
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development and field testing of a miniaturized sampling system for simultaneous sampling of vapours and droplets†
Dietmar
Breuer‡
*a,
George C.
Dragan‡
bc,
Claudia
Friedrich
a,
Carsten
Möhlmann
a and
Ralf
Zimmermann
bc
aInstitute for Occupational Safety and Health of the German Social Accident Insurance (IFA), 53757 Sankt Augustin, Germany. E-mail: Dietmar.Breuer@dguv.de
bJoint Mass Spectrometry Centre, Cooperation Group “Comprehensive Molecular Analytics”, Helmholtz Zentrum München, 85758 Neuherberg, Germany
cJoint Mass Spectrometry Centre, Chair of Analytical Chemistry, Institute of Chemistry, University of Rostock, 18057 Rostock, Germany
First published on 9th December 2014
Abstract
The sampling of semi volatiles (SV) in workplaces may lead to different results as measurements may be affected by sampling bias. The new European Standard EN 13936 defines “semi-volatiles” as substances with vapour pressures in the range between 0.001 and 100 Pa at room temperature. EN 13936 regulates the basic requirements for SV compounds that can occur as vapour and particle at the same time. Vapour and particles shall not be sampled separately and particles have to be sampled as inhalable fraction. Following EN 13936, the Institute for Occupational Safety and Health (Institut für Arbeitsschutz – IFA) has developed a miniaturized droplet–vapour sampler (GGP-Mini) which is designed to sample the inhalable aerosol fraction at low flow rates. The GGP-Mini uses 13 mm filters for particle sampling combined with adsorption tubes for vapour sampling. Laboratory tests were performed on 11 polar and non-polar compounds in a boiling point range from 188 °C to 318 °C. The substances were spiked directly on the filter followed by aspiration of 40 litres of air. Substances with boiling points below 230 °C were almost completely evaporated. Substances with boiling points above 230 °C up to 300 °C were found on both filter and charcoal tube. Lower-volatile compounds remained almost completely on the filter. For polar substances, the atmospheric humidity had a considerable influence upon the distribution of the liquid and vaporous components. A strong influence of the sampling temperature was found in the range from 0 °C to 50 °C. Droplet–vapour mixtures of n-hexadecane and diethylene glycol with droplet sizes between 1 μm and 4 μm were generated in a flow tube to verify the laboratory results. The aerosol concentrations were analysed on-line with a particle sizer and a flame ionisation detector, while parallel off-line samples were taken with the GGP-Mini. Evaporation losses from filters could be studied by comparing the on-line with off-line measurements. All sampling simulations, both spiking and tests on a droplet aerosol, have shown that the distribution between vapour and droplets is not constant and influenced e. g. by volatility, concentration, temperature and humidity. Only the sum of vapour and droplets constitutes a reproducible result.
Environmental impactIn the past, methods for the measurement of hazardous substances at workplaces were generally designed either for gases and vapours or for dusts. There are however substances with physical properties that cannot be readily assigned to these two distinct groups, as they are neither volatile nor non-volatile. The present paper focuses upon sampling of semi-volatile aerosols which are formed by mechanical processes, since these are more frequently encountered at workplaces and the development of a sampling system was geared to such aerosols. Based upon knowledge of the GSP sampler for inhalable particles, a particle sampling head was developed suitable for use at substantially lower flow rates than normally used for particulate sampling. |
Introduction
Sampling methods for the measurement of hazardous substances at workplaces are generally designed either for gases and vapours or for particulates. Classic solvents such as tetrachloroethene or acetone in vapour form are encountered at workplaces, as are particulates such as quartz. The vapour pressure of a compound usually dictates if the compound is to be found as vapour or as particulate in ambient air. The vapour pressures of typical solids are however so low that they are virtually impossible to measure. Copper for instance has a vapour pressure of 7.05 × 10−7 hPa at 810 °C. By contrast, the vapour pressures of some liquids are so high that they vaporize within a very short time at room temperature. Acetone for example has a vapour pressure of 233 hPa at room temperature; numerous other liquids such as toluene (26 hPa at 20 °C) also have a notable vapour pressure. Liquid droplets from such compounds can be expected to evaporate rapidly at room temperature.Beside volatiles and non-volatiles, there are substances with physical properties that cannot be readily assigned to these two distinct groups, as they are neither volatile nor non-volatile. Semi-volatile (SV) compounds like alkanolamines, inorganic acids,1,2 metalworking fluids (MWF)3,4 and many PAHs5 are included within this intermediary category. SV compounds can often be found as aerosols in workplaces, partitioned between the particle and vapour phases.6 Sampling of aerosols containing SV compounds using methods like filter sampling can prove to be challenging.7–9 The particles trapped on filters can readily evaporate10–12 or vapours can adsorb13 on filtration substrates, thus leading to measurement errors as a consequence.
Aerosols from SV compounds are highly dynamic, with a perpetual mass transfer taking place from the particle to the gas phase and vice versa. Depending on the ambient temperature, droplets can evaporate and vapours can condense on droplets or other surfaces. Droplets can have lifetimes ranging from less than one second up to several hours.14 The main factors that influence droplets lifetime in workplaces are compound volatility, concentration, droplet diameter, ambient temperature and relative humidity (RH). Submicron droplets can swiftly evaporate as they carry less mass, while larger droplets can have longer lifetimes as more time is required for them to completely evaporate. Coarse particles however are heavier and more likely to sediment, while fine ones can remain suspended in the air for longer periods.
Aerosols can be formed in workplaces either by mechanical15 or condensation processes. Examples for mechanically generated aerosols include working processes in which liquids are sprayed, droplets form on rotating parts, or particles are transported into the air by rising gas bubbles. Such droplets are generally large and evaporate when the particle–vapour equilibrium is not reached. The second important process is the condensation of vapours during hot processes. Known examples are the formation of bitumen fumes16 and oil mists.17 Bitumen fumes are created in a condensation process: very small particles are initially formed which gradually increase in size as further vapour condenses on the particles.
Dragan et al.14 conducted comprehensive studies focusing on aerosol evaporation dynamics in a flow tube (FT), with longer-chained alkanes (C14 to C20) serving as model substances. N-Alkanes are particularly suitable for modelling purposes, since they constitute a homologous series that also represents a continual progression with regard to their volatility. Special attention was given to the vaporization rates of droplets of differing size and volatility. The results obtained with monodisperse droplet aerosols demonstrate that the alkanes with boiling points in the range from approximately 200 °C to approximately 300 °C vaporize at different rates depending mainly on volatility and droplet size. The droplets' dwell time in the air ranges from a few seconds to several minutes. For lower-volatility alkanes from octadecane upwards, even longer dwell times are possible before they evaporate completely. In addition, the particle–vapour fractionation of an aerosol varies – up to the equilibrium vapour pressure – according to the residence time in the sampling area, i.e. the particle–vapour ratio is not constant. Therefore, vapour and particles are extremely difficult to determine independently of each other.
