
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modelling the interaction processes between nanoparticles and biomacromolecules of variable hydrophobicity: Monte Carlo simulations†
Fabrice
Carnal
,
Arnaud
Clavier
and
Serge
Stoll
*
Environmental Physical Chemistry, F.-A. Forel Institute, University of Geneva, 10 Route de Suisse, 1290 Versoix, Switzerland. E-mail: serge.stoll@unige.ch
First published on 24th June 2015
Abstract
The conformational properties and formation of a complex between a weak flexible biomacromolecule chain of variable hydrophobicity and one negatively charged nanoparticle in the presence of explicit counterions are investigated here using Monte Carlo simulations. The influence of the charge distribution and hydrophobicity, monomer distribution of the chain as well as the pH of the solution are systematically investigated. It is shown that the isolated chain conformations, built with random and block distribution of carboxylic, amino and hydrophobic groups, are the result of the subtle competition between intrachain attractive and repulsive electrostatic interactions as well as intrachain attractive short-range interactions due to hydrophobic properties. Extended conformations are found at low and high pH and folded conformations at physiological pH when hydrophilic and block polymer chains are considered. On the other hand, hydrophobic chain conformations do not show pH dependency and remain folded. The intrachain attractive electrostatic interactions clearly promote the deprotonation of carboxylic groups at low pH and the protonation of amino groups at high pH with higher efficiency for hydrophilic chains. The additional set of electrostatic interactions due to the presence of one negatively charged nanoparticle limits the deprotonation of carboxylic groups at low pH. Moreover, the attractive interactions between the biomacromolecule and the nanoparticle allow to observe the formation of a complex considering intermediate and hydrophilic chains even close to the chain isoelectric point due to the charge inhomogeneity distribution. Hydrophobic chain segments are not affected by the presence of the nanoparticle and remain desorbed. In all cases, the presence of one nanoparticle influences the biomacromolecule structures and acid/base properties, leading to more stretched conformations.
Nano impactNowadays, research is still needed to develop conceptual models to understand the physico-chemical parameters governing the fate, interactions and transformations of nanoparticles in environmental systems and living organisms, and to optimize predictive and mechanistic appreciation in the generated nanoparticle activity. In such context, Monte Carlo simulations are used here to elucidate in detail the possible transformation and properties of nanoparticles and biomacromolecules (denaturation) in changing environments. This study constitutes a novel and original approach to predict key parameters influencing the nanoparticle reactivity and their possible interactions with biological and environmental molecules of variable hydrophobicity. |
1. Introduction
Nanoparticles are widely involved in our daily life and industrial processes today.1–4 They can achieve at the nanoscale level very useful and reactive structures such as spheres, tubes, platelets, needle-like structures, etc. Due to their large specific surface area, nanoparticle reactivity is high which allows specific physico-chemical properties such as adsorption, catalysis, changes in mechanical and optical properties of materials, etc. to be achieved. Their surrounding environment in natural or biological systems is also of main importance since nanoparticles have the tendency to form aggregates of larger dimensions.5 Nanoparticles with soft cores, such as dendrimers, are known to successfully encapsulate ligands and metal ions6,7 and multivalent salt concentration is known to play a key role since collapsed dendrimer conformations as well as effective charge inversion can be observed, opening the way to new applications in the area of gene delivery.8,9Research in predicting nanoparticle fate, transport and transformations in environmental aquatic and biological systems, reactivity and its link to toxicity remains challenging due to the wide variety of parameters, such as temperature, the presence of environmentally relevant macromolecules (protein, polysaccharides, natural organic matter, etc.), ionic concentration or pH, and control of the toxicity and transport towards living organisms.10–14 Nanoparticles can induce harmful biological behavior through a variety of mechanisms which are not fully understood or quantified. The binding process with proteins, such as human serum albumin (HSA), is a key parameter since conformational changes are expected to be observed, hence modifying protein activities.15 The binding efficiency is dependent on the nanomaterial. For example, it was found that HSA proteins are destabilized with silver nanoparticles, but remain stable with gold nanoparticles which can be used to stop HSA unfolding by ultraviolet radiation.16,17 Folding and unfolding of proteins is then crucial for the regulation of biological activity and targeting of proteins to different cellular locations.18 It has to be noted that many natural polyelectrolytes, such as proteins or polysaccharides, are considered as polyampholytes (PAs) which have rich conformational and adsorption behavior due to acidic, basic and hydrophobic groups present at the same time on their backbone.
The adsorption dynamics and conformational changes between proteins and nanoparticles are reported to be mainly driven by local electrostatic interactions and pH of solution.19–21 Moreover, stronger affinity is observed with higher protein charge anisotropy.22 However, van der Waals, hydrophobic, hydrophilic and structural interactions, as well as surface curvature, may also play a role in the adsorption (coating) process on nanoparticles, or more generally nanomaterials.23,24 Indeed, Fowler et al.25 showed that the hydrophobicity variation modifies the ionization behavior of short polypeptides, hence influencing their reactivity. The specific sequence of amino acids in peptides or proteins allows the selective binding of targets such as semiconductor or metal surfaces, opening the way to new biomedical applications.26,27 Nanoparticles based on or coated with polyampholytes are also of main importance in the complex formation with proteins whose isoelectric point is high, or in protein delivery acting as nanocarriers.28,29 On the other hand, polyampholyte-coated magnetite nanoparticles have shown promising properties for magnetic resonance angiography (MRA) by eliminating strong interactions with proteins.30
The fantastic field of protein/polysaccharide/polyampholyte conformation elucidation as well as complexation with nanoparticles and surfaces in aquatic and biological systems day after day contributes to the intense development of theories and analytical studies. The subtle behavior of such systems is fully dependent on the structural properties of biomacromolecules such as charge, contour length, monomer distribution or degree of ionization. For example, chains built from alternately charged segments exhibit the lowest selectivity in ion adsorption as described by Borówko et al.31 through density functional theory in the case of surfaces modified by tethered polyampholytes. The stabilization and association behavior can also be regulated by adjusting the solution properties. Electrostatic interactions are modified with salts, allowing the prediction of small electrostatic contribution under physiological conditions in the case of DNA cyclization.32 Within the functional surface area, salts can switch on or off the response of polyampholyte brushes to pH variation as calculated with the self-consistent field theory.33 Interesting trends related to the formation of complexes between chains and nanoparticles, or more generally surfaces, have been investigated. Electrostatic forces can be in competition with depletion interactions arising from chain exclusion from the particle interior.34 Moreover, the surface curvature and chain persistence length also influence the adsorption process as studied by Kampmann et al. for semi-flexible polymers.35 Scaling relationships were found between the boundary layer thickness, surface charge density of the particle and ionization degree of the chain monomers via the self-consistent field theory.36 All these theoretical studies are of main importance to explain, for instance, the length of wrapped DNA around histones in terms of the binding energy to histone cores and elastic energy penalty of DNA wrapping.37
Adsorption processes on nanoparticles or surfaces involving DNA-like chains are commonly studied by computer simulations. Recently, systematic analysis of the complex formation between linear chains and soft nanoparticles by Langevin molecular simulations has confirmed the role of electrostatics.38 Moreover, the abrupt transitions between adsorbed and desorbed states, occurring when the interactions between chains and particles are not strong enough to overcome the chain entropic penalty, can be associated with a first-order-like transition between both states.39 The presence of charged spheres improves chain condensation onto surfaces, but the chain ionization degree seems to control the structural properties despite the presence of restraint area represented by spherical surfaces.40 Proteins can also act as amphiphilic chameleons not only due to electrostatic and hydrophobic interactions, but also due to conformational arrangements and charge regulation controlling the adsorption processes on surface or membrane crossing.41–43 The self-organization of proteins adsorbing on surfaces is shown to cause density inhomogeneities in the surface distribution of proteins. Moreover, proteins can be tracked laterally within a certain distance due to the influence of pre-adsorbed proteins in order to reach the nearest available binding site.44 The nanoparticle shape is also a determinant in the interaction with DNA. Indeed, convex surfaces with high charge densities may stick to DNA irreversibly. On the other hand, concave nanoparticles are not so detrimental since they are prone to short-range repulsion.45 The charge distribution at the nanoparticle surface can also play a key role, hence influencing the global charge behavior, and thus the interactions with other charged molecules.46 Conformational phase diagrams, provided by Guo et al.47 for the adsorption of different mixtures of proteins on small nanoparticles by molecular simulations, confirm the structural change of proteins by enhancing the β-sheets attributed to their geometry and flexibility. Similarly, random coils connecting the α-helices in human serum albumin were found to be strongly affected by the presence of carbon nanotubes, altering their tertiary structure, and thus their activity.15 Hydrophobic interactions are known to play a key role in the formation of polyampholyte structures. Indeed, a discontinuous transition between extended and collapsed conformations varying the pH of solution can be achieved due to the first-order-like nature of the transition.48 When surfaces have hydrophobic behavior, protein adsorption is greatly affected leading to the formation of a denser adsorbed protein layer due to higher affinity between chain amino acids and surface interaction sites.49 Solution properties may also influence DNA complexation with other biomacromolecules such as proteins. Dissolved ions in solution are able to condense on the DNA backbone within the hydrated state, allowing the protein to slide along the DNA as shown by Dahirel et al.50 using Monte Carlo simulations and analytical calculations. Hydrogen bonding can also play a role in some situations due to different affinities for dissolved ions as reported in the case of collagen proteins interacting more strongly with calcium than phosphate ions.51 The solution environment can modify the affinity properties by inducing strong conformational changes and charge inversion.52
The rich DNA-like chain behavior area has been investigated in a systematic way by means of Monte Carlo simulations.53,54 It was found that the acid/base properties of such polyampholytes are dependent on the chain charge distribution with higher charge accumulation at the extremities. Moreover, flexible backbones allowed the observation of dense conformations with optimized ion pairing. The complex formation between weak polyampholytes and charged nanoparticles has shown a subtle interplay between attractive and repulsive electrostatic interactions (inter- and intra-molecular). The primary structure of polyampholytes was also found to play an important role in the adsorption process. In these previous studies, the sequence of blocks was alternated with various lengths, while counterions and hydrophobic interactions were not taken into account.
