
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards understanding the antibacterial activity of Ag nanoparticles: electron microscopy in the analysis of the materials-biology interface in the lung
M.
López-Heras
a,
I. G.
Theodorou
a,
B. F.
Leo
ab,
M. P.
Ryan
a and
A. E.
Porter
*a
aDepartment of Materials, Imperial College London, London, UK. E-mail: a.porter@imperial.ac.uk
bDepartment of Mechanical Engineering, Faculty of Engineering Building, University of Malaya, Kuala Lumpur 50603, Malaysia
First published on 29th June 2015
Abstract
Bacterial infections of the pulmonary system are increasing. With almost half of today's infections being caused by strains of bacteria that are resistant to existing conventional antibiotics, there is an urgent need for the development of novel therapeutic platforms. Silver nanoparticles (AgNPs) have been receiving increasing attention due to their unique antibacterial properties, and whilst the biological efficacy of silver is well known, the mechanisms by which AgNPs degrade within cells and how these processes correlate to their bioreactivity are poorly understood. This review summarises the current knowledge on the bactericidal pathways of AgNPs and discusses the challenges to be faced before we are able to develop efficient and safe antibacterial agents for the treatment of bacterial infections in the lung.
Nano impactWith increasing commercialisation of silver nanomaterials (AgNMs) comes a concomitant need to understand occupational health, public safety and environmental implications of these materials. However, along with concerns about toxic effects, there is potential for enormous benefit via highly efficacious antibacterial modality. Nanoscale studies of the complex bio-nano interface lie at the heart of technical challenges. Despite numerous reports, there is no consensus regarding biological mechanisms enacted by AgNMs. This paper reviews how the physicochemical properties of AgNMs are linked to antibacterial properties in the respiratory system, in particular their interactions with and within cells and bacteria. The literature is discussed in the context of what is known, and also what information is missing, to allow safe development of AgNM-therapeutics. |
Introduction
Recent advances in the research and development of engineered nanomaterials have enabled a wide range of novel applications for commercial and industrial uses. One field that nanoparticles are expected to revolutionise is medicine, through the design of complex structures that can be targeted to specific tissues, have mechanisms for controlled release and evade rapid clearance.1,2 The lung is a particularly attractive target for nanomedicine, to treat pulmonary as well as systemic disease. Its large surface area and circumvention of first pass metabolism allow for high drug absorption. Moreover, inhalable drugs are non-invasive and can be self-administered, while their nano-size allows them to penetrate in the deep lung and reach the alveolar region more efficiently than larger particles. There are currently several types of nanoparticles being developed for respiratory applications, which aim to overcome the limitations of conventional drugs and be used for the treatment of numerous lung diseases, such as asthma, bacterial infections, emphysema, cystic fibrosis, cancer and others (Table 1).3 A major part of research on drug delivery nanoformulations has previously been devoted on “soft” nanoparticles, such as solid lipid nanoparticles, organic polymers and liposomes.3 Recently there has also been a move toward the use of “hard” inorganic nanoparticles, like carbon nanotubes or noble metal nanoparticles.1 In both cases, nanoparticles have often been used as vehicles for the administration of active pharmaceutical ingredients in the lungs. Silver has been used as a disinfectant since antiquity;4 therefore silver nanoparticles are receiving increasing attention for their inherent antibacterial properties and have already been incorporated in several consumer products (e.g. wound dressings, antibacterial textiles, cosmetic and hygiene products, antibacterial sprays, nasal drops). Consequently, AgNPs represent an exciting opportunity for the development of platforms for the treatment of pulmonary bacterial infections.| Type of nanoparticle | Application | Ref. |
|---|---|---|
| Solid lipid nanoparticles | Asthma | 96 |
| Lung cancer | 97 | |
| Cystic fibrosis | 98 | |
| Tuberculosis | 99 | |
| Insulin delivery | 100 | |
| Pulmonary hypertension | 101 | |
| Polymeric nanoparticles | Pulmonary fibrosis | 102 |
| Lung cancer | 103 | |
| Anti-oxidant delivery | 104 | |
| Lung infections | 105 | |
| Anticoagulant delivery | 106 | |
| Pulmonary hypertension | 107 | |
| Gene delivery | 108 | |
| Liposomes | Asthma | 109 |
| Anti-oxidant delivery | 110 | |
| Bacterial infections | 111 | |
| Pulmonary arterial hypertension | 112 | |
| Surfactant therapy | 113 | |
| Polymeric liposomes | Gene delivery | 114 |
| Protein and peptide delivery | 115 | |
| Lipid nanoparticles | Asthma | 116 |
| Lung infections | 117 | |
| Dried nanoparticles | Anti-inflammatory | 118 |
| Lung cancer | 119 | |
| Pulmonary infections | 120 | |
| Self-assembly nanoparticles | Antibody delivery | 121 |
| Carbon nanotubes | Cancer | 122 |
| Silver nanoparticles | Anti-proliferative effects in cancer cells | 123, 124 |
| Gold nanoparticles | Lung cancer diagnosis | 125 |
| Anti-cancer drug delivery | 126 | |
| Simultaneous magnetic resonance imaging (MRI) and therapy | 127 | |
| Photodynamic therapy | 128 |
Among lung diseases, invasive microbial infections affecting the lungs are increasing in incidence and diversity.5 Although the lung is constantly exposed to potential infectious agents, the extraordinary natural defence mechanisms of the respiratory tract may, in most cases, prevent serious infections. The most common causes of bacterial lung infections include Streptococcus pneumoniae, Haemophilus species, Staphylococcus aureus and Mycobacterium tuberculosis.6 After infection, bacteria bound to the surface of macrophages are rapidly internalised into phagosomes, which engage with endosomal trafficking processes and gradually acquire the characteristics of terminal phagolysosomes, an event concomitant with the death of the microbe. Pulmonary macrophages have a key role in the defence against respiratory infections produced by viruses, bacteria, mycobacteria and fungi. Together with dendritic cells, they are active producers of cytokines and leukotrienes, and have important pro- and anti-inflammatory roles in the alveoli and airway submucosa, respectively.7 However, some pathogens, such as Legionella spp. and Mycobacteria spp., can alter lysosome trafficking7 and phagolysosome fusion8 or even block host macrophage apoptosis by a TNF receptor-dependent mechanism9,10 in order to survive inside cells.
In the last two decades, the rate at which bacteria are becoming resistant to current antibiotic treatments has increased substantially, leading to an urgent need for the development of new antibiotics (World Health Organisation, 2014). Silver nanoparticles are considered as potential therapeutic agents which could have a significant impact on respiratory research and medicine, by offering a multifunctional platform that could simultaneously diagnose and treat bacterial infections.11–13 The antibacterial properties of AgNPs have been shown to depend on several physicochemical properties of the particles, including their size, shape, chemistry and surface coating.14–17 Currently, the mechanisms by which AgNPs are able to reduce the activity of bacteria are believed to arise due to a synergistic effect, including both direct particle-specific biological effects and also the release of Ag+ ions.18,19 However, several discrepancies in the published literature need to be addressed in order to draw accurate links between the physicochemical properties of the particles and the observed biological effects. Moreover, the same properties that make AgNPs excellent antimicrobial agents may also exert adverse effects on pulmonary structure and function. In fact, Ag+ ions released from AgNP surfaces have been shown to damage proteins by desulphurisation, generate reactive oxygen species (ROS)20 or interfere with NO redox equilibria in airway smooth muscle cells in the lung.21 This oxidative potential may increase the permeability of the lung epithelium to AgNPs, resulting in DNA damage, lipid membrane damage, chromosomal aberrations and cell-cycle arrest.22,23
The effects of AgNPs on bacteria are commonly evaluated by various in vitro viability measurements, such as minimum inhibitory concentration (MIC), fractional inhibitory concentration (FIC), zone of inhibition test, and the LIVE/DEAD bacterial assays. These assays can provide valuable information about the antibacterial action and potential cellular toxicity of AgNPs. However, more detailed mechanistic investigations of the cellular interactions with AgNPs are necessary in order to provide insights about how to optimise the design of AgNPs for the treatment of bacterial infections. Several reports have illustrated that biophysical interactions may occur between AgNPs and bacteria, such as biosorption, AgNP breakdown or aggregation and cellular uptake.24,25 AgNPs can increase the permeability of bacterial cell membranes, penetrate into the cytoplasm and disturb cellular functions.26
The application of three-dimensional (3D) analytical electron microscopy in this area could be invaluable, because it can provide high spatial resolution information of both the morphology and chemistry of AgNPs. Since the physicochemical properties of AgNPs may change in the cellular environment, sufficient characterization should take place not only for as-synthesized particles but also at the point of exposure, in order to understand what agent of Ag actually interacts with the cells. Moreover, spatially resolved maps of the anatomical distribution and physico-chemistry of AgNPs at the sub-cellular level and observation of changes in the cellular ultrastructure could help deciphering the antibacterial mechanisms exerted by nano-Ag. Therefore, nanoscale imaging methods have the potential to provide vital information about these systems and, when used in conjunction with complementary quantitative studies or other techniques such as low resolution optical imaging, could provide a holistic understanding of the system interactions.
In this review, the current literature on the antibacterial mechanisms of AgNPs against Gram-positive and Gram-negative bacteria is summarized. The results of studies on cellular interactions of AgNPs are correlated to the thermodynamics driving the dissolution of AgNPs in complex physiological environments. Furthermore, we discuss the mechanistic information that a range of microscopy techniques (including transmission and scanning electron microscopy, 3D confocal microscopy and dark field microscopy) can provide about the key biological processes involved in bacterial infection of alveolar macrophages, as well as the cellular uptake of AgNPs and the changes to their physicochemical properties occurring within bacteria. Improving our understanding on the mode of biological action of AgNPs and the physicochemical properties by which this is driven, will enable us to design and implement efficient and safe AgNP antibacterial therapeutics for the treatment of lung infections.
