
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In-house-made capillary electrophoresis instruments coupled with contactless conductivity detection as a simple and inexpensive solution for water analysis: a case study in Vietnam†
Hong Anh
Duong
a,
Minh Duc
Le
a,
Kim Diem
Mai Nguyen
a,
Peter C.
Hauser
b,
Hung Viet
Pham
*a and
Thanh Duc
Mai
*a
aCentre for Environmental Technology and Sustainable Development (CETASD), Hanoi University of Science, Nguyen Trai Street 334, Hanoi, Vietnam. E-mail: maithanhduc83@gmail.com; phamhungviet@hus.edu.vn; Web: http://www.ce-vietnam.com/ Fax: +84 4 3858 8152; Tel: +33 651377949
bUniversity of Basel, Department of Chemistry, Spitalstrasse 51, 4056 Basel, Switzerland
First published on 24th September 2015
Abstract
A simple and inexpensive method for the determination of various ionic species in different water matrices is discussed in this study. The approach is based on the employment of in-house-made capillary electrophoresis (CE) instruments with capacitively coupled contactless conductivity detection (C4D), which can be realized even when only a modest financial budget and limited expertise are available. Advantageous features and considerations of these instruments are detailed following their pilot deployment in Vietnam. Different categories of ionic species, namely major inorganic cations (K+, Na+, Ca2+, Mg2+, and NH4+) and major inorganic anions (Cl−, NO3−, NO2−, SO42−, and phosphate), in different water matrices in Vietnam were determined using these in-house fabricated instruments. Inorganic trivalent arsenic (As(III)), which is the most abundant form of arsenic in reducing groundwater, was determined by CE-C4D. The effect of some interfering ions in groundwater on the analytical performance was investigated and is highlighted. The results from in-house-made CE-C4D-instruments were cross-checked with those obtained using the standard methods (AAS, AES, UV and IC), with correlation coefficients r2 ≥ 0.9 and deviations from the referenced results less than 15%.
Environmental impactThis work provides a simple and inexpensive analytical approach for water-quality monitoring which can be realized even when only a modest financial budget and limited expertise are available. This approach renders the aqueous environmental control activities more realizable even in local and decentralized areas. The case study was carried out in Vietnam where arsenic contamination in groundwater has been a serious problem. Inorganic major cations and anions, which are the primary indicators of water quality, together with abundantly present arsenite in groundwater were successfully monitored with the purpose-made instruments. Information on some water contaminants, i.e. ammonium and arsenic contamination, could be obtained without having recourse to expensive and sophisticated commercial instrumentation. |
1. Introduction
The concentrations of major ionic species, i.e. K+, Na+, Ca2+, Mg2+, NH4+, Cl−, NO3−, NO2−, SO42− and phosphate, are the primary indicators for the evaluation of water quality. Whether it is rain water, surface water or groundwater, only when the concentrations of these ions are determined to be within the regulated ranges, further analyses of other compounds (heavy metals, arsenic for instance) for confirmation of water quality are implemented. In Vietnam, groundwater has been used as an important source of drinking water, especially in rural areas.1,2 The quality of groundwater therefore is controlled periodically by monitoring the concentrations of different ionic species, of which those of major inorganic anions and cations are the first parameters to be determined. So far, inorganic cations usually have been determined by flame atomic absorption spectrometry (F-AAS, for earth alkali ions) or flame atomic emission spectrometry (F-AES, for alkali ions), whereas most of the inorganic anions have been determined by ion chromatography (IC).1,3–5 Ammonium and phosphate contents have been determined spectrophotometrically using respectively the nitroprusside and molybdenum blue methods.1,3–5 The need for these different instrumental methods accompanied by different sample storage strategies for subsequent in-lab analyses renders this water quality-control operation costly (with AAS, AES, and IC) and time consuming. This monitoring operation therefore could be implemented only by central/national institutions with sufficient infrastructure support and funding. Routine analyses of the ionic species of water in local laboratories with limited financial resources have hardly been possible so far.The aforementioned issue led to the desire for a more approachable alternative for water analysis. From our point of view, the most suitable option is the employment of a system based on capillary electrophoresis (CE) with capacitively coupled contactless conductivity detection (C4D). In this case, ionic species are electrophoretically separated by application of a high voltage along a narrow separation capillary and are detected based on the difference of their electrical conductivities from that of the background electrolyte (BGE). Fundamental aspects of C4D can be found in ref. 6–13, whereas applications of CE-C4D for water analyses can be found in several reviews.8,14–18 While commercial CE-C4D benchtop instruments for such operations are available, an option that is more suitable for modest infrastructures is the construction and utilization of CE-C4D instruments which were designed and developed in our laboratory. These systems are referred to as in-house-made CE-C4D throughout the text. As for CE-C4D both the separation and detection of ions are based on electronic principles and the method only requires low-pressure fluidic components; CE-C4D offers many advantageous features, including the possible translation into portable instrumentation, high configuration flexibility and ease of construction and operation. Since the launch of the first in-house-made (portable) CE instrument in 1998,19 different prototypes have been developed and introduced by Hauser and co-workers, ranging from instruments with manual injection and flushing,20–22 systems where this has been semi-automated23 to fully automated single-channel versions24–27 and dual-channel configurations using one common buffer.28,29 In parallel, significant contributions have been made by other groups to in-house-built (portable) CE instrumentation. The more recent studies were communicated by Breadmore et al. for automated pKa determination30 and for simultaneous separation of anions and cations,31 Kaljurand et al. for fingerprinting postblast explosive residues,32 Porto et al. for analysis of ecstasy tablets33 and Gaertner et al. for on-site food analysis.34 A review on all in-house made (portable) CE instruments up to 2013 could be found in ref. 35 and 36. The C4D design has also been refined to match this evolution of CE setups. The most recent versions were designed for ease of construction and were miniaturized by integrating the entire circuitry in the detection cell and built for battery operation.28,37 An alternative compact version of C4D-cells has also been described by Lago and co-workers.38 For good performance with CE-C4D, BGEs having low specific conductivities but high ionic strengths are preferable. Organic molecules with low/moderate mobilities are thus normally used to prepare the BGE solutions. More details on guidance for BGE selection for CE-C4D operation can be found in ref. 8, 17, and 39.
Herein, the feasibility of the employment of in-house-built CE-C4D-instruments for water analysis as an alternative to the standard techniques (i.e. AAS, AES, IC and UV) is evaluated. The case study was implemented in Vietnam where these systems have been deployed for systematic analyses of different surface water and groundwater matrices. Some considerations on the use of in-house-made CE-C4D as well as the effects of some interfering ions on the analytical performance are discussed. In addition to the determination of major inorganic cations and anions, the CE-C4D approach was applied to the direct analysis of the widely present inorganic tri-valent arsenic As(III) in anaerobic groundwater, taking into account the adverse effect of the groundwater matrix (i.e. the abundant presence of ferrous and bicarbonate/carbonate ions). Arsenic contamination in groundwater is a highly critical issue for the water supply arrangements in the Red River Delta of Vietnam.2,40 Sensitive determination of arsenic in groundwater is normally carried out by hydride vapor generation (HVG)-AAS.41 CE has also been coupled with different detection/preconcentration techniques for arsenic determination/speciation in water.42–50 In our pioneering work on arsenic determination by CE-C4D,42 it was possible to detect As(III) prepared in standard solutions down to 22 μg L−1. The direct determination of As(III) in groundwater samples at the time however was not successful due to the presence of interfering ferrous ions. As(III) had to be oxidized to As(V) to be analyzed under acidic conditions to eliminate the adverse effect of this ion. The groundwater matrix effect on the CE-C4D performance for the determination of As(III) was not considered in that work. In this study, As(III) in groundwater samples was directly quantified with the improved detection limit (LOD) of 5 μg L−1. This LOD is below the regulated level of 10 μg L−1 for As(III) in drinking water that was set by the Environmental Protection Agency (EPA) in 2008.51
2. Experimental
2.1. Chemicals and materials
All chemicals were of analytical or reagent grade and purchased from Fluka (Buchs, Switzerland) or Merck (Darmstadt, Germany). Stock solutions (10 mmol L−1) of chloride, nitrate, sulfate, nitrite, and phosphate were used for the preparation of the standards of inorganic anions, using their corresponding sodium or potassium salts. Those of the inorganic cations (ammonium, potassium, calcium, sodium and magnesium) were prepared from the chloride salts. Tri-valent arsenic solution was prepared from sodium (meta)arsenite (Fluka, Buchs, Switzerland). Chemicals used for preparation of CE-C4D buffers include: arginine (Arg), acetic acid, histidine (His), 18-crown-6, cetyltrimethylammonium bromide (CTAB), 2-(N-morpholino)ethanesulfonic acid (MES), 3-(N-morpholino)propanesulfonic acid (MOPS), N-cyclohexyl-2-aminoethanesulfonic acid (CHES), tris(hydroxymethyl)aminomethane (TRIS) and 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS).Fused silica capillaries of 50 μm ID and 365 μm OD were obtained from Polymicro Technologies (Phoenix, AZ, USA). Before use, the fused silica capillaries were pre-conditioned with 1 M NaOH for 10 min and deionized water for 10 min prior to flushing with the buffer. The capillaries were then used continuously for successive analyses. Deionized water purified using a water purification system from Millipore – model Simplicity UV (Bedford, MA, USA) was used for the preparation of all solutions and for sample dilution if required.