We propose here the extension of these models and development of a computational framework to investigate the effect of biomacromolecule hydrophobicity and charge distribution in the adsorption/desorption, conformational change (linked to protein denaturation) and interaction processes of simple protein-like structures with nanoparticles. This enables a novel and much needed approach to predict nanoparticle reactivity with biomacromolecules of variable hydrophobicity and of environmental and biological interest. Charged monomers related to amino and carboxylic groups, as well as hydrophobic monomers, are randomly distributed but block distributions are also considered for comparison. In addition, the effect of pH is also systematically investigated to get an insight into the conformational changes (and denaturation) of hydrophobic and hydrophilic chains as well as intermediate situations, and isolate the important parameters controlling the complexation processes between nanoparticles and ‘natural’ macromolecules. This has been achieved here by generalizing a model which we have so far developed to make it applicable to other biomacromolecules and to incorporate the role of hydrophobic interactions within the corona that surrounds nanoparticles in biological and environmental contexts.
2. Model
The system is described by an off-lattice three-dimensional coarse grained model. The simulation box is cubic and periodic (minimum image convention) with a size of a few hundred angstroms per side (600–800 Å). The temperature is fixed at 298 K, and the solvent is treated implicitly as a dielectric medium with a relative dielectric permittivity constant εr = 78.54 taken as that of water. Monte Carlo (MC) simulations are carried out in the grand canonical ensemble according to the Metropolis algorithm to find the conformations of lower energy.55Explicit objects evolving within the simulation boxes are one weak polyampholyte chain (PA) surrounded by its counterions, and one fixed nanoparticle (NP) with its counterions (when a NP is considered). All counterions (PAs and NPs) have fixed radii of 2 Å with charges situated at their center (+1 and −1 for PA counterions, +1 for NP counterions). The NPs have a radius RNP set to 100 Å with −313 centered elementary charges leading to a homogeneous surface charge density σ = −39.9 mC m−2, which is within the range of weakly charged particles at physiological pH. Weak polyampholyte chains are represented as a sequence of 100 freely jointed monomers of radius Rm = 2 Å. Three different types of monomers are considered: monomers A (monoA), B (monoB) and H (monoH). Monomers H stay uncharged during the whole MC steps and are hydrophobic. On the other hand, the charge of monomers A and B is pH-dependent, and can be 0 or −1 for monoA, and 0 or +1 for monoB. Monomers A and B simulate the mean acid/base properties of carboxylic and amino groups in amino acids, respectively. Their pKa values correspond then to pKAa = 2.17 for monoA and pKBa = 9.53 for monoB. The titration curve of the isolated monomers corresponds to an ideal system. The dissociation constant of monomers A introduced via the Henderson–Hasselbalch equation is defined as follows:
 | (1) |
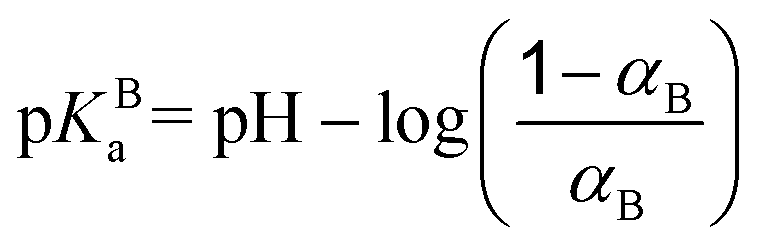 | (2) |
| β = αA,B − αidealA,B | (3) |
In these systems, all objects i and j interact with each other via a full Coulomb electrostatic and excluded volume potential which is positive or negative when repulsive or attractive interactions are considered. This potential is defined as:
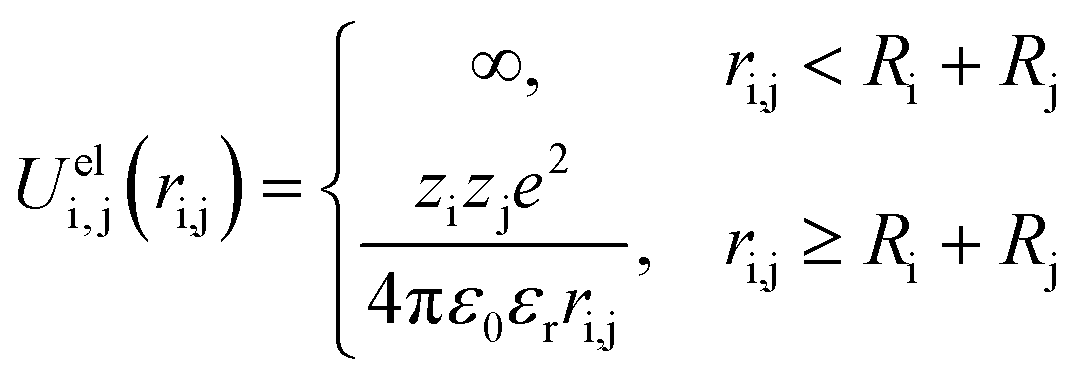 | (4) |
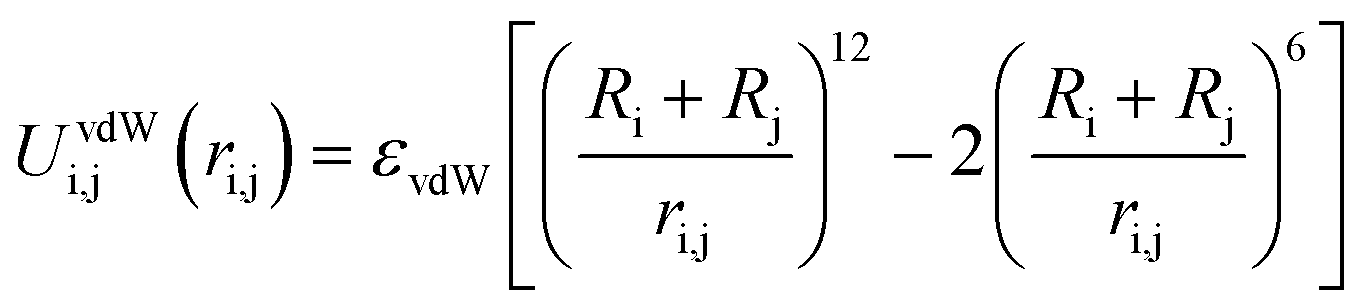 | (5) |
To reach conformations of low energy, PA and NP counterions move through the box by translational movements. The PA conformations are modified by well-known specific movements such as kink-jump, end-bond, reptation, partially clothed pivot and chain translation.57–59 After each conformation modification, ΔEtot = Efinaltot − Einitialtot is accepted or rejected according to the Metropolis algorithm.55 Since the monomer charge is pH-dependent, N/4 monomers are chosen randomly after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MC steps to achieve chain relaxation, and their charge states are modified if monoA or monoB is considered. Thus, the monomers are switched on or off depending on whether the monomers are neutral or charged. Consequently, one oppositely charged counterion is randomly inserted into the system if one charge appears on the PA backbone to keep the system electrostatically neutral. Similarly, a counterion is removed if a charged monomer becomes neutral. The acceptance of each protonation/deprotonation step of the PA monomers is related to the MC Metropolis selection criterion:60,61
000 MC steps to achieve chain relaxation, and their charge states are modified if monoA or monoB is considered. Thus, the monomers are switched on or off depending on whether the monomers are neutral or charged. Consequently, one oppositely charged counterion is randomly inserted into the system if one charge appears on the PA backbone to keep the system electrostatically neutral. Similarly, a counterion is removed if a charged monomer becomes neutral. The acceptance of each protonation/deprotonation step of the PA monomers is related to the MC Metropolis selection criterion:60,61
| ΔE = ΔEtot ± χkBTln10(pH − pKia) | (6) |
For random polyampholytes, the sequences of monoA, monoB and monoH are determined randomly at the beginning of the simulations and only the percentage of each monomer type is fixed. All observables are average values of five different random conformations. Due to the synthetic capability which nowadays allows the preparation of multiblock-type polymers, we also investigate the behavior of triblock and diblock polymers and a comparison was made with the random conformations.