Phagocytic mechanisms and morphological alterations in bacteria-infected macrophages
Since infection is a localized disease, the first step in the design of actively targeted nano-therapeutics would be to understand where bacilli reside in infected lungs. The use of microscopy techniques can provide crucial information about the topography and the biological processes taking place in infected lungs. Bacterial phagocytosis by immune cells is the crucial step in the host defence against microbial invaders.27 The phagosome is a central mediator of both the homeostatic and microbial functions of macrophages. This phagocytic vesicle is programmed to degrade internalized material with high efficiency, through rapid acidification and fusion with lysosomes, which are rich in proteolytic and lipolytic enzymes. However, bacilli have developed mechanisms to escape from phagosomes and enter into autophagosomes for survival and replication.28 For instance, Yuan et al. demonstrated that autophagosomes play a regulatory role in the clearance of Pseudomonas aeruginosa, a bacterium that frequently infects immunodeficient individuals.29 In their work, the accumulation of typical autophagosomes with double membranes in infected alveolar macrophages (MS-H) was identified using transmission electron microscopy (TEM) (Fig. 1A). Together with TEM imaging, the results of bio-analytical assays led to the conclusion that P. aeruginosa-induced autophagy represented a protective host mechanism.29 Recently, the kinetics and mechanical pathways by which macrophages may interact with, and internalize bacteria, was investigated in depth by Moller et al. using a combination of microscopy techniques, including scanning electron microscopy (SEM), differential interference contrast (DIC) microscopy with interference reflection (IRM) and 3D confocal microscopy (CM).30 Their findings revealed a multistep process in which macrophages used filopodia as phagocytic tentacles to sense and form long-lived interactions with surface-adhering bacteria (Escherichia coli). In the same work, fluorescence microscopy showed that Legionella pneumophila could enter macrophages through a cholesterol-sensitive pathway, thus avoiding immediate digestion inside lysosomes by interacting with the autophagy pathway of the host cells.30 | ||
Fig. 1 Electron microscopy images showing morphological alterations in bacteria infected macrophages. (A) Formation of autolysosomes (single arrows) and autophagosomes (double arrows) in Pseudomonas aeruginosa-infected MS-H cells after 1 h (MOI = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Journal of Cell Science by Company Of Biologists. Reproduced with permission of Company of Biologists Ltd.29 (B) Distribution of beads (b), lysosomes (Ly), lipid droplets (L), nuclei (N) and Mycobacterium tuberculosis bacilli (arrowheads) in infected human monocyte derived macrophages (HMDM) after 5 days (MOI = 5:1). Reprinted with permission from ref. 32. Copyright 2012 John Wiley & Sons Ltd. (C) Ultrastructural changes in the mitochondria of THP-1 cells infected with a virulent M.tb strain for 24 h (MOI = 10:1). Electron dense and elongated mitochondria, with prominent cristae were observed in infected cells, compared to uninfected THP-1 cells (D). Reprinted with permission from ref. 34. Copyright 2013 Nature Publishing Group. (E) Electron tomography images showing inter-mitochondrial fusion and breaches in the mitochondrial cell wall. Reprinted with permission from ref. 34. Copyright 2013 Nature Publishing Group. (F) Peripheral nuclear condensation and partial nuclear protrusion from condensed areas in bone marrow derived macrophages (BMDM) infected with M.tb (MOI = 25:1), after 3 h (F1); cells at the late phase of death with damaged plasma membrane (F2). Reprinted with permission from ref. 33. Copyright 2011 Lee et al. (G) Extensive loss of the plasma membrane, exposing the underlying cytoskeleton and intracellular organelles in M.tb-infected macrophages (G2) compared to uninfected cells (G1). Reprinted with permission from ref. 33. Copyright 2011 Lee et al. :1). Journal of Cell Science by Company Of Biologists. Reproduced with permission of Company of Biologists Ltd.29 (B) Distribution of beads (b), lysosomes (Ly), lipid droplets (L), nuclei (N) and Mycobacterium tuberculosis bacilli (arrowheads) in infected human monocyte derived macrophages (HMDM) after 5 days (MOI = 5:1). Reprinted with permission from ref. 32. Copyright 2012 John Wiley & Sons Ltd. (C) Ultrastructural changes in the mitochondria of THP-1 cells infected with a virulent M.tb strain for 24 h (MOI = 10:1). Electron dense and elongated mitochondria, with prominent cristae were observed in infected cells, compared to uninfected THP-1 cells (D). Reprinted with permission from ref. 34. Copyright 2013 Nature Publishing Group. (E) Electron tomography images showing inter-mitochondrial fusion and breaches in the mitochondrial cell wall. Reprinted with permission from ref. 34. Copyright 2013 Nature Publishing Group. (F) Peripheral nuclear condensation and partial nuclear protrusion from condensed areas in bone marrow derived macrophages (BMDM) infected with M.tb (MOI = 25:1), after 3 h (F1); cells at the late phase of death with damaged plasma membrane (F2). Reprinted with permission from ref. 33. Copyright 2011 Lee et al. (G) Extensive loss of the plasma membrane, exposing the underlying cytoskeleton and intracellular organelles in M.tb-infected macrophages (G2) compared to uninfected cells (G1). Reprinted with permission from ref. 33. Copyright 2011 Lee et al. | ||
Mycobacterium tuberculosis (M.tb) is the causative agent of tuberculosis, one of the most devastating infections in the lung.31 It is well-known that M.tb bacilli may spread infection by arresting phagosome maturation.31 Infected macrophages present an increase in the retention of host lipids, which provides a potential source of nutrients that could be accessed by bacteria. Confocal microscopy and TEM imaging revealed the presence of a great number of lipid droplets throughout the cytoplasm of infected macrophages (Fig. 1B).32 Apart from inhibiting phagosome maturation as a survival strategy, M.tb may interfere in processes linked to cell death, such as apoptosis. Apoptosis is considered as a mechanism whereby the host cell can contain the infection and eliminate it before it can spread further throughout the body.33 As shown in Fig. 1C–E, cells infected with virulent (H37Rv) M.tb strains present morphological perturbations in the ultrastructure of mitochondria, such as increased electron density, clear definition of the cristae and inter-mitochondrial fusion. These findings suggest an augmentation in the functional activity of mitochondria, which could be linked to an inhibition of the apoptotic response.34 Lee et al. investigated the characteristics and determinants of macrophage cell death caused by M.tb, using TEM and SEM (Fig. 1F and G).33 Their study provided evidence that cell death caused by virulent M.tb was distinct from classical apoptosis. The authors suggested a mechanism based on the permeabilization of lysosomal membranes, followed by the destruction of lipid bilayers and a concomitant degradation of several phospholipid species.33
In conclusion, the use of confocal and electron microscopy techniques can provide important information about key biological signals involved in bacterial infection. Cellular mechanisms taking place in infected host cells, such as autophagy or inhibition of phagosome maturation, can be observed using high resolution imaging. Together with data obtained from cytotoxicity assays (e.g. mitochondrial activity measurements), imaging revealing the ultrastructural morphology of cellular organelles, can provide a broader understanding about the pathways of cell death. Moreover, the use of techniques such as immunolabelling, could help identify molecular ligands involved in the cellular uptake of bacteria. This information could have a pivotal role in the identification of the appropriate targets for the development of antibacterial drugs based on AgNPs. Finally, in future studies, analytical TEM techniques will be indispensable in the study of the intracellular localization, and the morphological and chemical stability of AgNPs applied to bacteria-infected cells.