2.2. Instrumentation
Both manual and automated in-house-built CE instruments were used for analytical method development and sample analyses. Details on the construction and operation of different in-house-made CE prototypes can be found in ref. 20 and 21 for the single-channel version with manual siphoning injection,23 for the semi-automated single-channel setup,24–27 for the automated single-channel variants and28,29 for the multi-channel configurations. The automated CE instruments were controlled via a home-made computer program written with either LabView for Windows XP or Arduino. The controlling program has a graphical user interface that allows facile and intuitive operations.Detection was carried out with miniaturized high voltage (HV)-C4D built in-house according to the design reported previously.28 The resulting signals were recorded with a 12 V DC-powered E-corder 201 data acquisition system (eDAQ, Denistone East, NSW, Australia) connected to the USB-port of a personal computer. For data processing, the program Chart (version 5.1) developed by eDAQ was used. For powering the electrophoretic and fluidic parts, a lithium battery pack of 14.8 V and a capacity of 6.6 A h (CGR 18650CG 4S3P, Contrel, Hünenberg, Switzerland) fitted with a voltage regulator for production of a 12 V output was used. A separate pair of smaller Li-ion batteries with a capacity of 2.8 A h each (CGR 18659CG 4S1P, Contrel), which were fitted with positive and negative 12 V regulators, provided the split ±12 V supply for the C4D circuitry. Alternatively, main power can be utilized when available.
2.3. Field sampling
For groundwater samples used for the As(III) determination by CE-C4D, 1,10-phenanthroline was added into the sample after filtration to complex with ferrous ions in order to avoid aerobically induced precipitation of ferric hydroxide that may lead to co-precipitation of inorganic arsenic species. The samples were then flushed through cartridges containing a strong cation exchanger Wolfatit KPS 200 (VEB Farbenfabrik Wolfen, Germany) which released protons to react immediately with bicarbonate/carbonate in groundwater to form carbon dioxide. The sample was purged with nitrogen to completely remove the produced carbonic gas. Prior to each analysis, the sample was alkalized to pH 9.2 with arginine. The final sample solution contained 20 mM of Arg and 1.8 mM of 1,10-phenanthroline.
For subsequent cross-checking purpose, aqueous As(V) and As(III) ions were separated by filtering the water sample first through a 0.2 μm membrane filter (Sartorius, Göttingen, Germany) and then a disposable anion exchange cartridge,52 at a flow rate of 5–6 mL min−1 using a syringe. The anion exchange cartridge was mounted directly on the filter and the combination was carefully flushed with nitrogen before use. The cartridges contained 0.8 g aluminosilicate adsorbent that selectively adsorbs As(V) but not As(III).3 The sample containing only As(III) was then acidified with hydrochloric acid to pH < 2 and stored at 4 °C until subsequent in-lab analysis.
2.4. Analytical methods
All CE-C4D operations for the determination of major inorganic anions and cations in ground and surface water samples were carried out immediately upon conclusion of the sampling campaign to avoid/minimize sample nature modification. Samples were fed into the CE-C4D systems manually (without an autosampler). Hydrodynamic sample injection was carried out either via the siphoning effect (with the manual or semi-automated version) or via pressurization of the sample flow at the ground end of the capillary (with the automated systems). As thermostated chambers were not included in our CE-C4D instruments, on-site measurements were implemented only when the external temperature was moderate (25–35 °C) and the humidity was not too high (less than 90%). Otherwise these operations were done either in an air-conditioned room or in the lab. Determinations of inorganic anions, cations and phosphate were carried out independently on three different CE-C4D runs with the average time for each run of about 15 min. Unless otherwise stated, a BGE composed of 12 mM histidine and 2 mM 18-crown-6 adjusted to pH 3.7 with acetic acid and one composed of 12 mM histidine adjusted to pH 4 with acetic acid were employed for the determination of major inorganic cations and anions, respectively. The buffer used for phosphate determination was composed of 1 mM histidine adjusted to pH 3.5 with acetic acid. Samples collected from 5 lakes in Hanoi were analyzed with CE-C4D using the aforementioned BGE compositions. Samples were diluted with deionized water if needed to avoid peak overlaps with CE-C4D when the concentrations were out of the calibration ranges.The surface water and groundwater samples were also transported to the lab and stored at 4 °C for subsequent in-lab analysis and cross-checked with the standard methods. The cations i.e. earth alkali and alkali metals were analyzed by F-AAS or F-AES on a Shimadzu AAS 6800 instrument. Anions were analyzed by IC using a Shimadzu LC20AD/HIC-20ASuper instrument. Ammonium was determined spectro-photometrically using nitroprusside whereas phosphate was analyzed by the molybdenum blue method using a Shimadzu UV 3101 equipment. More details on analytical procedures can be found in ref. 1 and 3.
As(III) in groundwater samples was determined by CE-C4D using the standard addition method (3 points) with the average analysis time for each sample of 45 min. The final sample solution contained 20 mM of Arg and 1.8 mM of 1,10-phenanthroline. The optimized BGE for As(III) analysis was 12 mM MES/21 mM Arg/30 μM CTAB (pH 8.9). Samples were electrokinetically injected at −6 kV for 60 s and separated at −20 kV in a capillary of 60 cm total length. The C4D detector was situated at an effective length of 52 cm. For cross-checking purpose, arsenic was determined with a Shimadzu AAS 6800 instrument using a hydride vaporization generator (HVG) according to the standard method detailed in ref. 53.
3. Results and discussion
3.1. Instrumental aspects
A CE-C4D system should always be earthed because the high voltage supplies may lead to charging up of the (insulated) boxes of the instruments, which in turn can create a hazard to the user if the system is not earthed. This can however not always be done readily because in developing countries in general and in Vietnam in particular, the earth is often excluded from the main sockets of the electricity system for economical reasons. An alternative earth then has to be improvised. If available, a connection to a metallic water pipe may be made, or a metallic rod is buried in the ground.
Another challenge to overcome when working with CE-C4D in Vietnam is the high humidity. While a CE-C4D system may work fine for in-lab analyses in dry weather, noisy signals were at times observed when the humidity increased. The problem became more pronounced when the systems were set up outside the lab for mobile deployment. This is due to the discharge of the applied high voltage through humid air which renders the high voltages for separation not maintainable. The discharge could also produce pronounced noise on the C4D signal if the detector was not positioned far enough from the high-voltage electrode. To solve the problem, the first approach is to make the high voltage cage as large as possible and free of conducting parts inside. The vial containing the BGE, the capillary end and the electrode should be positioned in the center of this cage. Secondly, a reduction in the high voltage from 30 kV possible with conventional instruments may be necessary.
When possible the CE system should be operated in an air-conditioned room to avoid temperature variations and high humidity. This may not lead to additional expenses as the households in Vietnam are often equipped with air-conditioners for their own use. Application of the standard addition or internal standardization method may be needed for peak identification and quantification when there are significant drifts of the baseline and migration time due to ambient temperature fluctuations in the absence of an air-regulated condition.