The collection of vapour–droplet mixtures should only be done with an apparatus that can trap both phases. For example, droplets can be collected on a filter and vapours can be collected either on adsorber tubes or on denuders. Suitable sampling systems for chemically non-reactive substances are combined sample carriers comprising of a filter and an adsorption tube. For chemically reactive substances, filters with a reactive surface on which both droplets and vapours are sampled simultaneously are being used.
Regulatory authorities and occupational hygienists must give special consideration to the workplace occurrence of hazardous mixed phase aerosols. Failure to use an appropriate sampling strategy could lead to measurement errors in the field and to an exposure underestimation as a consequence. The European Committee for Standardization (CEN TC 137 WG2) recognised the necessity of adapting a standard for the distinctive issues raised by semi-volatiles. The essential parameters for the consideration of vapour–droplet mixtures are stated in Annex 2 of EN 13936, Workplace exposure – Procedures for measuring a chemical agent present as a mixture of airborne particles and vapour – Requirements and test methods.18 The provisions of this standard apply to the development of measurement methods for substances with vapour pressures in the range between 0.001 and 100 Pa at room temperature or boiling points in the temperature range from approximately 180 °C to 350 °C. In these methods, it is stated that droplets must be sampled as inhalable aerosols and that mixed phase aerosols can only be sampled as a sum of particles and vapours. The standard also contains provisions concerning suitable sampling systems. All sampling systems in which vapour and droplets are not collected together are deemed unsuitable, or the required effort not justifiable.
Considerations on the development of a miniaturised personal sampler for semi volatiles
The current sampling methods designed for the simultaneous sampling of aerosols and vapours have been available for only a small number of substances. The most well-known examples are methods for MWF,19 polycyclic aromatic hydrocarbons20 and varnish aerosols.21 In the methods described, the vapour collection technique was adapted to an existing sampling head for inhalable particles.The most well-known systems for the collection of particles are the closed-face cassette (Millipore cartridge), the IOM sampler and the GSP (Gesamt Staub Probenahme) inhalable dust sampling system used in Germany.22–25 The closed face cassette can be easily connected to an adsorption tube via a short piece of tubing. A modification of the GSP exists in the form of the GGP (Gesamtstaub-Gas-Probenahme = inhalable dust and vapour sampler), in which a cartridge can be connected directly to the GSP sampling head.19 The closed-face cassette and IOM sampler have a standard flow rate of 2 l min−1, the GSP of 3.5 l min−1. The measurement method employed in Germany for metalworking fluids for example is based upon collection of the aerosols by means of the GSP sampling head and a downstream XAD-2 adsorber resin layer.8
Depending on the adsorption tube type, vapour sampling is usually performed at flow rates of between 66 and 333 ml min−1. Particle sampling however is generally performed at substantially higher flow rates than vapour sampling. Increasing the flow rate for adsorption sampling in order to combine the common adsorption tubes with the available particle collectors is not possible, for a number of reasons. Because the pressure drop of the filter and the adsorption tube are added together when arranged in series, the backpressure exerted by the adsorption tubes increases substantially at higher flow rates. The battery-powered personal sampling pumps used for occupational measurement of hazardous substances26 are generally not powerful enough to overcome substantially higher backpressures. Furthermore, the adsorption volume for a given substance/tube combination is limited. Higher flow rates can therefore substantially reduce the sampling times for many substances. Long-term measurements, such as those required for evaluation of a mean shift value, would then no longer be possible. Larger tubes with a higher capacity and reduced flow resistance are not commercially available.
The lists of occupational exposure limit values worldwide27 contain numerous substances such as glycol ethers, amines, aromatic hydrocarbons (>C9), longer-chain aliphatic hydrocarbons (mineral oils) and longer-chain alcohols with physical properties falling directly within the range stated in EN 13936. To date, the methods for these substances have generally been geared for the sampling of the vapours, i.e. the substances are trapped on an adsorption tube. These tubes are designed for lower volumetric flow rates in the range of 0.066 to 0.5 l min−1, depending upon the tube type. For semi-volatile organic substances, it appeared more suitable to adapt the particle sampler to the vapour sampler. Based upon knowledge of the GSP, an inhalable particle sampling head suitable for use at substantially lower flow rates was developed. The first prototypes of the miniaturized GSP sampling head were designed for a volumetric flow of 0.333 l min−1. Nevertheless, the sampling head is of sufficiently flexible design that inhalable aerosols can be sampled at a flow rate of 0.066 l min−1 simply by substitution of the inlet cone.
Parallel to the flow rate reduction, filters of smaller diameter were required. The smallest commercially available filters measure 13 mm in diameter. Glass fibre, polytetrafluoroethylene (PTFE) and mixed cellulose ester (MCE) filters are usually available in this size. Compatibility with off-the-shelf accessories was very important, in order to keep the overall price as low as possible. Using smaller filters facilitated the design of a small, reusable sampling head that can be combined with most available adsorption tubes.
The GGP-Mini personal sampler was validated in three different test series. The first series involved laboratory tests in which different substances were spiked directly on the filters. The second test series employed aerosols of defined composition and were conducted in a flow tube at Helmholtz Zentrum München, the German Research Center for Environmental Health in Munich. The third series comprised of comparative tests employing collectors for inhalable dusts in the IFA's dust tunnel. The first two test series will be discussed in greater detail in the present publication.
The focus of the present study was to test the prototype GGP-Mini sampler for a series of organic compounds and to analyse the particle–vapour fractionation of several semi volatile compounds for differing concentrations, particle size distributions (PSD) and temperatures.