The thermodynamics of dissolution of AgNPs
In order to understand and predict how AgNPs behave in biological environments, it is also essential to understand the thermodynamics driving the dissolution of this material and how this is related to the physicochemical properties of the particles and environmental conditions. Several factors such as ionic strength and composition of the dispersion medium,35–38 pH,39,40 dissolved organic matter,41 relative humidity,42 dissolved oxygen concentration43 and temperature44 have been shown to alter the properties and dissolution rates of AgNPs and are consequently expected to affect their bioreactivity. Size, charge, and surface modifications of metallic AgNPs have been shown to affect their cytotoxicity.43,45–47Under environmental and physiological conditions, metallic silver is not thermodynamically stable but has been shown to oxidize and release Ag+ ions, which are often considered as the main active species in the biological action of Ag.48 Liu and Hurt have shown that the dissolution of AgNPs is a cooperative oxidation that involves both protons (H+) and dissolved oxygen (O2).39 They suggested that in simple aqueous solutions containing no other oxidants or reductants, the global reaction stoichiometry is:
 | (1) |
Recently, Zhang et al. developed a kinetic model to describe Ag+ release, based on the hard sphere theory.49 They proposed that the oxidation reaction can be expressed by first-order reaction kinetics and therefore, the Ag+ release rate can be described by the Arrhenius equation:
 | (2) |
 | (3) |
 | (4) |
 is the Ag+ release rate (mol L−1 h−1), k is the reaction rate constant (mol h−1), [AgNP], [O2] and [H+] are the molar concentrations (mol L−1) of AgNPs, dissolved oxygen and protons respectively, Eα is the activation energy (J), T is the temperature (298 K) and A is the frequency factor. Replacing the molar concentration of AgNPs ([AgNP]) by the mass-based concentration ([Ag]):
is the Ag+ release rate (mol L−1 h−1), k is the reaction rate constant (mol h−1), [AgNP], [O2] and [H+] are the molar concentrations (mol L−1) of AgNPs, dissolved oxygen and protons respectively, Eα is the activation energy (J), T is the temperature (298 K) and A is the frequency factor. Replacing the molar concentration of AgNPs ([AgNP]) by the mass-based concentration ([Ag]): | (5) |
 | (6) |
Unravelling the antibacterial mechanisms of AgNPs
Since AgNPs are known to dissolve and release Ag+ ions under physiological conditions, there has been much debate in the literature as to whether the reactivity of AgNPs arises due to an ionic35,45 or particulate effect,51–54 or both. The following section discusses the mechanisms by which the release of Ag+ ions can be correlated to their antibacterial activity and the influence of the AgNP-cell interface on these processes.Dissolution of AgNPs and effects of Ag+ ion release on cell membrane permeability
Recent evidence has suggested that AgNPs exert their antimicrobial properties by the localised release of Ag+ ions in the acidic and aerobic environment of E.coli.18 The bacterial membrane has an acidic pH and oxidising environment, which (according to eqn (1)) will accelerate AgNP dissolution and release of Ag+ ions. This release could disrupt the outer membrane of bacteria, enhancing AgNP and Ag+ ion uptake leading to a reduction in cell viability. This hypothesis has recently been supported by different authors, who used a sensor sensitive to Ag+ ion release to show that the toxicity of AgNP could be explained entirely by accumulation of intracellular Ag+ ions.16,18Silver salts alone, such as AgNO3, can also kill bacteria and damage the surrounding tissue.12,19 Therefore, it is assumed that one of the most important pathways through which AgNPs exert their antimicrobial properties is based on the effect of free Ag+ ions.18,19 Xiu et al. demonstrated that AgNPs themselves did not affect the biological activity of bacteria, only the ionic Ag+.45 In their study, the antibacterial properties of polyethylene glycol (PEG) coated AgNPs of three different diameters (2.8 ± 0.47 nm, 4.7 ± 0.20 nm and 10.5 ± 0.59 nm) were evaluated under aerobic and anaerobic conditions, that preclude Ag0 oxidation. Under anaerobic conditions, AgNPs did not exert any effects on the viability of E. coli, whereas prolonged air exposure induced higher toxicity. This work showed that AgNPs do not show antibacterial effects in anaerobic conditions that preclude Ag0 oxidation and Ag+ ion release.45 However, by removing the oxygen from the system, the redox active surface of AgNPs could lose its capability for generating reactive oxygen species (ROS), which has also been linked to their toxicity,55 as discussed in the following paragraphs. By not taking this factor into account, this study cannot completely isolate particulate and ionic effects. The above findings suggest that the antibacterial activity of AgNPs could be controlled by modulating Ag+ release, possibly through the manipulation of oxygen availability, coating agents or NP size.
The mechanism by which Ag+ ions permeate the cell wall, is thought to be due to a strong interaction between ionic Ag+ and thiol groups of cysteine residues of proteins present in the bacterial cell wall. This may lead to the disruption of these proteins56,57 and the accumulation of Ag+ ions in the cell wall.58 It has also been proposed that Ag+ ions can destabilize the membranes of bacteria, leading to the formation of pits in the bacterial cell wall,59,60 increasing its permeability and facilitating the internalization of AgNPs and Ag+ ions.18 Based on theoretically calculated and experimentally estimated Ag+ ions affinities, other amino acids such as arginine and histidine should be better ligands for Ag+ ions, but these interactions have not been widely investigated.12 The interaction of Ag+ ions with enzymes of the respiratory chain reaction, such as NADH dehydrogenase,61 could lead to the uncoupling of respiration from ATP synthesis. Ag+ ions may also bind to transport proteins, causing proton leakage and collapse of the proton motive force.62,63
Ag+ ions released from AgNPs or chemisorbed on its surface could also be responsible for the catalytic generation of ROS by serving as electron acceptor.64 ROS are natural by-products of the metabolism of respiring organisms and, while small levels can be controlled by the antioxidant defences of the cells, excess ROS production may produce oxidative stress.65 ROS mainly target lipids, DNA, RNA and proteins, causing severe consequences, such as membrane breakdown and impairment of the mitochondrial function or of the DNA replication machinery. In bacteria, Ag+ ions would likely induce ROS generation by impairing the respiratory chain enzymes, through direct interactions with thiol groups in these enzymes or with superoxide radical scavenging enzymes, such as superoxide dismutases.62,64,66
Some authors have also suggested ROS generation directly from the AgNPs. In one study, where the toxic effects of Ag+ ions, AgCl and AgNPs on nitrifying bacteria were investigated, the authors reported that all forms of Ag were able to induce intracellular ROS generation.55 The bacterial inhibition of each form of Ag was correlated with the corresponding concentration of intracellular ROS. However, at the same concentration of ROS, AgNPs were more toxic than the other forms of Ag, indicating that additional mechanisms contributed to the overall toxicity.55 The formation of the superoxide anion, but not of hydroxyl radicals, observed for Ag+ ions,64 was also observed for E. coli treated with AgNPs.67 In contrast to these findings, other authors claim that ROS production has a negligible contribution to the antimicrobial properties of AgNPs. Sintubin et al. showed that only a small amount of ROS were induced by the presence of AgNPs and an even smaller amount was produced in the presence of Ag+ ions alone.68
Taken together, these findings could support the hypothesis that the bacterial properties of AgNPs arise mainly due to Ag+ ion release from their surface. Morones et al. supported this hypothesis, demonstrating that Ag+ ions were more toxic than AgNPs to S. aureus bacteria, in relation to bacterial growth.24 TEM imaging revealed that the ultrastructural changes observed in AgNP-treated bacteria were similar to those induced by Ag+ ions: detachment of the cytoplasm membrane from the cell wall and formation of a low density region, rich in agglomerated DNA,69 which protects DNA molecules from damage induced by external stimuli. Similarly, low molecular weight proteins can also accumulate around the nucleus as a response to Ag+ ion attack.70
Direct particle–cell wall interaction
Although the principal mode of antibacterial activity of AgNPs involves the release of Ag+ ions, studies have demonstrated that the mechanism by which AgNPs interact with bacteria involves a combination of both chemical and physical interactions.16 The small size and extremely large surface area of AgNPs allows them to make strong contact with the bacterial cell wall.71 Several studies have reported that AgNPs are able to adhere to the plasma membrane, change its permeability and penetrate inside the cytoplasm.19,25,55,63,72 Studies have shown that a synergistic effect is required, where Ag+ ions are required for the antibacterial effects of AgNPs, but cell-NP contact increases Ag+ release and increases the amount of cellular uptake of particle associated Ag+ ions.16,18 A report by McQuillan et al. supports the hypothesis that dissolution at the cell membrane is the primary mechanism of action of silver nanoparticles.19Several other studies have demonstrated that AgNPs are more toxic to bacteria than the Ag+ ions released from the corresponding AgNPs in the extracellular media.15,40,53,73 Taglietti et al. pointed out that two factors need to be considered, the release of Ag+ ions in the exposure medium, referred to as a “long-distance mechanism”, but also a “short-distance mechanism” at the particle–bacterial membrane interface.74 Some authors suggested that this short-distance effect arises due to enhanced dissolution of the AgNPs close to the bacterial cell wall.16 Recent studies have provided new insight that may link the discrepancies existing on the relative effect of AgNPs and Ag+ ions on toxicity.16,18 Bondarenko et al. examined the effect of three types of AgNPs (uncoated, PVP-coated and protein-coated) against six bacterial strains (Gram-negative: E. coli, P. fluorescens, P. putida and P. aeruginosa and Gram-positive: B. subtilis and S. aureus). Their data demonstrated that direct contact between the bacterial cell and AgNPs enhanced the toxicity of AgNPs because the amount of internalized Ag+ ions released from the particle surface increased, which could be driven by the proton motive force of the bacterial membrane that decreases the local pH to as low as 3.18 Similarly, Ivask et al. analysed the amount of bioavailable Ag inside bacterial cells using bacterial bioreporters and showed that the concentrations of bioavailable Ag were substantially higher than the amount predicted based on the concentration of (abiotic) dissolved silver from the same NPs.16 These findings suggest that the particle–cell interface plays an essential role in the antibacterial action of AgNPs and therefore support a synergistic effect between AgNPs and Ag+ ions where the AgNPs can deliver a localised ionic dose to the cell.18 The low local pH around bacterial membranes is expected, according to eqn (6), to lead to higher AgNP dissolution rates. However, in our previous work, low pH values promoted the aggregation and coarsening of 20 nm citrate-capped AgNPs.75 As discussed in the section “Particle size,” this would compromise the ability of AgNPs to bind on bacterial cell walls.76 Therefore further investigation is necessary in order to deconvolute the effects of each parameter on the bioreactivity of AgNPs.