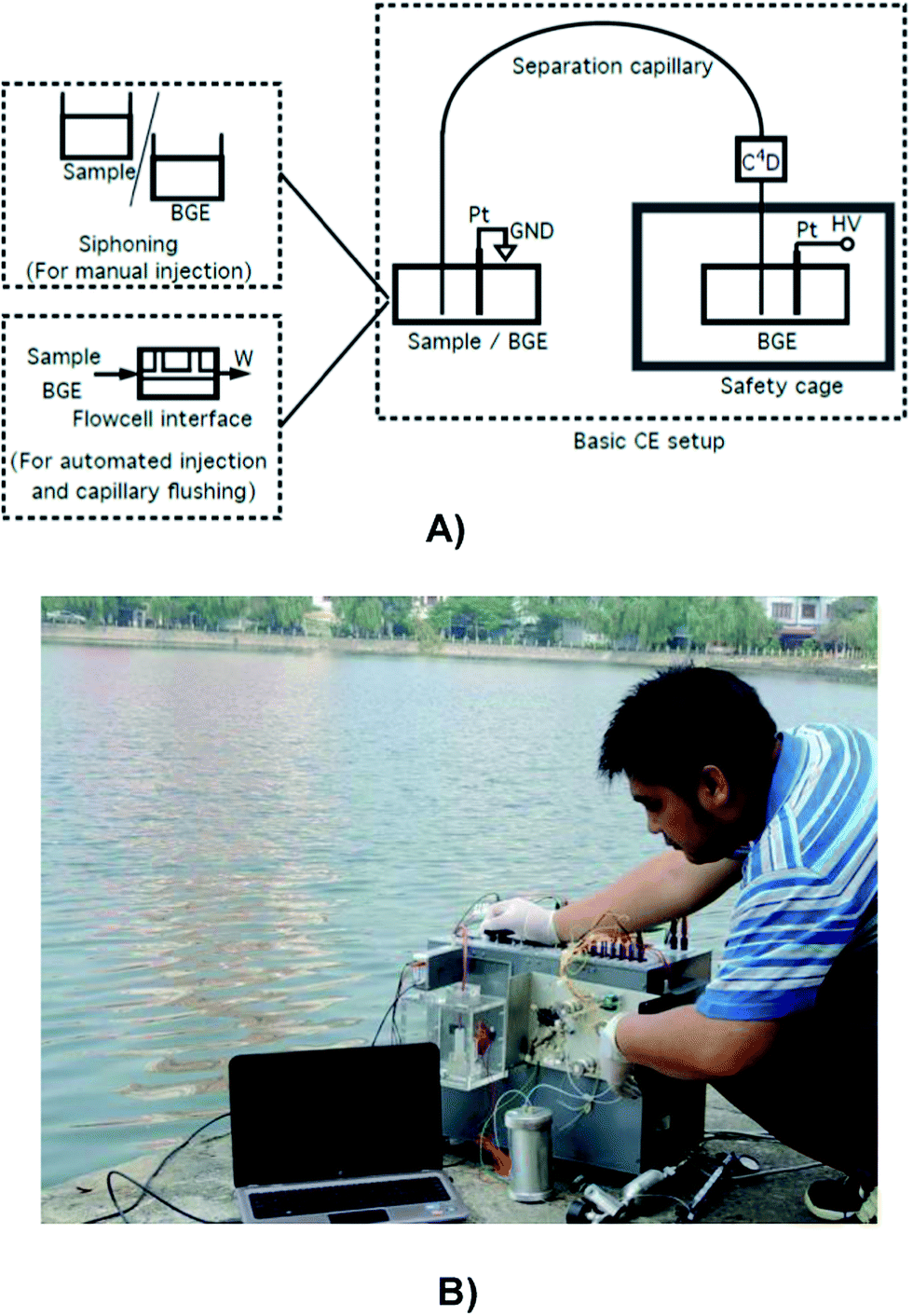 | ||
| Fig. 1 (A) A block-diagram demonstrating the basic set-up of a CE system with some optional automatable parts; (B) demonstration of an in-house-made CE system in operation. Pt: platinum electrode; W: waste; GND: ground electrode; BGE: background electrolyte; HV: high voltage; C4D: capacitively coupled contactless conductivity detector. | ||
The automated versions of in-house-built CE-C4D24,25,28 on the other hand can eliminate some aforementioned inconveniences encountered in manual and semi-automated versions. No capillary back and forth movement is needed and the systems can be operated in an unattended manner. In the case of the dual-channel CE system using the same buffer,28 different ionic categories, i.e. cationic and anionic species, can be simultaneously determined in a single run. Higher construction cost (up to 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–20000 Euros) and requirement of certain knowledge about operation of computer-based controlling programs however are two considerations to be taken into account when only modest financial funding and little expertise are available. These automated versions may be more vulnerable to the noisy signal problem when good grounding is not guaranteed. Commercial C4D and data acquisition units with software are available for some thousands of Euros. If the electronics expertise is available, a C4D can be built in-house for a few hundred Euros depending on the design. A good data acquisition system is essential and will have to be bought. The expenses for the mechanical hardware are minimal, but the workshop cost is possibly incurred.
000–20000 Euros) and requirement of certain knowledge about operation of computer-based controlling programs however are two considerations to be taken into account when only modest financial funding and little expertise are available. These automated versions may be more vulnerable to the noisy signal problem when good grounding is not guaranteed. Commercial C4D and data acquisition units with software are available for some thousands of Euros. If the electronics expertise is available, a C4D can be built in-house for a few hundred Euros depending on the design. A good data acquisition system is essential and will have to be bought. The expenses for the mechanical hardware are minimal, but the workshop cost is possibly incurred.
3.2. CE-C4D determination of major inorganic cations and anions in different water matrices
For separation of anions, 18-crown-6 was not needed and was removed from the electrolyte. At this low pH the electro-osmotic flow (EOF), which is the bulk movement of the liquid inside the capillary under an applied electrical field, is suppressed; therefore no EOF modification is needed. While a BGE consisting of 12 mM histidine adjusted to pH 4 with acetic acid resulted in good separations of major anions including Cl−, SO42−, NO32− and NO2−, it was found experimentally that this high content of histidine in the BGE hindered sensitive detection of phosphate (see Fig. S3 in the ESI† file). To improve the sensitivity for phosphate, the concentration of histidine in the BGE was reduced and the optimized BGE for phosphate determination was composed of 1 mM histidine adjusted to pH 3.5 with acetic acid.
The effect of bicarbonate on the performance of anion separation is illustrated in Fig. 2. By increasing the added concentration of bicarbonate from 0 to 600 mg L−1 (10 mM) into a standard solution containing inorganic anions of 100 μM, it was found that the optimized BGE at pH = 4 can tolerate the presence of bicarbonate up to 300 mg L−1 (5 mM). At bicarbonate concentrations higher than 5 mM, the peaks of Cl−, NO3−, SO42− and NO2− became distorted and were difficult to quantify. The reason for this problem however is not understood. A solution for avoiding this bicarbonate disturbance can be direct acidification of the sample with acetic acid prior to anion analysis by CE-C4D and the subsequent purging the samples by nitrogen stream.
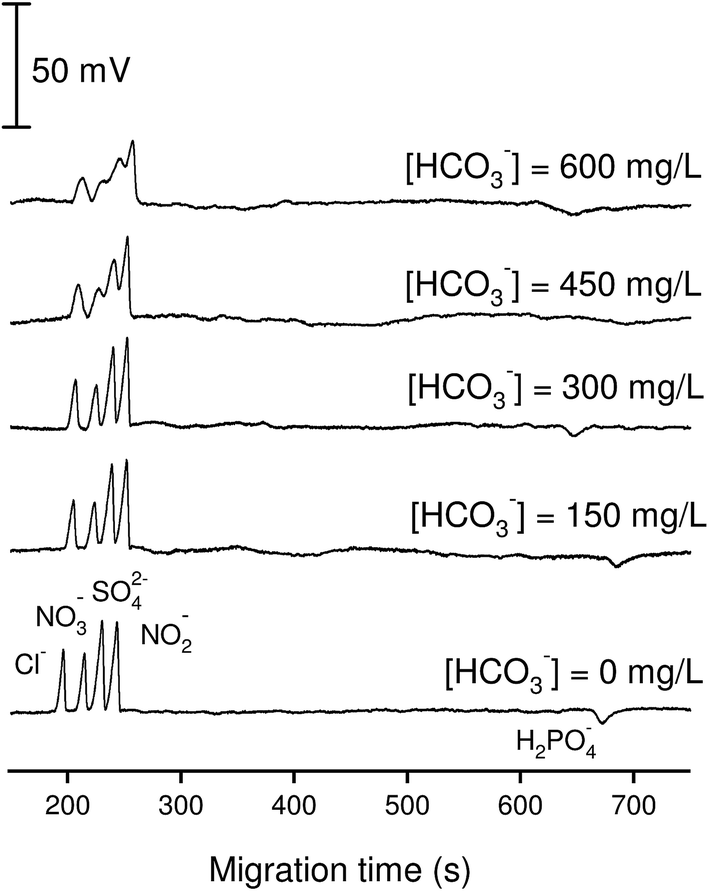 | ||
| Fig. 2 Effect of bicarbonate ions on the performance of anion separation by CE-C4D. Bicarbonate at different concentrations from 0–600 mg L−1 was spiked into the standard mixture solutions containing Cl− (100 μM), NO3− (100 μM), SO42− (50 μM), NO2− (100 μM) and phosphate (100 μM). Electrolyte solution: 12 mM histidine adjusted to pH 4 with CH3COOH; voltage: −15 kV; capillary: fused-silica, 25 μm id, Lt = 52 cm (Leff = 36 cm). | ||
An example of the analyses of standard mixtures of cations and anions with three different BGE solutions is shown in Fig. 3. The calibration data for the ions of interest are given in Table 1. The detection limits, determined from the peak heights corresponding to 3 times the baseline noise, were in the range of 2.5–10 μM. For inorganic cations, linear calibration curves were obtained up to 2000 μM whereas somewhat shorter linear ranges were achieved for the inorganic anions (up to 1000 μM). The obtained correlation coefficients were better than 0.99. The repeatability of the measurements of peak areas and migration times were better than 5% and around 2%, respectively.
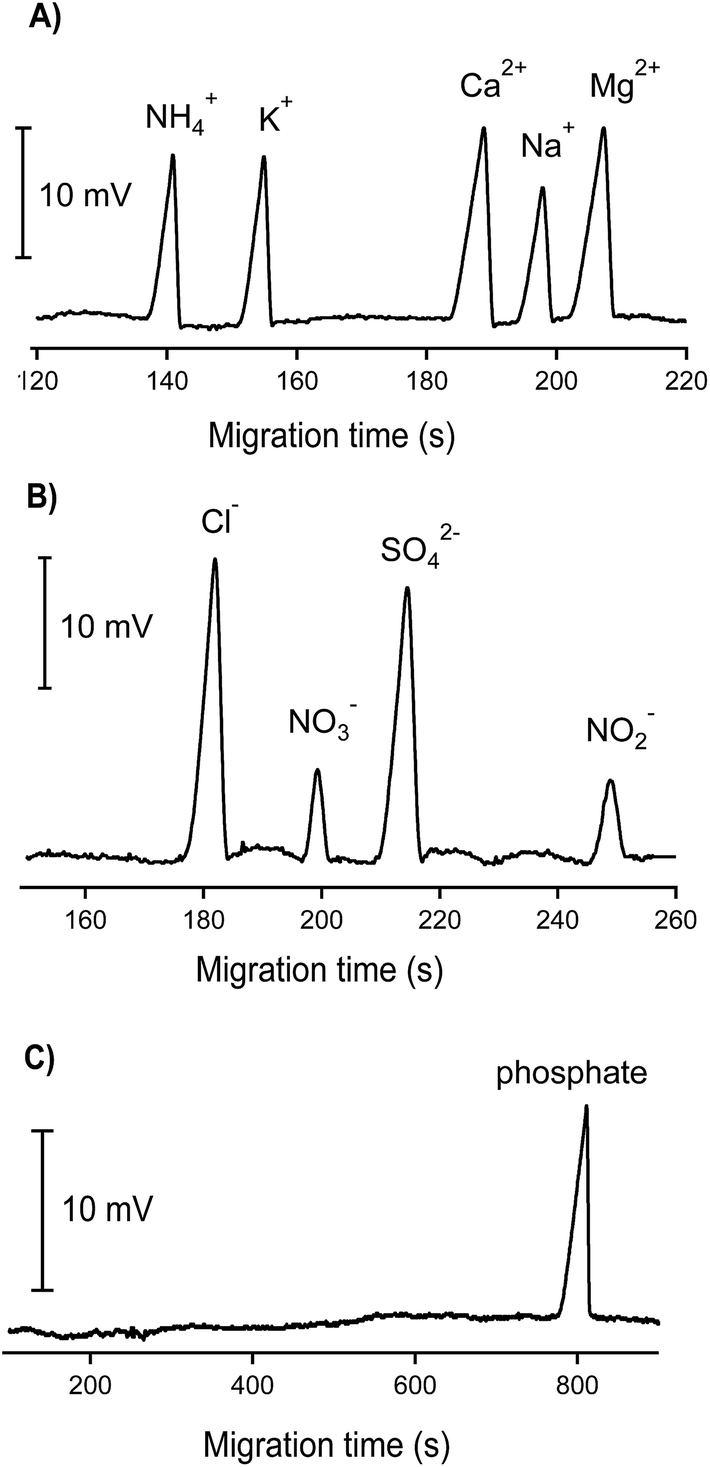 | ||
| Fig. 3 CE-C4D separations of inorganic anions and cations. (A) Cations: NH4+ (200 μM), K+ (200 μM), Ca2+ (200 μM), Na+ (200 μM), and Mg2+ (200 μM); electrolyte solution: 12 mM histidine and 2 mM 18-crown-6 adjusted to pH 3.7 with CH3COOH; voltage: 15 kV; capillary: fused-silica, 25 μm id, Lt = 65 cm (Leff = 49 cm); (B) anions: Cl− (200 μM), NO3− (50 μM), SO42− (100 μM), and NO2− (50 μM); electrolyte solution: 12 mM histidine adjusted to pH 4 with CH3COOH; voltage: −15 kV; capillary: fused-silica, 25 μm id, Lt = 52 cm (Leff = 36 cm); (C) phosphate (50 μM); electrolyte solution: 1 mM histidine adjusted to pH 3.5 with CH3COOH; voltage: −15 kV; capillary: fused-silica, 25 μm id, Lt = 52 cm (Leff = 36 cm). | ||
| Ion | Rangea (μM) | Correlation coefficient r2 | LODb (μM) | RSD% MTc (n = 4) | RSD% PAd (n = 4) |
|---|---|---|---|---|---|
| a 5 concentrations. b Based on peak heights corresponding to 3 times the baseline noise. c Migration time (measurement unit: second). d Peak area (measurement unit: mV s). e Determination of major inorganic cations: electrolyte solution: 12 mM histidine and 2 mM 18-crown-6 adjusted to pH 3.7 with CH3COOH. Voltage: +15 kV. Capillary: fused-silica, 25 μm id, Lt = 65 cm (Leff = 49 cm). f Determination of major inorganic anions: electrolyte solution: 12 mM histidine adjusted to pH 4 with CH3COOH. Voltage: −15 kV. Capillary: fused-silica, 25 μm id, Lt = 52 cm (Leff = 36 cm). g Determination of phosphate: electrolyte solution: 1 mM histidine adjusted to pH 3.5 with CH3COOH. Voltage: 15 kV. Capillary: fused-silica, 25 μm id, Lt = 52 cm (Leff = 36 cm). | |||||
| Channel 1 | |||||
| NH4+ | 20–400 | 0.999 | 5.5 | 0.7 | 4.2 |
| K+ | 20–400 | 0.997 | 6.0 | 1.4 | 2.7 |
| Ca2+ | 100–2000 | 0.998 | 4.5 | 1.6 | 2.8 |
| Na+ | 50–500 | 0.999 | 10.0 | 2.0 | 6.4 |
| Mg2+ | 50–1000 | 0.994 | 5.0 | 1.7 | 3.5 |
|
|||||
| Channel 2 | |||||
| Cl− | 50–1000 | 0.999 | 4.0 | 0.7 | 3.7 |
| SO42− | 10–1000 | 0.998 | 2.5 | 0.8 | 4.3 |
| NO2− | 20–200 | 0.994 | 4.5 | 1.0 | 2.6 |
| NO3− | 20–200 | 0.992 | 4.5 | 1.2 | 3.4 |
|
|||||
| Channel 3 | |||||
| PO43− | 10–100 | 0.997 | 5.0 | 2.7 | 3.8 |
Electropherograms of one groundwater sample are demonstrated in Fig. 4. As can be seen, Ca2+, Na+, Mg2+, Cl− and SO42− appeared in abundant amounts, reflected by high and large peaks whereas much smaller peaks of NH4+ and K+ were observed. No nitrogen-containing anions (NO3− and NO2−) were found in this groundwater sample. The concentration of phosphate (if any) was below the quantification limit; therefore the electropherogram of the CE channel used for phosphate determination is not shown. To verify the reliability of the results obtained by CE-C4D, all concentrations were cross-checked using the standard reference methods, i.e. AAS, IC and UV spectrometry. High correlation coefficients with r2 ≥ 0.9 were achieved showing very good agreement between the results obtained by CE-C4D and those by the standard reference techniques (see Tables S2 and S3 in the ESI† file). Among the 15 groundwater samples tested, NO3− and NO2− were not detected by our CE-C4D method whereas phosphate was found in only 1 sample. The concentration of phosphate determined by CE-C4D in this case fell in the range between 5 μM (LOD) and 10 μM (LOQ). Verification by IC revealed a phosphate concentration of 6 μM, which matched well with the result obtained by CE-C4D. Note that the concentrations of nitrogen-containing ions and phosphate can fluctuate significantly in different seasons and are also much dependent on the nature of the groundwater. Most groundwaters in the Red River Delta are anaerobic. As a result, dissolved inorganic nitrogen occurs mostly as NH4+ rather than NO3− and NO2. The phosphate concentration below the LOD in most of the collected groundwater samples can be reasonably explained that these groundwater resources are neither close to the domestic waste, agricultural activities nor natural mineral resource containing phosphate (e.g. apatite).
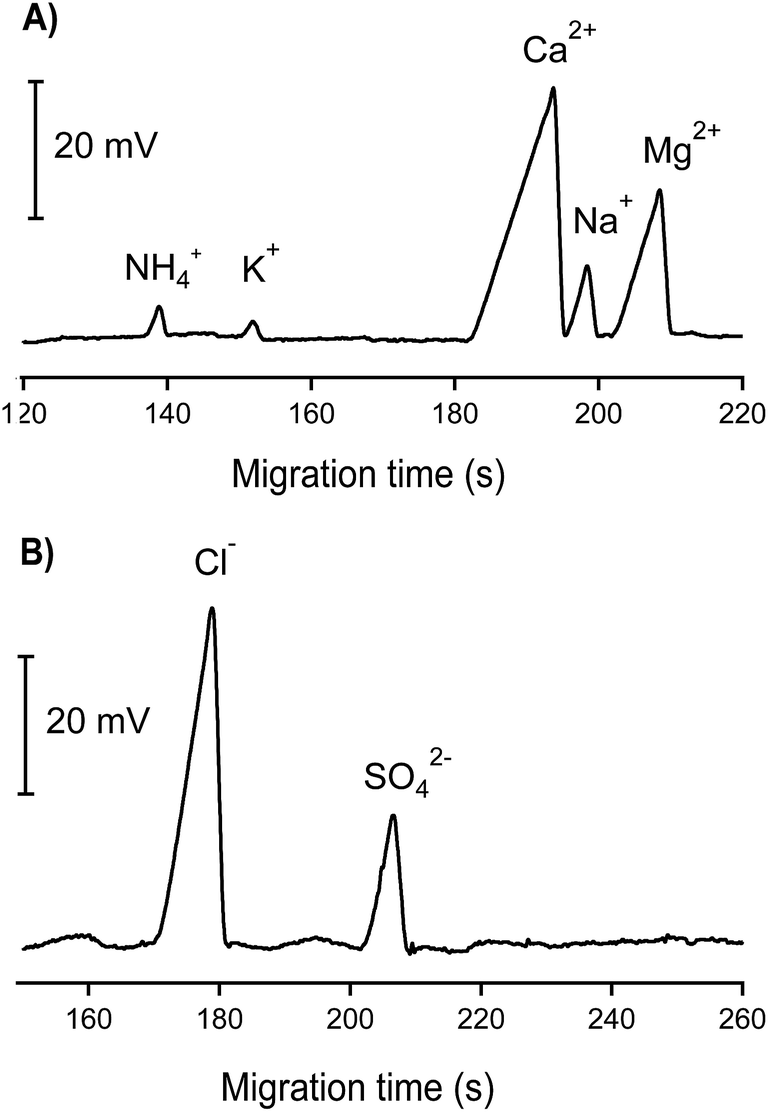 | ||
| Fig. 4 CE-C4D electropherograms of inorganic cations and anions in one groundwater sample. CE conditions as in Fig. 3. | ||
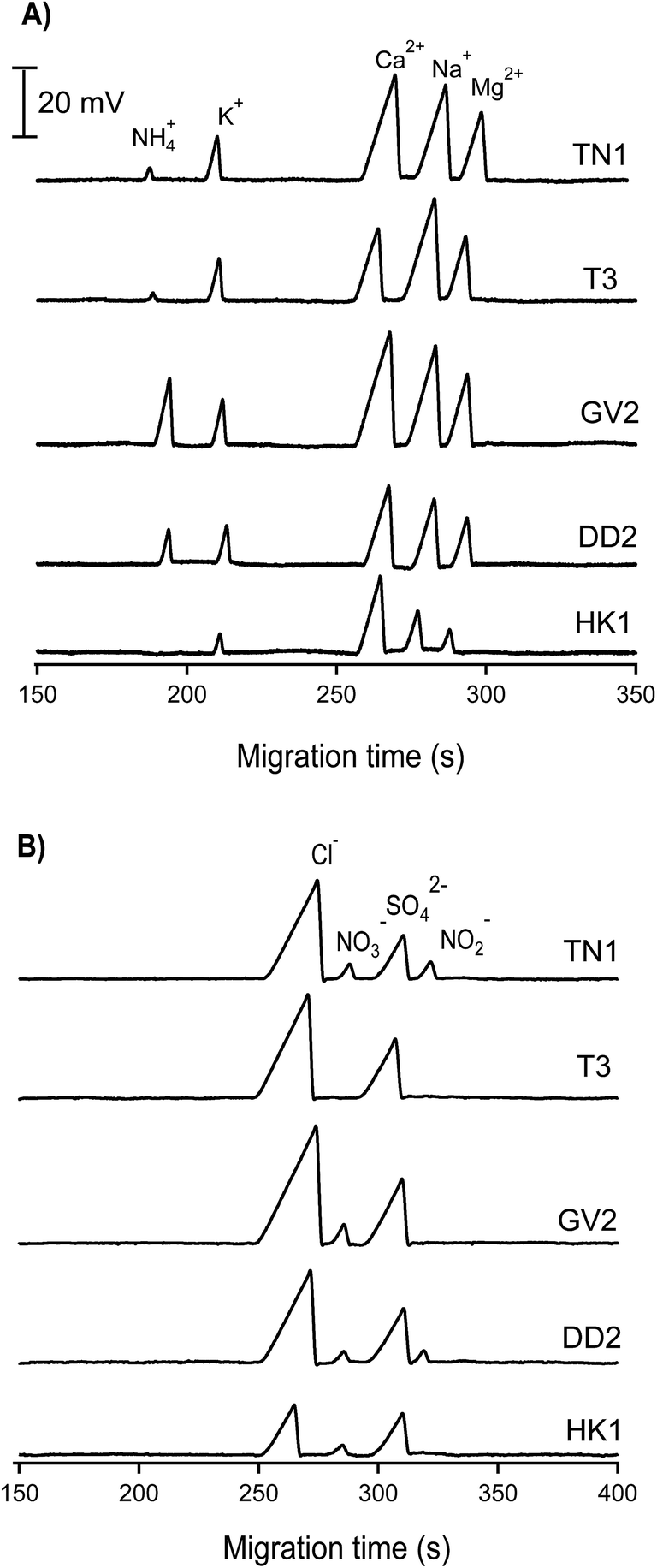 | ||
| Fig. 5 CE-C4D electropherograms of inorganic cations and anions in water samples collected from different lakes in Hanoi. CE conditions as in Fig. 3. | ||
3.3. Determination of As(III) in groundwater
Together with Bangladesh and India, Vietnam belongs to the countries in South and South East Asia suffering from a natural contamination of groundwater by arsenic. Under the reducing conditions of the Red River Delta groundwater, together with the abundant presence of microbacteria and organic matter, ferrous and As(III) ions can be released from sediments into groundwater.3,40 We showed earlier that As(III) in standard solutions could be determined by CE-C4D with a LOD of 22 μg L−1.42 The interfering effect of ferrous and bicarbonate/carbonate ions on CE-C4D determination of As(III) in groundwater samples however was not considered in this pioneering work. Herein, we propose a new CE-C4D method for direct quantification of As(III) species in groundwater taking into account various interfering parameters which may adversely affect its analytical performance. The detection limit for As(III) determination was improved to 5 μg L−1 (determined for standard solutions without recourse to the standard addition method), which is below the regulated level of arsenic in water (10 μg L−1).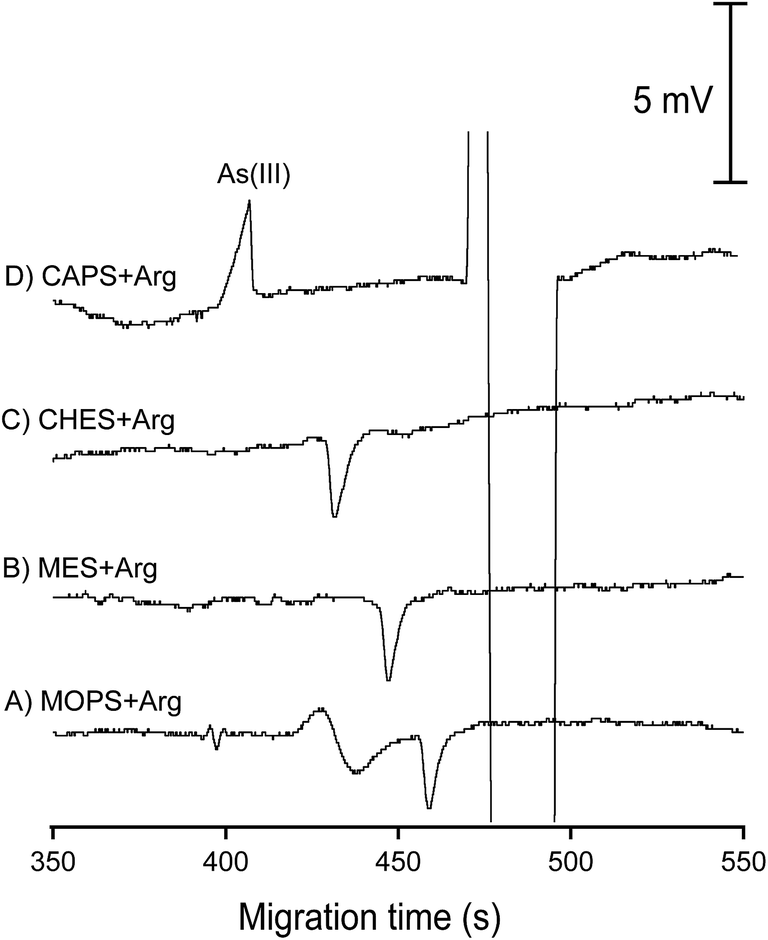 | ||
| Fig. 6 Electrophoretic separation of As(III) (5 mg L−1) using different BGEs. Samples were injected with the siphoning effect at a height of 20 cm for 45 seconds and separated at −20 kV over a capillary of 60 cm total length. The C4D detector was situated at an effective length of 52 cm. | ||
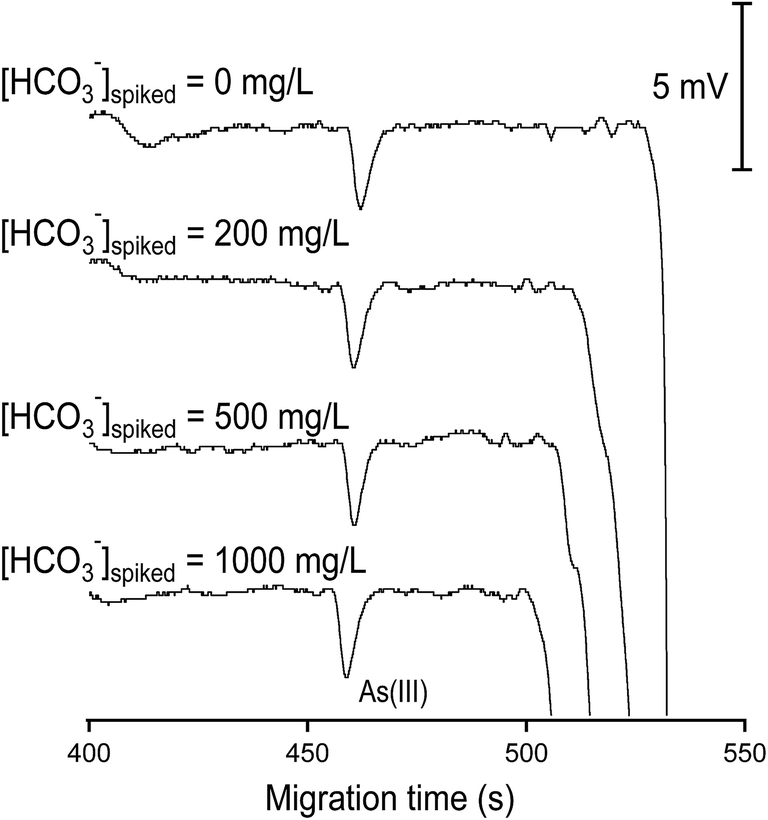 | ||
| Fig. 7 Bicarbonate removal capacity of the cation exchange resin KPS 200. This evaluation was made with As(III) of 100 μg L−1. Samples were electrokinetically injected at −6 kV for 60 s and separated at −20 kV over a capillary of 60 cm total length. The C4D detector was situated at an effective length of 52 cm. | ||
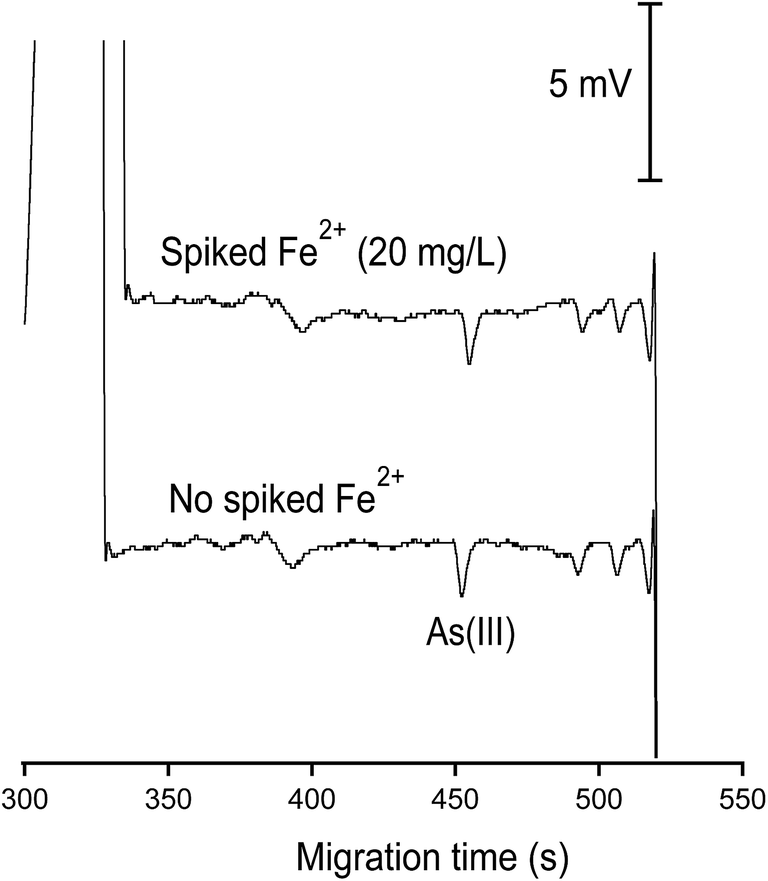 | ||
| Fig. 8 Demonstration of the prevention of the loss of As(III) in the abundant presence of Fe2+ (20 mM) by the addition of 1,10-phenanthroline (1.8 mM). This evaluation was made with As(III) of 100 μg L−1. CE conditions as in Fig. 7. | ||
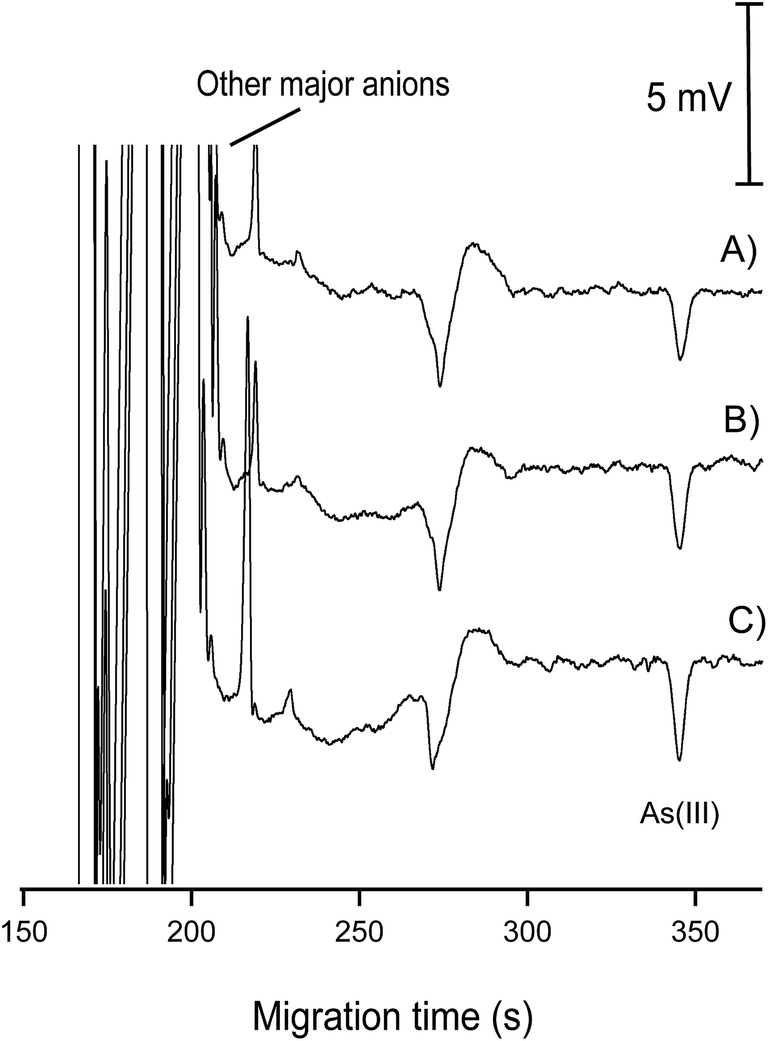 | ||
| Fig. 9 Electropherograms for the determination of As(III) in 1 groundwater sample. (A) Groundwater sample without As(III) spiking; (B) groundwater sample with spiked As(III) of 30 μg L−1; (C) groundwater sample with spiked As(III) of 50 μg L−1. CE conditions as in Fig. 7. | ||
4. Conclusions
In-house-made CE-C4D was found to be a simple and inexpensive solution for analysis of various inorganic ionic species in different water matrices. A simple CE-C4D setup can be built at the construction cost of 5000–7000 Euros. This approach eliminates the requirement of costly and sophisticated commercial instrumentation, thus rendering the water-quality monitoring activities feasible even at local laboratories where only modest budget and limited expertise are available. More laborious work with manual operations may nevertheless be encountered with in-house built CE-C4D instruments due to (i) exclusion of some/all automated options to reduce construction cost and (ii) lack of decent controlling software. Acceptable agreement between the results obtained with CE-C4D for determination of major inorganic ions as well as trivalent arsenic and those with the standard reference methods, i.e. AAS, AES, IC and UV, was achieved (with correlation coefficients r2 ≥ 0.9 and cross-check deviations less than 15%), demonstrating the reliability of the analytical data provided by CE-C4D. Extension of the CE-C4D application spectrum to other ionic water-quality indicator species (heavy metals for example) is envisaged.Acknowledgements
The authors are grateful for financial support from the National Foundation for Science and Technology Development of Vietnam (NAFOSTED, Grant No. 104.07-2010.21). Jorge Sáiz (University of Alcalá, Madrid, Spain), Israel Joel Koenka (University of Basel, Switzerland) and 3SAnalysis JSC (http://www.3SAnalysis.vn) are acknowledged for their instrumental support. We also would like to thank MSc. Thi Thanh Thuy Pham, MSc. Thi Mai Lan Vi, MSc. Thanh Dam Nguyen, MSc. Duy Chien Nguyen and MSc. Van Tang Nguyen (CETASD) for their help in sampling and cross-checking analysis operations.References
- K. S. Nitzsche, V. M. Lan, P. T. K. Trang, P. H. Viet, M. Berg, A. Voegelin, B. Planer-Friedrich, J. Zahoransky, S.-K. Muller, J. M. Byrne, C. Schroder, S. Behrens and A. Kappler, Sci. Total Environ., 2015, 502, 526–536 CrossRef CAS PubMed.
- A. van Geen, B. C. Bostick, P. Thi Kim Trang, V. M. Lan, N.-N. Mai, P. D. Manh, P. H. Viet, K. Radloff, Z. Aziz, J. L. Mey, M. O. Stahl, C. F. Harvey, P. Oates, B. Weinman, C. Stengel, F. Frei, R. Kipfer and M. Berg, Nature, 2013, 501, 204–207 CrossRef CAS PubMed.
- D. Postma, F. Larsen, N. T. Minh Hue, M. T. Duc, P. H. Viet, P. Q. Nhan and S. Jessen, Geochim. Cosmochim. Acta, 2007, 71, 5054–5071 CrossRef CAS.
- D. Postma, S. Jessen, N. T. M. Hue, M. T. Duc, C. B. Koch, P. H. Viet, P. Q. Nhan and F. Larsen, Geochim. Cosmochim. Acta, 2010, 74, 3367–3381 CrossRef CAS.
- S. Jessen, F. Larsen, D. Postma, P. H. Viet, N. T. Ha, P. Q. Nhan, D. D. Nhan, M. T. Duc, N. T. M. Hue, T. D. Huy, T. T. Luu, D. H. Ha and R. Jakobsen, Appl. Geochem., 2008, 23, 3116–3126 CrossRef CAS.
- T. D. Mai and P. C. Hauser, Chem. Rec., 2012, 12, 106–113 CrossRef CAS PubMed.
- W. K. T. Coltro, R. S. Lima, T. P. Segato, E. Carrilho, D. P. de Jesus, C. L. do Lago and J. A. F. da Silva, Anal. Methods, 2012, 4, 25–33 RSC.
- P. Kubáň and P. C. Hauser, Electrophoresis, 2009, 30, 176–188 CrossRef PubMed.
- P. Kubáň and P. C. Hauser, Electrophoresis, 2004, 25, 3387–3397 CrossRef PubMed.
- P. Kubáň and P. C. Hauser, Electrophoresis, 2004, 25, 3398–3405 CrossRef PubMed.
- J. G. A. Brito-Neto, J. A. F. da Silva, L. Blanes and C. L. do Lago, Electroanalysis, 2005, 17, 1198–1206 CrossRef CAS.
- J. G. A. Brito-Neto, J. A. F. da Silva, L. Blanes and C. L. do Lago, Electroanalysis, 2005, 17, 1207–1214 CrossRef CAS.
- A. J. Zemann, Electrophoresis, 2003, 24, 2125–2137 CrossRef CAS PubMed.
- P. Kuban and P. C. Hauser, Electrophoresis, 2014, 36, 195–211 CrossRef PubMed.
- P. Kubáň and P. C. Hauser, Electrophoresis, 2011, 32, 30–42 CrossRef PubMed.
- P. Kubáň and P. C. Hauser, Electrophoresis, 2013, 34, 55–69 CrossRef PubMed.
- P. Kubáň and P. C. Hauser, Anal. Chim. Acta, 2008, 607, 15–29 CrossRef PubMed.
- P. Kubáň and P. C. Hauser, Electroanalysis, 2004, 16, 2009–2021 CrossRef.
- T. Kappes and P. C. Hauser, Anal. Commun., 1998, 35, 325–329 RSC.
- P. Kubáň, H. T. A. Nguyen, M. Macka, P. R. Haddad and P. C. Hauser, Electroanalysis, 2007, 19, 2059–2065 CrossRef.
- N. T. Torres, P. C. Hauser, G. Furrer, H. Brandl and B. Mueller, Environ. Sci.: Processes Impacts, 2013, 15, 715–720 Search PubMed.
- N. T. Torres, L. M. Och, P. C. Hauser, G. Furrer, H. Brandl, E. Vologina, M. Sturm, H. Burgmann and B. Muller, Environ. Sci.: Processes Impacts, 2014, 16, 879–889 Search PubMed.
- T. A. H. Nguyen, T. N. M. Pham, T. T. Doan, T. T. Ta, J. Sáiz, T. Q. H. Nguyen, P. C. Hauser and T. D. Mai, J. Chromatogr. A, 2014, 1360, 305–311 CrossRef CAS PubMed.
- T. D. Mai, S. Schmid, B. Müller and P. C. Hauser, Anal. Chim. Acta, 2010, 665, 1–6 CrossRef CAS PubMed.
- T. D. Mai, T. T. T. Pham, J. Sáiz and P. C. Hauser, Anal. Chem., 2013, 85, 2333–2339 CrossRef CAS PubMed.
- J. Sáiz, T. D. Mai, P. C. Hauser and C. García-Ruiz, Electrophoresis, 2013, 34, 2078–2084 CrossRef PubMed.
- J. Sáiz, T. D. Mai, L. María López, C. Bartolomé, P. C. Hauser and C. García-Ruiz, Sci. Justice, 2013, 53, 409–414 CrossRef PubMed.
- T. T. T. Pham, T. D. Mai, T. D. Nguyen, J. Sáiz, H. V. Pham and P. C. Hauser, Anal. Chim. Acta, 2014, 841, 77–83 CrossRef CAS PubMed.
- J. Sáiz, M. T. Duc, I. J. Koenka, C. Martin-Alberca, P. C. Hauser and C. Garcia-Ruiz, J. Chromatogr. A, 2014, 1372, 245–252 CrossRef PubMed.
- J. M. Cabot, E. Fuguet, M. Rosés, P. Smejkal and M. C. Breadmore, Anal. Chem., 2015, 87, 6165–6172 CrossRef CAS PubMed.
- A. J. Gaudry, R. M. Guijt, M. Macka, J. P. Hutchinson, C. Johns, E. F. Hilder, G. W. Dicinoski, P. N. Nesterenko, P. R. Haddad and M. C. Breadmore, Anal. Chim. Acta, 2013, 781, 80–87 CrossRef CAS PubMed.
- E.-G. Kobrin, H. Lees, M. Fomitšenko, P. Kubáň and M. Kaljurand, Electrophoresis, 2014, 35, 1165–1172 CrossRef CAS PubMed.
- S. K. S. S. Porto, T. Nogueira, L. Blanes, P. Doble, B. D. Sabino, C. L. do Lago and L. Angnes, J. Forensic Sci., 2014, 59, 1622–1626 CrossRef CAS PubMed.
- C. Gaertner, R. Sewart, R. Klemm and H. Becker, Sensing Technologies for Global Health, Military Medicine, and Environmental Monitoring IV, 2014, vol. 9112 Search PubMed.
- A. P. Lewis, A. Cranny, N. R. Harris, N. G. Green, J. A. Wharton, R. J. K. Wood and K. R. Stokes, Meas. Sci. Technol., 2013, 24, 042001 CrossRef.
- M. Ryvolová, J. Preisler, D. Brabazon and M. Macka, TrAC, Trends Anal. Chem., 2010, 29, 339–353 CrossRef.
- M. Stojkovic, I. J. Koenka, W. Thormann and P. C. Hauser, Electrophoresis, 2014, 35, 482–486 CrossRef CAS PubMed.
- K. J. M. Francisco and C. L. do Lago, Electrophoresis, 2009, 30, 3458–3464 CrossRef CAS PubMed.
- T. D. Mai and P. C. Hauser, Electrophoresis, 2013, 34, 1796–1803 CrossRef CAS PubMed.
- M. Berg, H. C. Tran, T. C. Nguyen, H. V. Pham, R. Schertenleib and W. Giger, Environ. Sci. Technol., 2001, 35, 2621–2626 CrossRef CAS PubMed.
- D. Q. Hung, O. Nekrassova and R. G. Compton, Talanta, 2004, 64, 269–277 CrossRef CAS PubMed.
- H. T. A. Nguyen, P. Kubáň, V. H. Pham and P. C. Hauser, Electrophoresis, 2007, 28, 3500–3506 CrossRef CAS PubMed.
- N. Jia, M. Han, G. Zhao, L. Zhang, B. Liu and Y. Li, Asian J. Chem., 2014, 26, 5271–5274 CAS.
- L. Liu, B. He, Z. Yun, J. Sun and G. Jiang, J. Chromatogr. A, 2013, 1304, 227–233 CrossRef CAS PubMed.
- B. Deng, X. Qin, Y. Xiao, Y. Wang, H. Yin, X. Xu and C. Shen, Talanta, 2013, 109, 128–132 CrossRef CAS PubMed.
- K. Cheng, K. Choi, J. Kim, I. H. Sung and D. S. Chung, Microchem. J., 2013, 106, 220–225 CrossRef CAS.
- P. M. Flanigan, D. Ross and J. G. Shackman, Electrophoresis, 2010, 31, 3466–3474 CrossRef CAS PubMed.
- H. Zhang, J. Gavina and Y.-L. Feng, J. Chromatogr. A, 2011, 1218, 3095–3104 CrossRef CAS PubMed.
- O. S. Koshcheeva, O. V. Shuvaeva and L. I. Kuznetzova, Electrophoresis, 2009, 30, 1088–1093 CrossRef CAS PubMed.
- C. A. Suarez, G. C. L. Araujo, M. F. Gine, M. H. Kakazu and J. E. S. Sarkis, Spectrosc. Lett., 2009, 42, 376–382 CrossRef CAS.
- D. Melamed, Anal. Chim. Acta, 2005, 532, 1–13 CrossRef CAS.
- X. G. Meng and W. Wang, Book of posters of the 3rd Inter. Conf. on Arsenic Exposure and Health Effect, Society of Envir. Geochemistry and Health, University of Colorado at Oenner, USA, University of Colorado at Oenner, USA, 1998 Search PubMed.
- APHA, 22nd Edition of Standard Methods for the Examination of Water and Wastewater, American Public Health Association (APHA), Method 3113 B – 3114 B, 2012 Search PubMed.
- L. Blanes, W. K. Tomazelli Coltro, R. M. Saito, A. van Gramberg, C. L. do Lago and P. Doble, Electrophoresis, 2012, 33, 893–898 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5em00362h |
| This journal is © The Royal Society of Chemistry 2015 |