Materials and methods
GGP-Mini sampler
The GGP-Mini personal sampler (Fig. 1 and 2) comprises of a sampling head for inhalable aerosols with 13 mm filters for the separation of particulates and an adsorber tube for vapour trapping as main elements. The sampling head features a conical inlet and a filter holder, joined by a threaded connection. The sampling head and adsorber tube are connected by means of PTFE or silicone tubing. Munktell MG 160 glass fibre filters (diameter 13 mm, thickness 0.4 mm, Macherey-Nagel, Germany) and Type BIA activated carbon tubes (length 125 mm, OD 7 mm, ID 5 mm, coconut charcoal 300 mg/700 mg, Dräger KGaA, Germany) were used for the measurements presented in this study. A sampling pump is needed to operate the GGP-Mini sampler, usually at a flow rate of 0.333 l min−1. | ||
| Fig. 1 Picture of the GGP-Mini sampler sampling head (left) and the complete GGP-Mini sampler (right). | ||
 | ||
| Fig. 2 Mechanical drawing of the GGP-Mini prototype designed for the airflow of 0.5 l min−1. (1) – Conical inlet; (2) – filter with support grid; (3) – hose nozzle; (4) – braking protection tube. | ||
The sampling head is manufactured from X8CrNiS18-0 high-grade steel (material no. 1.4305) as it is adequate for organic substances and relatively easy to machine. The sampling head is around the size of a thumb and weighs 46 g (Fig. 1). It has an outer diameter of 22 mm and a length of 40 mm (Fig. 2). The filter used for particle separation is supported by a 13 mm high-grade steel flat mesh with a thickness of 0.1 mm that also serves as protection against puncturing. The conical inlet used for the flow rate of 0.333 l min−1 has a diameter of 2.4 mm.
Laboratory tests involving filter spiking
For the laboratory tests, 11 substances which cover the volatility range stated in EN 13936 (see Table 1) were selected. The substances were uniformly spiked on the GGP-Mini samplers' filters by means of a microliter syringe. After spiking, the inlet cone was screwed on the filter holder and then connected to the adsorber tube. A personal sampling pump (LFS 113DC, Gilian, USA) was then connected to the GGP-Mini sampler and adjusted to operate at a flow rate of 0.333 l min−1. The flow rate passing through the sampler was measured before and directly after each 2 hour experiment using a TSI 4100 flow meter (TSI Inc., USA). Following application, the collectors were connected to a gas stream28 of purified air with a RH of approximately 40% and in some tests 80%. The purified air was passed through the collectors for two hours. The filters and tubes were extracted separately. Non-polar compounds were extracted in CS2 (Promochem, Germany) while polar compounds were extracted in a CH2Cl2/CH3OH 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture (Merck, Germany). Extracts were analysed by gas chromatography with flame ionisation detection (GC-FID), following the standard IFA procedure. All tests were performed three times. The GC-FIDs used in the analysis are Clarus 500 (Perkin-Elmer, USA) with polar 60 m StabilWax (FD 0.5 μm, ID 0.25 mm) columns (Restek, Germany) for the analysis of polar compounds and AutoSysXL3 (Perkin Elmer, USA) with 30 m DB-5 column (FD 5 μm, ID 0.53 mm) for non-polar compounds.
:3 mixture (Merck, Germany). Extracts were analysed by gas chromatography with flame ionisation detection (GC-FID), following the standard IFA procedure. All tests were performed three times. The GC-FIDs used in the analysis are Clarus 500 (Perkin-Elmer, USA) with polar 60 m StabilWax (FD 0.5 μm, ID 0.25 mm) columns (Restek, Germany) for the analysis of polar compounds and AutoSysXL3 (Perkin Elmer, USA) with 30 m DB-5 column (FD 5 μm, ID 0.53 mm) for non-polar compounds.
| Substance | Boiling pointa [°C] | Proportion in percent | Proportion in percent | ||
|---|---|---|---|---|---|
| Low concentration (∼0.2 mg) | High concentration (∼1 mg) | ||||
| Filter | Tube | Filter | Tube | ||
| a GESTIS database on hazardous substances (http://www.dguv.de/dguv/ifa/Gefahrstoffdatenbanken/GESTIS-Stoffdatenbank/index-2.jsp). b ∼400 μg. | |||||
| 1-Ethoxy-2-propanol | 130 | — | — | N. d. | 100 |
| Ethoxypropyl acetate | 159 | — | — | N. d. | 100 |
| Propylene glycol | 188 | 4 | 96 | 6 | 94 |
| Diethylene glycol monomethyl ether | 193 | N. d. | 100 | 2 | 98 |
| Ethylene glycol | 197 | N. d. | 100 | 6 | 94 |
| Diethylene glycol monoethyl ether | 202 | 1 | 99 | 2 | 98 |
| Diethylene glycol monobutyl ether | 231 | N. d. | 100 | 8 | 92 |
| Diethylene glycol | 244 | 36 | 64 | 63 | 37 |
| Triethylene glycol monomethyl ether | 249 | N. d. | 100 | 9 | 91 |
| n-Hexadecane | 287 | 54b | 46 | 54 | 46 |
| n-Octadecane | 318 | 95b | 5 | 94 | 6 |
A temperature controlled chamber was used to study the temperature influence on the substances' liquid–vapour fractionation. The chamber was operated at temperatures between 0 and 50 °C, in 10 °C increments. After spiking, the sampling head was connected to the adsorber tube and placed inside the temperature controlled chamber. Conditioned air at 80% RH was drawn through the GGP-Mini sampler inside the chamber using personal sampling pumps.