Particle size is considered as one of the main physicochemical properties that affect the bactericidal behaviour of AgNPs, as explained by eqn (6).15 Metal particles with smaller diameters present a very high surface area compared to larger particles, potentially increasing their overall reactivity.77 Morones et al., for instance, demonstrated that the binding strength of particles to bacteria was dependent on the surface of interaction.24 Four types of Gram-negative bacteria (E. coli, V. cholera, P. aeruginosa and S. typhus) were exposed to AgNPs in the range of 1–100 nm and examined by complementary analytical electron microscopy techniques, including TEM in combination with elemental mapping analysis and high angle annular dark field (HAADF) scanning transmission electron microscopy (STEM). The combination of these techniques provided spatially resolved maps showing the morphological changes of bacteria following incubation with AgNPs, and the anatomical position of AgNPs inside the cell. Only the smallest particles, with diameters of 1–10 nm, were found on the surface of the cell wall and inside the bacteria. The presence of very small (∼1 nm) AgNPs was detected using HAADF-STEM, a technique in which images are formed by electrons that have been scattered to high angles, due to Rutherford scattering. As a result, the intensity of the images is related to Z2, which provides high contrast between metallic silver and organic material.78 The authors suggested that the mechanism through which AgNPs were able to penetrate into E. coli was mediated by changes to the structure of the cell wall, which led to an increase in its permeability.25 In another report, however, AgNPs from 20 nm up to 80 nm were able to penetrate through the inner and outer membrane of P. aeruginosa, suggesting that bacteria may also internalize larger particles.26 The data produced in both studies did not provide enough details about the structural damage induced to the cell membrane or possible differences in the uptake mechanism of AgNPs of different sizes. Moreover, the aggregation states of AgNPs in the media used in the two studies were not characterized, which could be another factor that affects their uptake. The effect of aggregation was demonstrated for E. coli exposed to 20–30 nm citrate-coated AgNPs, which were compared to citrate- and polycyclic aromatic hydrocarbon (PAH)-coated gold nanoparticles (AuNPs). TEM and field emission scanning electron microscopy (FEG-SEM) revealed that the binding of Ag or AuNPs to bacteria was different depending on their aggregation state. PAH-coated AuNPs were found to be more strongly bound to cell walls and penetrated more readily inside bacteria than citrate-capped AgNPs, which tended to aggregate more readily.76 Consequently, further research is needed in order to understand if the interaction of AgNPs with bacterial membranes or their internalization is size-dependent and what is the role of aggregation. Determining the optimal particle size for enhanced interaction between AgNPs and bacteria will enable an increase of their antibacterial activity while keeping the dose as low as possible. The use of 3D imaging techniques (e.g. electron tomography, focused ion beam scanning electron microscopy) would be beneficial for this purpose, as they can provide valuable information about the interaction dynamics of NPs with and within the cells, which can be limited or lost in the case of 2D imaging.
Positively charged surface coatings have been shown to play a key role in accelerating cell wall breakdown and cytoplasm release.76 For example, cationic polymer-stabilized AgNPs bind to the negatively charged bacterial surface via electrostatic interactions.81 However, the potential of using these coatings for biomedical applications remains an obstacle due to the high cytotoxicity of the polymers.82,83 One alternative to overcome this problem would be the use of peptides or polysaccharides containing amine groups.84 In one study, a pronounced destruction of the bacterial cell wall was observed in P. aeruginosa exposed to chitosan-coated AgNPs using SEM.84 Bacitracin A (BA) and polymyxin E (PE) are polypeptides with cationic macrocyclicamido groups, which are known as antimicrobial peptides (AMPs). These peptides were used by Mei et al. in the synthesis of functionalized AgNPs for the treatment of both Gram-negative (E. coli and P. aeruginosa) and Gram-positive bacteria (S. aureus and B. amyloliquefaciens).85 The immobilization of both peptides on the AgNP surface increased their antibacterial activity up to a 10-fold without symptoms of bacterial resistance. Fluorescence microscopy and TEM revealed that BA/PE-AgNPs were localized on the surface of bacteria and inside the cytoplasm. The membrane appeared seriously damaged, showing an undulating appearance and vesicles emanating from the cell wall, while some specimens were more severely damaged and showed cytoplasmic release (Fig. 2). The major target sites for antimicrobial agents are often present at the outermost layers of the bacterial cell. The cell wall of Gram-positive bacteria is rich in teichoic acids, whose main function is to provide rigidity by attracting cations such as Mg2+.86 AMPs are considered excellent antimicrobial agents87,88 because their macrocyclicamido groups can participate in metal–ligand p-bonding, producing a complex with Ca2+ and Mg2+ cations. Therefore, it has been suggested that BA and PE on the AgNP surface can chelate Mg2+ and Ca2+ and extract them from the original binding sites in teichoic acids, thus damaging the bacterial cell wall.85 Moreover, the same study showed the presence of BA/PE-NPs in the bacterial nucleoid. These findings suggested that AgNPs could bind to ribosomes and chromosomes, resulting in an inhibition of ribosomal function and a suppression of DNA replication, ultimately causing cell death.24 The important role that organic surface coronas surrounding nanoparticles may play towards controlling nanomaterial properties for biological applications has also been demonstrated by Daima et al.17 AgNPs were modified by the surface corona of biologically active polyoxometalates (POMs), which enhanced the damage to bacterial cells due to a synergistic antibacterial action of AgNPs and POMs. SEM micrographs revealed significant physical damage to treated bacterial cells, such as big holes in the bacterial structure or even complete disintegration. The modified AgNPs were toxic to both Gram positive and Gram negative bacteria, while being biocompatible with PC3 epithelial mammalian cells, suggesting their potential towards specific antimicrobial targeting.17
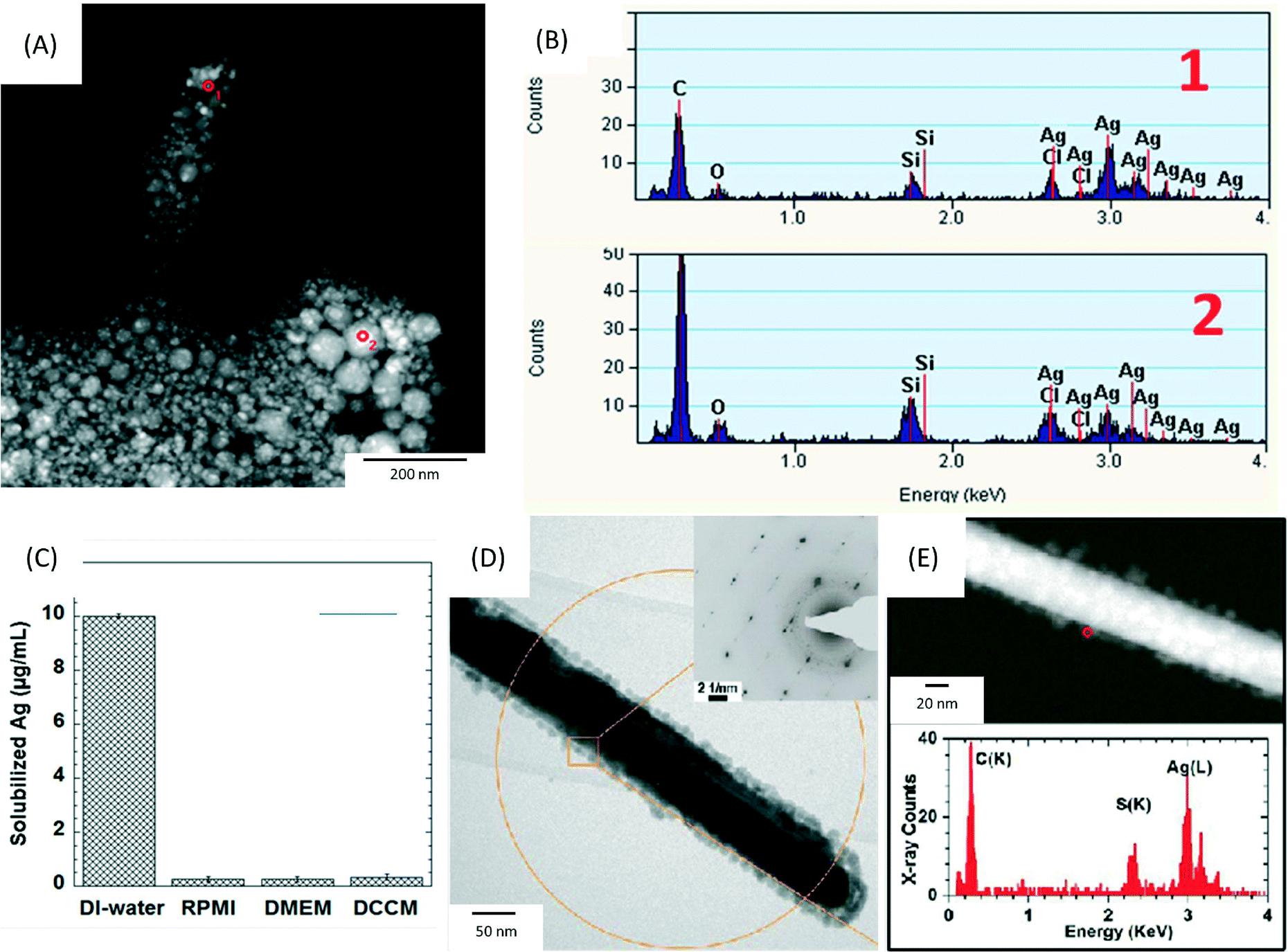 | ||
| Fig. 2 (A) High angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) image of precipitates formed after the incubation of 17.0 μg mL−1 AgNO3 (equal to a Ag concentration of 10 μg mL−1) in RPMI-1640 at 37 °C for 0.5 h. The precipitates were collected by filtering the solution through a 2 kDa filter membrane and washed 3 times by DI-water. (B) The corresponding energy-dispersive X-ray spectroscopy (EDX) spectra collected from the areas marked 1–2 in (A). (C) Inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis of solubilized silver concentrations following incubation at 37 °C for 0.5 h of 17.0 μg mL−1 AgNO3 in DI-water, RPMI-1640, DMEM and DCCM-1 cell culture media (n = 3). (D, E) Physicochemical characterization of silver nanowires (AgNWs) incubated for 6 h at 37 °C in small molecule solutes and salts extracted from DCCM-1. (D) Representative bright field transmission electron microscopy (BF-TEM) image of the AgNWs. The inset is a selected area electron diffraction (SAED) pattern obtained from the circled area (aperture size ∼550 nm). (E) HAADF-STEM image (top) obtained from the same area shown in (D), and EDX spectrum (bottom) collected from the edge of the AgNW (circled in E, top). Reprinted with permission from ref. 89. Copyright 2013 American Chemical Society. | ||
Therefore, surface coatings will affect the dissolution rates, bioavailability and aggregation state of AgNPs in vitro and in vivo. AgNPs aggregation or dispersion in cell culture media can be controlled by modifying their surface properties (i.e. hydrophobic or hydrophilic coatings), altering their uptake inside cells by changing their contact with the cell membrane. In fact, some publications have demonstrated how polymer stabilizers may play an important role in determining the toxicity of AgNPs by reducing the exposure of cells to particles.46 The surface properties of AgNPs may govern to a large extent their interaction with bacterial cell walls via electrostatic attraction, by complexation of metal cations localized on the cell wall, or through the interaction with sulphur-containing membrane proteins. Regardless of the exact mechanism involved, it is clear that AgNP and bacterial cell wall interactions increase the permeability of the cell wall and consequently lead to its disruption.