3. Results and discussion
3.1. Polyampholyte chain behavior as a function of pH and monomer properties
The case of a random isolated weak flexible PA chain (100 monomers) surrounded by positively and negatively charged monovalent counterions is first investigated. Three monomer distributions from highly hydrophobic to highly hydrophilic chains, randomly determined at the beginning of the simulations, are taken into account: (1) 10–10–80% (monoA–monoB–monoH), (2) 30–30–40%, and (3) 50–50–0%. Carboxylic and amino groups are represented by monoA and monoB, respectively, and hydrophobic monomers with a neutral charge are represented by monoH. The influence of pH is systematically investigated. Equilibrated conformations for the three model structures and various pH values are presented in Table 1. The chains are positively charged at low pH due to protonation of amino (positive) and carboxylic groups (neutral). Similarly, the amino and carboxylic groups remain neutral and negatively charged at high pH leading to a global negative PA charge. At intermediate pH, both groups are charged at the same time resulting in globally neutral and more folded chains. During the titration process, several competitive electrostatic interactions occur, such as attractive carboxylic–amino, or repulsive carboxylic–carboxylic and amino–amino interactions, hence modifying the chain global charge and conformation. These effects, in addition with hydrophobic interactions, are of main importance as shown in Table 1. Considering the hydrophobic backbones (80% monoH), PA conformations are not pH-dependent even if the global charge varies from positive at low pH to negative at high pH. With the decrease of chain hydrophobicity (40% and 0% monoH), the electrostatic interactions become more important and chain conformations are fully pH-dependent. Thus, stretched PA chains are observed when only one type of monomer is charged (extreme pH values), and more folded structures are observed at intermediate pH values due to the presence of charged carboxylic and amino groups at the same time. For a given pH value, PA rigidity increases with the decrease of hydrophobicity and then the increase of repulsive monomer–monomer electrostatic interactions (80% to 0% monoH).

|
The triblock and diblock PA conformations as a function of pH are presented in Table 2. Two chain structures are investigated: (1) triblocks with 30% monoA (carboxylic group)–30% monoB (amino group)–40% monoH (hydrophobic and neutral), and (2) diblocks with 50% monoA and 50% monoB. It is shown that the primary structure has an important effect on the equilibrated PA conformations. In particular, when the diblock conformation is considered, the presence of both negative and positive charges promotes the formation of more compact conformation at intermediate pH (via ion pairing), whereas at low and high pH, more extended conformations are achieved due to the stretching of the charged block resulting from the electrostatic repulsion between monomers of same signs. These results are also supported by the variations in the radius of gyration as a function of pH which are presented in Fig. 2a.

|
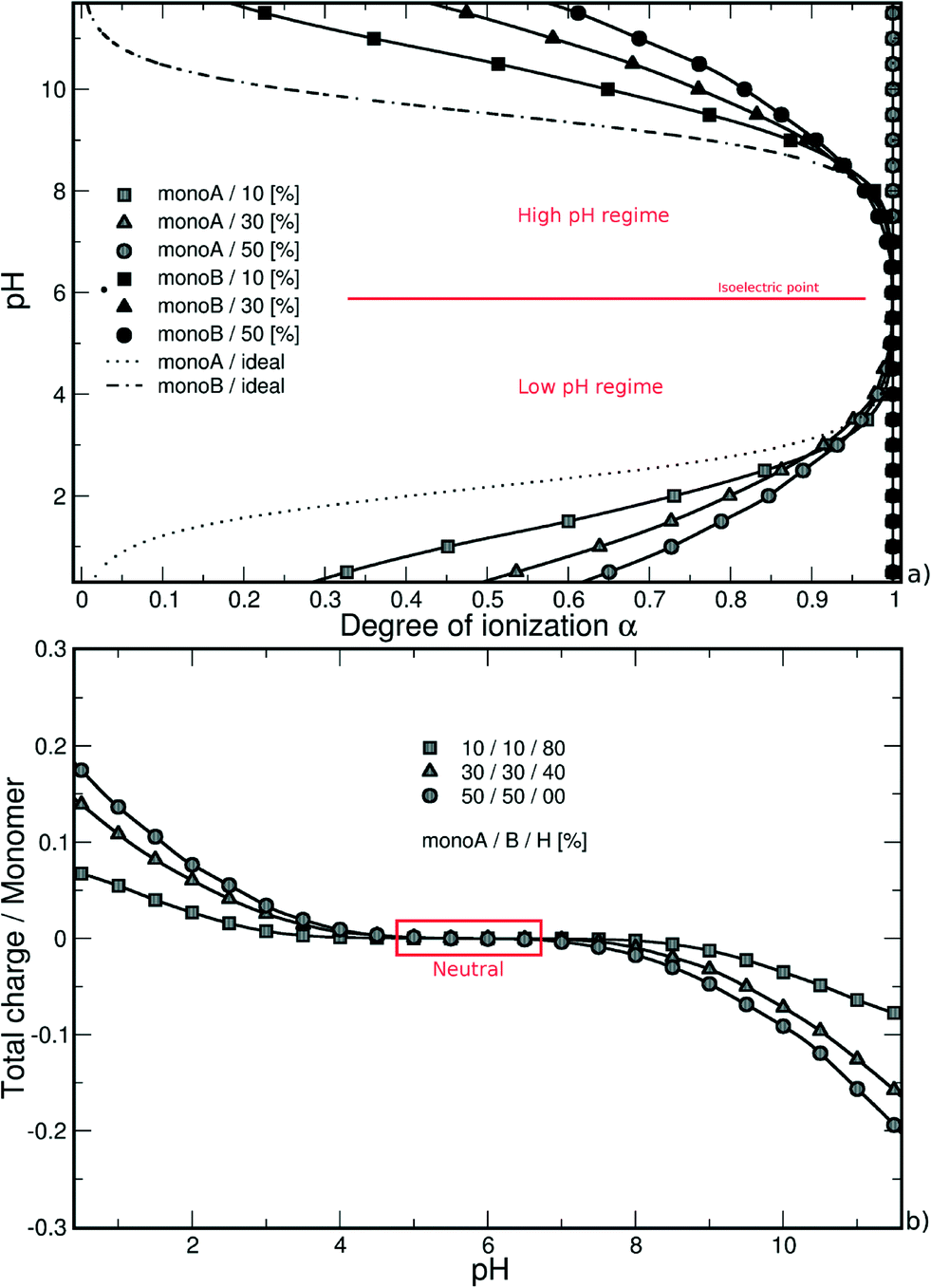 | ||
| Fig. 1 (a) Monte Carlo titration curves calculated as a function of the degree of ionization α, and (b) total charge per monomer as a function of pH of a weak random chain surrounded by monovalent counterions. The deprotonation of monomers A (or B) is promoted by the presence of charged monomers B (or A), and is more efficient in the case of strong attractive electrostatic interactions without hydrophobic monomers (0% monoH). Titration curves exhibit an intermediate pH range where chains are globally neutral with all monomers A and B charged. | ||
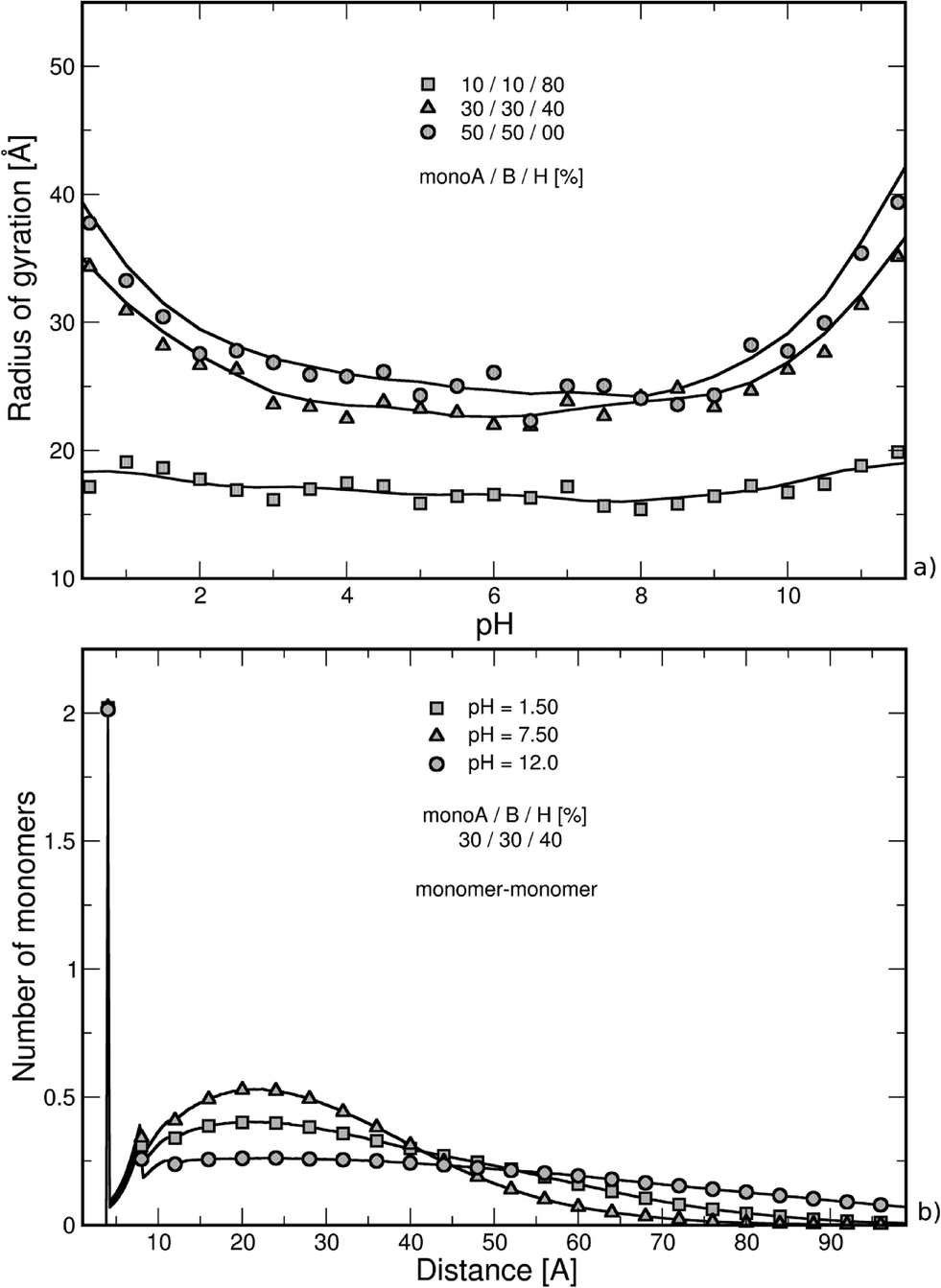 | ||