Flow tube tests involving monodisperse droplets
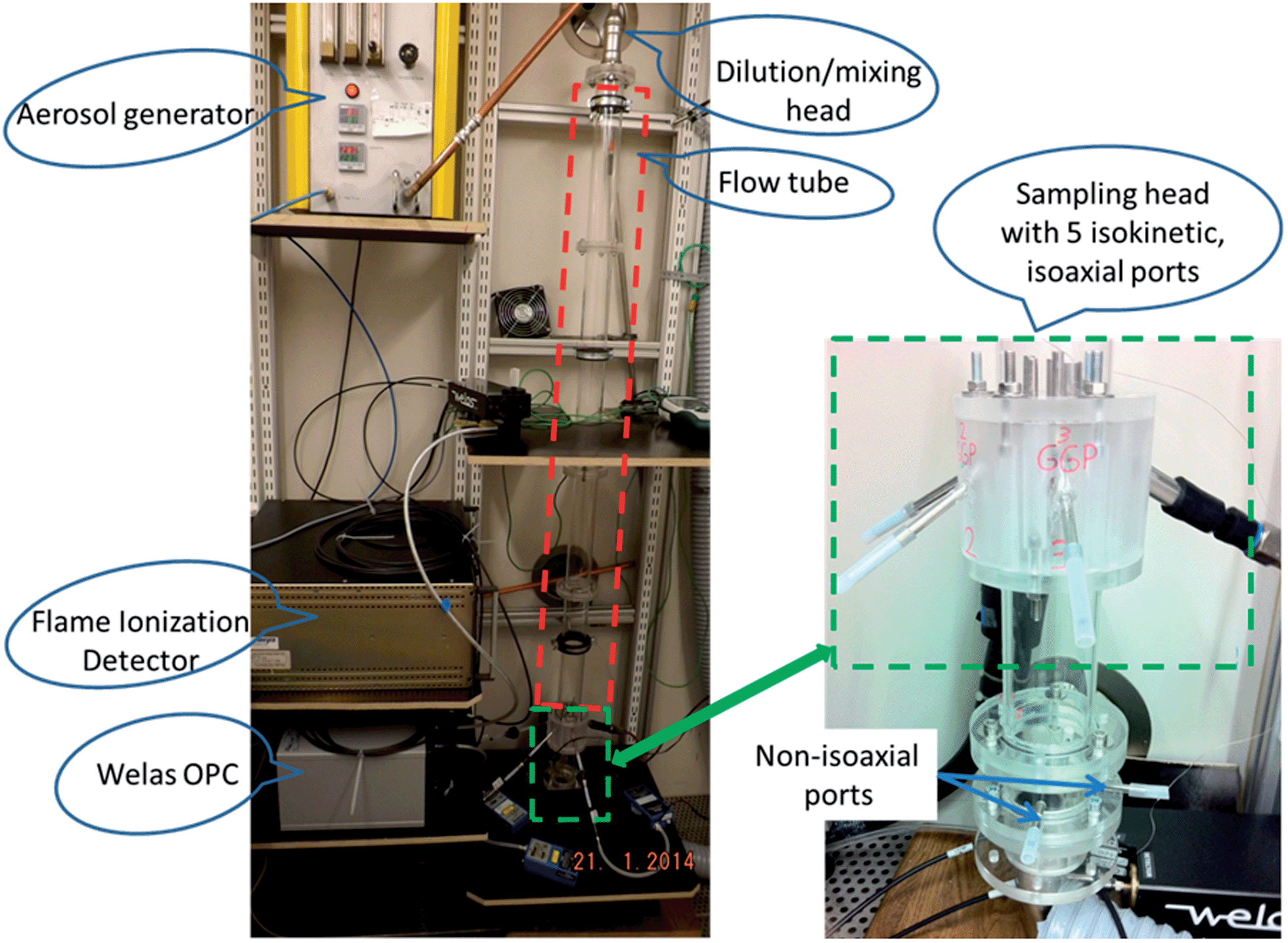 | ||
| Fig. 3 Flow tube experimental facility (left) and sampling head (right). | ||
The experimental facility was set-up in a temperature controlled chamber with a set temperature of 24.7 °C. The temperature was recorded with type K thermocouples and found to be stable (±0.2 °C) throughout the measurements.
A Topas SLG 270 aerosol generator (Topas GmbH, Germany) was chosen to produce monodisperse droplets of adjustable PSD. The generator's operation principle was described in detail by Altmann and Peters29,30 and was found to be suitable for oil mist generation by Dragan et al.8,14 For this study monodisperse droplets in the size range of 1 to 4 μm were generated at a flow rate of 5 l min−1.
A dilution and mixing unit was used to dilute the initial aerosol with 80 l min−1 particle free nitrogen (N2), for a total flow of 85 l min−1. The relative humidity of the make-up nitrogen was adjusted by passing the N2 stream through impingers filled with water. The RH in the FT was monitored using a FHAD 462 humidity sensor (Ahlborn GmbH, Germany). The dilution of the initial aerosol led to a decrease in vapour saturation that triggered the evaporation of droplets inside the FT.
A sampling head (Fig. 3) was designed and employed to obtain identical aerosol samples for each device connected to it. The sampling head is connected to the FT and consists of five isoaxial tubes of different diameters. The tubes diameters were chosen to achieve isokinetic sampling (same flow velocity in the tubes) for devices of varying flow rates. Isokinetic and isoaxial sampling ensures an equal distribution of the aerosol in its five ports.
For the on-line analysis the apparatus consisting of an FID (JUM 109A, JUM Engineering GmbH, Germany) and an Optical Particle Counter (OPC, Welas digital 3000, Palas GmbH, Germany) was set up to quantify the total analyte mass concentration (TM) and particle mass concentration (PM) respectively. The vapour mass concentration (VM) was calculated as the difference between FID and OPC measurements (VM = TM − PM).
The FID was used to continuously monitor the total concentration of the semi-volatile substance present in the aerosol. A transfer line heated to 180 °C was coupled to the FID inlet in order to evaporate all droplets within the aerosol. The FID was operated at a flow rate of 0.5 l min−1.
An OPC was used as a particle sizer to determine the aerosols' PSD and calculate the PM. The OPC software also allows adjusting the calibration curve for a refractive index of 1.45. Density corrections of 0.777 and 1.118 (kg m−3) were used for n-hexadecane (C16) and diethylene glycol (DEG) respectively in order to calculate the particle mass. OPC measurements were taken with a 10 minute time resolution and at a flow rate of 5 l min−1.
The analysis of the off-line filter and adsorber samples was performed similarly to the procedure described in the previous chapter for the spiking tests.
Results and discussion
Laboratory tests with spiked filters
In the laboratory tests, the compounds were applied only to the filter, and air was then drawn through the filter–adsorber combination for 2 hours. In principle, this corresponds to the “spiking” technique9 widely used in the development of measurement methods. The spiking experiments can show if a compound is likely to remain on the filters or evaporate partially or completely during the sampling.For the low concentration measurements, only three compounds were partially found on filters, namely diethylene glycol (DEG), hexadecane (C16) and octadecane (C18). The other compounds were only found as vapours in the adsorber tubes. This shows that components with boiling points below 240 °C are most likely to be sampled as vapours.
For the high concentration measurements (25 mg m−3) five compounds were still found on filters, namely Diethylene Glycol Butyl Ether (DEGBE), DEG, Triethylene Glycol Monomethyl Ether (TEGME), C16 and C18. For all other substances, less than 10% of the initial spiked amount remained on filters. For higher concentrations, the substances with boiling points above 200 °C can be partially found on filters. It can also be seen that for the higher concentrations more substance remains on the filter.
The individual results for the storage test can be seen in the ESI.† It was observed that storage up to 28 days did not have a notable effect on the samples, provided that the sampling heads and adsorber tubes were properly sealed. The samples can be therefore sealed and transported to be analysed at a later time, without the need of solvent extraction on the sampling site.