In summary, published literature has provided enough evidence that the release of Ag+ ions from the surface of AgNPs may be regarded as the main mechanism of antibacterial activity of AgNPs. However, it is becoming increasingly clear that dissolution of AgNPs in the exposure medium cannot wholly account for the observed antimicrobial effects. Additional effects at the particle–bacterial membrane interface and inside the cells seem to play a role in the action of AgNPs. Therefore, a synergistic effect between Ag+ ions and AgNPs must be considered in order to obtain accurate conclusions on the antibacterial pathways. Recent findings illustrate the importance of membrane interactions in the antimicrobial activity of AgNPs, as they can translocate across, and become internalized, directly through the cytoplasm or inside vesicles, where their subsequent dissolution driven by an oxidising and acidic pH at the plasma membrane could lead to very high local concentrations of Ag+ ions.16 Further research is necessary in order to understand the exact physicochemical and cellular mechanisms underlying these interactions and how they can be exploited in the design of therapeutic AgNPs.
Importance of materials characterization
Given that several physicochemical properties of AgNPs have been shown to influence their bioreactivity, characterisation of these properties in biological systems and in cell culture media at the point of exposure will be paramount in order to draw accurate conclusions about their biological activity.35,37 Inductively coupled plasma mass spectrometry and optical emission spectroscopy (ICP-MS and ICP-OES) or atomic absorption spectroscopy (AAS) techniques are commonly used to correlate the dissolution rate of AgNPs with their toxicity profiles. Table 2 shows that a wide variety of experimental setups and analytical methodologies are employed in in vitro studies to quantify the amount of Ag+ ions released from AgNPs. However, there are some discrepancies in the data provided by different authors, probably due to the use of inconsistent methodologies, an improper NP characterisation or the lack of dose response considerations in in vitro systems. This might explain, to some extent, why inconsistent dissolution rates are observed even when the same types of AgNPs are used. For instance, in the case of 10 nm citrate-AgNPs, different dissolution percentages were measured by UV-vis absorption spectroscopy and AAS, which required a previous centrifugation step.16 Due to the small size of AgNPs, they could not be effectively separated from solution by centrifugation and the values obtained by AAS were anomalously high.| Type of AgNPs | Experimental setup | Analytical methodology and analysis technique | Released Ag + ions | Ref. |
|---|---|---|---|---|
| n.q.: not quantified. ICP-MS: Inductively coupled plasma mass spectrometry. ICP-OES: Inductively coupled plasma optical emission spectroscopy. AAS: Atomic absorption spectroscopy. | ||||
| 10.5 ± 4.3 nm PVP-AgNPs | 10 μg ml−1 of AgNPs incubated in test media (half-strength NaCl-free lysogeny broth (LB)) for 4 h at 30 °C | Ultracentrifugation (1 h at 390000 g) |
4.4% | 18 |
| AAS | ||||
| 14 ± 1.6 nm Citrate-AgNPs | 25 μg ml−1 of AgNPs incubated in non-interacting perchlorate (ClO4) buffer solutions at pH 5 for 336 h | Ultrafiltration through 2 KDa filter membranes (13000 rpm) |
~2% | 75 |
| ICP-OES | ||||
| 20 nm Citrate-AgNPs | 300 μg ml−1 of AgNPs incubated in quarter-strength Hoagland media for 24 h and 336 h | Ultrafiltration through 3 kDa filter membranes (40min at 5000 g) |
17% (24 h) 40% (336 h) | 50 |
| ICP-MS | ||||
| 20 nm Citrate-AgNPs | 12.5 μg ml−1 of AgNPs incubated in BEGM cell media for 24 h at 37 °C | Centrifugation (1 h at 15000 rpm) |
~4.3% | 95 |
| ICP-OES | ||||
| 40 nm Citrate-AgNPs | 10 μg ml−1 of AgNPs incubated in BEGM cell media at pH 4.5 for 24 h at 37 °C | Centrifugation (1 h at 15000 rpm) |
~7% | 129 |
| AAS | ||||
| PVP-AgNWs 129 ± 74 nm in diameter and a bimodal length of 2.8 ± 2.4 μm and 7.0 ± 2.0 μm | 10 μg ml−1 of AgNPs incubated in DMEM, RPMI1640 and DCCM-1cell media for 168 h at 37 °C | Ultrafiltration through 2 KDa filter membranes (13000 rpm) |
n.q. | 89 |
| ICP-OES | ||||
| 10 nm Citrate-AgNPs | 12.5 μg ml−1 of AgNPs incubated in DI water or bacterial growth media ( pH 7) for 24 h at 37 °C | UV-vis | 3.5% (DI) 7% (bacterial growth media) | 16 |
| Centrifugation (1 h at 21000 g) AAS |
16.1% (DI) 26% (bacterial growth media) | |||
Furthermore, our group has recently revealed limitations in using ICP-OES for analysing Ag+ ion release in cell culture media.89 This study demonstrated that Ag+ ions bind with complex protein thiol groups or other anions in the medium such as Cl−, PO43−, S2− and SO42−, forming insoluble silver compounds, e.g. silver oxide and silver chloride (Fig. 3A and B). These compounds would reduce the bioavailability of released silver ions and could confound the interpretation of the Ag+ ion release rate via ICP studies. The authors compared the amount of free Ag+ ions that can be measured when AgNO3 is added in DI water compared to different culture media (RPMI, DMEM and DCCM), as shown in Fig. 3C. Although 100% of Ag+ ions were measured by ICP-OES in DI-water, the recovery of Ag+ was less than 5% in cell media. Therefore, non-interacting buffers (e.g. perchlorate buffer solutions) are needed when studying the dissolution rates of AgNPs in in vitro studies, with an understanding that significant sequestering of free Ag+ ion will occur in the in vivo environment. The same study also investigated how different sources of sulphur in the cellular environment can lead to transformations of the surface chemistry of AgNWs, using a set of spatially resolved analytical TEM techniques. The formation of silver sulphide (Ag2S) crystals on the surface of AgNWs within 1 h of incubation in DCCM-1 was identified by high resolution TEM (Fig. 3D and E). In contrast, incubation of AgNWs in RPMI-1640 or DMEM did not lead to sulfidation. These findings highlight the need to consider the effects of cell culture media in the analysis of toxicity assays, as well as the potential of high resolution analytical TEM for the detection of insoluble silver species, in contrast to ICP-OES.89
 | ||
| Fig. 3 TEM images of Gram-positive bacteria (S. areus) (A–E) and Gram-negative bacteria (P. aeruginosa) (F), before (A) and after (B–F) exposure to AgNPs, showing ultrastructural damage in bacterial cells induced by AgNPs. Reprinted with permission from ref. 85. Copyright 2013 Elsevier Ltd. | ||
Determining the fundamental physicochemical properties of AgNPs (e.g. size, coating and surface properties) in biological systems will be vital to understand their bioreactivity and antibacterial activity. When performing biological assays, the oxidation kinetics of AgNPs must be studied under the same biological conditions employed in the assay. To achieve this, standardized protocols need to be developed, that combine different techniques such as ICP-MS, UV-vis and high resolution imaging. There is also an urgent need to develop methods for the quantification of oxidation rates of AgNPs inside cells. In most of the published results, it is difficult to distinguish between the effects induced by AgNPs and/or Ag+ ions, because it is unclear whether NP dissolution occurs in the extracellular medium before uptake or intracellularly following ingestion. Recently, our group used ion-sensitive fluorescent dyes combined with confocal microscopy to visualize the amount of ionic Zn2+ released from ZnO NWs inside human macrophages.90 On the other hand, the development of a sensitive and selective colorimetric Ag+ detection method is still under research. Application of these methods could provide fundamental insight into the mechanisms by which AgNPs exert their biological effects by deconvoluting the effects of Ag+ ions and particles.
Conclusions and perspectives
The information presented in this review provides strong evidence to conclude that AgNPs present an exciting opportunity as antibacterial agents in biomedical applications. Further research is required to improve our understanding on the stability of AgNPs and the kinetics of Ag+ ion release in biological environments. AgNPs are highly dynamic systems, whose properties can undergo dramatic changes when incubated in biological media, leading for example to aggregation, the formation of biomolecule coronas or the precipitation of insoluble Ag species. Consequently, characterization of as-synthesized AgNPs alone is not enough to predict their biological activity. Appropriate characterization should also take place in the cell culture media and under the same conditions (e.g. temperature, time) as used in in vitro studies. Experimental techniques which have been commonly used in the past for AgNP characterization, such as atomic absorption spectroscopy or dynamic light scattering, have been proven to possess limitations in detecting transformations in the physicochemistry of AgNPs in different biological environments; therefore several complementary techniques need to be used. Moreover, characterization of AgNPs at the particle–cell interface to investigate possible transformations of the particles at the cellular environment can provide valuable information about the interactions of AgNPs with cellular components.Additionally, results on the mechanism of antibacterial action of AgNPs remain inconclusive, with further studies necessary to investigate the synergistic effect between AgNPs and Ag+ ions, in order to provide a holistic understanding of the system interactions and elucidate how the physicochemical properties of AgNPs affect bacterial responses. Similar discrepancies also exist in cytotoxicity studies evaluating the biological effects of AgNPs in host cells. Therefore, the focus should be placed on bridging the gap between toxicological studies and analytical techniques. While traditional cytotoxicity assays are extremely useful to evaluate the biological activity of AgNPs, new approaches based on the correlative application of high spatial and energy resolution analytical microscopy techniques may offer an improved understanding of the mechanisms by which AgNPs interact with cells, and can guide the selection of particular toxicological assays to test. For example, cytoplasmic localization of AgNPs could be correlated with plasma membrane permeability and reactive oxygen species (ROS) generation; mitochondrial AgNP localization could be related to measurements of mitochondrial membrane potential; or nuclear localization of AgNPs could be correlated with genotoxicity assays.16 Imaging and analysis also allows us to determine whether the toxicological findings relate directly to the location of AgNPs in the cell, or whether they are more general. The development of new methods for the quantification of Ag+ ions released intracellularly will also prove invaluable in discriminating between the effects of AgNPs and Ag+ ions. Understanding which properties of AgNPs drive their bioreactivity will enable their optimization as antimicrobial agents and will guide the development of these materials in nanomedicine and for environmental applications. For instance, the antibacterial effects of AgNPs may be strongly improved by using AgNPs coated with specific groups that enable their binding to cations or sulphur containing proteins present on the bacterial membrane, or by increasing the electrostatic interaction with the negatively charged cell wall. Moreover, treatment of bacterial infections is difficult due to the lack of known cellular targets of infected cells. Further research to identify these targets could lead to the design of novel antibacterial agents based on AgNPs coated with ligands that target specific receptors.