| Fig. 2 (a) Radius of gyration as a function of pH, and (b) nearest neighbor distance distribution function between chain monomers (center-to-center) in the presence of monovalent counterions (intermediate hydrophobicity: 40% monoH). Chain stretching is fully dependent on electrostatic and hydrophobic interactions. Strong hydrophobic chains (80% monoH) remain folded within the entire pH range, and extended conformations are observed at extreme pH values when monomers A and B are predominant. Nearest neighbor distance distribution function curves cause higher values to be obtained at intermediate pH due to more compact structures (presence of both charged monomers A and B). The first peak corresponds to the distance between consecutive monomers. | ||
Similarly, considering the low pH regime, the deprotonation of carboxylic groups (from 0 to −1) is promoted by positively charged amino groups and the titration curves are situated above the ideal case. The introduction of a charged monomer A becomes more difficult with increasing pH due to stronger repulsive monoA–monoA interactions, and smaller differences between the simulated and ideal curves are observed.
In both pH regimes, the degree of ionization α (monomer A or B) is dependent on the PA model structure. When more titration groups are available, the attractive electrostatic interactions are stronger between monomers A and B, hence favoring the deprotonation process for monoA (low pH regime) and the protonation process for monoB (high pH regime). For a given pH value, the degrees of ionization then decrease with the increase of chain hydrophobicity.
It has to be noted that the titration curves in Fig. 1a show the usual Henderson–Hasselbalch curve shape. In our case, each pKa value of the titration sites (considering monomer A or B) is not strictly identical due to the inhomogeneity in the charge distribution on the chains. However, the difference between them is expected to be limited. As suggested by Onufriev et al.62 who decomposed complex titration curves, the titration sites with very different pKa values would generate titration curves with unusual shapes.
The nearest neighbor distance distribution functions are given in Fig. 2b for the intermediate PA hydrophobicity case (40% monoH) at extreme pH values when the chains are globally neutral. The y-axis represents the mean local number of monomers relative to the other monomers. In all curves, a large correlation distance is found at 4 Å which corresponds to the separation distance of two consecutive monomers taking into account the excluded volume. We can also notice that monomers are more organized up to the second monomer layer (peak at 8 Å). Beyond this distance, values regularly increase and decrease with the distance of separation between monomers. Moreover, the monomer density is higher at pH equal to 7.5, which is in good agreement with more folded structures observed in Fig. 2a (both charged monoA and monoB present at the same time). At extreme pH values, chain conformations are extended with a lower nearest neighbor distance distribution function at short distances.
3.2. Formation of complexes between nanoparticles and polyampholyte chains
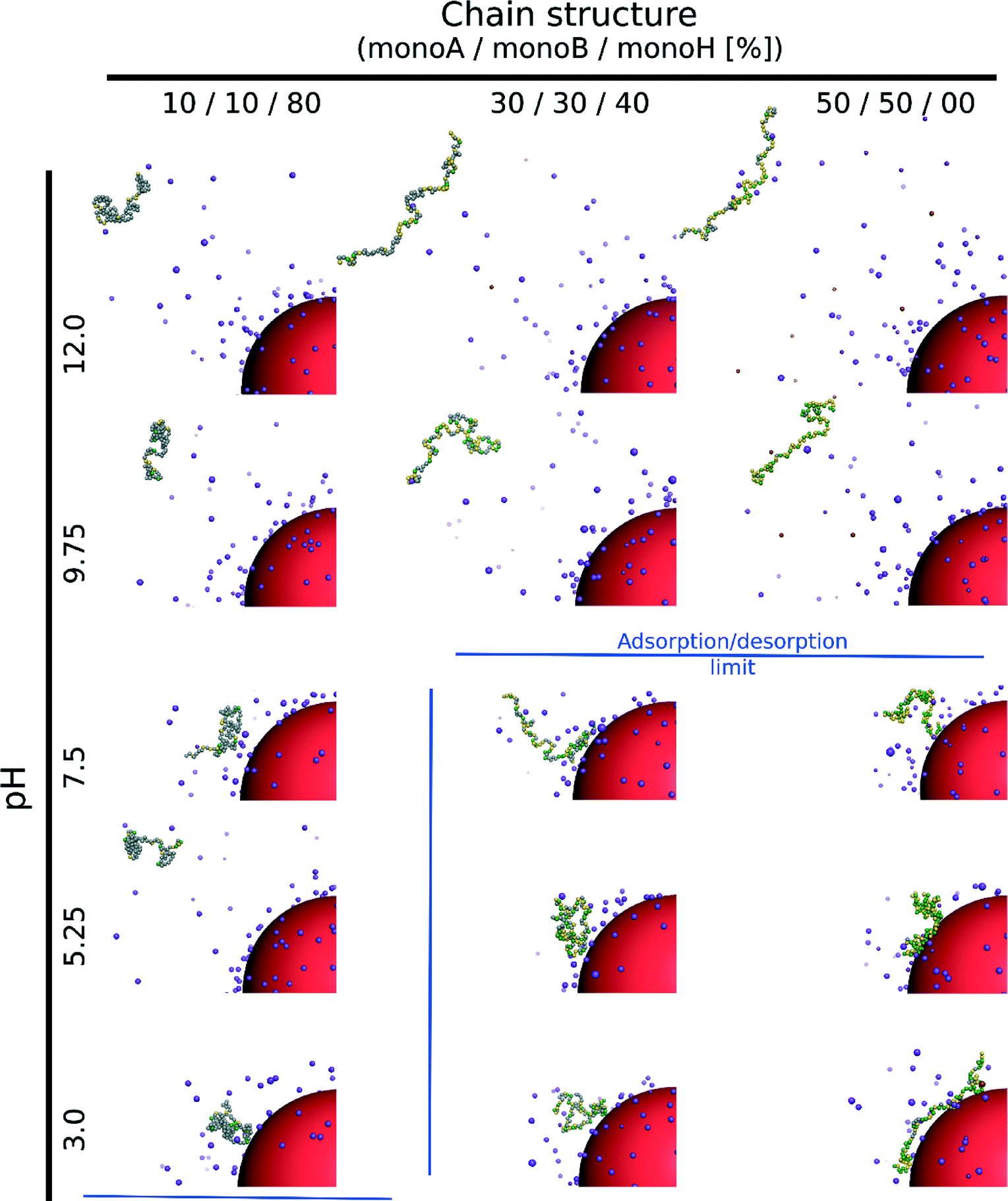
We investigate here the case of one isolated weak flexible PA chain (100 monomers) in the presence of one negatively charged NP (σ = −39.9 mC m−2) surrounded by positively and negatively charged monovalent counterions (PA and NP) for three random PA monomer structures composed of carboxylic groups (monoA), amino groups (monoB) and hydrophobic monomers (monoH) with percentages of 10–10–80% (monoA–monoB–monoH), 30–30–40% and 50–50–0%. Table 3 presents the equilibrated conformations of the PA chain and NP at various pH values.