The temperature effect on liquid–vapor partitioning was studied on a mixture of TEGME and DEG. The mixture was applied following the spiking procedure previously described. The tests took place inside a climate chamber at temperatures between 0 and 50 °C, in 10 °C increments. The RH was set at approximately 80%, and air was drawn in the chamber through the samplers for 2 hours. As can be seen in Fig. 4, the resulting influence is substantial. At the lowest temperature (0 °C), less than 5% volatilised away from the filter, whereas at the highest temperature (50 °C), around 95% of the liquid evaporates.
 | ||
| Fig. 4 Temperature dependence of the filter vs. sorbent tube distribution – filter portion. | ||
In a further test series, the influence of the temperature was studied on propylene glycol and ethylene glycol. Both of these polar substances vaporize almost completely at 25 °C, even at higher atmospheric humidities (RH ≈ 80%). However, when the same test is performed at 10 °C up to 75% of each substance remains on the filter (Table 2).
| Compound | Spiked mass [mg] | Temperature [°C] | Recovery filter ± st. dev. [%] | Recovery tube ± st. dev. [%] | Total recovery ± st. dev. [%] |
|---|---|---|---|---|---|
| Propylene glycol | 1 | 25 | 2 ± 100 | 95 ± 7 | 97 ± 4 |
| 1 | 10 | 62 ± 17 | 33 ± 18 | 95 ± 6 | |
| Ethylene glycol | 1 | 25 | N. d. | 84 ± 3 | 84 ± 3 |
| 1 | 10 | 74 ± 13 | 15 ± 26 | 92 ± 8 | |
| Hexadecane | 0.4 | 40 | N. d. | 96 ± 11 | 96 ± 11 |
| 1 | 40 | 4 ± 100 | 87 ± 10 | 91 ± 4 | |
| 8 | 40 | 65 ± 14 | 20 ± 42 | 85 ± 2 | |
| 0.4 | 10 | 96 ± 7 | 4 ± 30 | 100 ± 7 | |
| 1 | 10 | 95 ± 3 | 2 ± 34 | 97 ± 3 | |
| 8 | 10 | 97 ± 1 | 0.3 ± 31 | 97 ± 1 |
As expected, a pronounced influence of the temperature is also observed for the non-polar n-hexadecane. At lower temperatures, only a small amount of the substance is vaporized during sampling, irrespective of the substance quantity applied, whereas at a temperature of 40 °C, the vaporized component may rise to up to 100% (Table 2).
Sampling of both polar substances showed the atmospheric humidity to have a pronounced effect upon the distribution between the component remaining on the filter and the vaporized component determined on the tube (Table 3). Even though the results show some scatter, considerably more of the polar substances remained on the filter at an RH of 80% in all cases. For all three concentrations the most mass left on the filter was found at the higher RH value. The RH however did not influence the evaporation of the non-polar n-hexadecane. Nearly identical values were found for C16 at both RH levels for all three studied concentrations.
| Compound | Spiked Mass [mg] | Humidity [% RH] | Recovery filter ± st. dev. [%] | Recovery tube ± st. dev. [%] | Total recovery ± st. dev. [%] |
|---|---|---|---|---|---|
| Diethylene glycol | 0.2 | 40 | 36 ± 56 | 63 ± 23 | 99 ± 6 |
| 1 | 40 | 62 ± 21 | 36 ± 29 | 98 ± 4 | |
| 3.5 | 40 | 89 ± 9 | 9 ± 48 | 97 ± 5 | |
| 0.2 | 80 | 70 ± 14 | 30 ± 12 | 100 ± 10 | |
| 1 | 80 | 76 ± 2 | 24 ± 18 | 100 ± 1 | |
| 3.5 | 80 | 95 ± 7 | 5 ± 4 | 100 ± 6 | |
| Triethylene glycol monomethyl ether | 0.26 | 40 | N. d. | 77 ± 13 | 77 ± 13 |
| 1 | 40 | 9 ± 51 | 91 ± 8 | 100 ± 4 | |
| 5 | 40 | 77 ± 30 | 22 ± 21 | 99 ± 26 | |
| 0.26 | 80 | N. d. | 76 ± 10 | 76 ± 10 | |
| 1 | 80 | 39 ± 10 | 50 ± 7 | 89 ± 4 | |
| 5 | 80 | 84 ± 26 | 5 ± 21 | 89 ± 23 | |
| n-Hexadecane | 0.2 | 40 | 53 ± 39 | 47 ± 21 | 100 ± 13 |
| 1 | 40 | 50 ± 18 | 44 ± 6 | 94 ± 4 | |
| 5 | 40 | 92 ± 10 | 6 ± 3 | 98 ± 8 | |
| 0.2 | 80 | 49 ± 14 | 41 ± 17 | 90 ± 7 | |
| 1 | 80 | 60 ± 12 | 36 ± 12 | 96 ± 1 | |
| 5 | 80 | 92 ± 6 | 4 ± 48 | 96 ± 3 |
Flow tube tests involving monodisperse droplets
Once the laboratory spiking tests had confirmed the provisions of EN 13639, it was necessary to validate the results with further tests conducted on well-defined aerosols. Although spiking tests serve as good indicators for filter evaporation losses, they may not fully reflect reality. For the development of measurement methods, it is clearly preferable for sampling tests to be performed in a setup that is as realistic as possible.Contrary to the laboratory tests where the analysed compounds are spiked directly on filters, the flow tube tests offer a constant input of aerosols over the 2 hours of sampling. The droplets are uniformly collected on the filters, thus avoiding errors caused by non-uniform spiking. The GGP-Mini's filters will therefore gradually collect droplets. The evaporation flux downstream of the filters will also differ from spiking experiments, as it gradually increases when more droplets accumulate on the filters and will be capped when the incoming gas reaches vapour saturation.
Due to the primary aerosols dilution, the generated droplets will evaporate in the flow tube until they reach equilibrium with the gas phase. However, at the point where the on-line and off-line samples are being taken, equilibrium is in most cases not reached. As a result, the droplets separated on the off-line GGP-Mini will continue to evaporate until the passing air stream reaches vapour saturation. On the other hand, the on-line measurement report the aerosols' particle–vapour fractionation at the moment at which the measurement took place, without being influenced by evaporation artefacts. The extent of filter evaporative losses can be quantified by comparing the on-line with off-line measurements.
N-Hexadecane (C16) and diethylene glycol (DEG) were chosen as model substances to test the effect of aerosol concentration, particles size distribution and RH on the samplers' particle–vapour fractionation.
Fig. 5 and 6 depict the percentage particle–vapour partitioning of C16 aerosols for four different TM concentrations and two RH values (<5% and 70%) measured by on-line (FID + OPC) as well as off-line (GGP-Mini) methods. Also displayed is a “predicted” GGP-Mini particle–vapour fractionation, assuming that the air stream passing through the filters reached vapour saturation (18.2 mg m−3 at 24.7 °C).