One of the reasons which make AgNPs attractive as antibacterial agents is the fact that bacteria are not likely to become resistant to silver like antibiotics, due to the wide range of possible interactions of Ag+ ions with biomolecules. The biggest challenge will be to determine the “therapeutic window” where AgNPs can be used as antibacterial therapeutics for infected cells without affecting the metabolism of healthy cells.57 AgNPs have been identified as a possible exposure hazard91 and they have been shown to cause inflammation in the lung following inhalation. In inhalation studies using rodents, AgNPs induce pulmonary inflammation with mixed cellular infiltrates and decreased minute volumes, leading to the development of pulmonary function abnormalities.92–94 Recently, Wang et al. compared the effects of 20 and 110 nm AgNPs and demonstrated more acute pulmonary effects of the smaller particles, whereas the 110 nm particles induced mild pulmonary fibrosis.95 The authors suggested that these differences arose due to a difference in the rate at which Ag+ ions were released from the particles. Consequently, controlling the reactivity of AgNPs will depend on controlling the size and surface chemistry of AgNPs.
Undeniably, AgNPs hold a great potential for the development of novel treatment options for bacterial infections. There are certainly still several challenges to overcome in the design of AgNPs for medical applications, but a high control over their properties, an improved fundamental understanding of their bioreactivity pathways and the application of advanced and accurate nanometrology may hold the key that will open the door for the development of safe AgNP platforms for therapeutic treatment and bio-imaging applications.
Acknowledgements
This work was funded in part by a grant from the US EPA/NERC (EPA STAR RD83469301 and NERC). AEP acknowledges an ERC starting grant (Project number 257182; CNTBBB). AEP also acknowledges an Elsie Widdowson Fellowship from Imperial College London for support. BFL is grateful for a PhD scholarship from the Ministry of Higher Education of Malaysia.Notes and references
- A. J. Thorley and T. D. Tetley, Pharmacol. Ther., 2013, 140, 176–185 CrossRef CAS PubMed.
- A. Babu, A. K. Templeton, A. Munshi and R. Ramesh, J. Nanomater., 2013, 2013, 11 Search PubMed.
- M. Paranjpe and C. C. Muller-Goymann, Int. J. Mol. Sci., 2014, 15, 5852–5873 CrossRef CAS PubMed.
- J. W. Alexander, Surg. Infect., 2009, 10, 289–292 CrossRef PubMed.
- P. V. Dasaraju and C. Liu, in Medical Microbiology, ed. S. Baron, The University of Texas Medical Branch at Galveston, Galveston TX, 1996 Search PubMed.
- D. P. Speert, Novartis Found. Symp., 2006, 279, 42–51 CAS , disussion 51–45, 216–219.
- T. Lawrence and G. Natoli, Nat. Rev. Immunol., 2011, 11, 750–761 CrossRef CAS PubMed.
- D. L. Clemens and M. A. Horwitz, J. Exp. Med., 1995, 181, 257–270 CrossRef CAS.
- J. Keane, H. G. Remold and H. Kornfeld, J. Immunol., 2000, 164, 2016–2020 CrossRef CAS.
- C. Loeuillet, F. Martinon, C. Perez, M. Munoz, M. Thome and P. R. Meylan, J. Immunol., 2006, 177, 6245–6255 CrossRef CAS.
- S. Chernousova and M. Epple, Angew. Chem., Int. Ed., 2013, 52, 1636–1653 CrossRef CAS PubMed.
- S. Eckhardt, P. S. Brunetto, J. Gagnon, M. Priebe, B. Giese and K. M. Fromm, Chem. Rev., 2013, 113, 4708–4754 CrossRef CAS PubMed.
- G. B. Braun, T. Friman, H.-B. Pang, A. Pallaoro, T. H. D. Mendoza, A.-M. A. Willmore, V. R. Kotamraju, A. P. Mann, Z.-G. She, K. N. Sugahara, N. Reich, T. Teesalu and E. Ruoslahti, Nat. Mater., 2014, 3982, 1–8 Search PubMed.
- S. Pal, Y. K. Tak and J. M. Song, Appl. Environ. Microbiol., 2007, 73, 1712–1720 CrossRef CAS PubMed.
- G. A. Sotiriou and S. E. Pratsinis, Environ. Sci. Technol., 2010, 44, 5649–5654 CrossRef CAS PubMed.
- A. Ivask, A. ElBadawy, C. Kaweeteerawat, D. Boren, H. Fischer, Z. Ji, C. H. Chang, R. Liu, T. Tolaymat, D. Telesca, J. I. Zink, Y. Cohen, P. A. Holden and H. A. Godwin, ACS Nano, 2013, 8, 374–386 CrossRef PubMed.
- H. K. Daima, P. R. Selvakannan, A. E. Kandjani, R. Shukla, S. K. Bhargava and V. Bansal, Nanoscale, 2014, 6, 758–765 RSC.
- O. Bondarenko, A. Ivask, A. Käkinen, I. Kurvet and A. Kahru, PLoS One, 2013, 8, e64060 CAS.
- J. S. McQuillan, H. Groenaga Infante, E. Stokes and A. M. Shaw, Nanotoxicology, 2012, 6, 857–866 CrossRef CAS PubMed.
- S. Kim and D. Y. Ryu, J. Appl. Toxicol., 2013, 33, 78–89 CrossRef PubMed.
- M. A. Ramírez-Lee, H. Rosas-Hernández, S. Salazar-García, J. M. Gutiérrez-Hernández, R. Espinosa-Tanguma, F. J. González, S. F. Ali and C. González, Toxicol. Lett., 2014, 224, 246–256 CrossRef PubMed.
- J. Park, D.-H. Lim, H.-J. Lim, T. Kwon, J.-s. Choi, S. Jeong, I.-H. Choi and J. Cheon, Chem. Commun., 2011, 47, 4382–4384 RSC.
- R. de Lima, A. B. Seabra and N. Duran, J. Appl. Toxicol., 2012, 32, 867–879 CrossRef PubMed.
- J. R. Morones, J. L. Elechiguerra, A. Camacho, K. Holt, J. B. Kouri, J. T. Ramirez and M. J. Yacaman, Nanotechnology, 2005, 16, 2346–2353 CrossRef CAS PubMed.
- I. Sondi and B. Salopek-Sondi, J. Colloid Interface Sci., 2004, 275, 177–182 CrossRef CAS PubMed.
- X.-H. N. Xu, W. J. Brownlow, S. V. Kyriacou, Q. Wan and J. J. Viola, Biochemistry, 2004, 43, 10400–10413 CrossRef CAS PubMed.
- S. E. Autenrieth, P. Warnke, G. H. Wabnitz, C. Lucero Estrada, K. A. Pasquevich, D. Drechsler, M. Günter, K. Hochweller, A. Novakovic, S. Beer-Hammer, Y. Samstag, G. J. Hämmerling, N. Garbi and I. B. Autenrieth, PLoS Pathog., 2012, 8, e1002552 CAS.
- M. I. Colombo, Cell Death Differ., 2005, 12 Suppl(2), 1481–1483 CrossRef PubMed.
- K. Yuan, C. Huang, J. Fox, D. Laturnus, E. Carlson, B. Zhang, Q. Yin, H. Gao and M. Wu, J. Cell Sci., 2012, 125, 507–515 CrossRef CAS PubMed.
- J. Moller, T. Luhmann, M. Chabria, H. Hall and V. Vogel, Sci. Rep., 2013, 3 Search PubMed.
- D. G. Russell, Mol. Cell. Biol., 2001, 2, 569–586 CAS.
- M. Podinovskaia, W. Lee, S. Caldwell and D. G. Russell, Cell. Microbiol., 2013, 15, 843–859 CrossRef CAS PubMed.
- J. Lee, T. Repasy, K. Papavinasasundaram, C. Sassetti and H. Kornfeld, PLoS One, 2011, 6, e18367 CAS.
- S. Jamwal, M. K. Midha, H. N. Verma, A. Basu, K. V. S. Rao and V. Manivel, Sci. Rep., 2013, 3 Search PubMed.
- X. Yang, A. P. Gondikas, S. M. Marinakos, M. Auffan, J. Liu, H. Hsu-Kim and J. N. Meyer, Environ. Sci. Technol., 2011, 46, 1119–1127 CrossRef PubMed.