|
Considering the hydrophobic chains (80% monoH), no complex formation between the PA and NP occurs within the entire pH range although the chain is globally positively charged at low pH and the NP negatively charged. Indeed, electrostatic interactions between the charged monomers are very low and do not allow the observation of neither elongated PA conformations nor adsorption processes at the NP surface. The chain then moves freely within the whole simulation box for all pH values.
The increase of electrostatic interactions with the decrease of chain hydrophobicity (40% and 0% monoH) results in elongated and desorbed chains at high pH due to repulsive interactions between the negatively charged monomers and the nanoparticle. For both cases, an adsorption/desorption limit (blue lines) can be found at intermediate pH when the PAs are globally neutral. Indeed, the chain charge distribution is determined randomly at the beginning of the simulations, and some sections remain locally positively charged, hence favoring the adsorption process at the negatively charged NP surface. It has to be noted that the adsorption/desorption limit is based on the RDFs between the monomers and the NP, and the chain radii of gyration. This criterion gives the monomer distribution around the NP and allows the distinction between a fully adsorbed, a locally adsorbed and a non-adsorbed biomacromolecule to be made. We consider here a chain to be adsorbed if the integrated RDF value is higher than 50% (of the total) at the limit determined by the radius of gyration (from the NP surface). In the adsorption domain, the monomers can be distributed in tails, trains or loops. Attractive hydrophobic interactions between monoH lead to less elongated chain conformations. As a result, chains of intermediate hydrophobicity (40% monoH) cause adsorbed PAs to appear at the NP surface with the majority of monomers situated in tails. In the hydrophilic case, loops and trains are predominant at low pH due to the adsorption of most of the monomers at the NP surface.
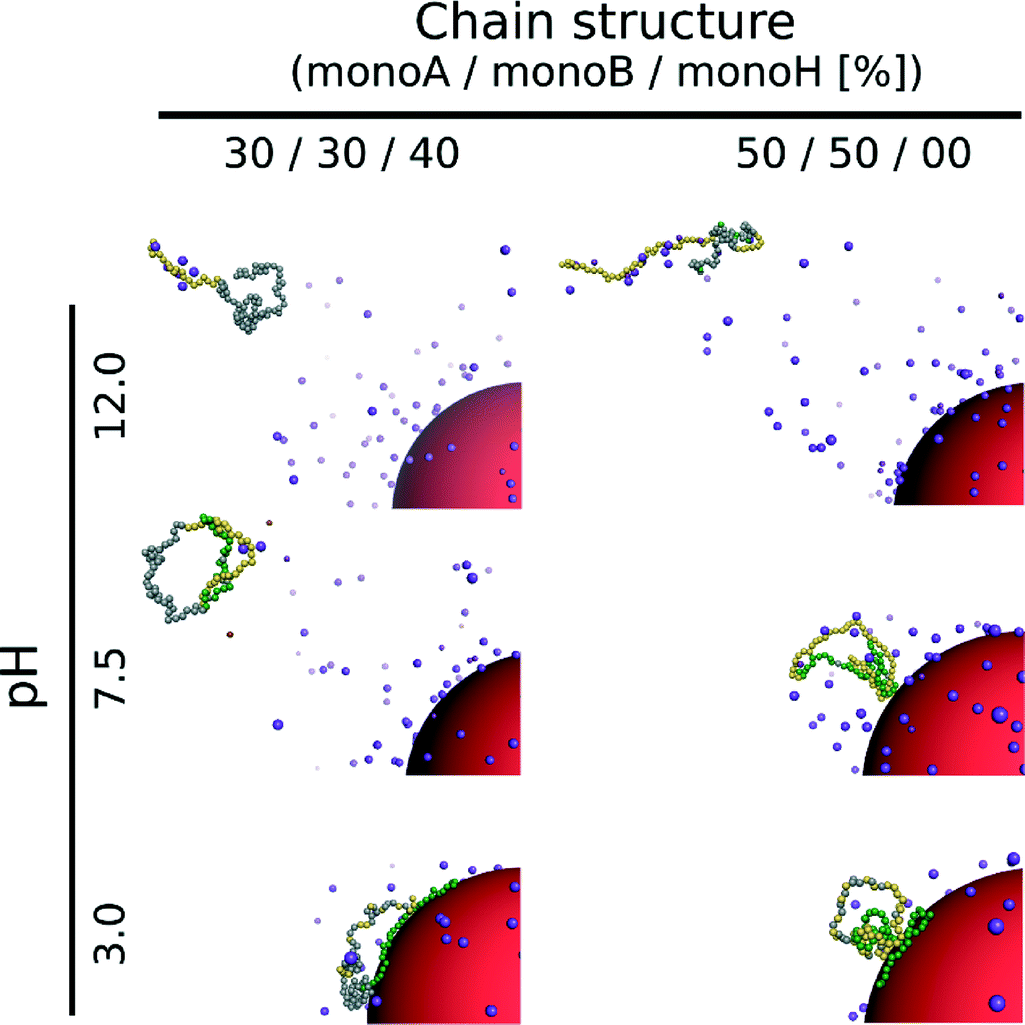
The triblock and diblock PA conformations as a function of pH in the presence of the NP are presented in Table 4. It is shown that the primary structure has an important effect on the adsorbed PA conformations. Indeed, the oppositely charged blocks here are found to be strongly adsorbed at the NP surface at low pH values (strong ion pairing with the NP). At intermediate pH values, due to the competition between the negatively charged NP and the negative monomers, and between the positive and negative monomers, the PA is poorly adsorbed at the NP surface. At high pH, the situation is similar to that of the random PA and no adsorption is observed.
|
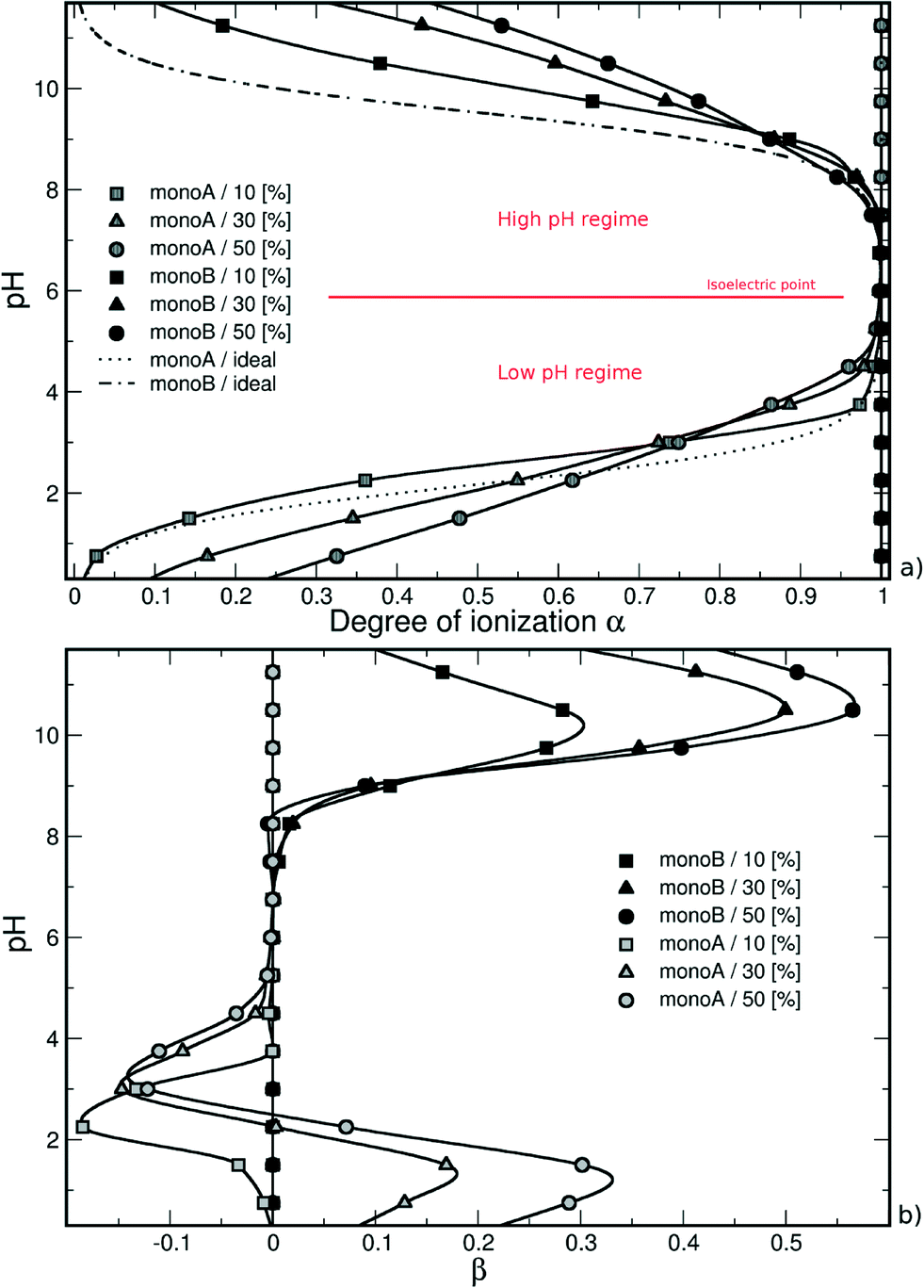 | ||
| Fig. 3 (a) Titration curves and (b) β = αA,B − αidealA,B variations of one flexible chain in the presence of one NP as well as monovalent counterions. Within the low pH regime, the deprotonation process of monomers A is less efficient due to the presence of the negatively charged NP and an intersection with the ideal curve is observed. The monoA deprotonation remains more efficient for low hydrophobic chains. The asymmetry, or change in the acid/base properties, due to the presence of NP is shown with the parameter β. Consequently, a pH range appears where β is negative. For strong hydrophobic chains, the monoA deprotonation is mainly driven by repulsive interactions with the NP (β < 0). | ||
In the high pH regime, the same behavior is observed compared with single PA titration curves, i.e., a more efficient protonation process of monomers B (amino groups) compared to isolated monomers due to the presence of fully charged carboxylic groups in this pH domain. As a result, the titration curves of the three model structures are found above the ideal case due to attractive electrostatic interactions between monomers A and B, hence favoring the monoB deprotonation process. The monoB charging behavior is more effective for the hydrophilic chains. It has to be noted that the nanoparticle has only a very limited effect in this pH regime compared with isolated chains resulting in a slight change of the apparent pKBa value of the PA.
The pH decrease in the low pH regime significantly modifies the subtle competition between repulsive monoA–NP, attractive monoB–NP and attractive monoA–monoB electrostatic interactions. Considering the hydrophilic chains (40% and 0% monoH) at low pH values, the PA is adsorbed at the NP surface resulting in strong interactions between charged amino groups and the nanoparticle. Thus, attractive monoA–monoB interactions, which are in direct competition with attractive monoB–NP interactions, are weaker. Moreover, repulsive monoA–NP interactions remain important due to the proximity with the negatively charged NP surface. The charging process of carboxylic groups then becomes less efficient compared with that of isolated PA chains resulting in higher apparent pKAa values. It has to be noted that an intersection of the ideal curve occurs for the two hydrophilic chains studied here.
In the case of hydrophobic backbones (80% monoH), the behavior is not exactly the same. On one hand, the apparent pKAa value of the PA is also modified, having higher values, but on the other hand, the shape of the titration curve is quite similar to that of the ideal curve and no intersection is observed. Indeed, attractive and repulsive electrostatic interactions remain limited within the entire monoA α range. Consequently, the deprotonation process of monoA is less efficient at low α compared with that of hydrophilic chains, and is favored at high α due to weaker attractive and repulsive electrostatic interactions with the NP at low and high α, respectively.
A convenient way to investigate the acid/base properties of monomers A and B is to calculate the β variations (Fig. 3b). Within the high pH regime, the curves remain positive for the three model structures mainly due to attractive interactions between monoA and monoB. We can see that these interactions increase with the decrease of PA hydrophobicity. Considering the low pH regime, the subtle competition between attractive and repulsive interactions resulting from the presence of the NP highlights the different behaviors between hydrophobic and hydrophilic chains. For hydrophobic chains, the deprotonation process of carboxylic groups remains less efficient compared to the ideal case due to limited intra- and inter-chain electrostatic interactions (negative values of β). The case of hydrophilic chains shows the subtle change of attractive/repulsive electrostatic interactions with pH. Indeed, β is positive at low pH due to strong attractive electrostatic interactions with the NP, and becomes negative with the increase of pH and repulsive intra- and inter-chain electrostatic interactions.
 | ||
| Fig. 4 (a) Radius of gyration of random chains in the presence of one NP and monovalent counterions (black symbols). For comparison, the curves of isolated chains are also given (grey symbols). The case with a NP results in the same behavior as that with single chains, i.e., the radius of gyration decreases with chain hydrophobicity, but the values are higher due to a change in electrostatic interactions. At low pH, chains are electrostatically attracted to the NP surface, of which the size is large compared to the chains. At high pH, the NP induces repulsive electrostatic interactions with charged chain monomers, hence promoting less compact conformations. (b) Radius of gyration of block polymer chains in the presence of one NP and monovalent counterions. More folded structures are obtained for the triblock due to the presence of hydrophobic monomers. An asymmetric shape is observed due to ion pairing within the block structures and nanoparticle. | ||
Considering the hydrophilic chains (40% and 0% monoH), the chain radii of gyration are found to be more pH-dependent in comparison to those of the isolated PA case (grey symbols) due to the competition between attractive and repulsive electrostatic interactions in the system. More folded conformations are found at intermediate pH when attractive interactions between charged carboxylic and amino groups are maximum, but in all cases, more stretched structures appear with higher radii of gyration due to the presence of repulsive interactions with the nanoparticle. Moreover, monomer adsorption at the NP surface is more effective in the low pH regime for hydrophilic backbones (0% monoH). In this case, the chains remain more open since the NP surface curvature is large compared to the chain size. The addition of these two effects, the repulsive electrostatic interactions with the NP and the curvature effect, results in higher variations of the hydrophilic chain. The radius of gyration of PA is presented in Fig. 4b for the diblock and triblock distributions in the presence of the NP. Globally, the variation of the radius of gyration with pH is accentuated with regard to the block structures. The strong ion pairing results in smaller values compared with that in the random structures in Fig. 4a (black symbols). At the isoelectric points (pH = 5), the strong intra-chain attractive interactions result in values in the same range as that for hydrophobic random chains (Fig. 4a, square symbols).
The study of the nearest neighbor distance distribution function between monomers in Fig. 5a supports the behavior observed in Fig. 4. The case of intermediate PA hydrophobicity (40% monoH) in the presence of a NP is shown in black, and the curves of the isolated PA chains are illustrated with grey symbols. The minimum distance between two monomers at 4 Å is represented here by the highest peak for all curves. Both cases with and without NP show the same tendencies, only a switch of the curves is observed. We have seen in Fig. 2b for isolated chains that nearest neighbor distance distribution function values increase with monomer density. Thus, the monomer density is lower here when a NP is present (black symbols) due to less folded PA conformations. Moreover, the variation between nearest neighbor distance distribution function values at low, intermediate and high pH is more limited when a nanoparticle is present, confirming the strong impact of NP on stretching PA conformations due to repulsive electrostatic interactions and NP curvature effects.
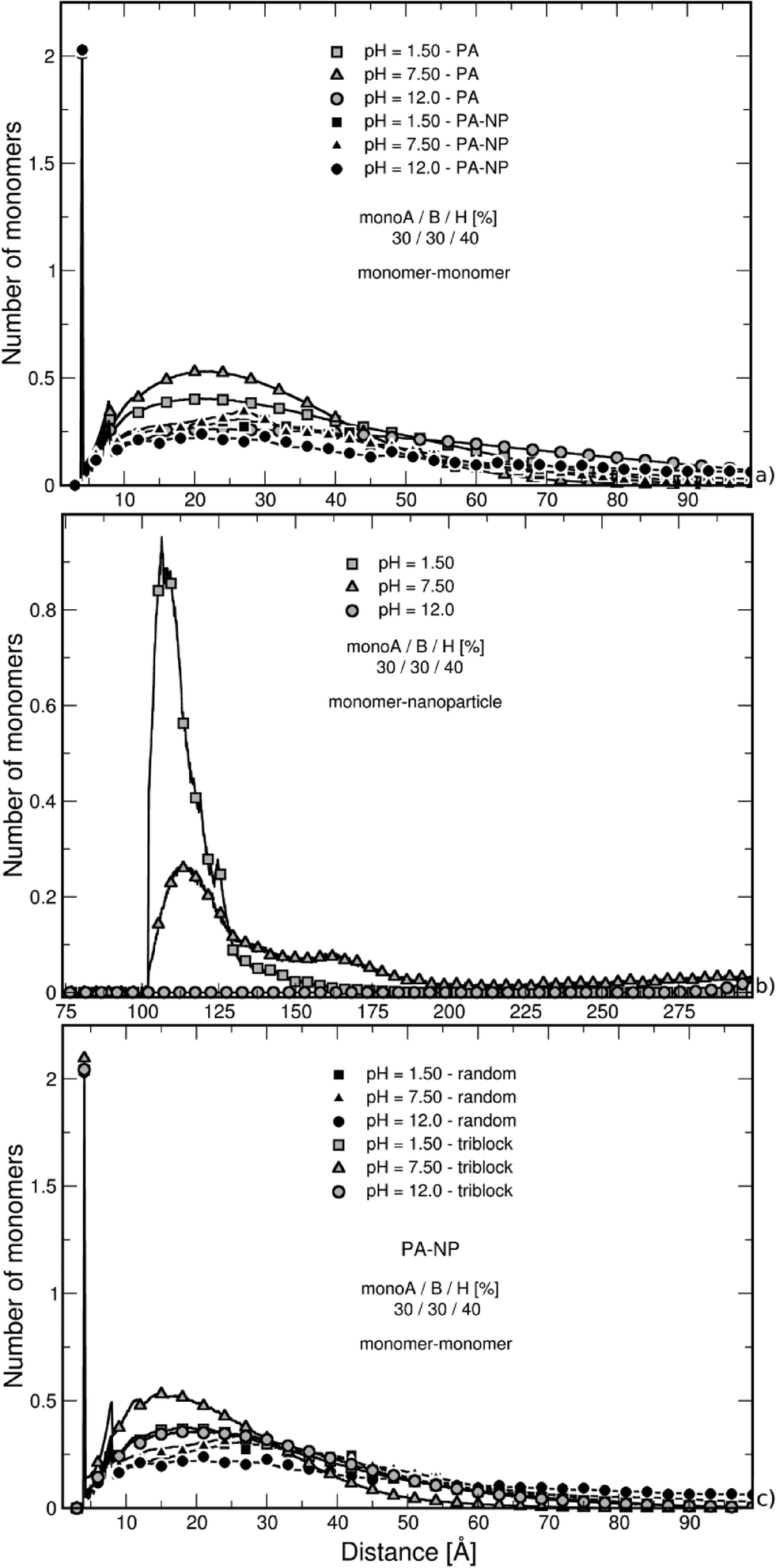 | ||
| Fig. 5 Nearest neighbor distance distribution function between (a) random chain monomers–monomers, (b) random chain monomers–NP surface, and (c) random and triblock chain monomers–monomers surrounded by counterions in the case of intermediate chain hydrophobicity (40% monoH). In part (a), the curves of isolated chains are also given. The cases with and without the NP show the same behavior, i.e., higher nearest neighbor distance distribution values due to more compact structures at intermediate pH values. The presence of NP results in lower values due to more open conformations. At low pH in part (b), the peak indicates more efficient monomer adsorption at the NP surface. At high pH value, chain monomers are situated at the box limit due to strong repulsive electrostatic interactions with the NP. In (c), it is shown that the triblock adopts more compact structures due to ion pairing. | ||
The nearest neighbor distance distribution functions of monomers around the NP in Fig. 5b show the evolution of monomer distribution around the NP surface. At low pH, the chain is strongly adsorbed at the surface resulting in a high peak at 102 Å which is the excluded volume limit. In contrast, PA monomers are situated at the box limit at high pH since both the NP and the chain carry a globally negative charge. In the intermediate state, the PA is globally neutral, but locally adsorbed due to an inhomogeneous charge distribution at its surface. As a result, monomers are found in the whole box with higher density in the first half of the box.
The nearest neighbor distance distribution functions between monomers shown in Fig. 5c for the triblock structure in the presence of a NP clearly confirm the strong ion pairing. Thus, the nearest neighbor distance distribution functions show higher values at short distances compared with the random chain distribution (in black) which is the consequence of more folded conformations.
4. Conclusions
Metropolis Monte Carlo simulations were carried out to investigate the conformational properties of simple DNA-like chains and estimate the impact of one negatively charged nanoparticle. The effects of chain hydrophobicity and pH were systematically investigated to get an insight into the role of the charge distribution as well as their ability to undergo large conformational changes (denaturation) considering pH variations and the presence of one negatively charged nanoparticle. We focused on three chain models and their properties (hydrophilic, intermediate, and hydrophobic) as well as different monomer distributions (random and block polymers).We found that pH modifies the acid/base properties of the chains, and thus their charge. The backbones remain globally neutral at physiological pH. On the other hand, they become negatively and positively charged at high and low pH, respectively, due to the protonation/deprotonation process of carboxylic (monoA) and amino groups (monoB). In the case of isolated chains, electrostatic repulsive monoA–monoA/monoB–monoB, electrostatic attractive monoA–monoB as well as attractive short-range interactions between hydrophobic monomers are in competition. Considering hydrophobic chains, short-range interactions are strong and folded conformations are observed within the entire pH range. On the other hand, pH variations affect the global chain conformation when hydrophilic and intermediate cases are considered leading to extended conformations at extreme pH due to repulsive interactions between monomers. This trend is confirmed by the chain radii of gyration which are smaller at physiological pH. The presence of a negatively charged nanoparticle induces extra electrostatic repulsive interactions with charged carboxylic groups and attractive interactions with protonated amino groups. Consequently, chain adsorption at the NP surface occurs only at low pH for intermediate and hydrophilic chains. In the case of hydrophobic chains, attractive short-range interactions are too strong to destabilize the chain structure. Moreover, the NP attractive/repulsive interactions result in modification of the chain acid/base properties. Indeed, the deprotonation of carboxylic groups was found to be less efficient in the low pH regime. The deprotonation process was then driven by the repulsive interactions with the nanoparticle and attractive interactions with the protonated amino groups. In addition, charge inhomogeneity along the chain backbone allowed the observation of less dense conformations at physiological pH due to strong repulsive interactions of chain segments with the nanoparticle even if the chains remain globally adsorbed. Concerning the effect of the monomer distribution (random, diblock and triblock), our results indicated significant conformational differences with and without the NP and the importance of ion pairing for the block monomer distribution.
Using Monte Carlo simulations, we demonstrated that the presence of one nanoparticle can significantly modify the acid/base properties of biomacromolecules and induce important conformational changes, in particular, when hydrophilic chains are considered. Overall, our results clearly indicate that computer simulation can isolate the important, subtle and key parameters when biomacromolecules and nanoparticle mixtures are investigated, and characterize the structure and composition of the corona formed at the surface of NPs.
Acknowledgements
The work leading to these results received funding from the European Union's Seventh Framework Programme (FP7/2007–2013) under agreement no. NMP4-LA-2013-310451. The authors are also grateful to F. Loosli, O. Oriekhova and S. Blanco-Ameijeiras for their encouragement and stimulating discussions.References
- T. M. Allen and P. R. Cullis, Science (Washington, DC, U. S.), 2004, 303, 1818–1822 CrossRef CAS PubMed.
- J.-F. Berret, N. Schonbeck, F. Gazeau, D. El Kharrat, O. Sandre, A. Vacher and M. Airiau, J. Am. Chem. Soc., 2006, 128, 1755–1761 CrossRef CAS PubMed.
- J. Buffle, Environ. Chem., 2006, 3, 155–158 CrossRef CAS.
- G. Han, C. T. Martin and V. M. Rotello, Chem. Biol. Drug Des., 2006, 67, 78–82 CAS.
- J. R. Lead and K. J. Wilkinson, Environ. Chem., 2006, 3, 159–171 CrossRef CAS.
- M. S. Diallo, W. Arasho, J. H. Johnson and W. A. Goddard III, Environ. Sci. Technol., 2008, 42, 1572–1579 CrossRef CAS.
- A. Kulczynska, T. Frost and L. D. Margerum, Macromolecules, 2006, 39, 7372–7377 CrossRef CAS.
- J. Lyklema, Colloids Surf., A, 2006, 291, 3–12 CrossRef CAS PubMed.
- W. Tian and Y. Ma, J. Phys. Chem. B, 2009, 113, 13161–13170 CrossRef CAS PubMed.
- S.-R. Chae, Y. Xiao, S. Lin, T. Noeiaghaei, J.-O. Kim and M. R. Wiesner, Water Res., 2012, 46, 4053–4062 CrossRef CAS PubMed.
- R. Grillo, A. H. Rosa and L. F. Fraceto, Chemosphere, 2015, 119, 608–619 CrossRef CAS PubMed.
- F. Loosli, P. L. Coustumer and S. Stoll, Environ. Sci.: Nano, 2014, 1, 154–160 RSC.
- A. Philippe and G. E. Schaumann, Environ. Sci. Technol., 2014, 48, 8946–8962 CrossRef CAS PubMed.
- N. von Moos and V. I. Slaveykova, Nanotoxicology, 2014, 8, 605–630 CrossRef CAS PubMed.
- J.-W. Shen, T. Wu, Q. Wang and Y. Kang, Biomaterials, 2008, 29, 3847–3855 CrossRef CAS PubMed.
- L. Calzolai, S. Laera, G. Ceccone, D. Gilliland, R. Hussain, G. Siligardi and F. Rossi, J. Nanopart. Res., 2013, 15, 1–5 CrossRef.
- S. Laera, G. Ceccone, F. Rossi, D. Gilliland, R. Hussain, G. Siligardi and L. Calzolai, Nano Lett., 2011, 11, 4480–4484 CrossRef CAS PubMed.
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS PubMed.
- M. Aramesh, O. Shimoni, K. Ostrikov, S. Prawer and J. Cervenka, Nanoscale, 2015, 7, 5726–5736 RSC.
- B. Mahltig, R. Jerome and M. Stamm, J. Polym. Res., 2003, 10, 219–223 CrossRef CAS.
- B. Wang, P. Wu, R. A. Yokel and E. A. Grulke, Appl. Surf. Sci., 2012, 258, 5332–5341 CrossRef CAS PubMed.
- K. Chen, Y. Xu, S. Rana, O. R. Miranda, P. L. Dubin, V. M. Rotello, L. Sun and X. Guo, Biomacromolecules, 2011, 12, 2552–2561 CrossRef CAS PubMed.
- T. Mesarič, L. Baweja, B. Drašler, D. Drobne, D. Makovec, P. Dušak, A. Dhawan and K. Sepčić, Carbon, 2013, 62, 222–232 CrossRef PubMed.
- S. Patil, A. Sandberg, E. Heckert, W. Self and S. Seal, Biomaterials, 2007, 28, 4600–4607 CrossRef CAS PubMed.
- M. Fowler, B. Siddique and J. Duhamel, Langmuir, 2013, 29, 4451–4459 CrossRef CAS PubMed.
- K.-I. Sano and K. Shiba, J. Am. Chem. Soc., 2003, 125, 14234–14235 CrossRef CAS PubMed.
- S. R. Whaley, D. S. English, E. L. Hu, P. F. Barbara and A. M. Belcher, Nature, 2000, 405, 665–668 CrossRef CAS PubMed.
- S. Ahmed, F. Hayashi, T. Nagashima and K. Matsumura, Biomaterials, 2014, 35, 6508–6518 CrossRef CAS PubMed.
- H. Lei, M. Wang, Z. Tang, Y. Luan, W. Liu, B. Song and H. Chen, Langmuir, 2014, 30, 501–508 CrossRef CAS PubMed.
- T. Zhao, K. Chen and H. Gu, J. Phys. Chem. B, 2013, 117, 14129–14135 CrossRef CAS PubMed.
- M. Borówko, S. Sokołowski, T. Staszewski, Z. Sokołowska and J. M. Ilnytskyi, J. Chem. Phys., 2012, 137, 074707 CrossRef PubMed.
- A. G. Cherstvy, J. Phys. Chem. B, 2011, 115, 4286–4294 CrossRef CAS PubMed.
- L.-J. Qu, X. Man, C. C. Han, D. Qiu and D. Yan, J. Phys. Chem. B, 2012, 116, 743–750 CrossRef CAS PubMed.
- V. Pryamitsyn and V. Ganesan, Macromolecules, 2014, 47, 6095–6112 CrossRef CAS.
- T. A. Kampmann, H.-H. Boltz and J. Kierfeld, J. Chem. Phys., 2013, 139, 034903 CrossRef PubMed.
- C. Tong, J. Chem. Phys., 2013, 139, 084903 CrossRef PubMed.
- A. G. Cherstvy and V. B. Teif, Phys. Biol., 2014, 11, 044001 CrossRef PubMed.
- Q. Cao and M. Bachmann, Chem. Phys. Lett., 2013, 586, 51–55 CrossRef CAS PubMed.
- V. M. de Oliveira and S. J. de Carvalho, Eur. Phys. J. E, 2014, 37, 1–7 CrossRef CAS PubMed.
- M. Mella, L. Mollica and L. Izzo, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 650–663 CrossRef CAS PubMed.
- C. H. J. Evers, T. Andersson, M. Lund and M. Skepö, Langmuir, 2012, 28, 11843–11849 CrossRef CAS PubMed.
- A. Kurut, J. Henriques, J. Forsman, M. Skepö and M. Lund, Proteins: Struct., Funct., Bioinf., 2014, 82, 657–667 CrossRef CAS PubMed.
- M. Lund and B. Jönsson, Biochemistry, 2005, 44, 5722–5727 CrossRef CAS PubMed.
- M. Rabe, D. Verdes and S. Seeger, J. Phys. Chem. B, 2010, 114, 5862–5869 CrossRef CAS PubMed.
- F. Paillusson, V. Dahirel, M. Jardat, J.-M. Victor and M. Barbi, Phys. Chem. Chem. Phys., 2011, 13 Search PubMed.
- A. Clavier, M. Seijo, F. Carnal and S. Stoll, Phys. Chem. Chem. Phys., 2015, 17, 4346–4353 RSC.
- X. Guo, J. Wang, J. Zhang and W. Wang, Mol. Simul., 2014, 1–12 Search PubMed.
- A. K. N. Nair, S. Uyaver and S. Sun, J. Chem. Phys., 2014, 141, 134905 CrossRef PubMed.
- M. Skepö, J. Chem. Phys., 2008, 129, 185101 CrossRef PubMed.
- V. Dahirel, F. Paillusson, M. Jardat, M. Barbi and J.-M. Victor, Phys. Rev. Lett., 2009, 102, 228101 CrossRef.
- N. Almora-Barrios and N. H. De Leeuw, Cryst. Growth Des., 2012, 12, 756–763 CAS.
- P.-Y. Hsiao, J. Phys. Chem. B, 2008, 112, 7347–7350 CrossRef CAS PubMed.
- S. Ulrich, M. Seijo and S. Stoll, J. Phys. Chem. B, 2007, 111, 8459–8467 CrossRef CAS PubMed.
- S. Ulrich, M. Seijo, F. Carnal and S. Stoll, Macromolecules, 2011, 44, 1661–1670 CrossRef CAS.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, J. Chem. Phys., 1953, 21, 1087–1092 CrossRef CAS PubMed.
- M. Voicescu, S. Ionescu and D. G. Angelescu, J. Nanopart. Res., 2012, 14, 1–13 CrossRef.
- H. L. Gordon and J. P. Valleau, Mol. Simul., 1995, 14, 361–379 CrossRef CAS PubMed.
- P. H. Verdier and W. H. Stockmayer, J. Chem. Phys., 1962, 36, 227–235 CrossRef CAS PubMed.
- F. T. Wall and F. Mandel, J. Chem. Phys., 1975, 63, 4592–4595 CrossRef CAS PubMed.
- C. E. Reed and W. F. Reed, J. Chem. Phys., 1992, 96, 1609–1620 CrossRef CAS PubMed.
- M. Ullner, B. Jönsson and P. O. Widmark, J. Chem. Phys., 1994, 100, 3365–3366 CrossRef CAS PubMed.
- A. Onufriev, D. A. Case and G. M. Ullmann, Biochemistry, 2001, 40, 3413–3419 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5en00054h |
| This journal is © The Royal Society of Chemistry 2015 |