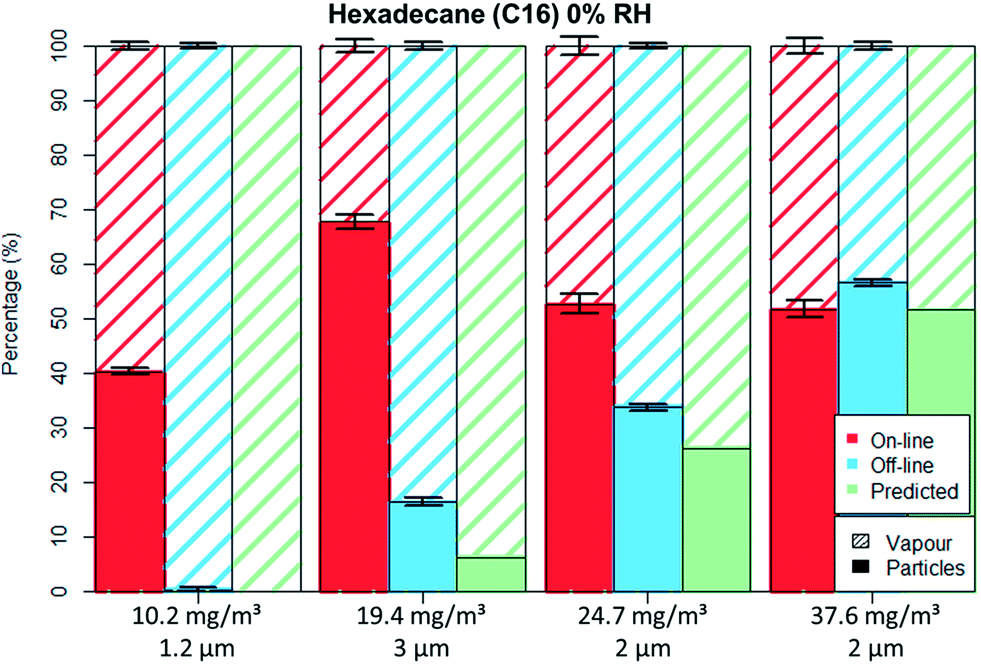 | ||
| Fig. 5 C16 aerosol fractionation between vapour and particle phase at 0% RH for on-line and off-line methods as well as the predicted off-line fractionation. Error bars represent one standard deviation. | ||
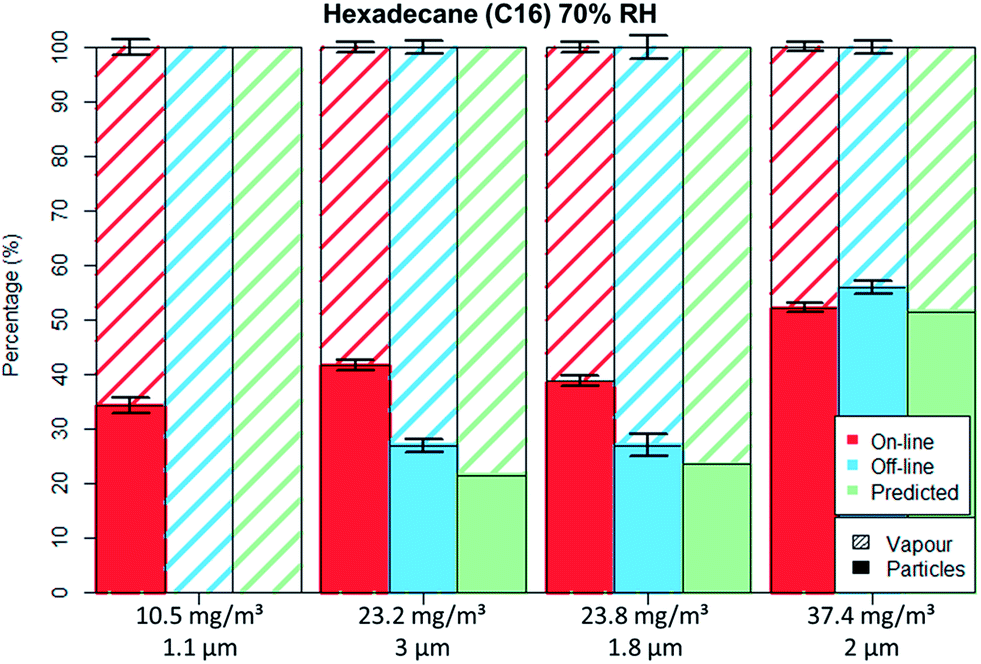 | ||
| Fig. 6 C16 aerosol fractionation between vapour and particle phase at 70% RH for on-line and off-line methods as well as the predicted off-line fractionation. Error bars represent one standard deviation. | ||
The C16 aerosol experiments show, similarly to the spiking experiments, that the mass percentage found on filters increases with concentration. By using aerosol concentrations in the range of 10 to 38 mg m−3 one can clearly observe the transition from total evaporation losses to no filter evaporation at all.
For the lowest tested concentration (10 mg m−3) the entire aerosol mass was found by the GGP-Mini sampler as vapour, even though more than 3.5 mg m−3 were detected by the on-line measurements. The predicted results match the off-line data, as the TM was below the saturation concentration. This shows that measurement of semi-volatiles should only be done with a combination of particle and vapour samplers, as filter sampling alone will undoubtedly lead to measurement errors caused by evaporation.
For the medium concentrations measured (20 to 25 mg m−3), C16 was detected on both filters and adsorber tubes. The evaporation of droplets from filters was limited mainly because the aerosol concentration was higher than the saturation concentration. The off-line data shows lower particle and higher vapour values than on-line data, clearly indicating a mass transfer from filters to the adsorbers. The predicted off-line distribution is to some degree lower than the actual measurement, however within a 10% deviation. Adsorption of vapours onto the glass fibre filters as well as some inaccuracies in vapour pressure data may explain this difference. Even though PM was still found on filter the evaporation bias is clearly seen. For the concentration of 23 mg m−3 nearly identical results were measured even though different droplet sizes were generated (1.8 and 3 μm), an indication that the filter evaporation losses were not influenced by the PSD.
In the case of high concentration measurements (37.5 mg m−3) almost no difference was seen for the particle–vapour fractionation, independent of sampling method. The on-line measurements revealed that the gas phase had already reached vapour saturation at the sampling point. Therefore, the predicted off-line concentration is identical with the on-line values. The measured off-line filter fraction is slightly higher, most probably due to some adsorption on the filters.