- M. A. Chappell, L. F. Miller, A. J. George, B. A. Pettway, C. L. Price, B. E. Porter, A. J. Bednar, J. M. Seiter, A. J. Kennedy and J. A. Steevens, Chemosphere, 2011, 84, 1108–1116 CrossRef CAS PubMed.
- L. Stebounova, E. Guio and V. Grassian, J. Nanopart. Res., 2011, 13, 233–244 CrossRef CAS.
- J. Liu, D. A. Sonshine, S. Shervani and R. H. Hurt, ACS Nano, 2010, 4, 6903–6913 CrossRef CAS PubMed.
- J. Liu and R. H. Hurt, Environ. Sci. Technol., 2010, 44, 2169–2175 CrossRef CAS PubMed.
- J. Liu, Z. Wang, F. D. Liu, A. B. Kane and R. H. Hurt, ACS Nano, 2012, 6, 9887–9899 CrossRef CAS PubMed.
- S.-J. Yu, Y.-G. Yin, J.-B. Chao, M.-H. Shen and J.-F. Liu, Environ. Sci. Technol., 2013, 48, 403–411 CrossRef PubMed.
- R. D. Glover, J. M. Miller and J. E. Hutchison, ACS Nano, 2011, 5, 8950–8957 CrossRef CAS PubMed.
- A. K. Suresh, D. A. Pelletier, W. Wang, J. L. Morrell-Falvey, B. Gu and M. J. Doktycz, Langmuir, 2012, 28, 2727–2735 CrossRef CAS PubMed.
- S. Kittler, C. Greulich, J. Diendorf, M. Köller and M. Epple, Chem. Mater., 2010, 22, 4548–4554 CrossRef CAS.
- Z.-m. Xiu, Q.-b. Zhang, H. L. Puppala, V. L. Colvin and P. J. J. Alvarez, Nano Lett., 2012, 12, 4271–4275 CrossRef CAS PubMed.
- J. J. Lin, W. C. Lin, R. X. Dong and S. H. Hsu, Nanotechnology, 2012, 23, 065102 CrossRef PubMed.
- M. V. Park, A. M. Neigh, J. P. Vermeulen, L. J. de la Fonteyne, H. W. Verharen, J. J. Briede, H. van Loveren and W. H. de Jong, Biomaterials, 2011, 32, 9810–9817 CrossRef CAS PubMed.
- B. Reidy, A. Haase, A. Luch, K. Dawson and I. Lynch, Materials, 2013, 6, 2295–2350 CrossRef CAS PubMed.
- W. Zhang, Y. Yao, N. Sullivan and Y. Chen, Environ. Sci. Technol., 2011, 45, 4422–4428 CrossRef CAS PubMed.
- A. Pratsinis, P. Hervella, J.-C. Leroux, S. E. Pratsinis and G. A. Sotiriou, Small, 2013, 9, 2576–2584 CrossRef CAS PubMed.
- E. Navarro, F. Piccapietra, B. Wagner, F. Marconi, R. Kaegi, N. Odzak, L. Sigg and R. Behra, Environ. Sci. Technol., 2008, 42, 8959–8964 CrossRef CAS.
- O. Choi and Z. Hu, Environ. Sci. Technol., 2008, 42, 4583–4588 CrossRef CAS.
- J. Fabrega, S. R. Fawcett, J. C. Renshaw and J. R. Lead, Environ. Sci. Technol., 2009, 43, 7285–7290 CrossRef CAS.
- L. Yin, Y. Cheng, B. Espinasse, B. P. Colman, M. Auffan, M. Wiesner, J. Rose, J. Liu and E. S. Bernhardt, Environ. Sci. Technol., 2011, 45, 2360–2367 CrossRef CAS PubMed.
- O. Choi, K. K. Deng, N.-J. Kim, L. Ross Jr, R. Y. Surampalli and Z. Hu, Water Res., 2008, 42, 3066–3074 CrossRef CAS PubMed.
- X. Chen and H. J. Schluesener, Toxicol. Lett., 2008, 176, 1–12 CrossRef CAS PubMed.
- J. R. Morones-Ramirez, J. A. Winkler, C. S. Spina and J. J. Collins, Sci. Transl. Med., 2013, 5(190), 190ra181 Search PubMed.
- C. Marambio-Jones and E. V. Hoek, J. Nanopart. Res., 2010, 12, 1531–1551 CrossRef CAS.
- W.-R. Li, X.-B. Xie, Q.-S. Shi, H.-Y. Zeng, Y.-S. Ou-Yang and Y.-B. Chen, Appl. Microbiol. Biotechnol., 2010, 85, 1115–1122 CrossRef CAS PubMed.
- F. Mirzajani, A. Ghassempour, A. Aliahmadi and M. A. Esmaeili, Res. Microbiol., 2011, 162, 542–549 CrossRef CAS PubMed.
- Y. Matsumura, K. Yoshikata, S.-I. Kunisaki and T. Tsuchido, Appl. Environ. Microbiol., 2003, 69, 4278–4281 CrossRef CAS.
- K. B. Holt and A. J. Bard, Biochemistry, 2005, 44, 13214–13223 CrossRef CAS PubMed.
- C.-N. Lok, C.-M. Ho, R. Chen, Q.-Y. He, W.-Y. Yu, H. Sun, P. K.-H. Tam, J.-F. Chiu and C.-M. Che, J. Proteome Res., 2006, 5, 916–924 CrossRef CAS PubMed.
- H.-J. Park, J. Y. Kim, J. Kim, J.-H. Lee, J.-S. Hahn, M. B. Gu and J. Yoon, Water Res., 2009, 43, 1027–1032 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- O. Gordon, T. Vig Slenters, P. S. Brunetto, A. E. Villaruz, D. E. Sturdevant, M. Otto, R. Landmann and K. M. Fromm, Antimicrob. Agents Chemother., 2010, 54, 4208–4218 CrossRef CAS PubMed.
- E. T. Hwang, J. H. Lee, Y. J. Chae, Y. S. Kim, B. C. Kim, B.-I. Sang and M. B. Gu, Small, 2008, 4, 746–750 CrossRef CAS PubMed.
- L. Sintubin, B. De Gusseme, P. Van der Meeren, B. G. Pycke, W. Verstraete and N. Boon, Appl. Microbiol. Biotechnol., 2011, 91, 153–162 CrossRef CAS PubMed.
- Q. L. Feng, J. Wu, G. Q. Chen, F. Z. Cui, T. N. Kim and J. O. Kim, J. Biomed. Mater. Res., 2000, 52, 662–668 CrossRef CAS.
- L. Nover and K. D. Scharf, Cell. Mol. Life Sci., 1997, 53, 80–103 CrossRef CAS.
- K. K. Y. Wong and X. Liu, MedChemComm, 2010, 1, 125–131 RSC.
- M. Raffi, F. Hussain, T. M. Bhatti, J. I. Akhter, A. Hameed and M. M. Hasan, J. Mater. Sci. Technol., 2008, 24, 192–196 CAS.
- X. Jin, M. Li, J. Wang, C. Marambio-Jones, F. Peng, X. Huang, R. Damoiseaux and E. M. V. Hoek, Environ. Sci. Technol., 2010, 44, 7321–7328 CrossRef CAS PubMed.
- A. Taglietti, Y. A. Diaz Fernandez, E. Amato, L. Cucca, G. Dacarro, P. Grisoli, V. Necchi, P. Pallavicini, L. Pasotti and M. Patrini, Langmuir, 2012, 28, 8140–8148 CrossRef CAS PubMed.
- B. F. Leo, S. Chen, Y. Kyo, K. L. Herpoldt, N. J. Terrill, I. E. Dunlop, D. S. McPhail, M. S. Shaffer, S. Schwander, A. Gow, J. Zhang, K. F. Chung, T. D. Tetley, A. E. Porter and M. P. Ryan, Environ. Sci. Technol., 2013, 47, 11232–11240 CrossRef CAS PubMed.
- Y. Zhou, Y. Kong, S. Kundu, J. D. Cirillo and H. Liang, J. Nanobiotechnol., 2012, 10, 19 CrossRef CAS PubMed.
- F. Raimondi, G. G. Scherer, R. Kötz and A. Wokaun, Angew. Chem., Int. Ed., 2005, 44, 2190–2209 CrossRef CAS PubMed.
- A. Porter and E. McGuire, in Encyclopedia of Nanotechnology, ed. B. Bhushan, Springer Netherlands, 2012, ch. 176, pp. 741–749 Search PubMed.
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, a000414 Search PubMed.
- G. D. Shockman and J. F. Barrett, Annu. Rev. Microbiol., 1983, 37, 501–527 CrossRef CAS PubMed.
- Y. Zhang, H. Peng, W. Huang, Y. Zhou, X. Zhang and D. Yan, J. Phys. Chem. C, 2008, 112, 2330–2336 CAS.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- J. Cai, Y. Yue, D. Rui, Y. Zhang, S. Liu and C. Wu, Macromolecules, 2011, 44, 2050–2057 CrossRef CAS.
- P. Jena, S. Mohanty, R. Mallick, B. Jacob and A. Sonawane, Int. J. Nanomed., 2012, 7, 1805–1818 CAS.
- L. Mei, Z. Lu, W. Zhang, Z. Wu, X. Zhang, Y. Wang, Y. Luo, C. Li and Y. Jia, Biomaterials, 2013, 34, 10328–10337 CrossRef CAS PubMed.
- C. Weidenmaier and A. Peschel, Nat. Rev. Microbiol., 2008, 6, 276–287 CrossRef CAS PubMed.
- L. Liu, K. Xu, H. Wang, P. K. Jeremy Tan, W. Fan, S. S. Venkatraman, L. Li and Y.-Y. Yang, Nat. Nanotechnol., 2009, 4, 457–463 CrossRef CAS PubMed.
- H. Wang, K. Xu, L. Liu, J. P. K. Tan, Y. Chen, Y. Li, W. Fan, Z. Wei, J. Sheng, Y.-Y. Yang and L. Li, Biomaterials, 2010, 31, 2874–2881 CrossRef CAS PubMed.
- S. Chen, I. G. Theodorou, A. E. Goode, A. Gow, S. Schwander, J. Zhang, K. F. Chung, T. D. Tetley, M. S. Shaffer, M. P. Ryan and A. E. Porter, Environ. Sci. Technol., 2013, 47, 13813–13821 CrossRef CAS PubMed.
- K. H. Muller, J. Kulkarni, M. Motskin, A. Goode, P. Winship, J. N. Skepper, M. P. Ryan and A. E. Porter, ACS Nano, 2010, 4, 6767–6779 CrossRef CAS PubMed.
- I. Theodorou, M. Ryan, T. Tetley and A. Porter, Int. J. Mol. Sci., 2014, 15, 23936–23974 CrossRef PubMed.
- J. H. Sung, J. H. Ji, J. D. Park, J. U. Yoon, D. S. Kim, K. S. Jeon, M. Y. Song, J. Jeong, B. S. Han, J. H. Han, Y. H. Chung, H. K. Chang, J. H. Lee, M. H. Cho, B. J. Kelman and I. J. Yu, Toxicol. Sci., 2009, 108, 452–461 CrossRef CAS PubMed.
- J. H. Sung, J. H. Ji, J. U. Yoon, D. S. Kim, M. Y. Song, J. Jeong, B. S. Han, J. H. Han, Y. H. Chung, J. Kim, T. S. Kim, H. K. Chang, E. J. Lee, J. H. Lee and I. J. Yu, Inhalation Toxicol., 2008, 20, 567–574 CrossRef CAS PubMed.
- K. S. Song, J. H. Sung, J. H. Ji, J. H. Lee, J. S. Lee, H. R. Ryu, J. K. Lee, Y. H. Chung, H. M. Park, B. S. Shin, H. K. Chang, B. Kelman and I. J. Yu, Nanotoxicology, 2013, 7, 169–180 CrossRef CAS PubMed.
- X. Wang, Z. Ji, C. H. Chang, H. Zhang, M. Wang, Y.-P. Liao, S. Lin, H. Meng, R. Li, B. Sun, L. V. Winkle, K. E. Pinkerton, J. I. Zink, T. Xia and A. E. Nel, Small, 2014, 10, 385–398 CrossRef CAS PubMed.
- W. Wang, R. Zhu, Q. Xie, A. Li, Y. Xiao, K. Li, H. Liu, D. Cui, Y. Chen and S. Wang, Int. J. Nanomed., 2012, 7, 3667–3677 CrossRef CAS PubMed.
- P. Xu, Q. Yin, J. Shen, L. Chen, H. Yu, Z. Zhang and Y. Li, Int. J. Pharm., 2013, 454, 21–30 CrossRef CAS PubMed.
- J. Varshosaz, S. Ghaffari, S. F. Mirshojaei, A. Jafarian, F. Atyabi, F. Kobarfard and S. Azarmi, BioMed Res. Int., 2013, 2013, 136859 CAS.
- J. Chuan, Y. Li, L. Yang, X. Sun, Q. Zhang, T. Gong and Z. Zhang, J. Nanopart. Res., 2013, 15, 1–9 CrossRef.
- Y. Z. Zhao, X. Li, C. T. Lu, Y. Y. Xu, H. F. Lv, D. D. Dai, L. Zhang, C. Z. Sun, W. Yang, X. K. Li, Y. P. Zhao, H. X. Fu, L. Cai, M. Lin, L. J. Chen and M. Zhang, Acta Diabetol., 2012, 49, 315–325 CrossRef CAS PubMed.
- M. Paranjpe, V. Neuhaus, J. H. Finke, C. Richter, T. Gothsch, A. Kwade, S. Buttgenbach, A. Braun and C. C. Muller-Goymann, Inhalation Toxicol., 2013, 25, 536–543 CrossRef CAS PubMed.
- R. Trivedi, E. F. Redente, A. Thakur, D. W. Riches and U. B. Kompella, Nanotechnology, 2012, 23, 505101 CrossRef PubMed.
- T. Zhao, H. Chen, Y. Dong, J. Zhang, H. Huang, J. Zhu and W. Zhang, Int. J. Nanomed., 2013, 8, 1947–1957 Search PubMed.
- D. Yoo, K. Guk, H. Kim, G. Khang, D. Wu and D. Lee, Int. J. Pharm., 2013, 450, 87–94 CrossRef CAS PubMed.
- B. Sinha, B. Mukherjee and G. Pattnaik, Nanomed.: Nanotechnol., Biol. Med., 2013, 9, 94–104 CrossRef CAS PubMed.
- A. Trapani, S. Di Gioia, N. Ditaranto, N. Cioffi, F. M. Goycoolea, A. Carbone, M. Garcia-Fuentes, M. Conese and M. J. Alonso, Int. J. Pharm., 2013, 447, 115–123 CrossRef CAS PubMed.
- J. Varshosaz, S. Taymouri and H. Hamishehkar, J. Appl. Polym. Sci., 2014, 131 Search PubMed.
- K. Sharma, S. Somavarapu, A. Colombani, N. Govind and K. M. Taylor, Int. J. Pharm., 2013, 455, 241–247 CrossRef CAS PubMed.
- A. M. A. Elhissi, M. A. Islam, B. Arafat, M. Taylor and W. Ahmed, Micro Nano Lett., 2010, 5, 184–188 CAS.
- L. M. Hoesel, M. A. Flierl, A. D. Niederbichler, D. Rittirsch, S. D. McClintock, J. S. Reuben, M. J. Pianko, W. Stone, H. Yang, M. Smith, J. V. Sarma and P. A. Ward, Antioxid. Redox Signaling, 2008, 10, 973–981 CAS.
- C. Liu, J. Shi, Q. Dai, X. Yin, X. Zhang and A. Zheng, Drug Dev. Ind. Pharm., 2015, 41, 272–278 CrossRef CAS PubMed.
- E. Kleemann, T. Schmehl, T. Gessler, U. Bakowsky, T. Kissel and W. Seeger, Pharm. Res., 2007, 24, 277–287 CrossRef CAS.
- L. Willis, D. Hayes Jr. and H. M. Mansour, Lung, 2012, 190, 251–262 CrossRef CAS PubMed.
- A. M. Ribeiro, A. C. Souza, A. C. Amaral, N. M. Vasconcelos, M. S. Jeronimo, F. P. Carneiro, L. H. Faccioli, M. S. Felipe, C. L. Silva and A. L. Bocca, J. Biomed. Nanotechnol., 2013, 9, 221–230 CrossRef CAS PubMed.
- M. Murata, T. Yonamine, S. Tanaka, K. Tahara, Y. Tozuka and H. Takeuchi, J. Pharm. Sci., 2013, 102, 1281–1289 CrossRef CAS PubMed.
- C. Jaafar-Maalej, V. Andrieu, A. Elaissari and H. Fessi, J. Nanosci. Nanotechnol., 2011, 11, 1841–1851 CrossRef CAS PubMed.
- X. Wu, D. Hayes Jr., J. B. Zwischenberger, R. J. Kuhn and H. M. Mansour, Drug Des., Dev. Ther., 2013, 7, 59–72 CAS.
- R. Ali, G. Mittal, R. Ali, M. Kumar, R. Kishan Khar, F. J. Ahmad and A. Bhatnagar, J. Microencapsulation, 2013, 30, 546–558 CrossRef CAS PubMed.
- W. H. Roa, S. Azarmi, M. H. Al-Hallak, W. H. Finlay, A. M. Magliocco and R. Lobenberg, J. Controlled Release, 2011, 150, 49–55 CrossRef CAS PubMed.
- C. W. Park, X. Li, F. G. Vogt, D. Hayes Jr., J. B. Zwischenberger, E. S. Park and H. M. Mansour, Int. J. Pharm., 2013, 455, 374–392 CrossRef CAS PubMed.
- A. R. Srinivasan and S. A. Shoyele, AAPS PharmSciTech, 2013, 14, 200–210 CrossRef CAS PubMed.
- S. R. Datir, M. Das, R. P. Singh and S. Jain, Bioconjugate Chem., 2012, 23, 2201–2213 CrossRef CAS PubMed.
- R. Govender, A. Phulukdaree, R. M. Gengan, K. Anand and A. A. Chuturgoon, J. Nanobiotechnol., 2013, 11, 5 CrossRef CAS PubMed.
- G. Zhou and W. Wang, Orient. J. Chem., 2012, 28, 651 CrossRef CAS.
- G. Peng, U. Tisch, O. Adams, M. Hakim, N. Shehada, Y. Y. Broza, S. Billan, R. Abdah-Bortnyak, A. Kuten and H. Haick, Nat. Nanotechnol., 2009, 4, 669–673 CrossRef CAS PubMed.
- Y. H. Chen, C. Y. Tsai, P. Y. Huang, M. Y. Chang, P. C. Cheng, C. H. Chou, D. H. Chen, C. R. Wang, A. L. Shiau and C. L. Wu, Mol. Pharmaceutics, 2007, 4, 713–722 CrossRef CAS PubMed.
- L. L. Ma, J. O. Tam, B. W. Willsey, D. Rigdon, R. Ramesh, K. Sokolov and K. P. Johnston, Langmuir, 2011, 27, 7681–7690 CrossRef CAS PubMed.
- B. Lkhagvadulam, J. H. Kim, I. Yoon and Y. K. Shim, BioMed Res. Int., 2013, 2013, 10 Search PubMed.
- A. R. Gliga, S. Skoglund, I. O. Wallinder, B. Fadeel and H. L. Karlsson, Part. Fibre Toxicol., 2014, 11, 11 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |