1 |
| PCB 28 |
0.052 |
|
0.019 |
|
| PCB 52 |
0.045 |
0.254 |
0.014 |
|
| PCB 101 |
0.052 |
|
0.014 |
|
| PCB 153 |
0.045 |
0.212 |
0.008 |
|
|
o.p′-DDE |
0.048 |
|
|
0.005 |
|
p,p′-DDE |
0.034 |
|
|
0.006 |
|
o,p′-DDD |
0.038 |
|
|
0.005 |
4.5 Offload kinetics
The PRC concept presents a possibility to adjust the lab-derived uptake rates for the effects of biofouling, temperature or hydrodynamic conditions during field exposure.38 Five compounds were tested in the present study as possible PRCs for highly hydrophobic organic compounds: PCB 202, PCB 54, D10-Benz(a)antracene, 13C6-HCB and D10-Fluoranthene. 2 μg of each PRC were added to the receiving phase before exposure. The offload of the PRCs were observed during the 14 days experiments. The characteristic offload curves are presented in supplementary data (Fig. S2 and S3†). The offload rates were obtained by nonlinear regression analysis using eqn (4) and were between 0.017 d−1 to 0.027 d−1 in fresh water and 0.034 d−1 to 0.051 d−1 in 25 PSU NaCl water (see Table 6). In the presence of NaCl the elimination constant is higher than in fresh water. However, due to the high variation of the mass of PRC remaining in the Empore disk in fresh water, these elimination rates are relatively uncertain. These higher uncertainties may result from the fact that the PRCs were not loaded homogeneously to the receiving phases of Chemcatchers under investigation. Another explanation was given by Huckins et al.39 who found out that the detecting of the elimination of PRCs with logKow values higher than >5.5 from SPMDs could be problematic unless they were deployed in warm and highly turbulent environment.
Table 6 Elimination constants of studied PRCs in tap water and 2.5% salt water
| PRC |
k
ex fresh water [d−1] |
k
ex salt water [d−1] |
| D10-Fluoranthene |
0.024 |
0.051 |
|
13C6-HCB |
0.022 |
0.042 |
| D10-Benz(a)anthracene |
0.017 |
0.035 |
| PCB 202 |
0.024 |
0.034 |
| PCB 54 |
0.027 |
0.034 |
The PRCs values determined in this laboratory study can be used together with offload rates of PRCs measured during future field campaigns to obtain more realistic uptake rates for the target substances with the Chemcatcher by calculating field sampling rates Rs-f according to eqn (5).
5. Conclusion
This study demonstrates again the suitability of the nonpolar Chemcatcher® device for time-integrated monitoring of highly hydrophobic organic substances in different aqueous environments. Further laboratory studies and especially field campaigns are necessary to investigate more detailed the effect of dissolved salt(s) in the water matrix on both, the uptake of target analytes in the sampler and the elimination of preloaded performance reference compounds from the device. As the refined characterization of the water flow (pattern) along the Chemcatcher surface showed, it is very important to consider the design of the exposure tank used in laboratory studies. The authors used, for example, in subsequent studies a stainless steel tank with a square cross section instead of a round glass cylinder because this modification can prevent the dragging of the water phase by the rotating carousel (at least to a large extent). Also the exposure of inexpensive passive flow monitors (based on the determination of mass loss from a cast of calcium sulfate) alongside the Chemcatcher devices in tank experiments and field trials (as successfully demonstrated by O'Brien et al.40) can help to consider the influence of the hydrodynamic and thus to reduce the uncertainty of sampling rates and of final TWA concentrations. This approach we prefer in our ongoing application of the nonpolar Chemcatcher for time-integrated monitoring of organic micropollutants in German coastal waters and selected rivers.
Acknowledgements
The authors want to thank U. Schroeter (UFZ) for his skillful assistance in the experimental work. Furthermore we are grateful to Dr. B. Vrana (Water Research Institute, Bratislava, Slovakia) for helpful information and to the University of Portsmouth (UK) for the provided Chemcatcher bodies.
References
-
European Commission, Directive 2000/60/EC of the European Parliament and of the Council of 23 October 2000 establishing a framework for Community action in the field of water policy, Off. J. Eur. Communities, L 327, 22/12/2000, pp. 1–77 Search PubMed.
-
Passive Sampling Techniques in Environmental Monitoring, ed. R. Greenwood, G. A. Mills and B. Vrana, vol. 48 of Wilson & Wilson’s Comprehensive Analytical Chemistry (ed. D. Barceló), Elsevier, Amsterdam, The Netherlands, 2007 Search PubMed.
- B. Vrana, I. J. Allan, R. Greenwood, G. A. Mills, E. Dominiak, K. Svensson, J. Knutsson and G. Morrison, TrAC, Trends Anal. Chem., 2005, 24, 845–868 CrossRef CAS PubMed.
- European Communities, Common Implementation Strategy for the Water Framework Directive (2000/60/EC), Guidance Document No. 19 – Guidance on surface water chemical monitoring under the water framework directive, Technical Report 2009-025 (ISBN 978-92-79-11297-3).
- ISO-5667-23, International organization for standardization, 2011.
- J. N. Huckins, G. K. Manuweera, J. D. Petty, D. Mackay and J. A. Lebo, Environ. Sci. Technol., 1993, 27, 2489–2496 CrossRef CAS.
- J. N. Huckins, H. F. Prest, J. D. Petty, J. A. Lebo, M. M. Hodgins, R. C. Clark, D. A. Alvarez, W. R. Gala, A. Steen, R. Gale and C. G. Ingersoll, Environ. Sci. Technol., 2004, 23, 1617–1628 CAS.
- H. Zhang and W. Davison, Anal. Chem., 1995, 67, 3391–3400 CrossRef CAS.
- D. A. Alvarez, J. D. Petty, J. N. Huckins, T. L. Jones-Lepp, D. T. Getting, J. P. Goddard and S. E. Manahan, Environ. Toxicol. Chem., 2004, 23, 1640–1648 CrossRef CAS.
- B. Vrana, P. Popp, A. Paschke and G. Schüürmann, Anal. Chem., 2001, 73, 5191–5200 CrossRef CAS.
-
A. Paschke, B. Vrana, P. Popp, L. Wennrich, H. Paschke and G. Schüürmann, ch. 10 in ref. 2, pp. 231–249.
- K. Booij, F. Smedes and E. M. Van Weerlee, Chemosphere, 2002, 46, 1157–1161 CrossRef CAS.
- J. K. Kingston, R. Greenwood, G. A. Mills, G. M. Morrison and L. B. Persson, J. Environ. Monit., 2000, 2, 487–495 RSC.
- B. Vrana, G. A. Mills, E. Dominiak and R. Greenwood, Environ. Pollut., 2006, 142, 333–343 CrossRef CAS PubMed.
- B. Vrana, G. Mills, R. Greenwood, J. Knutsson, K. Svensson and G. Morrison, J. Environ. Monit., 2005, 7, 612–620 RSC.
-
K. Booij, B. Vrana and J. N. Huckins, ch.7 in ref. 2, pp. 141–169.
-
J. N. Huckins, J. D. Petty and K. Booij, Monitors of organic chemicals in the environment: semipermeable membrane devices, Springer Verlag, New York, 2006 Search PubMed.
-
European Commission, Directive 2008/105/EC of the European Parliament and of the Council of 16 December 2008 on environmental quality standards in the field of water policy, Off. J. Eur. Union, L 348, 24/12/2008, pp. 84–97 Search PubMed.
- A. Paschke and P. Popp, J. Chromatogr. A, 2003, 999, 35–42 CrossRef CAS.
- E. Braekevelt, S. A. Tittlemier and G. T. Tomy, Chemosphere, 2003, 51, 563–567 CrossRef CAS.
- A. Leo, P. Jow, C. Silipo and C. Hansch, J. Med. Chem., 1975, 18, 865–868 CrossRef CAS.
- W. M. Meylan and P. H. Howard, J. Pharm. Sci., 1995, 84, 83–92 CrossRef CAS.
- A. Finizio, M. Vighi and D. Sandroni, Chemosphere, 1997, 34, 131–161 CrossRef CAS.
- W. Dongbin, T. Yongrui, L. Xinhui, W. Liansheng and H. Hong-Ying, J. Liq. Chromatogr. Relat. Technol., 2002, 25, 627–637 CrossRef PubMed.
- C. T. Chiou, V. H. Freed, D. W. Schmedding and R. L. Kohnert, Environ. Sci. Technol., 1977, 11, 475–478 CrossRef CAS.
- S. H. Yalkowsky and R. Pinal, Chemosphere, 1993, 26, 1239–1261 CrossRef CAS.
- S. A. Tittlemier, T. Halldorson, G. A. Stern and G. T. Tomy, Environ. Toxicol. Chem., 2002, 21, 1804–1810 CrossRef CAS.
- University of Portsmouth, The non-polar Chemcatcher sampling device-Handling protocol, 2009 Search PubMed.
- A. de la Cal, M. Kuster, M. L. de Alda, E. Eljarrat and D. Barceló, Talanta, 2008, 76, 327–332 CrossRef CAS PubMed.
- E. Baltussen, P. Sandra, F. David and C. Cramers, J. Microcolumn Sep., 1999, 11, 737–747 CrossRef CAS.
- B. Kolahgar, A. Hoffmann and A. C. Heiden, J. Chromatogr. A, 2002, 963, 225–230 CrossRef CAS.
-
E. Pfannkoch, J. Whitecavage and A. Hoffmann, GERSTEL Appl Notes, 2002, vol. 4 Search PubMed.
- R. Gunold, R. B. Schäfer, A. Paschke, G. Schüürmann and M. Liess, Environ. Pollut., 2008, 155, 52–60 CrossRef CAS PubMed.
- R. Lohmann, K. Booij, F. Smedes and B. Vrana, Environ. Sci. Pollut. Res., 2012, 19, 1885–1895 CrossRef CAS PubMed.
- K. Booij, H. E. Hofmans, C. V. Fischer and E. M. Van Weerlee, Environ. Sci. Technol., 2002, 37, 361–366 CrossRef.
- J. C. Meadows, K. R. Echols, J. N. Huckins, F. A. Borsuk, R. F. Carline and D. E. Tillitt, Environ. Sci. Technol., 1998, 32, 1847–1852 CrossRef CAS.
-
J. P. Petty, J. N. Huckins, H. F. Prest, R. C. Clark, D. A. Alvarez, C. E. Oranzio, J. A. Lebo, W. L. Cranor and B. T. Johnson, A guide for the use of semipermeable membrane devices (SPMDs) as sampler of waterborne hydrophobic organic contaminants, Publication No. 4690, American Petroleum Institute, Washington DC, 2000 Search PubMed.
- T. Lobpreis, B. Vrana, E. Dominiak, K. Dercová, G. A. Mills and R. Greenwood, Environ. Pollut., 2008, 153, 706–710 CrossRef CAS PubMed.
- J. N. Huckins, J. D. Petty, J. A. Lebo, F. V. Almeida, K. Booij, D. A. Alvarez, W. L. Cranor, R. C. Clark and B. B. Mogensen, Environ. Sci. Technol., 2001, 36, 85–91 CrossRef.
- D. O'Brien, M. Bartkow and J. F. Mueller, Chemosphere, 2011, 83, 1290–1295 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ew00043a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.