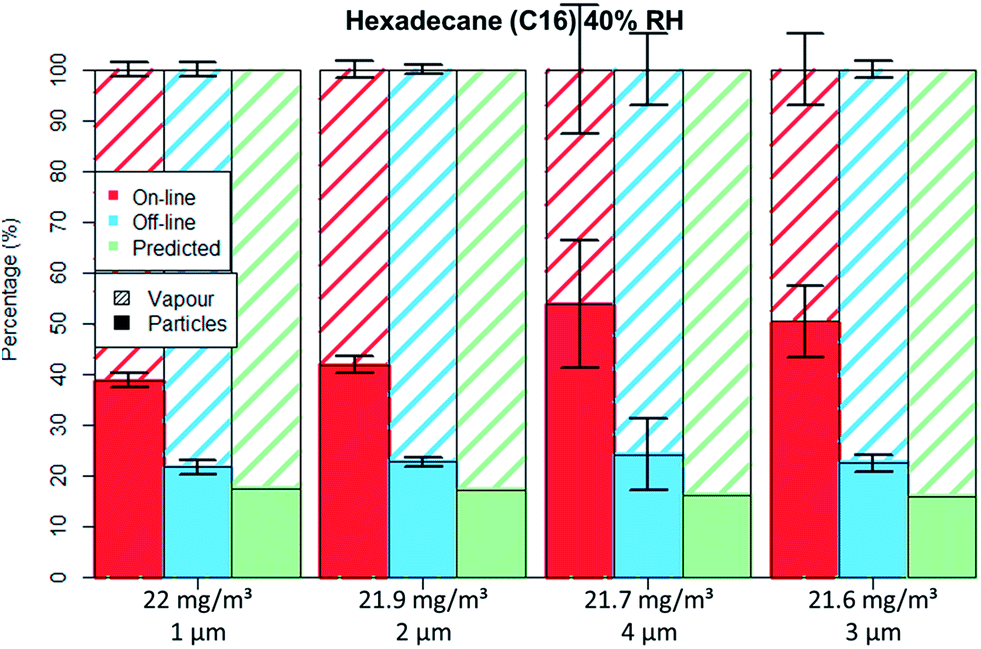 | ||
| Fig. 7 C16 aerosol fractionation between vapour and particle phase for four different particle size distributions at similar aerosol concentration. Error bars represent one standard deviation. | ||
The droplets' diameter did not influence the droplet–vapour distribution for the polar compound either (Table 4), no differences were seen for the GGP-Mini measurements even though different droplet diameters were generated.
| Relative humidity (RH) | Concentration/droplet diameter | Filter mass [mg m−3] | Tube mass [mg m−3] | Total mass filter plus tube [mg m−3] |
|---|---|---|---|---|
| <5% | Low/1 μm | 0.3 ± 0.1 | 6.3 ± 0.4 | 6.6 ± 0.4 |
| Medium/1 μm | 0.5 ± 0.3 | 14.8 ± 0.7 | 15.3 ± 0.8 | |
| Medium/1.5 μm | 0.5 ± 0.1 | 16.0 ± 1.0 | 16.5 ± 1.0 | |
| High/2 μm | 25.3 ± 1.1 | 16.9 ± 0.7 | 42.2 ± 1.4 | |
| 70% | Low/1.5 μm | 4.0 ± 0.6 | 3.3 ± 0.1 | 7.2 ± 0.6 |
| Medium/2 μm | 12 ± 1.3 | 4.5 ± 0.4 | 16.5 ± 1.0 | |
| Medium/1.1 μm | 12.1 ± 0.7 | 3.8 ± 0.2 | 15.9 ± 0.6 | |
| High/2 μm | 38.2 ± 1.6 | 3.9 ± 0.2 | 42.1 ± 1.5 |
For the non-polar C16 three levels of RH were tested, namely 0%, 40% and 70%. It can be seen by comparing Fig. 5 to 6 that only the aerosol concentration influences the partitioning on the off-line GGP-Mini. No significant differences were observed for the different levels of RH.
On the other hand, RH had a substantial effect on the droplet–vapour distribution of DEG (Table 4). With dry air, the results were as anticipated; with increasing concentration, the droplet mass remaining on filters also increased. The size of the droplets was however of only minor significance. Except at the highest concentration, the greater part of the diethylene glycol was recovered on the activated carbon tube. At the low concentrations, the droplet percentage was below 10%; while at a high concentration, the distribution was approximately 60% droplets and 40% vapour.
Conversely, at an elevated atmospheric humidity of 70%, the picture was quite different. As in the laboratory spiking tests previously presented, the RH had a considerable influence on the particle–vapour distribution. The observed particle–vapour ratios for humid air were fully reversed compared to the ratios found in dry air. In this test series, the DEG was recovered primarily on the filter. The values differed fundamentally from those with dry air. At the lowest concentration the ratio was 60% found on filter to 40% measured as vapour. At medium concentrations a filter to vapour ratio of approximately 75–25% was found while at high concentrations the vapour component is only 10%.
Altogether, it can be stated that the flow tube aerosol tests clearly confirm the laboratory results obtained by spiking. Although different distributions were observed in some cases, the influences and effects determined are closely related.
Conclusions and recommendations
Owing to the change in the legal situation in Germany and to recent publication of a new European standard, a need had arisen to develop a system specifically suitable for the sampling of vapour–droplet mixtures of low-volatility substances in the boiling range from 180 to 350 °C. Since conventional particle sampling and conventional vapour sampling cannot be readily combined, a sampling head was developed which adapts the sampling of particles/droplets to the vapour sampling apparatus.At the present state of development, the new GGP-Mini sampling system is suitable for the sampling of droplet aerosols. The GGP-Mini can combine the 13 mm filters employed for particle sampling with all off-the-shelf adsorption tubes. The total analyte concentration doesn't substantially change if the sample carriers are tightly sealed immediately after sampling. Therefore, GGP-Mini samples can be sealed and transported to be analysed at a later point, without substantial losses.
All sampling tests, both simple reproduction in the laboratory and tests on a droplet aerosol have shown that the distribution between vapour and droplets is not constant and that only the sum of vapour and droplets constitutes a reproducible result. The distribution is dependent upon numerous influencing factors. The quantity or concentration of the aerosol, the temperature, and – in the case of polar substances – the atmospheric humidity appear to be especially important. The aerosol's PSD did not influence the droplet–vapor distribution inside the GGP-Mini.
The sampling of semi-volatiles using just filters could lead to considerable exposure underestimation. The flow tube aerosol tests have shown that droplets can completely evaporate from filters. Filter-only sampling will only be accurate if the gas phase is saturated with vapour. All semi-volatiles should be therefore sampled by a method that combines droplet as well as vapour measurements. Occupational exposure limits should only be considered for the sum of particles and vapours.
The system is expected to be tested for use on further classes of substances. The IFA is currently revising methods for low-volatility amines such as dicyclohexylamine and n-ethyl pyrrolidone. Here again, the atmospheric humidity is expected to have a considerable influence.
References
- D. Breuer and A. Howe, J. Environ. Monit., 2006, 8, 120–126 RSC.
- D. Breuer, P. Heckmann, K. Gusbeth, G. Schwab, M. Blaskowitz and A. Moritz, J. Environ. Monit., 2012, 14, 440–445 RSC.
- A. Simpson, M. Stear, J. A. Groves, M. Piney, S. D. Bradley, S. Stagg and B. Crook, Ann. Occup. Hyg., 2003, 47, 17–30 CrossRef CAS.
- P. W. Wilsey, J. H. Vincent, M. J. Bishop, L. M. Brosseau and I. A. Greaves, Am. Ind. Hyg. Assoc. J., 1996, 57, 1149–1153 CrossRef CAS PubMed.
- D. Breuer, J.-U. Hahn, D. Hober, C. Emmel, U. Musanke, R. Ruhl, A. Spickenheuer, M. Raulf-Heimsoth, R. Bramer, A. Seidel, B. Schilling, E. Heinze, B. Kendzia, B. Marczynski, P. Welge, J. Angerer, T. Bruning and B. Pesch, Arch. Toxicol., 2011, 85(suppl. 1), S11–S20 CrossRef PubMed.
- J. Volckens and D. Leith, Ann. Occup. Hyg., 2003, 47, 157–164 CrossRef CAS.
- D. Breuer, J. Environ. Monit., 1999, 1, 299–305 RSC.
- G. C. Dragan, D. Breuer, M. Blaskowitz, E. Karg, J. Schnelle-Kreis, J. M. Arteaga-Salas, H. Nordsieck and R. Zimmermann, Environ. Sci.: Processes Impacts, 2014 10.1039/C4EM00468J.
- A. Simpson, J. A. Groves, J. Unwin and M. Piney, Ann. Occup. Hyg., 2000, 44, 165–172 CAS.
- M. Furuuchi, H. Fissan and J. Horodecki, Powder Technol., 2001, 118, 171–179 CrossRef CAS.
- B. Sutter, D. Bémer, J.-C. Appert-Collin, D. Thomas and N. Midoux, Aerosol Sci. Technol., 2010, 44, 395–404 CrossRef CAS.
- S. J. Cooper, P. C. Raynor and D. Leith, Appl. Occup. Environ. Hyg., 1996, 11, 1204–1211 CrossRef CAS.
- E. Galarneau and T. F. Bidleman, Atmos. Environ., 2006, 40, 4258–4268 CrossRef CAS PubMed.
- G. C. Dragan, E. Karg, H. O. Nordsieck, J. Schnelle-Kreis, D. Breuer, J. M. Arteaga-Salas, G. A. Ferron and R. Zimmermann, Environ. Eng. Manage. J., 2014, 13, 1775–1785 Search PubMed.
- A. Atmadi, D. A. Stephenson and S. Y. Liang, Int. J. Adv. Manuf. Tech., 2001, 17, 238–243 CrossRef.
- L.-G. Ekstrom, A. Kriech, C. Bowen, S. Johnson and D. Breuer, J. Environ. Monit., 2001, 3, 439–445 RSC.
- M. R. Chen, P. J. Tsai, C. C. Chang, T. S. Shih, W. J. Lee and P. C. Liao, J. Hazard. Mater., 2007, 146, 393–398 CrossRef CAS PubMed.
- European_Committe_for_Standardisation, Workplace exposure – Procedures for measuring a chemical agent present as a mixture of airborne particles and vapour – Requirements and test methods, 2014.
- D. Breuer and M. Blaskowitz, IFA Arbeitsmappe, Blatt 7750-1, Messung von Gefahrstoffen, published by: Deutsche Gesetzliche Unfallversicherung (DGUV), Berlin: Erich Schmidt (loose-leaf).
- B. IFA Arbeitsmappe, Messung von Gefahrstoffen, IFA Arbeitsmappe, Blatt 6272, Messung von Gefahrstoffen, published by: Deutsche Gesetzliche Unfallversicherung (DGUV), Berlin: Erich Schmidt (loose-leaf).
- C. Friedrich, M. Hennig and N. Lichtenstein, The MAK Collection for Occupational Health and Safety, Air Monitoring Methods, 2003, published online: 31 January 2012, http://onlinelibrary.wiley.com/doi/10.1002/3527600418.am0lackaerd0013a/abstract.
- IFA Arbeitsmappe, Messung von Gefahrstoffen, Blatt 3010, published by: Deutsche Gesetzliche Unfallversicherung (DGUV), Berlin: Erich Schmidt (loose-leaf) .
- A. Zugasti, N. Montes, J. M. Rojo and M. J. Quintana, J. Environ. Monit., 2012, 14, 375–382 RSC.
- L. C. Kenny, R. Aitken, C. Chalmers, J. F. Fabriès, E. Gonzalez-Fernandez, H. Kromhout, G. Lidén, D. Mark, G. Riediger and V. Prodi, Ann. Occup. Hyg., 1997, 41, 135–153 CrossRef CAS.
- K. S. Galea, A. Searl, A. Sánchez-Jiménez, T. WoldbæK, K. Halgard, S. Thorud, K. Steinsvåg, K. Krüger, L. MacCalman, J. W. Cherrie and M. van Tongeren, Ann. Occup. Hyg., 2011, 56, 1–9 Search PubMed.
- D. Breuer, J. Occup. Environ. Hyg., 2012, 9, D25–D32 CrossRef PubMed.
- GESTIS, in GESTIS International limit values for chemical agents: Occupational exposure limits (OELs), DGUV, 2014, http://limitvalue.ifa.dguv.de/Webform_gw.aspx Search PubMed.
- A. Moritz and D. Breuer, J. Environ. Monit., 2008, 10, 1454–1459 RSC.
- J. Altmann and C. Peters, J. Aerosol Sci., 1992, 23, 277–280 CrossRef.
- C. Peters and J. Altmann, J. Aerosol Med., 1993, 6, 307–315 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4em00602j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |